

Abstract
Chronic visceral pain, altered motility and bladder dysfunction are common, yet poorly managed symptoms of functional and inflammatory disorders of the gastrointestinal and urinary tracts. Recently, numerous human channelopathies of the voltage‐gated sodium (NaV) channel family have been identified, which induce either painful neuropathies, an insensitivity to pain, or alterations in smooth muscle function. The identification of these disorders, in addition to the recent utilisation of genetically modified NaV mice and specific NaV channel modulators, has shed new light on how NaV channels contribute to the function of neuronal and non‐neuronal tissues within the gastrointestinal tract and bladder. Here we review the current pre‐clinical and clinical evidence to reveal how the nine NaV channel family members (NaV1.1–NaV1.9) contribute to abdominal visceral function in normal and disease states.
Keywords: colon, bladder, sensory afferents, nociceptors, pain, inflammation, dorsal root ganglia
Introduction
Chronic visceral pain, altered intestinal motility and bladder dysfunction remain poorly managed symptoms of functional and inflammatory disorders of the gastrointestinal and urinary tracts. A lack of suitable treatments for these disorders is a major contributing factor to their debilitating nature and the large socio‐economic cost accrued by patients, their families and society (NIH, 2009; Gaskin & Richard, 2012; Enck et al. 2016). Conventional analgesics, such as opioids and non‐steroidal anti‐inflammatory drugs (NSAIDs), are unsuitable for treating chronic pain originating in the gastrointestinal and lower urinary tract, as they are associated with severe side effects. This includes tolerance, a lack of efficacy and importantly for some inflammatory gastrointestinal disorders the potential to exacerbate the disease (Sikandar & Dickenson, 2012; Farrell et al. 2014). The colon, rectum and bladder are innervated by specialised sensory afferents travelling via the splanchnic and pelvic nerves that terminate within the dorsal horn of the thoracolumbar and lumbosacral spinal cord, respectively (Brierley et al. 2004; Harrington et al. 2012; Brierley & Linden, 2014). These neurons detect both non‐noxious physiological stimuli, including muscle stretch during organ distension, and noxious mechanical and chemical stimuli such as bloating, intense distension/contraction, or the presence of inflammatory mediators (Brierley & Linden, 2014; Brierley, 2016). To encode for such wide‐ranging stimuli, visceral organs rely on an array of stimuli‐activated primary ‘sentinel’ transducers, including transient receptor potential (TRP) channels, acid‐sensing ion channels (ASIC), mechanosensitive two‐pore domain K (K2P) channels and Piezo channels (Grundy, 2002; Brierley, 2010; Christianson & Davis, 2010; La & Gebhart, 2011; Brierley, 2016; Alcaino et al. 2017). Furthermore, primary transducers and ion channels involved in sensory signalling can be modulated and controlled by G‐protein coupled receptors (GPCRs) and regulators of GPCR signalling proteins, in response to endogenous mediators (Geppetti et al. 2015; Salaga et al. 2016).
Voltage‐gated sodium (NaV) channels are secondary in the neuronal response to non‐noxious or noxious stimuli. They perform the crucial role of regulating neuronal excitability and the key function of amplifying cation influx generated by the primary transducers to generate and propagate action potentials (Catterall, 2012; King & Vetter, 2014). Voltage‐gated potassium (KV) channels repolarise the membrane potential following Na+ influx and modulate firing frequency, and have been reported to contribute to visceral hypersensitivity in peripheral neurons in animal models (Hirano et al. 2007; Qian et al. 2009; Luo et al. 2011; Du & Gamper, 2013); however, this family of ion channels is not covered within the scope of this review.
The NaV channel family contains nine isoforms (NaV1.1–NaV1.9), which are encoded by nine SCN genes (SCN1A, SCN2A, SCN3A, SCN4A, SCN5A, SCN8A, SCN9A, SCN10A, SCN11A). Functionally, these channels are historically categorised as either tetrodotoxin‐sensitive (TTX‐S: NaV1.1‐NaV1.4, NaV1.6 and NaV1.7), or tetrodotoxin‐resistant (TTX‐R: NaV1.5, NaV1.8 and NaV1.9). Anatomically, these channels display wide and diverse expression patterns across neuronal and smooth muscle cells throughout the body (Table 1), as well as cells of the immune system (including macrophages and mast cells) where they are involved in migration and phagocytosis (Bradding et al. 2003; Roselli et al. 2006; Carrithers et al. 2011; Black & Waxman, 2013). NaV1.1, NaV1.2, NaV1.3 and NaV1.6 are traditionally considered to be the predominant isoforms expressed in the brain and spinal cord, whilst NaV1.7, NaV1.8 and NaV1.9 are preferentially expressed in the peripheral nervous system (PNS). NaV1.4 is found predominantly within skeletal muscle and NaV1.5 is the major isoform in cardiac myocytes (Catterall et al. 2005). Furthermore, NaV channels are regulated by a range of enzymes and structural proteins, including auxiliary β‐subunits (β1, β1B, β2, β3, β4) (Qin et al. 2003; Tseng et al. 2007), kinases and ubiquitin‐protein ligases (Feng et al. 2012; Savio‐Galimberti et al. 2012; Laedermann et al. 2015), which collectively regulate NaV channel biophysical properties and expression.
Table 1.
Expression of NaV isoforms in neuronal and non‐neuronal cells in different species relevant for visceral sensation and processing
| NaV isoform | Species | System or tissue | Found in: | Not found in: | Reference |
|---|---|---|---|---|---|
| NaV1.1 | Human | CNS | Cerebral cortex, cerebellum, hypothalamus, caudate, hippocampus, amygdala, C1 level spinal cord | GTEx Consortium et al. 2017) | |
| Rat | CNS | Hippocampus, cerebellum, spinal cord (dorsal horn, ventral horn, primarily grey matter restricted) | Embryonic brain and spinal cord | (Beckh et al. 1989; Westenbroek et al. 1989) | |
| Mouse | CNS | Cerebral cortex, cerebellum, hippocampus, thalamus, central grey, pons, medulla | Fimbria, corpus callosum | (Duflocq et al. 2008) | |
| Human | PNS | L3–L5 | (Chang et al. 2018) | ||
| Rat | PNS | L4–L5; L5 | (Black et al. 1996; Fukuoka et al. 2008; Wang et al. 2011) | ||
| Mouse | PNS | Colonic neurons in T10–L1 and L5–S1; T10–L1 | L3–L6 dorsal and ventral roots | (Duflocq et al. 2008; Osteen et al. 2016; Hockley et al. 2017) | |
| Human | ENS | Colonic myenteric plexus | (Hetz et al. 2014) | ||
| Guinea pig | ENS | Duodenal myenteric plexus | (Sage et al. 2007) | ||
| NaV1.2 | Human | CNS | Cerebral cortex, cerebellum, hypothalamus, caudate, hippocampus, amygdala, C1 level spinal cord | (GTEx Consortium et al. 2017) | |
| Rat, cat | CNS | Cortex, hippocampus, cerebellum, hypothalamus, spinal cord grey matter | (Jarnot & Corbett, 2006) | ||
| Rat | CNS | Hippocampus and cerebellum; embryonic brain and spinal cord | (Beckh et al. 1989; Westenbroek et al. 1989) | ||
| Human | PNS | L3–L5 | (Chang et al. 2018) | ||
| Rat | PNS | L4–L5; L5 | (Black et al. 1996; Fukuoka et al. 2008) | ||
| Mouse | PNS | Colonic neurons in T10–L1 and L5–S1 | (Chang et al. 2018; Hockley et al. 2017) | ||
| Human | ENS | Colonic myenteric plexus | (Hetz et al. 2014) | ||
| Guinea pig | ENS | Duodenal myenteric plexus | (Sage et al. 2007) | ||
| NaV1.3 | Human | CNS | Caudate, cerebellum, cerebral cortex, hippocampus, hypothalamus, amygdala, C1 level spinal cord | (GTEx Consortium et al. 2017) | |
| Rat | CNS | Embryonic brain and spinal cord | Adult brain and spinal cord | (Beckh et al. 1989) | |
| Human | PNS | L3–L5 | (Chang et al. 2018) | ||
| Rat | PNS | L4–L5 | L5 | (Black et al. 1996; Fukuoka et al. 2008) | |
| Mouse | PNS | Colonic neurons in T10–L1 and L5–S1; DRG | (Chang et al. 2018; Hockley et al. 2017) | ||
| Human | ENS | Colonic myenteric plexus | (Hetz et al. 2014) | ||
| Guinea pig | ENS | Duodenal myenteric plexus | (Sage et al. 2007) | ||
| Human, mouse | Neuroendocrine | Jejunal and colonic enterochromaffin cells | (Bellono et al. 2017; Strege et al. 2017a,b) | ||
| NaV1.4 | Human | CNS | Brain | (GTEx Consortium et al. 2017) | |
| Rat | PNS | L5 | (Fukuoka et al. 2008) | ||
| Mouse | PNS | Colonic neurons in T10–L1 and L5–S1 | (Hockley et al. 2017) | ||
| Human | ENS | Colonic myenteric plexus | (Hetz et al. 2014) | ||
| Human | Muscle | Oesophageal smooth muscle | (Deshpande et al. 2002) | ||
| NaV1.5 | Human | CNS | Brain | (GTEx Consortium et al. 2017) | |
| Mouse | PNS | Colonic neurons in T10–L1 and L5–S1 | (Hockley et al. 2017) | ||
| Human | ENS | Colonic myenteric plexus | (Hetz et al. 2014) | ||
| Mouse | ENS | Duodenal myenteric plexus | (Osorio et al. 2014) | ||
| Human | Interstitial cells | Jejunal interstitial cells of Cajal | (Strege et al. 2003) | ||
| Human, dog, rat | Muscle | Jejunal circular smooth muscle | (Holm et al. 2002; Ou et al. 2002; Strege et al. 2007; Beyder et al. 2016) | ||
| Human, rat | Muscle | Colonic circular smooth muscle | (Strege et al. 2003; Beyder et al. 2016) | ||
| Human, mouse | Muscle | Jejunal longitudinal smooth muscle | (Ou et al. 2002; Strege et al. 2007) | ||
| Pig, guinea pig | Muscle | Jejunal circular smooth muscle | (Strege et al. 2007) | ||
| Human | Macrophages | Macrophages | (Carrithers et al. 2007, 2011; Black & Waxman, 2013) | ||
| NaV1.6 | Human | CNS | Cerebral cortex, cerebellum, hypothalamus, caudate, hippocampus | (Whitaker et al. 1999; GTEx Consortium et al. 2017) | |
| Rat | CNS | Cerebellum, hippocampus, spinal cord (white and grey matter) | (Tzoumaka et al. 2000) | ||
| Mouse | CNS | Spinal cord white and grey matter | (Duflocq et al. 2008) | ||
| Human | PNS | L3–L5 | (Chang et al. 2018) | ||
| Rat | PNS | L4–L5; L5 | (Tzoumaka et al. 2000; Fukuoka et al. 2008) | ||
| Mouse | PNS | L3–L6 dorsal and ventral roots; DRG | (Duflocq et al. 2008; Chang et al. 2018) | ||
| Mouse | PNS | Colonic neurons in T10–L1 and L5–S1; T10–L1; T9–T13; L6 | (King et al. 2009; Feng et al. 2015; Hockley et al. 2017; Inserra et al. 2017) | ||
| Human | ENS | Colonic myenteric plexus | (Hetz et al. 2014) | ||
| Guinea pig | ENS | Duodenal myenteric plexus | (Sage et al. 2007) | ||
| Human | Macrophages | Macrophages | (Carrithers et al. 2007, 2011; Black & Waxman, 2013) | ||
| NaV1.7 | Human | CNS | Hypothalamus | (GTEx Consortium et al. 2017) | |
| Rat | CNS | Hypothalamus, subfornical organ, intermediolateral cell column | Cerebellum, cerebral cortex, hippocampus, striatum, septum, thalamic nuclei | (Morinville et al. 2007) | |
| Mouse | CNS | Hypothalamus | (Branco et al. 2016) | ||
| Human | PNS | L3–L5; DRG | (Flegel et al. 2015; Chang et al. 2018) | ||
| Rat | PNS | L5 | (Fukuoka et al. 2008) | ||
| Mouse | PNS | Colonic neurons in T10–L1 and L5–S1; T10–L1; L6 | (Feng et al. 2015; Hockley et al. 2017; Inserra et al. 2017) | ||
| Human | ENS | Colonic myenteric plexus | (Hetz et al. 2014) | ||
| Guinea pig | ENS | Duodenal myenteric plexus | (Sage et al. 2007) | ||
| NaV1.8 | Human | CNS | Brain | (Flegel et al. 2015; GTEx Consortium et al. 2017) | |
| Human | PNS | L3–L5; DRG | (Flegel et al. 2015; Chang et al. 2018) | ||
| Rat | PNS | Colonic neurons T13–L2 | (Hu et al. 2013a, 2016; Lin et al. 2017) | ||
| Mouse | PNS | Colonic neurons in T10–L1 and L5–S1; T10–L1; T9–T13; T9–L1; L6 | (Beyak et al. 2004; Hillsley et al. 2006; King et al. 2009; Feng et al. 2015; Hockley et al. 2017; Inserra et al. 2017) | ||
| Human | ENS | Colonic myenteric plexus | (Hetz et al. 2014) | ||
| Mouse | ENS | Duodenal myenteric plexus | (Osorio et al. 2014) | ||
| NaV1.9 | Human | CNS | Brain | (GTEx Consortium et al. 2017) | |
| Human | PNS | L3–L5; DRG | (Flegel et al. 2015; Chang et al. 2018) | ||
| Rat | PNS | L4–L5 | (Dib‐Hajj et al. 1998) | ||
| Mouse | PNS | Colonic neurons T9–T13; trigeminal ganglia | (Beyak et al. 2004; Padilla et al. 2007; King et al. 2009) | ||
| Human | ENS | Colonic submucosal and myenteric plexus | (Hetz et al. 2014; O'Donnell et al. 2016) | ||
| Rat | ENS | Duodenal myenteric plexus | (Rugiero et al. 2003) | ||
| Mouse | ENS | Sensory, Dogiel type II, myenteric and submucosal neurons | (Padilla et al. 2007; Osorio et al. 2014) | ||
| Guinea pig | ENS | Duodenal intrinsic primary afferent neurons; duodenal myenteric plexus | (Rugiero et al. 2003; Copel et al. 2009) | ||
| Human | Muscle | Colonic smooth muscle | (O'Donnell et al. 2016) | ||
| Human | Mast cells | lung, skin and cord blood‐derived mast cells | (Bradding et al. 2003) |
Recently, numerous studies have reported NaV isoform channelopathies, including for NaV1.7 (SCN9A), NaV1.8 (SCN10A) and NaV1.9 (SCN11A) as the primary cause of increased pain or loss of pain phenotypes in humans (Yang et al. 2004; Cox et al. 2006; Fertleman et al. 2006; Klein et al. 2013; Leipold et al. 2013; Huang et al. 2014, 2017; Waxman et al. 2014; Dib‐Hajj et al. 2015; Han et al. 2015). Pharmacological modulation of NaV channels supports these genetic observations, including the finding that activation of all NaV channels by Pacific ciguatoxin 1 (P‐CTX‐1) or veratridine due to accidental consumption manifests as acute and severe gastrointestinal disturbances associated with abdominal pain in humans (Schep et al. 2006; Stewart et al. 2010). Intracolonic administration of purified P‐CTX‐1 also causes pain behaviour in mice (Inserra et al. 2017). On the other hand, TTX (which blocks NaV1.1–NaV1.4, NaV1.6 and NaV1.7) poisoning in humans is associated with paralysis rather than pain (Lago et al. 2015). Whilst potentially fatal upon consumption, administration of NaV‐selective agents such as TTX and neosaxitoxin has been shown to decrease pain responses in a range of pain modalities including visceral pain in humans (Hagen et al. 2011, 2017; Manriquez et al. 2015) and rodents (Marcil et al. 2006; Gonzalez‐Cano et al. 2017). Similarly, intrarectal administration of lidocaine (lignocaine) in irritable bowel syndrome (IBS) patients reduces rectal sensitivity and abdominal pain, suggesting NaV channels and activation of peripheral afferent endings in the colon play key roles in the pathogenesis of chronic visceral pain in IBS patients (Verne et al. 2005).
Human genetic studies have triggered widespread investigation into the therapeutic potential of NaV channels in the treatment of acute and chronic pain and also prompted studies to identify the wider roles of these channels throughout the body. It is also clear from most studies utilising inflammatory, nematode or bacterial models that gut‐ and bladder‐innervating neurons become hyperexcitable after the initial insult, which involves changes in TTX‐R and TTX‐S NaV currents, amongst others. This is apparent in neurons innervating the stomach (Gebhart et al. 2002; Bielefeldt et al. 2002a, b; Dang et al. 2004), small intestine (Moore et al. 2002; Stewart et al. 2003; Hillsley et al. 2006; Keating et al. 2008), the colon (Beyak et al. 2004; Ibeakanma et al. 2009; King et al. 2009) and the bladder (Yoshimura & deGroat, 1997). This review presents recent evidence on the specific roles of NaV1.1–NaV1.9 in transmitting sensation and nociception from the distal gut and bladder in healthy and pathological states.
NaV1.1
NaV1.1 is predominantly expressed in cell bodies, axon initial segments and at the nodes of Ranvier in the central nervous system (CNS) (Westenbroek et al. 1989; Duflocq et al. 2008; Carithers et al. 2015; Uhlen et al. 2015; GTEx Consortium et al. 2017). It is also expressed in human, rat and mouse PNS (Fukuoka et al. 2008; Wang et al. 2011; Osteen et al. 2016; Chang et al. 2018), and in human, but not guinea pig, myenteric plexus (Sage et al. 2007; Hetz et al. 2014) (Table 1). In thoracolumbar (T10–L1) and lumbar (L5) dorsal root ganglia (DRG) neurons, which contain the cell bodies of sensory neurons innervating the colon, rectum, bladder and skin, NaV1.1 is expressed in 15–35% of all neurons. Expression is predominantly in Tropomyosin‐related kinase C (TrkC)‐ and Tropomyosin‐related kinase A (TrkA)‐expressing myelinated A‐fibres of medium to large diameter and nearly absent in C‐fibre small diameter neurons innervating the skin (Fukuoka et al. 2008; Wang et al. 2011; Osteen et al. 2016). However, NaV1.1 mRNA transcript is detected in approximately half of thoracolumbar (T10–L1) and lumbosacral (L5–S1) mouse DRG neurons innervating the colon (Osteen et al. 2016; Hockley et al. 2017). As colonic afferents are predominantly peptidergic C‐fibres, there are clearly key differences in the populations of afferent neurons expressing NaV1.1 when comparing between the colon and the skin. In colon‐innervating DRG neurons, NaV1.1 is frequently co‐localised with NaV1.2, NaV1.3, NaV1.6, NaV1.7, NaV1.8 and NaV1.9 (Osteen et al. 2016; Hockley et al. 2017). Functional studies of colonic afferents reveal that NaV1.1 plays a crucial role in the signalling of mechanical pain from the colon (Osteen et al. 2016). Application of the selective NaV1.1 agonist, δ‐theraphotoxin‐Hm1a (Hm1a), enhances mechanically evoked firing in a subpopulation of high‐threshold colonic nociceptors. Notably, the mechanical hypersensitivity evoked by Hm1a was blocked by incubation with the NaV1.1/NaV1.3 antagonist ICA‐121431 (Table 2) (Osteen et al. 2016). Furthermore, Hm1a also induces hyperexcitability of isolated colon‐innervating DRG neurons from healthy control mice (Osteen et al. 2016). Notably, the percentage of colon‐innervating afferents/neurons affected by Hm1a is similar to the percentage of colon‐innervating DRG neurons expressing NaV1.1, as determined by single cell PCR (Osteen et al. 2016; Hockley et al. 2017). Importantly, colon‐innervating DRG neurons isolated from mice with chronic visceral hypersensitivity (CVH) show significantly enhanced responsiveness to Hm1a compared to healthy control mice, suggesting that NaV1.1 may be essential for the development and maintenance of chronic visceral pain conditions (Osteen et al. 2016). As such, antagonism of NaV1.1 may be a future target for the treatment of disorders accompanied by chronic visceral pain originating from the colon. There are currently no reports on the expression profile or function of NaV1.1 in the bladder or bladder‐innervating sensory neurons.
Table 2.
Predominant NaV isoforms contributing functionally to visceral sensation
| A. Healthy states | |||
|---|---|---|---|
| Species | Test | Response | Reference |
| Human | Appendix distension (ex vivo extracellular recordings of mesenteric afferents) before and after exposure to PF‐5198007 (Nav1.7 antagonist) | No difference in mesenteric afferent peak firing | (Hockley et al. 2017) |
| Mouse | Colonic mechanical stimulation (ex vivo extracellular recordings of colonic afferents in the splanchnic nerve) after Hm1a (highly selective Nav1.1 agonist, mucosal application) | Increase in colonic nociceptor response to mechanical stimuli in a sub‐population of afferents. | (Osteen et al. 2016) |
| Colonic stretch (ex vivo extracellular recordings of colorectal afferents in the pelvic nerve) μ‐conotoxin GIIIa, and μ‐conotoxin PIIIa, serosal/mucosal application) | Reduced action potential firing of stretch‐sensitive afferent response | (Feng et al. 2015) | |
| Colonic stretch (ex vivo extracellular recordings of colorectal afferents in the pelvic nerve) ‐ ProTxII (Nav1.7 antagonist, serosal/mucosal application) | No difference in stretch‐sensitive afferent response | (Feng et al. 2015) | |
| Ciguatoxin (pan‐Nav agonist) (intracolonic) | Increased pain behavioural response | (Inserra et al. 2017) | |
| Colonic incubation with A‐803467 (Nav1.8 antagonist) (ex vivo extracellular recordings of colorectal nociceptors), followed by ciguatoxin | Inhibited afferent firing induced by ciguatoxin | ||
| Incubation with supernatant from colitis patients | Increased excitability of colonic DRG neurons associated with enhanced NaV1.8 currents | (Ibeakanma & Vanner, 2010) | |
| Tumour necrosis factor‐α incubation | |||
| B. Knock‐out and knock‐down models | ||||
|---|---|---|---|---|
| Model | Species | Test | Response | Reference |
| NaV1.7Nav1.8 | Mouse | Formalin (intraplantar) | Reduction in pain behavioural response in phase I and phase II of formalin response | (Nassar et al. 2004) |
| Complete Freund's adjuvant (intraplantar) | Reduction in thermal hyperalgesia and mechanical allodynia from day 1 to day 10 | |||
| Carrageenan (intraplantar) | Reduction in thermal hyperalgesia from 1 to 4 h | |||
| Nerve growth factor (intraplantar) | Absence of phase I thermal hyperalgesia and reduction in phase II | |||
| Colonic distension (ex vivo extracellular recordings of lumbar splanchnic nerve activity) | No difference in afferent firing in physiological range (0–80 mmHg) | (Hockley et al. 2017) | ||
| Reduction in firing in supramaximal range (80–145 mmHg) | ||||
| Capsaicin (intracolonic) | Normal pain behavioural response | |||
| Mustard oil (intracolonic) | ||||
| Cyclophosphamide‐induced cystitis | Normal level of referred mechanical hyperalgesia responses | |||
| NaV1.8−/− | Mouse | Whole‐cell patch clamp | Reduced action potential amplitude in retrogradely labelled neurons projecting to the peritoneal cavity (DRG, T9–T13) | (Hillsley et al. 2006) |
| Nippostrongylus brasiliensis post‐infectious stage, whole‐cell patch clamp | Absence of neuronal hyperexcitability 19–25 days post‐infection in retrogradely labelled neurons projecting to the peritoneal cavity (DRG, T9–T13) | |||
| Acetylcholine (intraperitoneal injection) | Normal pain behavioural response | (Laird et al. 2002) | ||
| Capsaicin (intracolonic) | Reduced pain behavioural response | |||
| Mustard oil (intracolonic) | ||||
| Cyclophosphamide‐induced cystitis | Normal pain and inflammatory responses | |||
| NaV1.8 knock‐down (L6–S1) | Rat | Cystometry (saline) | No change in intercontraction intervals | (Yoshimura et al. 2001) |
| Acetic acid (intravesical) | Hyper‐reflexia attenuated | |||
| NaV1.9−/− | Mouse | Whole‐cell patch clamp | Normal excitability and action potential characteristics in colonic neurons (DRG T9–T13) | (Hillsley et al. 2006) |
| Nippostrongylus brasiliensis post‐infectious stage, whole‐cell patch clamp | No change in neuronal hyperexcitability 19–25 days post‐infection in retrogradely labelled neurons projecting to the peritoneal cavity (DRG, T9–T13) | |||
| Colorectal distension | Normal pain behavioural response | (Martinez & Melgar, 2008) | ||
| R‐848 (toll‐like receptor 7 activator)‐induced colonic inflammation, colorectal distension | Reduced pain behavioural response | |||
| Colonic distension (ex vivo extracellular recordings of splanchnic nerve activity) | Reduced afferent discharge | (Hockley et al. 2014) | ||
| Ex vivo extracellular recordings of lumbar splanchnic nerve activity following inflammatory soup (bradykinin, ATP, histamine, PGE2 and 5‐HT), or inflammatory bowel disease patient colonic supernatant application | Reduced afferent fibre responses | |||
| Ex vivo extracellular recordings of lumbar splanchnic nerve activity following UTP (P2Y2 and P2Y4 agonist) or ADP (P2Y1, P2Y12 and P2Y13 agonist), application | Reduced afferent fibre responses | (Hockley et al. 2016a) | ||
| Cystometry (saline) | No change in basal urodynamics | (Ritter et al. 2009) | ||
| Cyclophosphamide‐induced cystitis | ||||
| Bladder distension (ex vivo extracellular recordings of bladder nerve activity) following PGE2 bladder infusion application | Reduced afferent excitability | |||
| C. Inflammatory hypersensitivity models | ||||
|---|---|---|---|---|
| Model/disease | Species | Test | Response | Reference |
| Neonatal induced colitis | Rat | Protein expression | Increase in NaV1.7 and Nav1.8 protein in colonic (DRG, T13–L2) neurons post‐inflammation | (Qu et al. 2013) |
| Whole‐cell patch clamp | Increase in Na+ current in colonic neurons (DRG, T13–L2) 6 weeks post‐inflammation | |||
| No change in Na+ current in colonic neurons (DRG, T13–L2) 10 weeks post‐inflammation | ||||
| No change in Na+ current in non‐colonic neurons (DRG, L4–L5) 6 or 10 weeks post inflammation | ||||
| Acute TNBS‐induced colitis | Mouse | Whole‐cell patch clamp | Increased slow TTX‐R Na+ current in colonic neurons (DRG, T9–L1) 7–10 days post‐ induction | (Beyak et al. 2004) |
| No change in persistent TTX‐R Na+ currents in colonic neurons (DRG, T9–L1) 7–10 days post‐induction | ||||
| Gene and protein expression | No change in NaV1.7 mRNA or protein in retrogradely labelled colonic neurons (DRG, T9–T13) 1 week post‐induction | (King et al. 2009) | ||
| Gene expression | Tenfold reduction in NaV1.8 mRNA 2–4 days post‐induction, no change at day 7, in retrogradely labelled colonic neurons (DRG, T9–T13) | |||
| Protein expression | No change in NaV1.8 protein 2–4 days post‐induction, up‐regulation at day 7, in retrogradely labelled colonic neurons (DRG, T9–T13) 1 week post‐induction | |||
| No change in Nav1.9 protein in colonic neurons (DRG, T9–T13) day 7 post‐induction | ||||
| Post‐TNBS‐induced colitis | Mouse | Whole‐cell patch clamp in the presence of Hm1a (Nav1.7 agonist) | Pronounced increase in excitability of colonic DRG neurons: significant lowering of rheobase and a dramatic increase in the number of action potentials fired at 2× rheobase | (Osteen et al. 2016) |
| Gene expression | Up‐regulation of NaV1.7 mRNA in retrogradely labelled colonic neurons (DRG, L6–S1) 4 weeks post‐induction | (Campaniello et al. 2016) | ||
| Nippostrongylus brasiliensis post‐infectious stage | Mouse | Gene expression | No change in NaV1.8 or Nav1.9 mRNA 19–25 days post ‐infection in retrogradely labelled neurons projecting to the peritoneal cavity (DRG, T9–T13) | (Hillsley et al. 2006) |
| Interstitial cystitis/bladder pain syndrome | Human | Neosaxitoxin (blocker of TTX‐S Nav channels) (bladder infiltration) | Analgesia and reduced frequency lasting up to 90 days | (Manriquez et al. 2015) |
| Cyclophosphamide‐induced cystitis | Rat | A‐803467 administration (intraperitoneal) | No change in pain behavioural response | (Jarvis et al. 2007) |
| D. Non‐inflammatory hypersensitivity models | ||||
|---|---|---|---|---|
| Model/disease | Species | Test | Response | Reference |
| Clinical rectal hypersensitivity | Human | Protein expression (full thickness rectal biopsies) | Increased NaV1.7‐immunoreactive nerve fibres in mucosal, submucosal and muscle layers | (Yiangou et al. 2007) |
| Maternal separation model (visceral hypersensitivity) | Rat | Gene expression | No change in NaV1.8 mRNA in colonic neurons (DRG, T13–L2) | (Hu et al. 2013a) |
| Protein expression | Increase in NaV1.8 protein in colonic neurons (DRG, T13–L2) | |||
| Whole‐cell patch clamp | Increased TTX‐R Na+ current in colonic neurons (DRG, T13–L2) | |||
| Streptozotocin‐induced diabetes (visceral hypersensitivity) | Protein expression | Increase in NaV1.7 and NaV1.8 protein in colonic neurons (DRG, T13–L2) | (Hu et al. 2016) | |
| Whole‐cell patch clamp | Increased TTX‐R Na+ current in colonic neurons (DRG, T13–L2) | |||
| Partial colonic obstruction (visceral hypersensitivity) | Gene expression | Increase in NaV1.8 mRNA in colonic neurons (DRG, T13–L2) | (Lin et al. 2017) | |
| Whole‐cell patch clamp | Increased TTX‐R Na+ current in colonic neurons (DRG, T13–L2) | |||
| T8 spinal transection | Whole‐cell patch clamp | Reduced TTX‐R Na+ current in bladder neurons | (Yoshimura & deGroat, 1997) | |
NaV1.2
NaV1.2 is extensively expressed in the CNS (Jarnot & Corbett, 2006) but has also been detected at low levels in small‐diameter DRG neurons (Black et al. 1996; Fukuoka et al. 2008; Chang et al. 2018). Conversely, in colon‐innervating DRG neurons of the mouse, NaV1.2 mRNA transcript is present in 69% of thoracolumbar (T10–L1) neurons and at a similar level in lumbosacral (L5–S1) neurons (Hockley et al. 2017) (Table 1). Despite this mRNA expression, there is currently no functional data to support a role for NaV1.2 in colonic sensory signalling or pain. Similarly, there are currently no reports on the expression profile or function of NaV1.2 in the bladder or bladder‐innervating sensory neurons.
NaV1.3
NaV1.3 is highly expressed in sensory neurons during embryogenesis in rats, but its expression traditionally subsides in fully developed neurons (Beckh et al. 1989). The major body of NaV1.3 research in nociception focuses on its role in neuropathic pain, as NaV1.3 is re‐expressed following neuropathic injury in large diameter, myelinated A‐fibre neurons where it may contribute to ectopic discharge and painful neuropathy (Waxman et al. 1994; Zang et al. 2010). However, due to the limited expression of this channel in adult tissues and lack of channelopathy‐associated pain syndromes, studies investigating the role of NaV1.3 in other pain pathways are few. In relation to the viscera, NaV1.3 mRNA is detected in adult guinea‐pig enteric nervous system (ENS) neurons (Sage et al. 2007), but its functional role has yet to be determined. Initial experiments indicate that NaV1.3 expression is low in rat lumbar (L5) DRG neurons (Fukuoka et al. 2008). However, NaV1.3 mRNA transcripts are detected in approximately half of the colon‐innervating thoracolumbar (T10–L1) and lumbosacral (L5–S1) DRG neurons in the mouse (Hockley et al. 2017) (Table 1).
More recent studies show a key role for NaV1.3 in non‐neuronal tissues, specifically within enterochromaffin cells located within the epithelium from the small and large intestine of humans and mice (Bellono et al. 2017; Strege et al. 2017a,b). Voltage‐gated sodium currents generated by NaV1.3 likely allow enterochromaffin cells to respond to the detection of mechanical and chemical stimuli within the lumen of the intestine (Bellono et al. 2017; Strege et al. 2017b). In contrast, expression of the other eight NaV isoforms is very low, or indeed lacking from both intestinal enterochromaffin cells and the wider population of intestinal epithelial cells (Bellono et al. 2017). There are currently no reports on the role of NaV1.3 in the bladder or bladder‐innervating sensory neurons.
NaV1.4
NaV1.4 is the predominant NaV isoform in skeletal muscle (Trimmer et al. 1990) but is also found in human oesophageal smooth muscle tissue (Deshpande et al. 2002). In peripheral neurons, NaV1.4 transcripts are nearly absent in rat lumbar (L5) DRG (Fukuoka et al. 2008) and in colon‐innervating mouse DRG neurons (Hockley et al. 2017) (Table 1). In agreement with tissue distribution, NaV1.4 channelopathies appear to exclusively involve deficits in skeletal muscle function, and to date no involvement in colon or bladder function has been shown.
NaV1.5
NaV1.5 channels have been identified in circular smooth muscle of the jejunum of human, dog, rat and mouse but are absent in pig and guinea pig. NaV1.5 is also absent from human and mouse jejunal longitudinal smooth muscle (Holm et al. 2002; Ou et al. 2002; Strege et al. 2007; Beyder et al. 2016). NaV1.5 has been found in colonic circular smooth muscle of human and rat (Strege et al. 2003), in jejunal interstitial cells of Cajal in human (Strege et al. 2003), and in myenteric plexuses of human and mouse (Hetz et al. 2014; Osorio et al. 2014).
NaV1.5 in circular smooth muscle may contribute to normal intestinal motility through modulation of slow‐wave activity and muscle contractility (Ou et al. 2002; Strege et al. 2007). These findings are supported by data showing that ranolazine, a treatment for chronic angina, is able to inhibit NaV1.5 currents in human colonic smooth muscle cells (Neshatian et al. 2015), which is likely to be responsible for the constipation seen during long‐term ranolazine treatment (Nash & Nash, 2008). These data strongly point towards a primary role for NaV1.5 channels in mediating gastrointestinal motility and transit (Beyder & Farrugia, 2016). Similarly, several loss‐of‐function mutations in SCN5A, the gene encoding NaV1.5 channels, are associated with IBS and abdominal pain (Saito et al. 2009; Beyder et al. 2014; Strege et al. 2017c). Whether this is purely a consequence of reduced gastrointestinal contractility or whether NaV1.5 channels also play a direct role in visceral sensation remains unclear, as NaV1.5 mRNA transcripts are expressed in 18% of thoracolumbar and 51% of lumbosacral colon‐innervating DRG neurons (Hockley et al. 2017) (Table 1). Whether this translates into channel expression and a functional role remains to be determined. There are currently no reports on the expression profile or function of NaV1.5 in the bladder or bladder‐innervating sensory neurons.
NaV1.6
NaV1.6 is extensively expressed within the CNS and PNS (Whitaker et al. 1999; Tzoumaka et al. 2000; Catterall et al. 2005; Catterall, 2012; Chang et al. 2018), commonly located in clusters at the nodes of Ranvier (Duflocq et al. 2008), indicating that NaV1.6 may have a primary role in transmitting rather than initiating action potentials. In rat lumbar (L5) DRG neurons, NaV1.6 transcripts are detected in a third of all neurons and selectively expressed in TrkC‐ and TrkA‐expressing myelinated A‐fibre nociceptors (Fukuoka et al. 2008). In colon‐innervating mouse DRG neurons, NaV1.6 mRNA transcript is present in 63–87% of thoracolumbar (T10–L1) neurons, and in 51% of lumbosacral (L5–S1) neurons (Hockley et al. 2017; Inserra et al. 2017). Immunohistochemical and western blot analysis show that NaV1.6 protein is present in the cell bodies of sensory neurons and on sensory afferent nerve endings innervating the distal colon and rectum in mice (Feng et al. 2015) (Table 1). Antagonism of NaV1.6 reduces action potential firing of stretch‐sensitive colorectal afferents in vitro (Feng et al. 2015) (Table 2). Whether these effects are altered in animal models of inflammatory or chronic visceral pain remains to be investigated. It has, however, been reported that there is no change in NaV1.6 expression in colon‐innervating DRG neurons (T9–T13) during the acute inflammatory phase of the mouse model of trinitrobenzenesulphonic acid (TNBS)‐induced colitis (King et al. 2009). This corresponds with the phase when colorectal afferent hypersensitivity also occurs (Hughes et al. 2009). Activation of low‐threshold stretch‐sensitive afferents is essential for normal physiological function of the colon (Brierley et al. 2004; Kyloh et al. 2011) and NaV1.6 appears to play a key integrative role in this process. Whether NaV1.6 contributes to aberrant colonic afferent sensory signalling during chronic visceral hypersensitivity remains to be determined. There are currently no reports on the expression profile or function of NaV1.6 in the bladder or bladder‐innervating sensory neurons.
NaV1.7
NaV1.7 has become a key target of interest as several human mutations in the SCN9A gene, which encodes NaV1.7, lead to either a loss of pain or increased pain perception (Bennett & Woods, 2014). For example, a loss‐of‐function mutation of SCN9A results in a congenital insensitivity to pain (CIP) (Cox et al. 2006; Goldberg et al. 2007), whereas gain‐of‐function mutations produce distinct pain syndromes, such as erythromelalgia, small‐fibre neuropathy and paroxysmal extreme pain disorder (Fertleman et al. 2006). NaV1.7 is extensively expressed in sensory and sympathetic neurons of the PNS, as well as ENS neurons, and is highly restricted in the CNS (Klugbauer et al. 1995; Catterall et al. 2005; Morinville et al. 2007; Sage et al. 2007; Branco et al. 2016; Chang et al. 2018). In rat lumbar (L5) DRG neurons, NaV1.7 transcripts are preferentially expressed in TrkA‐expressing C‐fibre neurons, and in a subset of A‐fibre neurons (Fukuoka et al. 2008). Robust immunolabelling of NaV1.7 is present within the peripheral endings of sensory nerves in the skin (Black et al. 2012).
From mouse knock‐out studies, it appears that NaV1.7 in NaV1.8‐expressing cells (NaV1.7Nav1.8) does not contribute in the development of neuropathic pain, nor noxious cold or heat detection (Nassar et al. 2004, 2005; Minett et al. 2012, 2014; Hockley et al. 2017). However, NaV1.7Nav1.8 mice have significantly reduced behavioural responses to inflammatory mediators (formalin, complete Freund's adjuvant, carrageenan and nerve growth factor) when injected into the sole of the hind paw (Nassar et al. 2004) and impaired somatic noxious mechanosensation (Minett et al. 2012, 2014). Thus far, only the deletion of NaV1.7 in sympathetic and sensory (Wnt1‐expressing) neurons and the global NaV1.7 knock‐out have been able to significantly reduce pain responses to a range of stimuli and recapitulate the human SCN9A‐associated CIP phenotype (Gingras et al. 2014; Minett et al. 2014). Recent studies also show that endogenous opioids contribute to pain insensitivity in both humans and mice lacking NaV1.7, as the opioid antagonist naloxone reverses analgesia associated with the loss of NaV1.7 expression (Minett et al. 2015). This suggests that NaV1.7 channel blockers alone may not replicate the analgesic phenotypes of NaV1.7 null mutants, but may be potentiated with exogenous opioids. NaV1.7‐selective inhibitors are currently in clinical trial for different types of pain (Pennington et al. 2017; Yekkirala et al. 2017).
In relation to visceral sensation, NaV1.7 is highly abundant in human lumbar DRG, and is expressed in 100% of mouse colon‐innervating thoracolumbar (T10–L1) DRG neurons, and in most colon‐innervating lumbosacral (L5–S1) DRG neurons (Chang et al. 2018; Hockley et al. 2017; Inserra et al. 2017) (Table 1). Accordingly, NaV1.7 constitutes the most prevalent TTX‐S isoform within colon‐innervating DRG neurons. It is of interest to note that ‘paroxysmal extreme pain disorder’, caused by the human gain of function SCN9A mutation, was originally called ‘familial rectal pain syndrome’. As the name implies, this disorder is characterised by excruciating rectal and abdominal pain commonly associated with defecation (Fertleman et al. 2006), suggesting a key role for NaV1.7 in visceral pain. Moreover, pain perception in a subset of patients with interstitial cystitis/bladder pain syndrome (IC/BPS) is shown to correlate with a polymorphism in SCN9A (Reeder et al. 2013). IC/BPS patients treated with a bladder infiltration of neosaxitoxin, a blocker of TTX‐S NaV channels, resulted in significant analgesia and reduced bladder overactivity for 90 days after the treatment (Manriquez et al. 2015). Normal physiological function of the bladder, however, appears to be independent of NaV1.7, as SCN9A‐associated CIP individuals have normal bladder control, and no increased incidence of urinary infections, incontinence, or retention (Cox et al. 2006).
Despite these studies, the initial promise of NaV1.7's contribution to visceral pain is somewhat tempered by experimental studies showing that NaV1.7Nav1.8 mice exhibit normal nocifensive responses to intracolonic administration of capsaicin (TRPV1 agonist) and mustard oil (TRPA1 agonist), indicating that NaV1.7 is not crucial for acute visceral pain signalling (Hockley et al. 2017). Low‐threshold stretch‐sensitive pelvic afferents are unaffected by the NaV1.7 antagonist ProTX‐II (Feng et al. 2015) (Table 2). Similarly, ex vivo extracellular recordings of mesenteric afferents from resected human appendices show that peak firing before and after exposure to a novel NaV1.7‐selective antagonist, PF‐5198007, is unchanged during repeat noxious ramp distensions (Hockley et al. 2017). Afferent responses in mouse ex vivo colorectal recordings are attenuated by application of TTX (Feng et al. 2015), indicating that TTX‐S channels other than NaV1.7 may be important in responding to innocuous and noxious mechanical stimuli. Accordingly, intracolonic co‐administration of TTX and P‐CTX‐1 did not significantly alter the pain response induced by P‐CTX‐1 (Inserra et al. 2017).
Ex vivo extracellular recordings of splanchnic nerve activity from the distal colon of NaV1.7Nav1.8 mice show no difference in peak firing between NaV1.7Nav1.8 and littermate control afferents in the physiological and supraphysiological pressure range (0–80 mmHg) (Hockley et al. 2017). However, significantly less action potential firing in afferents from NaV1.7Nav1.8 mice at distension pressures in the supramaximal range (80–145 mmHg) is observed, suggesting that NaV1.7 in NaV1.8‐positive colonic afferent neurons may be involved in transducing non‐physiological extremes of pressure. This may be important and more relevant to chronic visceral pain states, when splanchnic afferents show mechanical hypersensitivity and decreased activation thresholds to mechanical stimuli (Hughes et al. 2009; Castro et al. 2013, 2017; de Araujo et al. 2014; Osteen et al. 2016). In the bladder, NaV1.7Nav1.8 mice have comparable levels of referred hyperalgesia in an acute cyclophosphamide‐induced cystitis model compared to littermates (Hockley et al. 2017). Overall, these findings suggest that NaV1.7 has a role in mediating acute inflammatory pain in somatic but not visceral pathways. While studies on visceral nociception using NaV1.7Nav1.8 mice have provided valuable insight, replication of these studies in mice with sensory neuron‐specific deletion of NaV1.7 (e.g. NaV1.7Advill) will be beneficial to strengthen conclusions concerning NaV1.7 in visceral pain signalling.
Diseases that have a significant visceral pain component are commonly chronic and have unmet needs in terms of clinical treatment. Therefore, further investigations into the role of NaV1.7 in long term and chronic visceral pain models, which are more clinically relevant to pathological chronic visceral pain states, are critical. For example, significant up‐regulation of NaV1.7 mRNA occurs 4 weeks after induction of colitis in colon‐innervating DRG (L6–S1) neurons (Campaniello et al. 2016). Similarly, rats with streptozotocin‐induced diabetes show hypersensitivity to colonic distension, which corresponds with the up‐regulation of NaV1.7 protein in thoracolumbar (T13–L2) DRG neurons 4 weeks post‐induction (Hu et al. 2016). In support of these findings, rat neonatal colitis‐induced visceral hypersensitivity induces up‐regulation of NaV1.7 protein levels in DRG from higher spinal levels (T13–L2), but not lower spinal levels (L4–L5) at 6 weeks post‐colitis compared to control animals (Qu et al. 2013). Taken together, these findings suggest that NaV1.7 may have an acquired role during chronic visceral pain states. It is well documented that inflammation, tissue damage and healing of visceral organs can induce structural, synaptic or intrinsic neuroplasticity, altering neuronal and gastrointestinal function in the long term (Brierley & Linden, 2014). For example, rectal samples from patients with physiologically characterised rectal hypersensitivity show significantly increased numbers of NaV1.7‐immunoreactive nerve fibres in the mucosal, submucosal and muscle layers compared to control tissues (Yiangou et al. 2007). In addition to these findings, changes in the ratios of NaV1.7 to a pan‐neuronal structural marker, PGP9.5, indicate that increased NaV1.7 expression and nerve sprouting occurs in rectal mucosa, which may contribute to enhanced sensitivity in these patients (Yiangou et al. 2007).
Overall, the role of NaV1.7 in pain sensation is complicated, and species differences in expression, assumed translatability of isoform‐compound interaction, and effects of NaV knock‐out on other genes may confound overall conclusions. Furthermore, few studies using human tissue have been completed, and healthy tissue is often obtained from patients with colonic or rectal carcinoma (Yiangou et al. 2007; Hetz et al. 2014; Hockley et al. 2017). In addition to species‐dependent differences in tissue distributions (Table 1), there are also differences in relative isoform distributions, for example, NaV1.7 is the most abundant isoform in human lumbar DRG, whereas NaV1.8 is more abundant in mouse (Chang et al. 2018). NaV1.7 isoforms from different species can also have different compound selectivity in heterologous expression systems that should be carefully considered during experimental design. For example, human, monkey, dog and mouse NaV1.7 isoforms were found to be largely insensitive to a small molecule inhibitor of NaV1.1/NaV1.3 (ICA‐121431) and potently inhibited by a small molecule inhibitor of NaV1.7 (PF‐04856264), whereas rat NaV1.7 was potently inhibited by ICA‐121431, but largely insensitive to PF‐04856264 (McCormack et al. 2013). A ProTxII analogue, JNJ63955918, on the other hand was equipotent at human and rat NaV1.7 (Flinspach et al. 2017). Single cell studies have shown that NaV channel expression is heterologous across cells, and there is high co‐localisation of NaV1.7 with NaV1.6, NaV1.8 and NaV1.9 in colon‐innervating thoracolumbar and lumbosacral neurons in mice (Hockley et al. 2017). However, functional relationships of co‐expression and investigations of redundancy between NaV channels are unclear. In knock‐out models, deletion of one NaV gene can lead to a change in expression levels of over 190 genes (Minett et al. 2015). Studies investigating NaV channel contribution to pain signalling using knock‐out models or pharmacological modification may benefit from collecting data on regulation of other NaV family genes and auxiliary β‐subunits in parallel, and other key genes where possible. Furthermore, inducible knock‐out models offer the advantage of normal development and being able to compare NaV channel contribution pre‐ and post‐induction of visceral hypersensitivity in the adult, thereby increasing therapeutic potential of these findings.
NaV1.8
NaV1.8 mediates slowly inactivating TTX‐R Na+ currents and carries the majority of the current underlying the upstroke of the action potential in nociceptive neurons. Hence, they are considered to play an important role in action potential electrogenesis (Renganathan et al. 2001). NaV1.8‐null mice display reduced sensitivity to noxious mechanical stimuli (tail pressure) and noxious thermal stimuli (radiant heat), but normal sensitivity to acute noxious colonic distension by isotonic saline and intraperitoneal acetylcholine (Akopian et al. 1999; Laird et al. 2002). NaV1.8 is the most abundant isoform expressed in mouse lumbar DRG (Chang et al. 2018), and is prevalently expressed in thoracolumbar (96%) and lumbosacral (91%) colonic sensory DRG neurons, with almost complete co‐expression with NaV1.7 (Hockley et al. 2017). Consistent with this, knock‐down of NaV1.8 in DRG neurons results in action potentials with reduced peak amplitude and slower rise times, but similar baseline excitability (Renganathan et al. 2001; Hillsley et al. 2006). Similarly, A‐803467, a selective NaV1.8 antagonist, does not significantly affect the frequency of action potential firing from low‐threshold mechanosensitive colonic afferent nerve endings (Feng et al. 2015). Together, these data seem to suggest that NaV1.8 channels do play a major role in mediating visceral sensations and pain under physiological conditions.
NaV1.8 channels also have a major role in visceral signalling under pathophysiological conditions. Several studies support increased expression of NaV1.8 protein in colon‐innervating sensory DRG neurons in murine models of visceral hypersensitivity (Beyak et al. 2004; Hillsley et al. 2006; King et al. 2009; Qu et al. 2013; Hu et al. 2013a,b; Inserra et al. 2017; Lin et al. 2017) (Table 2). In most studies, increased channel expression correlates with enhanced TTX‐R Na+ current density in colon‐innervating DRG neurons in vitro, and with visceral hypersensitivity in vivo, as the visceromotor response to noxious colonic distension in rats is significantly reduced following intraperitoneal administration of the NaV1.8‐specific antagonist A‐803467 (Jarvis et al. 2007). Similarly, colonic co‐administration of A‐803467 with P‐CTX‐1 significantly reduces P‐CTX‐1‐induced nocifensive behaviours in mice (Inserra et al. 2017). These findings are consistent with studies in NaV1.8‐null mice, which do not develop visceral hypersensitivity after intracolonic administration of sensitising agents such as capsaicin (TRPV1 agonist) and mustard oil (TRPA1 agonist). Furthermore, unlike their wild‐type littermates, DRG neurons from NaV1.8‐null mice do not display enhanced neuronal hyperexcitability following intestinal infection with Nippostrongylus brasiliensis (Laird et al. 2002; Hillsley et al. 2006).
Inflammatory mediators acting via GPCRs are powerful modulators of NaV1.8 currents, and are believed to underlie increased excitability of nociceptive DRG neurons and associated hyperalgesia (Beyak et al. 2004). In this regard, colon‐innervating DRG neurons incubated with supernatant from colonic biopsies from patients with active ulcerative colitis (a chronic inflammatory bowel disease) show increased action potential discharge and enhanced NaV1.8 currents (Ibeakanma & Vanner, 2010). These effects were replicated by incubation with tumour necrosis factor α (TNFα), whose levels are enhanced in the ulcerative colitis supernatant. Similar sensitising effects have been reported for prostaglandin E2 (PGE2), adenosine, serotonin (5‐HT), ATP, as well as nerve growth factor (NGF), which may persist during and possibly after the inflammation as a result of increased expression of NaV1.8 channels (Gold, 1999; Gold et al. 2002; Beyak et al. 2004). Recent data, however, indicate that these effects are not limited to inflammatory conditions, but may extend to non‐inflammatory chronic pain states. Partial colonic obstruction is associated with an increase in NaV1.8 mRNA expression, as well as enhanced TTX‐R Na+ currents and referred in vivo hyperalgesia, effects that were abolished by anti‐NGF treatment (King et al. 2009; Ibeakanma & Vanner, 2010).
Decreased TTX‐R currents occur in bladder‐innervating DRG neurons from T8 spinal transected rats (Yoshimura & deGroat, 1997), which has since been attributed to a down‐regulation of NaV1.8 (Black et al. 2003). This change is only seen in bladder‐innervating DRG neurons, and is accompanied by an up‐regulation of TTX‐S current, which may also enhance the excitability of these afferent neurons. Knock‐down of NaV1.8 in rats at spinal levels L6–S1, known to contain the majority of bladder sensory terminals, does not have an effect on intercontraction intervals following cystometry with saline; however, intravesical acetic acid‐induced hyper‐reflexia is attenuated in knock‐down rats (Yoshimura et al. 2001). NaV1.8‐null mice develop normal pain and inflammatory responses during cyclophosphamide‐induced cystitis compared to littermates (Laird et al. 2002), and pain behaviours are sustained in rats with cyclophosphamide‐induced cystitis following intraperitoneal administration of A‐803467 (Jarvis et al. 2007).
Cross‐organ sensitisation of the gastrointestinal and lower urinary tract is evident clinically and in animal models (Malykhina et al. 2004, 2012; Lei & Malykhina, 2012), highlighting the importance of understanding the mechanisms of viscero‐visceral crosstalk. Several studies report increases in TTX‐resistant Na+ current in bladder‐innervating DRG neurons following colitis, implicating some involvement of TTX‐R channels in bladder pain as a consequence of gastrointestinal tract inflammation. C‐fibre bladder‐innervating DRG neurons, involved in the transduction of noxious stimuli signalling (Fowler et al. 2008), in the majority express TTX‐R currents (Yoshimura & deGroat, 1997). Collectively, experimental findings to date indicate that NaV1.8 is not crucial for visceral pain signalling from the bladder in response to several noxious stimuli, but it may have an important role during referred hyperalgesia and in response to certain irritants.
NaV1.9
Several human NaV1.9 channelopathies are associated with congenital episodic pain syndromes, painful neuropathy, and an insensitivity to pain (Huang et al. 2014, 2017). NaV1.9 channels are preferentially expressed in small‐diameter nociceptors (Dib‐Hajj et al. 1998; Tate et al. 1998), and mediate ultraslow or persistent TTX‐R Na+ currents. Due to their kinetic properties, NaV1.9 channels are unlikely to contribute to action potential generation, but instead regulate neuronal excitability by setting the resting membrane potential closer to threshold (Dib‐Hajj et al. 1998, 2002; Tate et al. 1998). In colonic afferents, action potential firing in response to colonic ramp distension is reduced in NaV1.9−/− mice, and accompanied by a run‐down of responses to repeated phasic distension (Hockley et al. 2014). Similar to NaV1.8 channels, several studies indicate that NaV1.9 currents can be enhanced via GPCRs (Maingret et al. 2008; Ostman et al. 2008; Vanoye et al. 2013; Hockley et al. 2016b). Colonic afferent excitatory responses to the application of multiple inflammatory mediators (applied at once, either in the form of supernatants from chronically inflamed human bowel or as an experimental inflammatory soup containing ATP, PGE2, bradykinin, histamine and 5‐HT) are significantly reduced in visceral afferents from NaV1.9−/− mice (Hockley et al. 2014, 2016a) (Table 2).
NaV1.9−/− mice have similar baseline visceromotor responses to colonic distension to wild‐type littermates, but reduced visceral hypersensitivity in vivo after colonic inflammation induced by activation of toll‐like receptor 7 (Martinez & Melgar, 2008). Neuronal hyperexcitability following Nippostrongylus bransiliensis infection is unchanged in NaV1.9−/− mice compared to wild‐type littermates, reporting similar action potential characteristics and excitability of colon‐innervating DRG neurons (Hillsley et al. 2006). Likewise, others do not see changes in NaV1.9 protein expression in colon‐innervating DRG neurons, nor differences in either the numbers of neurons expressing persistent TTX‐R (NaV1.9) currents or the magnitude of these currents in acute TNBS‐induced colitis (Beyak et al. 2004; King et al. 2009). It is unclear whether these discrepancies in the contribution of NaV1.9 to neuronal (hyper)excitability relates to differences in knock‐out constructs and mice strains, or to differences in the inflammatory insult studied. The latter may be of considerable importance as inflammatory mediators such as bradykinin, ATP, histamine, PGE2 and noradrenaline (norepinephrine), potentiate NaV1.9 channel activity when applied conjointly, but fail to modulate NaV1.9 currents when applied separately (Maingret et al. 2008).
NaV1.9 channels are also present in myenteric plexus neurons in human, mouse, rat and guinea‐pig (Rugiero et al. 2003; Padilla et al. 2007; Copel et al. 2009; Osorio et al. 2014) pointing towards an additional role in intestinal motor function. In line with this, colonic migrating motor complex patterns are altered in NaV1.9−/− mice (Copel et al. 2013). Moreover, expression of NaV1.9 channels is decreased in submucosal and myenteric plexus neurons (most likely intrinsic primary afferent neurons) in Hirschsprung's disease (O'Donnell et al. 2016). Interestingly, these findings apply not only to aganglionic bowel sections, but in some patients extend to those sections containing normal ganglia numbers, which could explain some of the post‐surgery bowel dysmotility issues frequently encountered by these patients (O'Donnell et al. 2016). Conversely, a gain‐of‐function mutation (L811P) in the NaV1.9 gene, SCN11A, identified in three unrelated individuals with congenital insensitivity to pain, is associated with severe gastrointestinal dysmotility, including alternating episodes of diarrhoea and constipation (Leipold et al. 2013; Woods et al. 2015). In contrast, other gain‐of‐function mutations are predominantly linked to chronic pain syndromes such as autosomal‐dominant episodic pain and small fibre neuropathy (Zhang et al. 2013; Huang et al. 2014; Han et al. 2015).
No difference is observed in basal urodynamics between wild‐type and NaV1.9−/− mice; however, the change of urodynamic parameters associated with cyclophosphamide‐induced cystitis is absent in NaV1.9−/− mice, as well as attenuation of PGE2‐induced afferent excitability during bladder distension (Ritter et al. 2009). It remains to be investigated whether this involvement of NaV1.9 in bladder nociception is due to functional up‐regulation of NaV1.9 in bladder afferents, or whether NaV1.9 has a role in central processing of bladder nociceptive pathways.
Conclusion
Recent findings highlight the diversity in expression patterns of NaV isoforms in abdominal visceral organs. This diversity extends across neurons (enteric, extrinsic sensory DRG innervating the intestine or bladder) and non‐neuronal cells (intestinal enterochromaffin cells, intestinal smooth muscle cells, and interstitial cells of Cajal). NaV channels have a range of functions in health and disease and we are only now, with the development of novel pharmacological and genetic tools, beginning to unpick their complex physiological and pathophysiological interactions. NaV1.1, NaV1.6, NaV1.8 and NaV1.9, contribute to visceral hypersensitivity, particularly within colonic pathways, and respond to inflammatory mediators in pathophysiological models (Fig. 1).
Figure 1. Current understanding of how specific voltage‐gated sodium channels (NaV) contribute to the functioning of neurons and non‐neuronal cells within visceral organs.
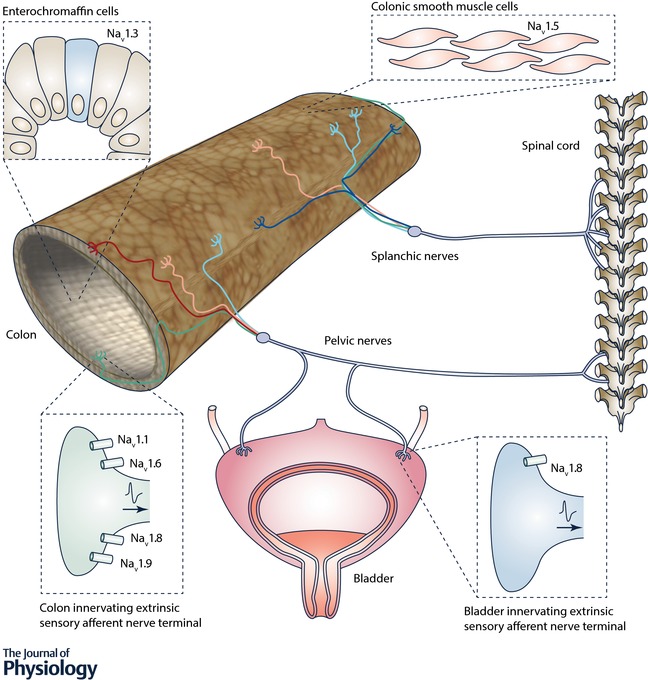
Whilst NaV1.3 contributes to enterochromaffin cell function and NaV1.5 contributes to intestinal smooth muscle cells and interstitial cells of Cajal function, there is currently no determined function in visceral afferents for NaV1.2, NaV1.3, NaV1.4 or NaV1.5, despite significant mRNA expression of NaV1.2 and NaV1.5 in visceral afferent pathways. NaV1.7 is one of the most extensively expressed and studied NaV channels, but a role in visceral pain, like that attributed to NaV1.7 in somatic pain studies is currently unclear. Although many of these NaV channels have been investigated under physiological conditions or in models of acute pain, chronic visceral pain models are necessary for the determination of a precise role in long term pathological visceral pain. Future studies would benefit from the further development of novel, specific agonists and antagonists, as we have seen with recent advances in the role of NaV1.1 in mechanical pain. Likewise, selective NaV modulators with low systemic uptake for in vivo studies will advance our understanding of NaV channels in visceral pain signalling and the suitability of targeting NaV channels in the treatment of pain originating in the distal gut and bladder.
Additional information
Competing interests
In relation to the content covered within this review, the authors have nothing to declare.
Author contributions
All authors contributed to searching the published literature and to writing the review. All authors approved the final version and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
S.M.B. is a National Health and Medical Research Council of Australia (NHMRC) R.D. Wright Biomedical Research Fellow (APP1126378) and is funded by NHMRC Australia Project Grants 1083480, 1139366 and 1140297. A.M.H. receives funding via the Australian Research Council (ARC) Discovery Early Career Research Award. A.M.H. and S.M.B. receive funding via the ARC Discovery Project DP180101395.
Biographies
Joel Castro, Andrea Harrington, Luke Grundy, Annemie Deiteren and Sonia Garcia‐Caraballo are postdoctoral research fellows within the Visceral Pain Research Group, whilst Andelain Erickson and Ashlee Caldwell are PhD students enrolled via the University of Adelaide. Our research comprises pre‐clinical and translational science investigating the causes and cures of chronic abdominal and pelvic pain associated with highly prevalent gastrointestinal disorders such as irritable bowel syndrome and inflammatory bowel disease, and bladder disorders such as interstitial cystitis/painful bladder syndrome.

Stuart M. Brierley is an NHMRC R.D. Wright Fellow and Matthew Flinders Research Fellow in Gastrointestinal Neuroscience. He is Head of the Visceral Pain Research Group located at Flinders University and the South Australian Health and Medical Research Institute (SAHMRI) in Adelaide, Australia.
Edited by: Ole Petersen & Yasushi Okamura
This is an Editor's Choice article from the 1 March 2018 issue.
References
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH & Wood JN (1999). The tetrodotoxin‐resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci 2, 541–548. [DOI] [PubMed] [Google Scholar]
- Alcaino C, Farrugia G & Beyder A (2017). Mechanosensitive piezo channels in the gastrointestinal tract. Curr Top Membr 79, 219–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckh S, Noda M, Lübbert H & Numa S (1989). Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. EMBO J 8, 3611–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellono NW, Bayrer JR, Leitch DB, Castro J, Zhang C, O'Donnell TA, Brierley SM, Ingraham HA & Julius D (2017). Enterochromaffin cells are gut chemosensors that couple to sensory neural pathways. Cell 170, 185–198.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DL & Woods CG (2014). Painful and painless channelopathies. Lancet Neurol 13, 587–599. [DOI] [PubMed] [Google Scholar]
- Beyak MJ, Ramji N, Krol KM, Kawaja MD & Vanner SJ (2004). Two TTX‐resistant Na+ currents in mouse colonic dorsal root ganglia neurons and their role in colitis‐induced hyperexcitability. Am J Physiol Gastrointest Liver Physiol 287, G845–G855. [DOI] [PubMed] [Google Scholar]
- Beyder A & Farrugia G (2016). Ion channelopathies in functional GI disorders. Am J Physiol Gastrointest Liver Physiol 311, G581–G586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyder A, Gibbons SJ, Mazzone A, Strege PR, Saravanaperumal SA, Sha L, Higgins S, Eisenman ST, Bernard CE, Geurts A, Kline CF, Mohler PJ & Farrugia G (2016). Expression and function of the Scn5a‐encoded voltage‐gated sodium channel Nav1.5 in the rat jejunum. Neurogastroenterol Motil 28, 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyder A, Mazzone A, Strege PR, Tester DJ, Saito YA, Bernard CE, Enders FT, Ek WE, Schmidt PT, Dlugosz A, Lindberg G, Karling P, Ohlsson B, Gazouli M, Nardone G, Cuomo R, Usai‐Satta P, Galeazzi F, Neri M, Portincasa P, Bellini M, Barbara G, Camilleri M, Locke GR III, Talley NJ, D'Amato M, Ackerman MJ & Farrugia G (2014). Loss‐of‐function of the voltage‐gated sodium channel Nav1.5 (channelopathies) in patients with irritable bowel syndrome. Gastroenterology 146, 1659–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielefeldt K, Ozaki N & Gebhart GF (2002a). Experimental ulcers alter voltage‐sensitive sodium currents in rat gastric sensory neurons. Gastroenterology 122, 394–405. [DOI] [PubMed] [Google Scholar]
- Bielefeldt K, Ozaki N & Gebhart GF (2002b). Mild gastritis alters voltage‐sensitive sodium currents in gastric sensory neurons in rats. Gastroenterology 122, 752–761. [DOI] [PubMed] [Google Scholar]
- Black JA, Cummins TR, Yoshimura N, de Groat WC & Waxman SG (2003). Tetrodotoxin‐resistant sodium channels Nav1.8/SNS and Nav1.9/NaN in afferent neurons innervating urinary bladder in control and spinal cord injured rats. Brain Res 963, 132–138. [DOI] [PubMed] [Google Scholar]
- Black JA, Dib‐Hajj S, McNabola K, Jeste S, Rizzo MA, Kocsis JD & Waxman SG (1996). Spinal sensory neurons express multiple sodium channel alpha‐subunit mRNAs. Brain Res Mol Brain Res 43, 117–131. [DOI] [PubMed] [Google Scholar]
- Black JA, Frezel N, Dib‐Hajj SD & Waxman SG (2012). Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol Pain 8, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JA & Waxman SG (2013). Noncanonical roles of voltage‐gated sodium channels. Neuron 80, 280–291. [DOI] [PubMed] [Google Scholar]
- Bradding P, Okayama Y, Kambe N & Saito H (2003). Ion channel gene expression in human lung, skin, and cord blood‐derived mast cells. J Leukoc Biol 73, 614–620. [DOI] [PubMed] [Google Scholar]
- Branco T, Tozer A, Magnus CJ, Sugino K, Tanaka S, Lee AK, Wood JN & Sternson SM (2016). Near‐perfect synaptic integration by Nav1.7 in hypothalamic neurons regulates body weight. Cell 165, 1749–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley SM (2010). Molecular basis of mechanosensitivity. Auton Neurosci 153, 58–68. [DOI] [PubMed] [Google Scholar]
- Brierley SM (2016). Altered ion channel/receptor expression and function in extrinsic sensory neurons: the cause of and solution to chronic visceral pain? Adv Exp Med Biol 891, 75–90. [DOI] [PubMed] [Google Scholar]
- Brierley SM, Jones RCW, Gebhart GF & Blackshaw LA (2004). Splanchnic and pelvic mechanosensory afferents signal different qualities of colonic stimuli in mice. Gastroenterology 127, 166–178. [DOI] [PubMed] [Google Scholar]
- Brierley SM & Linden DR (2014). Neuroplasticity and dysfunction after gastrointestinal inflammation. Nat Rev Gastroenterol Hepatol 11, 611–627. [DOI] [PubMed] [Google Scholar]
- Campaniello MA, Harrington AM, Martin CM, Ashley Blackshaw L, Brierley SM & Hughes PA (2016). Activation of colo‐rectal high‐threshold afferent nerves by interleukin‐2 is tetrodotoxin‐sensitive and upregulated in a mouse model of chronic visceral hypersensitivity. Neurogastroenterol Motil 28, 54–63. [DOI] [PubMed] [Google Scholar]
- Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, Compton CC, DeLuca DS, Peter‐Demchok J, Gelfand ET, Guan P, Korzeniewski GE, Lockhart NC, Rabiner CA, Rao AK, Robinson KL, Roche NV, Sawyer SJ, Segre AV, Shive CE, Smith AM, Sobin LH, Undale AH, Valentino KM, Vaught J, Young TR & Moore HM; GTEx Consortium (2015). A novel approach to high‐quality postmortem tissue procurement: The GTEx Project. Biopreserv Biobank 13, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrithers LM, Hulseberg P, Sandor M & Carrithers MD (2011). The human macrophage sodium channel NaV1.5 regulates mycobacteria processing through organelle polarization and localized calcium oscillations. FEMS Immunol Med Microbiol 63, 319–327. [DOI] [PubMed] [Google Scholar]
- Carrithers MD, Dib‐Hajj S, Carrithers LM, Tokmoulina G, Pypaert M, Jonas EA & Waxman SG (2007). Expression of the voltage‐gated sodium channel NaV1.5 in the macrophage late endosome regulates endosomal acidification. J Immunol 178, 7822–7832. [DOI] [PubMed] [Google Scholar]
- Castro J, Harrington AM, Garcia‐Caraballo S, Maddern J, Grundy L, Zhang J, Page G, Miller PE, Craik DJ, Adams DJ & Brierley SM (2017). α‐Conotoxin Vc1.1 inhibits human dorsal root ganglion neuroexcitability and mouse colonic nociception via GABAB receptors. Gut 66, 1083–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro J, Harrington AM, Hughes PA, Martin CM, Ge P, Shea CM, Jin H, Jacobson S, Hannig G, Mann E, Cohen MB, MacDougall JE, Lavins BJ, Kurtz CB, Silos‐Santiago I, Johnston JM, Currie MG, Blackshaw LA & Brierley SM (2013). Linaclotide inhibits colonic nociceptors and relieves abdominal pain via guanylate cyclase‐C and extracellular cyclic guanosine 3′,5′‐monophosphate. Gastroenterology 145, 1334–1346. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2012). Voltage‐gated sodium channels at 60: structure, function and pathophysiology. J Physiol 590, 2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL & Waxman SG (2005). International Union of Pharmacology. XLVII. Nomenclature and structure‐function relationships of voltage‐gated sodium channels. Pharmacol Rev 57, 397–409. [DOI] [PubMed] [Google Scholar]
- Chang W, Berta T, Kim YH, Lee S, Lee S‐Y & Ji R‐R (2018). Expression and role of voltage‐gated sodium channels in human dorsal root ganglion neurons with special focus on Nav1.7, species differences, and regulation by paclitaxel. Neurosci Bull 34, 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JA & Davis BM (2010). The role of visceral afferents in disease In Translational Pain Research: From Mouse to Man, chap. 3, ed. Kruger L. & Light AR. CRC Press/Taylor & Francis, Boca Raton, FL, USA. [Google Scholar]
- Copel C, Clerc N, Osorio N, Delmas P & Mazet B (2013). The Nav1.9 channel regulates colonic motility in mice. Front Neurosci 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copel C, Osorio N, Crest M, Gola M, Delmas P & Clerc N (2009). Activation of neurokinin 3 receptor increases Nav1.9 current in enteric neurons. J Physiol 587, 1461–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al‐Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM & Woods CG (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang K, Bielefeldt K & Gebhart GF (2004). Gastric ulcers reduce A‐type potassium currents in rat gastric sensory ganglion neurons. Am J Physiol Gastrointest Liver Physiol 286, G573–G579. [DOI] [PubMed] [Google Scholar]
- de Araujo AD, Mobli M, Castro J, Harrington AM, Vetter I, Dekan Z, Muttenthaler M, Wan J, Lewis RJ, King GF, Brierley SM & Alewood PF (2014). Selenoether oxytocin analogues have analgesic properties in a mouse model of chronic abdominal pain. Nat Commun 5, 1–12. [DOI] [PubMed] [Google Scholar]
- Deshpande MA, Wang J, Preiksaitis HG, Laurier LG & Sims SM (2002). Characterization of a voltage‐dependent Na+ current in human esophageal smooth muscle. Am J Physiol Cell Physiol 283, C1045–C1055. [DOI] [PubMed] [Google Scholar]
- Dib‐Hajj S, Black JA, Cummins TR & Waxman SG (2002). NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci 25, 253–259. [DOI] [PubMed] [Google Scholar]
- Dib‐Hajj SD, Black JA & Waxman SG (2015). NaV1.9: a sodium channel linked to human pain. Nat Rev Neurosci 16, 511–519. [DOI] [PubMed] [Google Scholar]
- Dib‐Hajj SD, Tyrrell L, Black JA & Waxman SG (1998). NaN, a novel voltage‐gated Na channel, is expressed preferentially in peripheral sensory neurons and down‐regulated after axotomy. Proc Natl Acad Sci USA 95, 8963–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X & Gamper N (2013). Potassium channels in peripheral pain pathways: expression, function and therapeutic potential. Curr Neuropharmacol 11, 621–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duflocq A, Le Bras B, Bullier E, Couraud F & Davenne M (2008). Nav1.1 is predominantly expressed in nodes of Ranvier and axon initial segments. Mol Cell Neurosci 39, 180–192. [DOI] [PubMed] [Google Scholar]
- Enck P, Aziz Q, Barbara G, Farmer AD, Fukudo S, Mayer EA, Niesler B, Quigley EM, Rajilic‐Stojanovic M, Schemann M, Schwille‐Kiuntke J, Simren M, Zipfel S & Spiller RC (2016). Irritable bowel syndrome. Nat Rev Dis Primers 2, 16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell KE, Callister RJ & Keely S (2014). Understanding and targeting centrally mediated visceral pain in inflammatory bowel disease. Front Pharmacol 5, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, Zhu Y, La JH, Wills ZP & Gebhart GF (2015). Experimental and computational evidence for an essential role of Nav1.6 in spike initiation at stretch‐sensitive colorectal afferent endings. J Neurophysiol 113, 2618–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Pflueger M, Lin SX, Groveman BR, Su J & Yu XM (2012). Regulation of voltage‐gated sodium current by endogenous Src family kinases in cochlear spiral ganglion neurons in culture. Pflugers Arch 463, 571–584. [DOI] [PubMed] [Google Scholar]
- Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM & Rees M (2006). SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 52, 767–774. [DOI] [PubMed] [Google Scholar]
- Flegel C, Schobel N, Altmuller J, Becker C, Tannapfel A, Hatt H & Gisselmann G (2015). RNA‐Seq analysis of human trigeminal and dorsal root ganglia with a focus on chemoreceptors. PLoS One 10, e0128951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinspach M, Xu Q, Piekarz AD, Fellows R, Hagan R, Gibbs A, Liu Y, Neff RA, Freedman J, Eckert WA, Zhou M, Bonesteel R, Pennington MW, Eddinger KA, Yaksh TL, Hunter M, Swanson RV & Wickenden AD (2017). Insensitivity to pain induced by a potent selective closed‐state Nav1.7 inhibitor. Sci Rep 7, 39662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ, Griffiths D & de Groat WC (2008). The neural control of micturition. Nat Rev Neurosci 9, 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka T, Kobayashi K, Yamanaka H, Obata K, Dai Y & Noguchi K (2008). Comparative study of the distribution of the α‐subunits of voltage‐gated sodium channels in normal and axotomized rat dorsal root ganglion neurons. J Comp Neurol 510, 188–206. [DOI] [PubMed] [Google Scholar]
- Gaskin DJ & Richard P (2012). The economic costs of pain in the United States. J Pain 13, 715–724. [DOI] [PubMed] [Google Scholar]
- Gebhart GF, Bielefeldt K & Ozaki N (2002). Gastric hyperalgesia and changes in voltage gated sodium channel function in the rat. Gut 51, i15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppetti P, Veldhuis NA, Lieu T & Bunnett NW (2015). G protein‐coupled receptors: dynamic machines for signaling pain and itch. Neuron 88, 635–649. [DOI] [PubMed] [Google Scholar]
- Gingras J, Smith S, Matson DJ, Johnson D, Nye K, Couture L, Feric E, Yin RY, Moyer BD, Peterson ML, Rottman JB, Beiler RJ, Malmberg AB & McDonough SI (2014). Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS One 9, e105895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS (1999). Tetrodotoxin‐resistant Na+ currents and inflammatory hyperalgesia. Proc Natl Acad Sci USA 96, 7645–7649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Zhang L, Wrigley DL & Traub RJ (2002). Prostaglandin E2 modulates TTX‐R I Na in rat colonic sensory neurons. J Neurophysiol 88, 1512–1522. [DOI] [PubMed] [Google Scholar]
- Goldberg YP, MacFarlane J, MacDonald ML, Thompson J, Dube MP, Mattice M, Fraser R, Young C, Hossain S, Pape T, Payne B, Radomski C, Donaldson G, Ives E, Cox J, Younghusband HB, Green R, Duff A, Boltshauser E, Grinspan GA, Dimon JH, Sibley BG, Andria G, Toscano E, Kerdraon J, Bowsher D, Pimstone SN, Samuels ME, Sherrington R & Hayden MR (2007). Loss‐of‐function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet 71, 311–319. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Cano R, Tejada MA, Artacho‐Cordon A, Nieto FR, Entrena JM, Wood JN & Cendan CM (2017). Effects of tetrodotoxin in mouse models of visceral pain. Mar Drugs 15, E188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2002). Neuroanatomy of visceral nociception: vagal and splanchnic afferent. Gut 51 Suppl. 1, i2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC) – Analysis Working Group; Statistical Methods groups – Analysis Working Group; Enhancing GTEx (eGTEx) groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site – NDRI; Biospecimen Collection Source Site – RPCI; Biospecimen Core Resource – VARI; Brain Bank Repository – University of Miami Brain Endowment Bank; Leidos Biomedical – Project Management; ELSI Study; Genome Browser Data Integration &Visualization – EBI; Genome Browser Data Integration &Visualization – UCSC Genomics Institute, University of California Santa Cruz; Lead analysts:; Laboratory, Data Analysis &Coordinating Center (LDACC):; NIH program management:; Biospecimen collection:; Pathology:; eQTL manuscript working group: Battle A, Brown CD, Engelhardt BE & Montgomery SB (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen NA, Cantin L, Constant J, Haller T, Blaise G, Ong‐Lam M, du Souich P, Korz W & Lapointe B (2017). Tetrodotoxin for moderate to severe cancer‐related pain: a multicentre, randomized, double‐blind, placebo‐controlled, parallel‐design trial. Pain Res Manag 2017, 7212713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen NA, Lapointe B, Ong‐Lam M, Dubuc B, Walde D, Gagnon B, Love R, Goel R, Hawley P, Ngoc AH & du Souich P (2011). A multicentre open‐label safety and efficacy study of tetrodotoxin for cancer pain. Curr Oncol 18, e109–e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Yang Y, de Greef BT, Hoeijmakers JG, Gerrits MM, Verhamme C, Qu J, Lauria G, Merkies IS, Faber CG, Dib‐Hajj SD & Waxman SG (2015). The domain II S4‐S5 linker in Nav1.9: a missense mutation enhances activation, impairs fast inactivation, and produces human painful neuropathy. Neuromolecular Med 17, 158–169. [DOI] [PubMed] [Google Scholar]
- Harrington AM, Brierley SM, Isaacs N, Hughes PA, Castro J & Blackshaw LA (2012). Sprouting of colonic afferent central terminals and increased spinal mitogen‐activated protein kinase expression in a mouse model of chronic visceral hypersensitivity. J Comp Neurol 520, 2241–2255. [DOI] [PubMed] [Google Scholar]
- Hetz S, Acikgoez A, Moll C, Jahnke H‐G, Robitzki AA, Metzger R & Metzger M (2014). Age‐related gene expression analysis in enteric ganglia of human colon after laser microdissection. Front Aging Neurosci 6, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillsley K, Lin JH, Stanisz A, Grundy D, Aerssens J, Peeters PJ, Moechars D, Coulie B & Stead RH (2006). Dissecting the role of sodium currents in visceral sensory neurons in a model of chronic hyperexcitability using Nav1.8 and Nav1.9 null mice. J Physiol 576, 257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano K, Kuratani K, Fujiyoshi M, Tashiro N, Hayashi E & Kinoshita M (2007). Kv7.2–7.5 voltage‐gated potassium channel (KCNQ2–5) opener, retigabine, reduces capsaicin‐induced visceral pain in mice. Neurosci Lett 413, 159–162. [DOI] [PubMed] [Google Scholar]
- Hockley JRF, Boundouki G, Cibert‐Goton V, McGuire C, Yip PK, Chan C, Tranter M, Wood JN, Nassar MA, Blackshaw LA, Aziz Q, Michael GJ, Baker MD, Winchester WJ, Knowles CH & Bulmer DC (2014). Multiple roles for NaV1.9 in the activation of visceral afferents by noxious inflammatory, mechanical, and human disease‐derived stimuli. Pain 155, 1962–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockley JRF, González‐Cano R, McMurray S, Tejada‐Giraldez MA, McGuire C, Torres A, Wilbrey AL, Cibert‐Goton V, Nieto FR, Pitcher T, Knowles CH, Baeyens JM, Wood JN, Winchester WJ, Bulmer DC, Cendán CM & McMurray G (2017). Visceral and somatic pain modalities reveal NaV1.7‐independent visceral nociceptive pathways. J Physiol 595, 2661–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockley JRF, Tranter MM, McGuire C, Boundouki G, Cibert‐Goton V, Thaha MA, Blackshaw LA, Michael GJ, Baker MD, Knowles CH, Winchester WJ & Bulmer DC (2016a). P2Y receptors sensitize mouse and human colonic nociceptors. J Neurosci 36, 2364–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockley JRF, Winchester WJ & Bulmer DC (2016b). The voltage‐gated sodium channel NaV1.9 in visceral pain. Neurogastroenterol Motil 28, 316–326. [DOI] [PubMed] [Google Scholar]
- Holm AN, Rich A, Miller SM, Strege P, Ou Y, Gibbons S, Sarr MG, Szurszewski JH, Rae JL & Farrugia G (2002). Sodium current in human jejunal circular smooth muscle cells. Gastroenterology 122, 178–187. [DOI] [PubMed] [Google Scholar]
- Hu J, Song Z‐Y, Zhang H‐H, Qin X, Hu S, Jiang X & Xu G‐Y (2016). Colonic hypersensitivity and sensitization of voltage‐gated sodium channels in primary sensory neurons in rats with diabetes. J Neurogastroenterol Motil 22, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Xiao Y, Zhu L, Li L, Hu CY, Jiang X & Xu GY (2013a). Neonatal maternal deprivation sensitizes voltage‐gated sodium channel currents in colon‐specific dorsal root ganglion neurons in rats. Am J Physiol Gastrointest Liver Physiol 304, G311–G321. [DOI] [PubMed] [Google Scholar]
- Hu S, Xu W, Miao X, Gao Y, Zhu L, Zhou Y, Xiao Y & Xu GY (2013b). Sensitization of sodium channels by cystathionine β‐synthetase activation in colon sensory neurons in adult rats with neonatal maternal deprivation. Exp Neurol 248, 275–285. [DOI] [PubMed] [Google Scholar]
- Huang J, Han C, Estacion M, Vasylyev D, Hoeijmakers JGJ, Gerrits MM, Tyrrell L, Lauria G, Faber CG, Dib‐Hajj SD, Merkies IS, Waxman SG & PROPANE Study Group (2014). Gain‐of‐function mutations in sodium channel Nav1.9 in painful neuropathy. Brain 137, 1627–1642. [DOI] [PubMed] [Google Scholar]
- Huang J, Vanoye CG, Cutts A, Goldberg YP, Dib‐Hajj SD, Cohen CJ, Waxman SG & George AL Jr (2017). Sodium channel Nav1.9 mutations associated with insensitivity to pain dampen neuronal excitability. J Clin Invest 127, 2805–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PA, Brierley SM, Martin CM, Brookes SJH, Linden DR & Blackshaw LA (2009). Post‐inflammatory colonic afferent sensitisation: different subtypes, different pathways and different time courses. Gut 58, 1333–1341. [DOI] [PubMed] [Google Scholar]
- Ibeakanma C, Miranda‐Morales M, Richards M, Bautista‐Cruz F, Martin N, Hurlbut D & Vanner S (2009). Citrobacter rodentium colitis evokes post‐infectious hyperexcitability of mouse nociceptive colonic dorsal root ganglion neurons. J Physiol 587, 3505–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibeakanma C & Vanner S (2010). TNFα is a key mediator of the pronociceptive effects of mucosal supernatant from human ulcerative colitis on colonic DRG neurons. Gut 59, 612–621. [DOI] [PubMed] [Google Scholar]
- Inserra MC, Israel MR, Caldwell A, Castro J, Deuis JR, Harrington AM, Keramidas A, Garcia‐Caraballo S, Maddern J, Erickson A, Grundy L, Rychkov GY, Zimmermann K, Lewis RJ, Brierley SM & Vetter I (2017). Multiple sodium channel isoforms mediate the pathological effects of Pacific ciguatoxin‐1. Sci Rep 7, 42810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarnot M & Corbett AM (2006). Immunolocalization of NaV1.2 channel subtypes in rat and cat brain and spinal cord with high affinity antibodies. Brain Res 1107, 1–12. [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Honore P, Shieh CC, Chapman M, Joshi S, Zhang XF, Kort M, Carroll W, Marron B, Atkinson R, Thomas J, Liu D, Krambis M, Liu Y, McGaraughty S, Chu K, Roeloffs R, Zhong C, Mikusa JP, Hernandez G, Gauvin D, Wade C, Zhu C, Pai M, Scanio M, Shi L, Drizin I, Gregg R, Matulenko M, Hakeem A, Gross M, Johnson M, Marsh K, Wagoner PK, Sullivan JP, Faltynek CR & Krafte DS (2007). A‐803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci USA 104, 8520–8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating C, Beyak M, Foley S, Singh G, Marsden C, Spiller R & Grundy D (2008). Afferent hypersensitivity in a mouse model of post‐inflammatory gut dysfunction: role of altered serotonin metabolism. J Physiol 586, 4517–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DE, Macleod RJ & Vanner SJ (2009). Trinitrobenzenesulphonic acid colitis alters Nav1.8 channel expression in mouse dorsal root ganglia neurons. Neurogastroenterol Motil 21, 880–e864. [DOI] [PubMed] [Google Scholar]
- King GF & Vetter I (2014). No gain, no pain: Nav1.7 as an analgesic target. ACS Chem Neurosci 5, 749–751. [DOI] [PubMed] [Google Scholar]
- Klein CJ, Wu Y, Kilfoyle DH, Sandroni P, Davis MD, Gavrilova RH, Low PA & Dyck PJ (2013). Infrequent SCN9A mutations in congenital insensitivity to pain and erythromelalgia. J Neurol Neurosurg Psychiatry 84, 386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer N, Lacinova L, Flockerzi V & Hofmann F (1995). Structure and functional expression of a new member of the tetrodotoxin‐sensitive voltage‐activated sodium channel family from human neuroendocrine cells. EMBO J 14, 1084–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyloh M, Nicholas S, Zagorodnyuk VP, Brookes SJ & Spencer NJ (2011). Identification of the visceral pain pathway activated by noxious colorectal distension in mice. Front Neurosci 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La JH & Gebhart GF (2011). Colitis decreases mechanosensitive K2P channel expression and function in mouse colon sensory neurons. Am J Physiol Gastrointest Liver Physiol 301, G165–G174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laedermann CJ, Abriel H & Decosterd I (2015). Post‐translational modifications of voltage‐gated sodium channels in chronic pain syndromes. Front Pharmacol 6, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lago J, Rodríguez LP, Blanco L, Vieites JM & Cabado AG (2015). Tetrodotoxin, an extremely potent marine neurotoxin: distribution, toxicity, origin and therapeutical uses. Mar Drugs 13, 6384–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird JM, Souslova V, Wood JN & Cervero F (2002). Deficits in visceral pain and referred hyperalgesia in Nav1.8 (SNS/PN3)‐null mice. J Neurosci 22, 8352–8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Q & Malykhina AP (2012). Colonic inflammation up‐regulates voltage‐gated sodium channels in bladder sensory neurons via activation of peripheral transient potential vanilloid 1 receptors. Neurogastroenterol Motil 24, 575–e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipold E, Liebmann L, Korenke GC, Heinrich T, Giesselmann S, Baets J, Ebbinghaus M, Goral RO, Stodberg T, Hennings JC, Bergmann M, Altmueller J, Thiele H, Wetzel A, Nuernberg P, Timmerman V, De Jonghe P, Blum R, Schaible H‐G, Weis J, Heinemann SH, Huebner CA & Kurth I (2013). A de novo gain‐of‐function mutation in SCN11A causes loss of pain perception. Nat Genet 45, 1399–1404. [DOI] [PubMed] [Google Scholar]
- Lin YM, Fu Y, Winston J, Radhakrishnan R, Sarna SK, Huang LM & Shi XZ (2017). Pathogenesis of abdominal pain in bowel obstruction: Role of mechanical stress‐induced upregulation of nerve growth factor in gut smooth muscle cells. Pain 158, 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo JL, Qin HY, Wong CK, Tsang SY, Huang Y & Bian ZX (2011). Enhanced excitability and down‐regulated voltage‐gated potassium channels in colonic DRG neurons from neonatal maternal separation rats. J Pain 12, 600–609. [DOI] [PubMed] [Google Scholar]
- McCormack K, Santos S, Chapman ML, Krafte DS, Marron BE, West CW, Krambis MJ, Antonio BM, Zellmer SG, Printzenhoff D, Padilla KM, Lin Z, Wagoner PK, Swain NA, Stupple PA, de Groot M, Butt RP & Castle NA (2013). Voltage sensor interaction site for selective small molecule inhibitors of voltage‐gated sodium channels. Proc Natl Acad Sci USA 110, E2724–E2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingret F, Coste B, Padilla F, Clerc N, Crest M, Korogod SM & Delmas P (2008). Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J Gen Physiol 131, 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malykhina AP, Qin C, Foreman RD & Akbarali HI (2004). Colonic inflammation increases Na+ currents in bladder sensory neurons. Neuroreport 15, 2601–2605. [DOI] [PubMed] [Google Scholar]
- Malykhina AP, Wyndaele J‐J, Andersson K‐E, De Wachter S & Dmochowski RR (2012). Do the urinary bladder and large bowel interact, in sickness or in health? Neurourol Urodyn 31, 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manriquez V, Caperan DC, Guzman R, Naser M, Iglesia V & Lagos N (2015). First evidence of neosaxitoxin as a long‐acting pain blocker in bladder pain syndrome. Int Urogynecol J 26, 853–858. [DOI] [PubMed] [Google Scholar]
- Marcil J, Walczak JS, Guindon J, Ngoc AH, Lu S & Beaulieu P (2006). Antinociceptive effects of tetrodotoxin (TTX) in rodents. Br J Anaesth 96, 761–768. [DOI] [PubMed] [Google Scholar]
- Martinez V & Melgar S (2008). Lack of colonic‐inflammation‐induced acute visceral hypersensitivity to colorectal distension in Nav1.9 knockout mice. Eur J Pain 12, 934–944. [DOI] [PubMed] [Google Scholar]
- Minett MS, Eijkelkamp N & Wood JN (2014). Significant determinants of mouse pain behaviour. PLoS One 9, e104458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minett MS, Nassar MA, Clark AK, Passmore G, Dickenson AH, Wang F, Malcangio M & Wood JN (2012). Distinct Nav1.7‐dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun 3, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minett MS, Pereira V, Sikandar S, Matsuyama A, Lolignier S, Kanellopoulos AH, Mancini F, Iannetti GD, Bogdanov YD, Santana‐Varela S, Millet Q, Baskozos G, MacAllister R, Cox JJ, Zhao J & Wood JN (2015). Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nat Commun 6, 8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore BA, Stewart TM, Hill C & Vanner SJ (2002). TNBS ileitis evokes hyperexcitability and changes in ionic membrane properties of nociceptive DRG neurons. Am J Physiol Gastrointest Liver Physiol 282, G1045–G1051. [DOI] [PubMed] [Google Scholar]
- Morinville A, Fundin B, Meury L, Juréus A, Sandberg K, Krupp J, Ahmad S & O'Donnell D (2007). Distribution of the voltage‐gated sodium channel Nav1.7 in the rat: Expression in the autonomic and endocrine systems. J Comp Neurol 504, 680–689. [DOI] [PubMed] [Google Scholar]
- Nash DT & Nash SD (2008). Ranolazine for chronic stable angina. Lancet 372, 1335–1341. [DOI] [PubMed] [Google Scholar]
- Nassar MA, Levato A, Stirling LC & Wood JN (2005). Neuropathic pain develops normally in mice lacking both Nav1.7 and Nav1.8. Mol Pain 1, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar MA, Stirling LC, Forlani G, Baker MD, Matthews EA, Dickenson AH & Wood JN (2004). Nociceptor‐specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci USA 101, 12706–12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neshatian L, Strege PR, Rhee PL, Kraichely RE, Mazzone A, Bernard CE, Cima RR, Larson DW, Dozois EJ, Kline CF, Mohler PJ, Beyder A & Farrugia G (2015). Ranolazine inhibits voltage‐gated mechanosensitive sodium channels in human colon circular smooth muscle cells. Am J Physiol Gastrointest Liver Physiol 309, G506–G512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH (2009). Opportunities & Challenges in Digestive Diseases Research: Recommendations of the National Commission on Digestive Diseases. United States Department of Health & Human Services, National Institute of Diabetes and Digestive and Kidney Diseases, NIH Publication No. 08–6514, Bethesda, MD, USA.
- O'Donnell AM, Coyle D & Puri P (2016). Decreased Nav1.9 channel expression in Hirschsprung's disease. J Pediatr Surg 51, 1458–1461. [DOI] [PubMed] [Google Scholar]
- Osorio N, Korogod S & Delmas P (2014). Specialized functions of Nav1.5 and Nav1.9 channels in electrogenesis of myenteric neurons in intact mouse ganglia. J Neurosci 34, 5233–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen JD, Herzig V, Gilchrist J, Emrick JJ, Zhang C, Wang X, Castro J, Garcia‐Caraballo S, Grundy L, Rychkov GY, Weyer AD, Dekan Z, Undheim EAB, Alewood P, Stucky CL, Brierley SM, Basbaum AI, Bosmans F, King GF & Julius D (2016). Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 534, 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostman JA, Nassar MA, Wood JN & Baker MD (2008). GTP up‐regulated persistent Na+ current and enhanced nociceptor excitability require NaV1.9. J Physiol 586, 1077–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Y, Gibbons SJ, Miller SM, Strege PR, Rich A, Distad MA, Ackerman MJ, Rae JL, Szurszewski JH & Farrugia G (2002). SCN5A is expressed in human jejunal circular smooth muscle cells. Neurogastroenterol Motil 14, 477–486. [DOI] [PubMed] [Google Scholar]
- Padilla F, Couble M‐L, Coste B, Maingret F, Clerc N, Crest M, Ritter AM, Magloire H & Delmas P (2007). Expression and localization of the Nav1.9 sodium channel in enteric neurons and in trigeminal sensory endings: Implication for intestinal reflex function and orofacial pain. Mol Cell Neurosci 35, 138–152. [DOI] [PubMed] [Google Scholar]
- Pennington MW, Czerwinski A & Norton RS (2017). Peptide therapeutics from venom: Current status and potential. Bioorg Med Chem; https://doi.org/10.1016/j.bmc.2017.1009.1029. [DOI] [PubMed] [Google Scholar]
- Qian AH, Liu XQ, Yao WY, Wang HY, Sun J, Zhou L & Yuan YZ (2009). Voltage‐gated potassium channels in IB4‐positive colonic sensory neurons mediate visceral hypersensitivity in the rat. Am J Gastroenterol 104, 2014–2027. [DOI] [PubMed] [Google Scholar]
- Qin N, D'Andrea MR, Lubin ML, Shafaee N, Codd EE & Correa AM (2003). Molecular cloning and functional expression of the human sodium channel β1B subunit, a novel splicing variant of the β1 subunit. Eur J Biochem 270, 4762–4770. [DOI] [PubMed] [Google Scholar]
- Qu R, Tao J, Wang Y, Zhou Y, Wu G, Xiao Y, Hu C‐Y, Jiang X & Xu G‐Y (2013). Neonatal colonic inflammation sensitizes voltage‐gated Na+ channels via upregulation of cystathionine β‐synthetase expression in rat primary sensory neurons. Am J Physiol Gastrointest Liver Physiol 304, G763–G772. [DOI] [PubMed] [Google Scholar]
- Reeder JE, Byler TK, Foster DC, Landas SK, Okafor H, Stearns G, Wood RW, Zhang Y & Mayer RD (2013). Polymorphism in the SCN9A voltage‐gated sodium channel gene associated with interstitial cystitis/bladder pain syndrome. Urology 81, 210e1–210e4. [DOI] [PubMed] [Google Scholar]
- Renganathan M, Cummins TR & Waxman SG (2001). Contribution of Nav1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol 86, 629–640. [DOI] [PubMed] [Google Scholar]
- Ritter AM, Martin WJ & Thorneloe KS (2009). The voltage‐gated sodium channel Nav1.9 is required for inflammation‐based urinary bladder dysfunction. Neurosci Lett 452, 28–32. [DOI] [PubMed] [Google Scholar]
- Roselli F, Livrea P & Jirillo E (2006). Voltage‐gated sodium channel blockers as immunomodulators. Recent Pat CNS Drug Discov 1, 83–91. [DOI] [PubMed] [Google Scholar]
- Rugiero F, Mistry M, Sage D, Black JA, Waxman SG, Crest M, Clerc N, Delmas P & Gola M (2003). Selective expression of a persistent tetrodotoxin‐resistant Na+ current and NaV1.9 subunit in myenteric sensory neurons. J Neurosci 23, 2715–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage D, Salin P, Alcaraz G, Castets F, Giraud P, Crest M, Mazet B & Clerc N (2007). Nav1.7 and Nav1.3 are the only tetrodotoxin‐sensitive sodium channels expressed by the adult guinea pig enteric nervous system. J Comp Neurol 504, 363–378. [DOI] [PubMed] [Google Scholar]
- Saito YA, Strege PR, Tester DJ, Locke GR, Talley NJ, Bernard CE, Rae JL, Makielski JC, Ackerman MJ & Farrugia G (2009). Sodium channel mutation in irritable bowel syndrome: evidence for an ion channelopathy. Am J Physiol Gastrointest Liver Physiol 296, G211–G218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaga M, Storr M, Martemyanov KA & Fichna J (2016). RGS proteins as targets in the treatment of intestinal inflammation and visceral pain: New insights and future perspectives. Bioessays 38, 344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savio‐Galimberti E, Gollob MH & Darbar D (2012). Voltage‐gated sodium channels: biophysics, pharmacology, and related channelopathies. Front Pharmacol 3, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep LJ, Schmierer DM & Fountain JS (2006). Veratrum poisoning. Toxicol Rev 25, 73–78. [DOI] [PubMed] [Google Scholar]
- Sikandar S & Dickenson AH (2012). Visceral pain – the Ins and Outs, the Ups and Downs. Curr Opin Support Palliat Care 6, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart I, Lewis RJ, Eaglesham GK, Graham GC, Poole S & Craig SB (2010). Emerging tropical diseases in Australia. Part 2. Ciguatera fish poisoning. Ann Trop Med Parasitol 104, 557–571. [DOI] [PubMed] [Google Scholar]
- Stewart T, Beyak MJ & Vanner S (2003). Ileitis modulates potassium and sodium currents in guinea pig dorsal root ganglia sensory neurons. J Physiol 552, 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strege PR, Knutson K, Eggers SJ, Li JH, Wang F, Linden D, Szurszewski JH, Milescu L, Leiter AB, Farrugia G & Beyder A (2017a). Sodium channel NaV1.3 is important for enterochromaffin cell excitability and serotonin release. Sci Rep 7, 15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strege PR, Knutson KR, Eggers SJ, Wang F, Li J, Leiter A, Farrugia G & Beyder A (2017b). SCN3A‐encoded voltage‐gated sodium channel NaV1.3 is specifically expressed in human and mouse gastrointestinal enterochromaffin cells and is important for enterochromaffin cell excitability. FASEB J 31, 1007–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strege PR, Mazzone A, Bernard CE, Neshatian L, Gibbons SJ, Saito YA, Tester DJ, Calvert ML, Mayer EA, Chang L, Ackerman MJ, Beyder A & Farrugia G (2017c). Irritable bowel syndrome (IBS) patients have SCN5A channelopathies that lead to decreased NaV1.5 current and mechanosensitivity. Am J Physiol Gastrointest Liver Physiol; https://doi.org/10.1152/ajpgi.00016.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strege PR, Mazzone A, Kraichely RE, Sha L, Holm AN, Ou Y, Lim I, Gibbons SJ, Sarr MG & Farrugia G (2007). Species dependent expression of intestinal smooth muscle mechanosensitive sodium channels. Neurogastroenterol Motil 19, 135–143. [DOI] [PubMed] [Google Scholar]
- Strege PR, Ou YJ, Sha L, Rich A, Gibbons SJ, Szurszewski JH, Sarr MG & Farrugia G (2003). Sodium current in human intestinal interstitial cells of Cajal. Am J Physiol Gastrointest Liver Physiol 285, G1111–G1121. [DOI] [PubMed] [Google Scholar]
- Tate S, Benn S, Hick C, Trezise D, John V, Mannion RJ, Costigan M, Plumpton C, Grose D, Gladwell Z, Kendall G, Dale K, Bountra C & Woolf CJ (1998). Two sodium channels contribute to the TTX‐R sodium current in primary sensory neurons. Nat Neurosci 1, 653–655. [DOI] [PubMed] [Google Scholar]
- Trimmer JS, Cooperman SS, Agnew WS & Mandel G (1990). Regulation of muscle sodium channel transcripts during development and in response to denervation. Dev Biol 142, 360–367. [DOI] [PubMed] [Google Scholar]
- Tseng TT, McMahon AM, Johnson VT, Mangubat EZ, Zahm RJ, Pacold ME & Jakobsson E (2007). Sodium channel auxiliary subunits. J Mol Microbiol Biotechnol 12, 249–262. [DOI] [PubMed] [Google Scholar]
- Tzoumaka E, Tischler AC, Sangameswaran L, Eglen RM, Hunter JC & Novakovic SD (2000). Differential distribution of the tetrodotoxin‐sensitive rPN4/NaCh6/Scn8a sodium channel in the nervous system. J Neurosci Res 60, 37–44. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J & Ponten F (2015). Proteomics. Tissue‐based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Vanoye CG, Kunic JD, Ehring GR & George AL Jr (2013). Mechanism of sodium channel Nav1.9 potentiation by G‐protein signaling. J Gen Physiol 141, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verne GN, Sen A & Price DD (2005). Intrarectal lidocaine is an effective treatment for abdominal pain associated with diarrhea‐predominant irritable bowel syndrome. J Pain 6, 493–496. [DOI] [PubMed] [Google Scholar]
- Wang W, Gu J, Li Y‐Q & Tao Y‐X (2011). Are voltage‐gated sodium channels on the dorsal root ganglion involved in the development of neuropathic pain? Mol Pain 7, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Kocsis JD & Black JA (1994). Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J Neurophysiol 72, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Merkies IS, Gerrits MM, Dib‐Hajj SD, Lauria G, Cox JJ, Wood JN, Woods CG, Drenth JP & Faber CG (2014). Sodium channel genes in pain‐related disorders: phenotype‐genotype associations and recommendations for clinical use. Lancet Neurol 13, 1152–1160. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Merrick DK & Catterall WA (1989). Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron 3, 695–704. [DOI] [PubMed] [Google Scholar]
- Whitaker W, Faull R, Waldvogel H, Plumpton C, Burbidge S, Emson P & Clare J (1999). Localization of the type VI voltage‐gated sodium channel protein in human CNS. Neuroreport 10, 3703–3709. [DOI] [PubMed] [Google Scholar]
- Woods CG, Babiker MOE, Horrocks I, Tolmie J & Kurth I (2015). The phenotype of congenital insensitivity to pain due to the NaV1.9 variant p.L811P. Eur J Hum Genet 23, 561–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X & Shen Y (2004). Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet 41, 171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekkirala AS, Roberson DP, Bean BP & Woolf CJ (2017). Breaking barriers to novel analgesic drug development. Nat Rev Drug Discov 16, 545–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiangou Y, Facer P, Chessell IP, Bountra C, Chan C, Fertleman C, Smith V & Anand P (2007). Voltage‐gated ion channel Nav1.7 innervation in patients with idiopathic rectal hypersensitivity and paroxysmal extreme pain disorder (familial rectal pain). Neurosci Lett 427, 77–82. [DOI] [PubMed] [Google Scholar]
- Yoshimura N & deGroat WC (1997). Plasticity of Na+ channels in afferent neurones innervating rat urinary bladder following spinal cord injury. J Physiol 503, 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura N, Seki S, Novakovic SD, Tzoumaka E, Erickson VL, Erickson KA, Chancellor MB & de Groat WC (2001). The involvement of the tetrodotoxin‐resistant sodium channel Nav1.8 (PN3/SNS) in a rat model of visceral pain. J Neurosci 21, 8690–8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang Y, He XH, Xin WJ, Pang RP, Wei XH, Zhou LJ, Li YY & Liu XG (2010). Inhibition of NF‐kappaB prevents mechanical allodynia induced by spinal ventral root transection and suppresses the re‐expression of Nav1.3 in DRG neurons in vivo and in vitro . Brain Res 1363, 151–158. [DOI] [PubMed] [Google Scholar]
- Zhang XY, Wen J, Yang W, Wang C, Gao L, Zheng LH, Wang T, Ran K, Li Y, Li X, Xu M, Luo J, Feng S, Ma X, Ma H, Chai Z, Zhou Z, Yao J, Zhang X & Liu JY (2013). Gain‐of‐function mutations in SCN11A cause familial episodic pain. Am J Hum Genet 93, 957–966. [DOI] [PMC free article] [PubMed] [Google Scholar]