

Abstract
Loss-of-function mutations in progranulin, a lysosomal glycoprotein, cause neurodegenerative disease. Progranulin haploinsufficiency causes frontotemporal dementia (FTD) and complete progranulin deficiency causes CLN11 neuronal ceroid lipofuscinosis (NCL). Progranulin replacement is a rational therapeutic strategy for these disorders, but there are critical unresolved mechanistic questions about a progranulin gene therapy approach, including its potential to reverse existing pathology. Here, we address these issues using an AAV vector (AAV-Grn) to deliver progranulin in Grn−/− mice (both male and female), which model aspects of NCL and FTD pathology, developing lysosomal dysfunction, lipofuscinosis, and microgliosis. We first tested whether AAV-Grn could improve preexisting pathology. Even with treatment after onset of pathology, AAV-Grn reduced lipofuscinosis in several brain regions of Grn−/− mice. AAV-Grn also reduced microgliosis in brain regions distant from the injection site. AAV-expressed progranulin was only detected in neurons, not in microglia, indicating that the microglial activation in progranulin deficiency can be improved by targeting neurons and thus may be driven at least in part by neuronal dysfunction. Even areas with sparse transduction and almost undetectable progranulin showed improvement, indicating that low-level replacement may be sufficiently effective. The beneficial effects of AAV-Grn did not require progranulin binding to sortilin. Finally, we tested whether AAV-Grn improved lysosomal function. AAV-derived progranulin was delivered to the lysosome, ameliorated the accumulation of LAMP-1 in Grn−/− mice, and corrected abnormal cathepsin D activity. These data shed light on progranulin biology and support progranulin-boosting therapies for NCL and FTD due to GRN mutations.
SIGNIFICANCE STATEMENT Heterozygous loss-of-function progranulin (GRN) mutations cause frontotemporal dementia (FTD) and homozygous mutations cause neuronal ceroid lipofuscinosis (NCL). Here, we address several mechanistic questions about the potential of progranulin gene therapy for these disorders. GRN mutation carriers with NCL or FTD exhibit lipofuscinosis and Grn−/− mouse models develop a similar pathology. AAV-mediated progranulin delivery reduced lipofuscinosis in Grn−/− mice even after the onset of pathology. AAV delivered progranulin only to neurons, not microglia, but improved microgliosis in several brain regions, indicating cross talk between neuronal and microglial pathology. Its beneficial effects were sortilin independent. AAV-derived progranulin was delivered to lysosomes and corrected lysosomal abnormalities. These data provide in vivo support for the efficacy of progranulin-boosting therapies for FTD and NCL.
Keywords: cathepsin D, frontotemporal dementia, gene therapy, lysosome, neuronal ceroid lipofuscinosis, progranulin
Introduction
Loss-of-function mutations in progranulin (GRN) cause neurodegenerative disease with a gene-dose effect. GRN mutations are among the leading causes of dominantly inherited frontotemporal dementia (FTD) (Baker et al., 2006; Cruts et al., 2006; Gass et al., 2006). These GRN mutations typically cause progranulin haploinsufficiency, with plasma progranulin levels reduced by >50% relative to controls (Finch et al., 2009). Individuals with mutations in both GRN alleles, resulting in nearly complete progranulin deficiency, develop the lysosomal storage disorder neuronal ceroid lipofuscinosis (NCL) (Smith et al., 2012; Canafoglia et al., 2014; Almeida et al., 2016). NCL due to GRN mutations has been termed CLN11-NCL and is characterized by seizures and retinal degeneration with onset in the early twenties (Smith et al., 2012; Canafoglia et al., 2014; Almeida et al., 2016). Despite very different clinical presentations, FTD patients with GRN mutations exhibit some pathological similarities to CLN11-NCL because brains of patients with both diseases exhibit gliosis, increased levels of lysosomal proteins, and lipofuscinosis (Götzl et al., 2014; Ward et al., 2017).
Progranulin is a widely expressed, secreted glycoprotein that performs a variety of functions (Bateman and Bennett, 1998; Eriksen and Mackenzie, 2008; Cenik et al., 2012; Nguyen et al., 2013). Progranulin acts as a trophic factor for many cell types, including neurons (Van Damme et al., 2008; Ryan et al., 2009; Gass et al., 2012; Beel et al., 2017). It also modulates inflammation and facilitates wound healing (Zhu et al., 2002; Yin et al., 2010). The development of NCL in patients with complete progranulin deficiency shows that progranulin is critical for proper lysosomal function (Smith et al., 2012). Recent data have shown that progranulin is critical for proper trafficking and function of lysosomal enzymes such as β-glucocerebrosidase and cathepsin D (CatD), providing a potential mechanism by which progranulin promotes lysosomal function (Jian et al., 2016; Beel et al., 2017; Valdez et al., 2017; Zhou et al., 2017b). Progranulin knock-out mice have been used to study the effects of progranulin insufficiency and model the gene-dose effect seen in humans. Grn−/− mice, but not Grn+/− mice, develop NCL-like pathology, including accumulation of lipofuscin and lysosomal proteins in the brain, astrogliosis, and microgliosis (Ahmed et al., 2010; Smith et al., 2012; Wils et al., 2012; Filiano et al., 2013; Tanaka et al., 2014).
Because most, if not all, disease-associated GRN mutations appear to be loss-of-function mutations, boosting progranulin levels, particularly in the brain, is a rational approach to preventing or treating FTD and NCL in GRN mutation carriers. Several progranulin-boosting approaches have been developed to increase progranulin levels from the intact GRN allele in heterozygous mutation carriers (Hu et al., 2010; Capell et al., 2011; Cenik et al., 2011; Lee et al., 2014). However, these approaches are not suitable for CLN11-NCL patients, who have mutations in both GRN alleles (Smith et al., 2012; Canafoglia et al., 2014; Almeida et al., 2016). For these CLN11-NCL patients, a gene therapy approach would be required to express progranulin. Viral vectors are an excellent tool for such an approach and have been tested in models of CLN1-NCL, CLN2-NCL, and CLN5-NCL (Griffey et al., 2006; Passini et al., 2006; Cabrera-Salazar et al., 2007; Sondhi et al., 2007; Macauley et al., 2012; Hughes et al., 2014; Katz et al., 2015). In addition, we have recently observed that restoration of progranulin with an adeno-associated virus (AAV) vector to Grn+/− mice corrects social behavior deficits (Arrant et al., 2017).
The goal of this study was to determine whether AAV-mediated expression of progranulin could improve NCL-like pathology in Grn−/− mice when administered after the onset of pathology and to elucidate mechanisms by which progranulin gene therapy might work. We infused an AAV vector expressing mouse progranulin (AAV-Grn) or an AAV-GFP control virus into the medial prefrontal cortex (mPFC) of wild-type or Grn−/− mice after the onset of pathology and collected brain tissue for analysis 8–10 weeks later.
Materials and Methods
Animals.
The line of progranulin-deficient mice used for this study was generated and crossed onto a C57BL/6J background as described previously (Martens et al., 2012; Filiano et al., 2013). The mice used for this study were obtained by breeding Grn+/− mice to produce wild-type, Grn+/−, and Grn−/− mice. The resulting wild-type and Grn−/− littermates were used for all experiments. Male and female mice were included in the study. The mice were bred and housed in a barrier facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. Mice were maintained on a 12 h:12 h light/dark cycle with lights on at 6:00 A.M. and were given ad libitum access to food (NIH-31 diet #7917; Envigo) and water in all phases of the study. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Antibodies.
The following antibodies were used for immunostaining and Western blot: CD68 (1:500 rat monoclonal, #MCA1957; Bio-Rad), Iba1 (1:500 rabbit polyclonal, #019-19741; Wako), MHCII (1:100 rat monoclonal, #14-5321-81; Thermo Fisher Scientific), GFAP (1:5000 rabbit polyclonal, #Z0334; Dako), NeuN (1:1000 mouse monoclonal, #MAB377; Millipore/Sigma-Aldrich), progranulin (1:500 for Western blot and chromogenic immunostaining, 1:200 for fluorescent immunostaining, sheep polyclonal, #AF2557; R&D Systems), GFP (1: 1000 rabbit monoclonal, #2956; Cell Signaling Technology), CatD (1:500 goat polyclonal, #sc-6486; Santa Cruz Biotechnology), LAMP-1 (1:250 rat monoclonal, #1D4B; Developmental Studies Hybridoma Bank), sortilin (1:1000 rabbit polyclonal, #ab16640; Abcam), α-tubulin (1:1000 mouse monoclonal, #T5168; Sigma-Aldrich), and SCMAS (1:300 rabbit polyclonal, provided by Dr. Yasuo Uchiyama, Juntendo University) (Koike et al., 2000).
AAV constructs and vectors.
An AAV2/1 progranulin vector (AAV-Grn, rAAV2-CBA-mGrn-Myc-WPRE-rBG) and GFP-expressing control vector (AAV-GFP, #AV-1-PV1963, AAV1-CB7-CI-eGFP-WPRE-rBG) were produced at the University of Pennsylvania Vector Core as described previously (Arrant et al., 2017). An N-terminal-tagged mouse progranulin AAV construct was also generated to serve as a positive control for sortilin immunoprecipitation. This N-terminal-tagged progranulin AAV construct was generated using a synthetic construct containing the RNA-coding sequence of mouse progranulin with an HA tag inserted after the signal peptide (GenScript). The N-terminal HA-tagged mouse progranulin sequence was into the same CIGW AAV2 vector used for the C-terminal myc-tagged progranulin vector to generate a similar N-terminal tagged mouse progranulin AAV construct (rAAV2-CBA-HA-mGrn-WPRE-rBG) (St Martin et al., 2007; Arrant et al., 2017).
AAV injection.
AAV-Grn or AAV-GFP were bilaterally infused into the mPFC of 10- to 12-month-old wild-type and Grn−/− mice using stereotaxic surgery (coordinates +1.9 mm anterior and ± 0.3 mm lateral from bregma, −2.2 mm from the surface of the skull) under isoflurane anesthesia as described previously (Arrant et al., 2017). Then, 1 μl of AAV (7.36 × 1011 genomes/ml) was infused into each hemisphere with a syringe pump (Harvard Apparatus) at a flow rate of 0.5 μl/min. After allowing 5 min for diffusion into the tissue, the injection needle (Hamilton) was withdrawn, the skull was sealed with bone wax, and the wound was closed with surgical staples.
Brain and plasma collection.
Mice were killed for collection of tissue samples 8–10 weeks after AAV injection. The mice were anesthetized with pentobarbital (100 mg/kg, Fatal Plus; Vortech Pharmaceuticals) and blood was collected by cardiac puncture in syringes containing EDTA (250 mm) to prevent clotting. The blood was kept on ice and later centrifuged at 5000 × g for 10 min at 4°C to separate plasma. The mice were then transcardially perfused with 0.9% saline. Brains were removed and bisected into hemibrains, one of which was immediately frozen on dry ice for biochemical analysis and the other postfixed for 48 h in 4% paraformaldehyde for histological analysis.
Immunostaining.
Fixed hemibrains were cryoprotected in 30% sucrose and cut into 30 μm sections on a sliding microtome (Leica Biosystems). The sections were then immunostained as described previously (Palop et al., 2011). For analysis of pathology and a qualitative assessment of progranulin immunoreactivity, the sections were incubated overnight in primary antibody and the following day were incubated with a species-matched biotinylated secondary antibody (Vector Laboratories), followed by avidin-biotin complex (Vectastain Elite; Vector Laboratories). Immunostaining was visualized with diaminobenzidine (MP Biomedicals).
For immunofluorescence, brain sections were sequentially immunostained for progranulin followed by an Alexa Fluor-488-conjugated anti-sheep antibody (Thermo Fisher Scientific), and then markers for neurons (NeuN) or microglia (Iba1) followed by species-matched Alexa Fluor-647-conjugated antibodies (Thermo Fisher Scientific). Subcellular localization of progranulin within neurons was assessed by sequential immunostaining for progranulin, NeuN, and LAMP-1, followed by species-matched Alexa Fluor-488, Alexa Fluor-647, and Alexa Fluor-594 antibodies (Thermo Fisher Scientific).
Microscopy and image analysis.
For pathology analysis, immunostained sections were imaged at 20× with a light microscope (Nikon) and CCD camera (Nikon). Low-magnification, high-resolution images of progranulin immunostaining were obtained with a slide scanner (PathScan Enabler IV; Meyer Instruments). Lipofuscinosis was measured by mounting unstained tissue sections on slides and coverslipping with a mounting medium containing DAPI (Vectashield; Vector Laboratories). Autofluorescent lipofuscin granules were imaged at 20× with an epifluorescent microscope (Nikon) in the red channel to avoid fluorescence from GFP and recorded with a CCD camera (Andor Technologies). All images were analyzed with ImageJ. The density of immunostaining or autofluorescent lipofuscin was determined by thresholding and measuring the area occupied by immunoreactive pixels. Iba1 soma size (see Fig. 4c,d) was measured by applying a uniform threshold to exclude background and cellular processes and measuring the average size of Iba1+ cell bodies with ImageJ's analyze particles function. Double- and triple-labeled immunofluorescent sections were imaged at 60× with a TCS-SP5 laser scanning confocal microscope (Leica Microsystems). Z-stacks of 0.5 μm were collected through the entire section.
Figure 4.
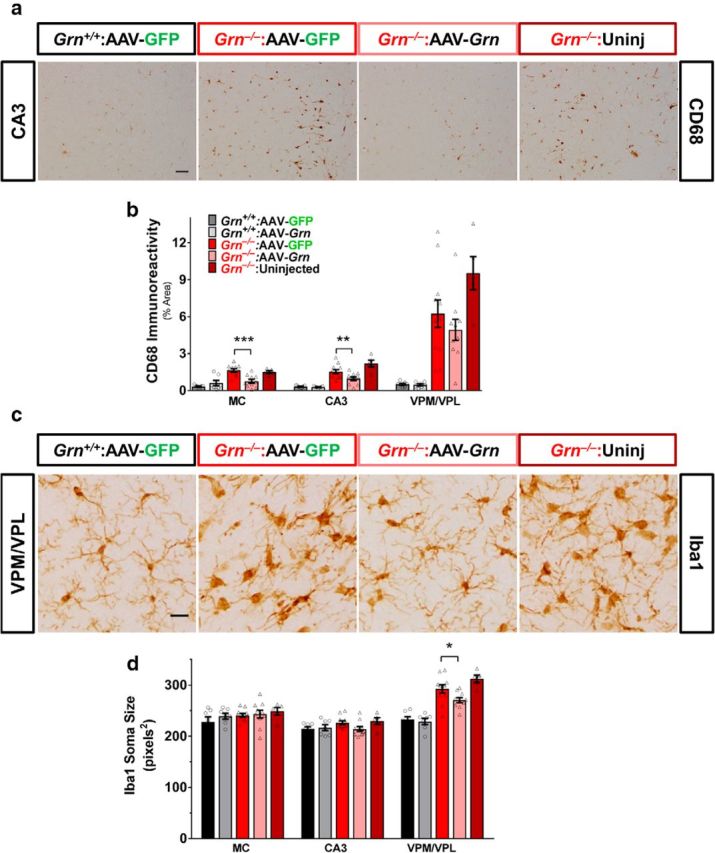
AAV-Grn improves microgliosis in Grn−/− mice. Microgliosis was assessed by CD68 immunoreactivity and the morphology of Iba1+ cells. a, b, AAV-Grn reduced CD68 immunoreactivity in the motor cortex (MC; p = 0.0007, ANOVA, genotype × virus, F(1,31) = 14.02) and CA3 (p = 0.0392, ANOVA, effect of virus, F(1,31) = 4.634). c, d, Grn−/− mice exhibited an increase in Iba1+ soma size only in the VPM/VPL thalamus (p < 0.0001, ANOVA effect of genotype, F(1,31) = 55.58). AAV-Grn significantly improved this phenotype because Iba1+ soma size was significantly reduced in AAV-Grn-treated Grn−/− mice compared with their AAV-GFP-treated controls (n = 7–11 mice per AAV treatment group and 5 uninjected Grn−/− mice; **p < 0.01, ***p < 0.001, Sidak's post hoc test). Scale bars: a, 50 μm; b, 20 μm.
Progranulin ELISA.
Brain progranulin levels were measured with a progranulin ELISA kit (Adipogen) using the manufacturer's protocol, as described previously (Arrant et al., 2015).
CatD activity assay.
CatD activity was determined by incubating brain tissue lysates with a fluorogenic CatD/E substrate (Yasuda et al., 1999). Brain tissue was homogenized in lysis buffer without protease inhibitors (50 mm Tris, pH 7.5, 150 mm NaCl, 5 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate) and centrifuged at 5000 × g for 10 min. The protein concentration of the supernatant was determined by Bradford assay (Coomassie Plus; Thermo Fisher Scientific) and samples were diluted with 50 mm sodium acetate buffer, pH 4.0, such that 5 μg of protein was loaded per well in a 96-well plate, with 40 μm fluorogenic CatD/E substrate (7-methoxycoumarin-4-yl)acetyl-GKPILF∼FRLK(2,4-dinitrophenyl)-d-R-NH2 (Millipore/Sigma-Aldrich). Immediately after the addition of substrate, samples were placed in a fluorescent plate reader (Synergy 2; Biotek Instruments) and maintained at 37°C. Fluorescence was measured every 2 min for 40 min with a 340/30 nm excitation filter and a 400/30 emission filter. Specific CatD activity was determined by subtracting fluorescence observed in the presence of the CatD inhibitor pepstatin A (15 μm; Fisher Scientific) from fluorescence observed without pepstatin A. Samples were run in duplicate under both conditions.
Western blot.
Brain lysates were run on 4–12% Bis-Tris gels (Thermo Fisher Scientific) and transferred to Immobilon-FL PVDF membranes (Millipore/Sigma-Aldrich). The membranes were blocked in 50% Odyssey blocking buffer (LI-COR Biotechnology) before overnight incubation with primary antibody at 4°C. The blots were then probed with IR-Dye-conjugated species-matched secondary antibodies (1:20 000; LI-COR Biotechnology) and scanned on an Odyssey scanner (LI-COR Biotechnology). Blots were quantitated with ImageStudio Lite software (LI-COR Biotechnology).
qRT-PCR.
Grn RNA levels were measured using Taqman qRT-PCR assay Mm01245914_g1 (Thermo Fisher Scientific) as described previously (Arrant et al., 2015). Grn was normalized to Actb (Mm00607939_s1; Thermo Fisher Scientific). qRT-PCR was performed on a Lightcycler 480 thermal cycler (Roche Diagnostics).
Assay for circulating anti-mouse progranulin and anti-GFP antibodies.
To detect circulating anti-mouse progranulin or anti-GFP antibodies in plasma from AAV-treated mice, 500 ng of recombinant mouse progranulin (#22557-PG; R&D Systems) or recombinant GFP (from A. victoria, #ab84191; Abcam) per lane was run on a 4–12% Bis-Tris gel (Thermo Fisher Scientific) and transferred to Immobilon-FL PVDF membranes (Millipore/Sigma-Aldrich). The membranes were blocked in 5% milk in Tris-buffered saline with 0.05% Tween 20 and incubated overnight in plasma from AAV-treated mice diluted 1:50 in 5% milk. The following day, the membranes were incubated in an IR-Dye-conjugated anti-mouse antibody (1:20 000; LI-COR Biotechnology). With each round of testing, one membrane was incubated with a sheep anti-mouse progranulin antibody (#AF2557; R&D Systems) or rabbit anti-GFP antibody (#2956; Cell Signaling Technology) as a positive control. The progranulin antibody was detected by incubation with a biotinylated anti-sheep antibody (Vector Laboratories), followed by IR-Dye 800-conjugated streptavidin (LI-COR Biotechnology), and the GFP antibody was detected with an IR-Dye 680LT-conjugated anti-rabbit secondary antibody (LI-COR Biotechnology). All blots were scanned on an Odyssey scanner (LI-COR Biotechnology). For comparison of anti-progranulin versus anti-GFP antibody titers, the same general protocol was used with 500 ng of either recombinant progranulin or GFP loaded per lane for SDS-PAGE. Plasma from AAV-Grn-treated Grn−/− mice was run in parallel with plasma from AAV-GFP-treated wild-type or Grn−/− mice at dilutions of 1:50, 1:100, 1:500, 1:1000, 1:2500, and 1:5000 in 5% milk. After detection with an IR-Dye-800-conjugated anti-mouse antibody (LI-COR Biotechnology), the blots were scanned at the same intensity setting on an Odyssey scanner (LI-COR Biotechnology).
Y-maze.
Y-maze testing was conducted as described previously (Li et al., 2014) using video-tracking software (Cleversys). Spontaneous alternations were tracked over a 5 min test period as a measure of PFC-dependent memory. Spontaneous alternations were defined as successive entries into all three arms of the maze without reentry into a previous arm.
Conditioned fear extinction.
Conditioned fear training, in which a 75 dB white noise cue was associated with a 0.5 mA foot shock, was conducted as described previously (Filiano et al., 2013). Conditioned fear extinction was performed using a previously described method (Arrant et al., 2017) adapted from (Izquierdo et al., 2006). For extinction, mice were placed in an altered context from training and presented with 40 30 s bursts of the white noise cue with 5 s between each cue. The extinction phase was divided into trial blocks of four presentations each for analysis of freezing behavior.
Cell culture and transfection.
HEK-293 cells (#CRL-1573; ATCC) were cultured in DMEM (Corning Life Sciences) with 10% FBS (Atlanta Biologicals) and 1% penicillin/streptomycin (Thermo Fisher Scientific). The cells were maintained at 37°C with 5% CO2. For immunoprecipitation experiments, HEK cells were transiently cotransfected with a plasmid expressing human sortilin (#SC118300; OriGene) and either the C-terminal myc-tagged or N-terminal HA-tagged progranulin CIGW constructs described above using Fugene HD transfection reagent (Promega). The cells were harvested 48 h after transfection for progranulin/sortilin coimmunoprecipitation.
Immunoprecipitation.
Transfected HEK-293 cells were lysed in Triton X-100 buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 5 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate) with Halt protease inhibitor mixture (Thermo Fisher Scientific) and centrifuged at 5000 × g for 10 min at 4°C. After determination of protein concentration by Bradford assay (Fisher Scientific), 150 μg of protein from each lysate was precleared with 20 μl of Protein G Dynabeads (Thermo Fisher Scientific) for 20 min at 4°C. The lysates were then incubated overnight with 2.5 μg of anti-sortilin antibody (rabbit polyclonal, #ab16640; Abcam) at 4°C. The following day, the lysates were incubated with 25 μl of Protein G Dynabeads for 4 h at 4°C. The beads were then pulled down and washed four times with Triton X-100 buffer. After the final wash, protein was eluted from the beads by adding LDS sample buffer (Thermo Fisher Scientific) and sample reducing agent (Thermo Fisher Scientific) at 1× in Triton X-100 buffer and heating for 10 min at 70°C. The beads were then pulled down a final time and the supernatant was run on a 4–12% Bis-Tris gel for Western blot.
Experimental design and statistical analysis.
This study was conducted on two independent cohorts of mice and included approximately equal numbers of male and female mice. A small group of uninjected Grn−/− mice (n = 5) was included in this study as a benchmark control to enable identification of any unexpected effects of the control AAV-GFP virus on pathology in Grn−/− mice. Except for the behavioral comparison in Figure 5, e and f, this group of uninjected mice was not included in statistical analyses due to its small size and the lack of a corresponding uninjected wild-type group. The uninjected Grn−/− mice were used in the behavioral comparison as a control for potential adverse functional effects of AAV administration. Uninjected wild-type mice were not included in the study because preliminary studies revealed no adverse effects of AAV-GFP in wild-type mice (Arrant et al., 2017 and additional preliminary data not shown).
Figure 5.
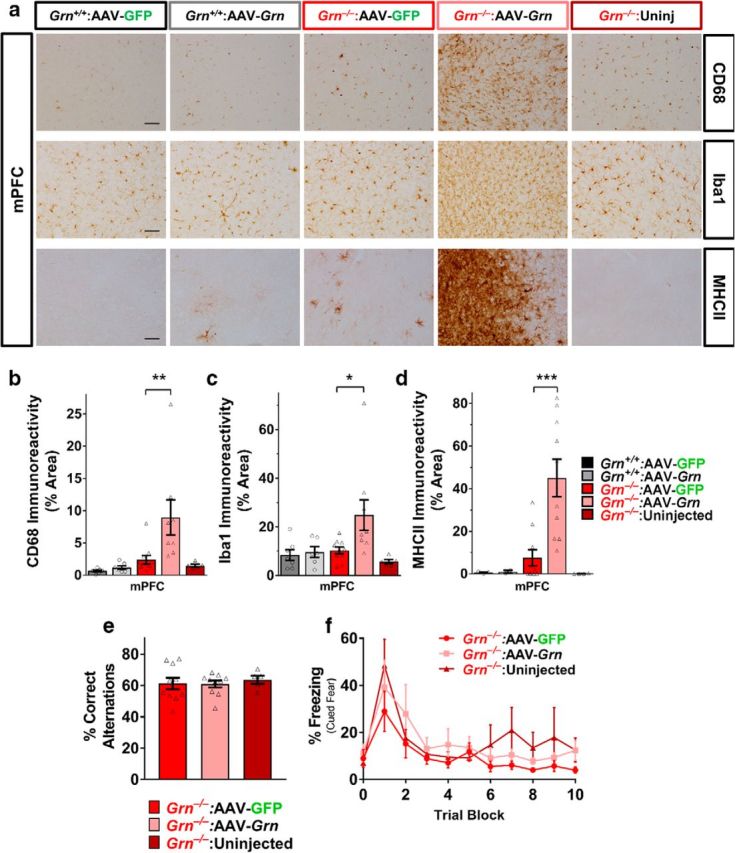
AAV-Grn causes inflammation in the medial PFC of Grn−/− mice, but does not impair mPFC-dependent behaviors. a–c, In contrast to its effects in other brain regions, AAV-Grn increased microgliosis in the mPFC of Grn−/− mice as assessed by CD68 (n = 7–11 mice per treatment group with 5 uninjected mice; ANOVA, effect of virus, F(1,28) = 5.95, p = 0.0213, genotype × virus, F(1,28) = 4.325, p = 0.0468, **p < 0.01, Sidak's post hoc test) and Iba1 immunoreactivity (n = 7–11 mice per treatment group with 5 uninjected mice; ANOVA, effect of genotype, F(1,28) = 4.655, p < 0.0001, *p < 0.05, Sidak's post hoc test). a, d, This increase in microgliosis was associated with immune activation because AAV-Grn-treated Grn−/− mice exhibited strongly elevated MHCII in the mPFC (n = 3 per group for wild-type mice, 10 per group for AAV-treated Grn−/− mice, and 4 uninjected Grn−/− mice, ANOVA, effect of genotype, F(1,21) = 6.444, p = 0.0191, ***p < 0.001, Sidak's post hoc test). e, f, However, the increased inflammation in the mPFC of AAV-Grn-treated Grn−/− mice did not impair Y-maze performance (p = 0.8502, ANOVA, effect of virus, F(2,22) = 0.1634) or extinction of cued fear conditioning (RM-ANOVA, effect of trial block, F(10,100) = 9.965, p < 0.0001), but no effect of virus (F(2,10) = 0.9968, p = 0.4029) or interaction of virus and trial block (F(20,100) = 0.6798, p = 0.8373) relative to AAV-GFP-treated Grn−/− mice (Y-maze, n = 11 mice per AAV group and 5 uninjected mice; fear extinction, n = 5–6 mice per AAV group and 2 uninjected mice). Representative 20× images in a are shown with 50 μm scale bars.
Each of the pathology datasets shown in Figures 3 and 4 (lipofuscinosis, CD68, Iba1, and MHCII) were analyzed first by three-way repeated-measures (RM)-ANOVA that included all four brain regions examined (with genotype, AA, and brain region as factors) to avoid multiple-comparisons errors. Datasets with significant effects of virus or interaction of virus with genotype or brain region were followed by two-way ANOVA (with genotype and AAV as factors) for each brain region. Significant main effects or interactions in two-way ANOVA were followed by Sidak's post hoc test to determine the effect of virus within each genotype. CatD activity for AAV-treated mice (see Fig. 9) was analyzed by three-way RM-ANOVA (factors of genotype, virus, and time), followed by two-way RM-ANOVA (factors of virus and time) to test the effects of virus within each genotype. CatD activity for uninjected mice (see Fig. 8) was analyzed by two-way RM-ANOVA (factors of time and genotype). For all CatD assays, significant effects of virus/genotype or an interaction of virus/genotype with time were followed by Fisher's LSD post hoc test. Immunoblots of CatD and LAMP-1, the ratio of mature/pro-CatD, and the relative CatD activity from AAV-treated mice were analyzed by two-way ANOVA (factors of genotype and virus) followed by Tukey's post hoc test. The presence or absence of progranulin antibodies (see Fig. 6) in mouse plasma was analyzed by χ2 test. Progranulin ELISA data (see Fig. 1) were analyzed by three-way ANOVA with factors of genotype, AAV treatment, and brain region. To determine the effect of virus within each genotype, the global analysis was followed by a two-way ANOVA for each genotype, with factors of virus and brain region. Progranulin qRT-PCR was analyzed by two-way ANOVA with factors of genotype and virus. For both ELISA and qRT-PCR data, significant main effects or interactions in ANOVA were followed by one-tailed t tests to determine the effect of virus within genotype in each brain region. We observed that wild-type AAV-Grn-treated mice appeared to have higher progranulin levels than AAV-Grn-treated Grn−/− mice, so we made this additional comparison by two-tailed t test. Conditioned fear extinction (see Fig. 5) was analyzed by two-way RM-ANOVA with factors of virus and trial block. Y-maze (see Fig. 5) was analyzed by one-way ANOVA with a factor of virus. CatD and LAMP-1 protein levels, relative CatD activity from untreated mice (see Fig. 8), and GFP Western blot (see Fig. 1) were analyzed by t test. The correlation between CatD protein levels and activity (see Fig. 9) was analyzed by Pearson correlation. Three-way ANOVA was performed with JMP Pro 12 (SAS Institute) and all other tests were performed with GraphPad Prism 7 software. Except where noted, two-tailed p-values were calculated and α was set at 0.05 for all analyses. Data are shown as mean ± SEM.
Figure 3.
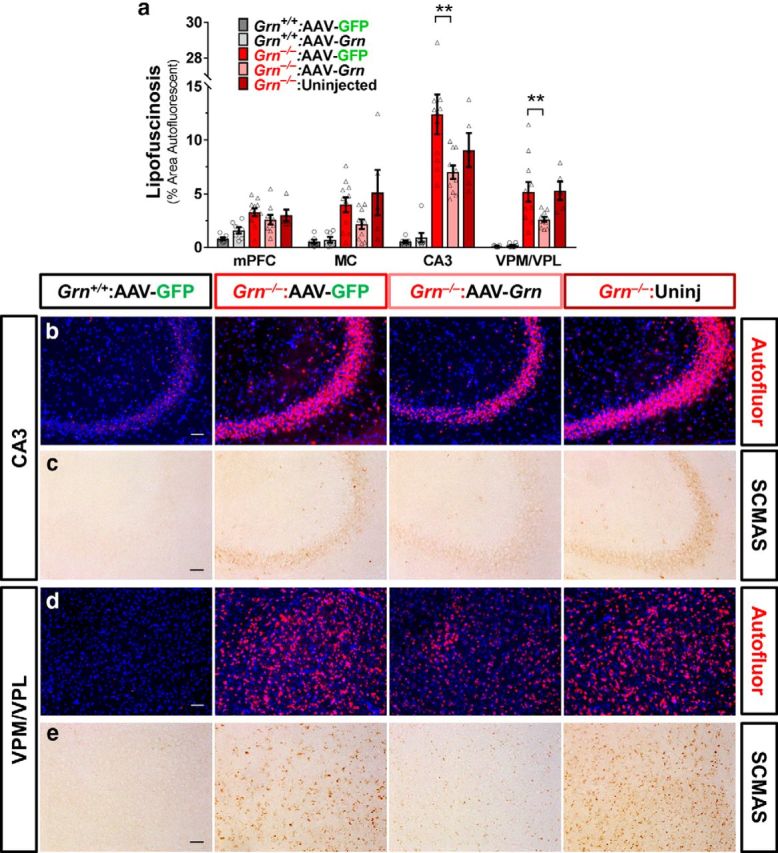
AAV-Grn improves lipofuscinosis in Grn−/− mice. a, Accumulation of autofluorescent lipofuscin was reduced in AAV-Grn-treated versus AAV-GFP-treated Grn−/− mice in CA3 (p = 0.0254, ANOVA, genotype × virus, F(1,32) = 5.502; b) and VPM/VPL (p = 0.0310, ANOVA, genotype × virus, F(1,32) = 5.902; d), with similar trends in the mPFC (ANOVA: genotype × virus, F(1,30) = 3.902, p = 0.0575) and motor cortex (MC, ANOVA: genotype × virus, F(1,31) = 3.43, p = 0.0736; n = 7–11 mice per AAV treatment group plus 5 uninjected Grn−/− mice; **p < 0.01, Sidak's post hoc test). c, e, Similarly, SCMAS accumulation in Grn−/− mice was reduced by AAV-Grn in CA3 and VPM/VPL thalamus. Representative 20× images show autofluorescent lipofuscin granules in red and nuclei stained with DAPI in blue or SCMAS immunoreactivity. Scale bars, 50 μm.
Figure 9.
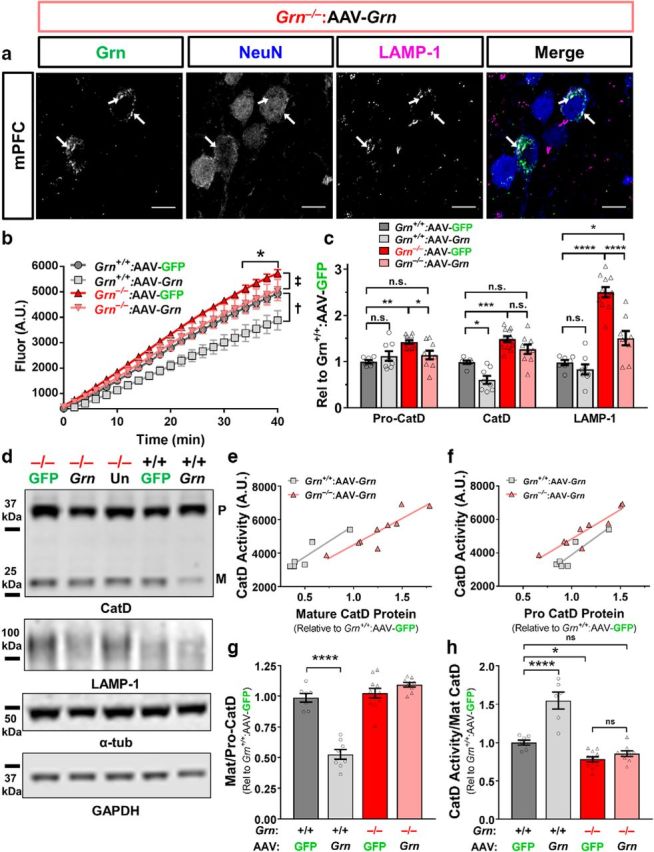
AAV-Grn normalizes CatD activity in Grn−/− mice. a, Progranulin-immunoreactive neurons exhibited bright puncta that were generally colabeled with the lysosomal marker LAMP-1 (white arrows), indicating lysosomal localization of progranulin. b, AAV-GFP-treated Grn−/− mice had elevated CatD activity in the ventral striatum (n = 6–11 mice per group, 3-way RM-ANOVA effect of genotype, F(1,30) = 12.14, p = 0.0015), which was normalized by AAV-Grn treatment (3-way RM-ANOVA effect of virus, F(1,30) = 7.47, p = 0.0104, ‡2-way RM-ANOVA of AAV-GFP-treated vs AAV-Grn-treated Grn−/− mice: virus × time interaction, F(20,380) = 2.07, p = 0.0047; * = AAV-Grn-treated Grn−/− mice < AAV-GFP-treated Grn−/− mice, p < 0.05, Fisher's post hoc test). AAV-Grn also reduced CatD activity in wild-type mice (†2-way RM-ANOVA of AAV-GFP-treated vs AAV-Grn-treated wild-type mice: effect of virus F(1,11) = 9.5, p = 0.0104, virus × time interaction F(20,220) = 6.432, p < 0.001). c, d, Consistent with their elevated CatD activity, Grn−/− mice exhibited elevated levels of pro-CatD (ANOVA effect of genotype F(1,31) = 8.758, p = 0.0059, **p < 0.01, Tukey's post hoc test) and mature CatD (ANOVA effect of genotype, F(1,31) = 51.83, p < 0.0001, ***p < 0.001, Tukey's post hoc test). AAV-Grn significantly reduced levels of pro-CatD in Grn−/− mice (ANOVA genotype × virus interaction F(1,31) = 7.102, p = 0.0121, *p < 0.05, Tukey's post hoc test), although levels of mature CatD were not significantly reduced. In contrast, the reduced CatD activity of AAV-Grn-treated wild-type mice was associated with reduced levels of mature CatD (ANOVA effect of virus F(1,31) = 13.99, p = 0.0007, *p < 0.05, Tukey's post hoc test), but not pro-CatD. Grn−/− mice also exhibited elevated levels of the lysosomal membrane protein LAMP-1 (ANOVA effect of genotype F(1,31) = 82.65, p < 0.0001, ****p < 0.0001, Tukey's post hoc test) that were significantly reduced by AAV-Grn (ANOVA effect of virus F(1,31) = 22.41, p < 0.0001, virus × genotype interaction F(1,31) = 12.39, p = 0.0014). LAMP-1 levels were not significantly reduced by AAV-Grn in wild-type mice. e, f, Both mature CatD and pro-CatD protein levels were strongly correlated with CatD activity in AAV-Grn-treated wild-type (n = 6 AAV-Grn-treated wild-type mice, Pearson correlation pro-CatD: r = 0.9135, r2 = 0.8345, p = 0.0109, mature CatD: r = 0.9259, r2 = 0.8527, p = 0.008) and Grn−/− mice (n = 9 AAV-Grn-treated Grn−/− mice, Pearson correlation pro-CatD: r = 0.9249, r2 = 0.8555, p = 0.0004, mature CatD: r = 0.8909, r2 = 0.7937, p = 0.0013), suggesting that protein levels were a major driver of CatD activity. g, AAV-Grn reduced maturation of CatD in wild-type mice, but not Grn−/− mice, because the ratio of mature/pro-CatD was significantly reduced in AAV-Grn-treated wild-type, but not Grn−/− mice (ANOVA effect of virus (F(1,31) = 16.34, p < 0.0001, virus × genotype interaction (F(1,31) = 30, p < 0.0001, ****p < 0.0001, Tukey's post hoc test). h, AAV-Grn also enhanced the relative activity of CatD (fluorescence at 40 min/mature CatD) in wild-type, but not Grn−/− mice (ANOVA effect of virus F(1,29) = 24.08, p < 0.0001, virus × genotype interaction F(1,29) = 14.38, p = 0.0002, ***p < 0.0001, Tukey's post hoc test). Representative images of Western blots are shown in c. The CatD blots show bands for pro-CatD (P) and mature CatD (M). Representative 60× images in a are shown with 10 μm scale bars.
Figure 8.
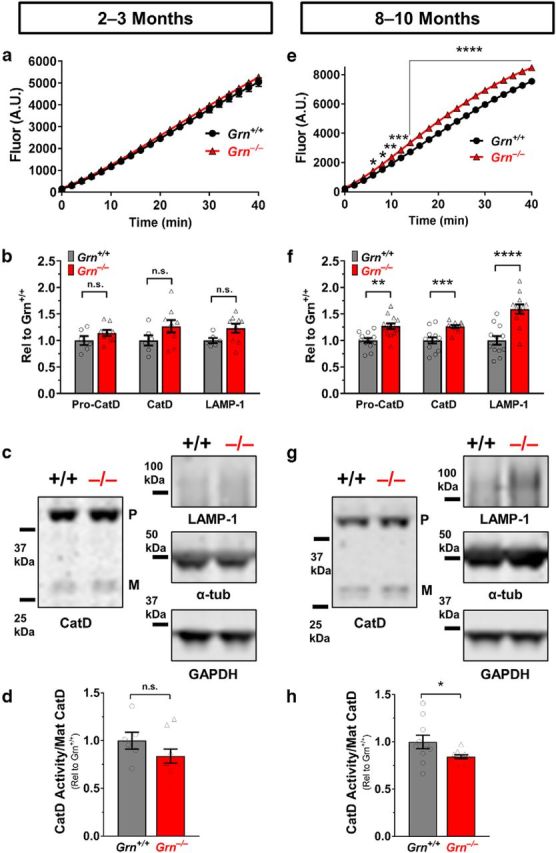
Grn−/− mice exhibit age-dependent increases in CatD and LAMP-1. a, CatD activity was not significantly increased at age 2–3 months (n = 6–7 mice per genotype) in the frontal cortex of 2- to 3-month-old Grn−/− mice. b, c, CatD and LAMP-1 protein levels were also not significantly increased at 2–3 months. d, In addition, the relative CatD activity of 2- to 3-month-old Grn−/− mice (fluorescence at 40 min/mature CatD) was not significantly different from wild-type. e, CatD activity was significantly increased in the frontal cortex of 8- to 10-month-old Grn−/− mice (n = 12–13 per genotype; activity, RM-ANOVA, genotype × time interaction, F(20,460) = 30.11, p < 0.0001; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 versus wild-type, Fisher's post hoc test). f, g, Both pro-CatD (t(23) = 3.748, p = 0.0011) and mature CatD (t(23) = 4.283, p = 0.0003) protein levels were also increased at 8–10 months (**p < 0.01 and ***p < 0.001, t test). LAMP-1 levels were also increased in the frontal cortex of 8- to 10-month-old Grn−/− mice (t(22) = 4.942, p < 0.0001), indicating a more general increase in lysosomal proteins (****p < 0.0001 vs wild-type, t test). h, Relative CatD activity was decreased in 8- to 10-month-old Grn−/− mice (t(19) = 2.231, p = 0.0379). Representative images of Western blots are shown in c and g. The CatD blots show bands for pro-CatD (P) and mature CatD (M).
Figure 6.

AAV-Grn-treated Grn−/− mice produce anti-progranulin antibodies and both AAV-GFP-treated wild-type and Grn−/− mice produce anti-GFP antibodies. a, b, Progranulin autoantibodies were detected by probing blots of recombinant mouse progranulin with plasma from AAV-Grn-treated Grn−/− mice, but not plasma from AAV-GFP-treated Grn−/− mice or AAV-Grn-treated wild-type mice (n = 11 AAV-Grn-treated wild-type, 7 AAV-GFP-treated Grn−/− mice, and 9 AAV-Grn-treated Grn−/− mice, χ2: df 2,22.74, p < 0.0001). c, In contrast, both wild-type and Grn−/− mice treated with AAV-GFP demonstrated circulating anti-GFP antibodies. d, AAV-Grn-treated Grn−/− mice generated more antibodies against progranulin than AAV-GFP-treated wild-type or Grn−/− mice generated against GFP because the immunoreactivity of AAV-Grn-treated Grn−/− plasma persisted at greater dilutions than either of the AAV-GFP-treated groups. MW, Molecular weight marker; mGrn, recombinant mouse progranulin. ****p < 0.0001, χ2 test.
Figure 1.
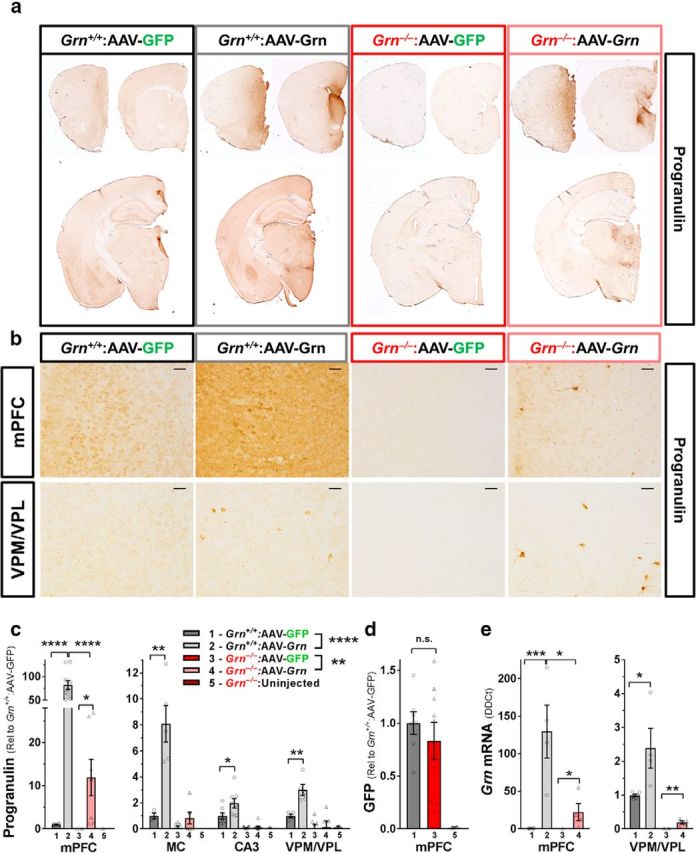
AAV-Grn transduced many cells in the mPFC, but also transduced small numbers of cells throughout the forebrain. a, Immunostaining for progranulin revealed strong increases in progranulin immunoreactivity in the mPFC, with smaller increases in the septum, striatum, and thalamus of mice treated with AAV-Grn. b, Higher-magnification (20×) images of progranulin immunostaining revealed large numbers of strongly progranulin-immunoreactive cells in the mPFC of both wild-type and Grn−/− mice treated with AAV-Grn, with smaller numbers of strongly progranulin-immunoreactive cells in regions away from the injection site such as the VPM/VPL thalamus. c, Progranulin protein levels as measured by ELISA were increased in AAV-Grn-treated wild-type (****p < 0.0001, ANOVA, effect of virus, F(1,40) = 28.57) and Grn−/− mice (**p = 0.0043, ANOVA, effect of virus, F(1,51) = 9.707) relative to their GFP-treated controls, with the highest progranulin expression in the mPFC. In addition, wild-type mice exhibited significantly higher progranulin overexpression in the mPFC than Grn−/− mice (t test, t(15) = 6.034, p < 0.0001; n = 3–11 mice per group. *p < 0.05, **p < 0.01, and ****p < 0.0001, t test). d, In contrast, we did not observe a significant genotype difference in expression of GFP in AAV-GFP-treated wild-type and Grn−/− mice by Western blot (p = 0.4629, t test). e, AAV-Grn also increased Grn RNA levels in the mPFC (ANOVA: effect of virus, F(1,14) = 21.26, p = 0.0004) and VPM/VPL thalamus (ANOVA: effect of virus, F(1,16) = 11.38, p = 0.0039). In both brain regions, wild-type AAV-Grn-treated mice had significantly higher Grn RNA levels than AAV-Grn-treated Grn−/− mice (mPFC ANOVA: effect of genotype, F(1,14) = 10.87, p = 0.0053, genotype × virus, F(1,14) = 10.59, p = 0.0058, VPM/VPL ANOVA: effect of genotype, F(1,16) = 46.09, p < 0.0001, genotype × virus, F(1,16) = 6.622, p = 0.0204). n = 4–6 mice per group; *p < 0.05, **p < 0.01, and ***p < 0.001, t test). Representative 20× images in b are shown with 50 μm scale bars.
Results
A key question for progranulin-boosting therapies in NCL and FTD is whether boosting progranulin after the onset of pathology could reduce lipofuscinosis and microgliosis. To address this question, we treated wild-type and Grn−/− mice with an AAV vector expressing mouse progranulin with a C-terminal myc tag (AAV-Grn) or a control AAV-GFP vector at age 10–12 months. Grn−/− mice exhibit robust NCL-like pathology at this age, characterized by lipofuscin accumulation and gliosis (Ahmed et al., 2010; Wils et al., 2012; Filiano et al., 2013; Petkau et al., 2016). The AAV vectors were intracranially injected into the mPFC, as in our prior study investigating the behavioral effects of AAV-Grn in Grn+/− mice (Arrant et al., 2017). A small additional group of uninjected Grn−/− mice was included in the study as a positive control for the expected abnormalities in Grn−/− mice to rule out nonspecific effects of the control AAV-GFP treatment.
Regional distribution of progranulin expression
We first characterized the pattern of progranulin expression produced by AAV-Grn. In a previous study, we demonstrated that this AAV-Grn vector strongly increased progranulin levels in wild-type and Grn+/− mice at the injection site in the mPFC, with smaller but significant increases in progranulin throughout the forebrain (Arrant et al., 2017). To determine whether AAV-Grn had similar effects in Grn−/− mice, we qualitatively screened AAV-mediated progranulin expression by immunohistochemistry (Fig. 1a,b) and measured progranulin protein levels by ELISA (Fig. 1c). As expected, progranulin immunoreactivity was strongly increased in the mPFC of wild-type and Grn−/− mice treated with AAV-Grn, and more modestly increased in the septum and medial regions of the striatum and thalamus (Fig. 1a). With higher-magnification imaging, we observed large numbers of strongly progranulin-immunoreactive cells in the mPFC (Fig. 1b). Brain regions more distal to the injection site, such as the ventral posteromedial/lateral (VPM/VPL) thalamus and CA3 of the hippocampus, had scattered strongly progranulin-immunoreactive cells, but far fewer than mPFC (Fig. 1b). Global analysis of progranulin levels by ELISA (Fig. 1c) revealed that AAV-Grn increased progranulin (RM-ANOVA: effect of virus, F(1,10) = 49.78, p < 0.001) in a genotype-specific (RM-ANOVA: genotype × virus, F(1,10) = 27.7, p = 0.0004) and region-specific (RM-ANOVA: region × virus, F(3,8) = 15.3, p = 0.0011, region × genotype × virus, F(3,8) = 8.183, p = 0.008) manner. In Grn+/+ mice, AAV-Grn increased progranulin in all brain regions examined (Fig. 1c). In Grn−/− mice, progranulin levels induced by AAV-Grn were lower than in Grn+/+ mice as measured by ELISA. In fact, the scattered progranulin-positive cells observed by immunohistochemistry in thalamus and hippocampus (Fig. 1b, bottom right) were not sufficient to yield a significant progranulin signal by ELISA (Fig. 1c). In this case, due to the low number of transduced cells, immunostaining was the more sensitive method of detection. We conclude that AAV-Grn-treated Grn−/− mice expressed lower levels of AAV-mediated progranulin than AAV-Grn-treated wild-type mice for the reasons discussed below. The lower progranulin levels in Grn−/− mice were specific to AAV-Grn because levels of GFP expression induced by AAV-GFP did not differ between Grn+/+ and Grn−/− mice (Fig. 1d).
We suspected that the scattered progranulin-positive cells in distal regions represented cells transduced by AAV-Grn spreading from the injection site; the alternative possibility is that these cells had taken up progranulin secreted from the axons of transduced cortical neurons (Petoukhov et al., 2013). To distinguish between these possibilities, we measured Grn RNA levels. We observed a definite increase in Grn RNA at the mPFC injection site and a much more modest but significant increase in Grn RNA in the thalamus of AAV-Grn-treated mice (Fig. 1e). As with progranulin protein levels, AAV-Grn-treated Grn−/− mice exhibited detectable increases in Grn RNA, but Grn RNA levels were lower than in AAV-Grn-treated wild-type mice. Based on these data, we conclude that AAV-Grn transduced small numbers of cells in regions distant from the mPFC, probably due to viral particles diffusing through the brain to a limited extent.
AAV-Grn transduces neurons, but not microglia
We next investigated which cell types were transduced by AAV-Grn. In our prior study with AAV-Grn in wild-type and Grn+/− mice, we found that AAV-Grn transduced neurons, but not microglia (Arrant et al., 2017). To confirm that AAV-Grn has similar effects in Grn−/− mice, we performed double immunostaining for progranulin and markers of neurons (NeuN) and microglia (Iba1) in AAV-Grn-treated Grn−/− mice (Fig. 2a,b) to determine whether microglia exhibited any progranulin immunoreactivity. Because this immunostaining was performed in Grn−/− mice, progranulin immunoreactivity was entirely composed of AAV-expressed progranulin. All of the strongly progranulin-immunoreactive cells observed were NeuN positive (Fig. 2a); no Iba1-positive cells exhibited progranulin immunoreactivity (Fig. 2b). Therefore, AAV-Grn only transduced neurons in Grn−/− mice, consistent with our prior study in wild-type and Grn+/− mice (Arrant et al., 2017) and the expected tropism of the AAV. The absence of progranulin from microglia is somewhat surprising because progranulin secreted from neurons could potentially be taken up by microglia. Although we do not have in vivo data on the secretion of AAV-expressed progranulin, cultured cells transfected with the AAV-Grn plasmid secrete myc-tagged progranulin (data not shown), indicating that AAV-expressed progranulin can be secreted by some cell types. Therefore, the AAV-expressed progranulin produced in neurons in vivo is either not secreted, not taken up by microglia, or is rapidly degraded within microglia.
Figure 2.

AAV-expressed progranulin is found primarily in neurons, but not microglia. a, b, The strongly progranulin-immunoreactive cells observed in the mPFC and other brain regions were also positive for the neuronal marker NeuN (a), whereas no progranulin immunoreactive microglia (Iba1) were observed (b). VS, Ventral striatum. Representative 60× images are shown with 20 μm scale bars.
AAV-Grn improves lipofuscinosis in Grn−/− mice
We tested the effects of AAV-Grn on lipofuscinosis by quantitating autofluorescence in the mPFC, motor cortex, hippocampus (CA3), and VPM/VPL thalamus of AAV-treated wild-type and Grn−/− mice (Fig. 3a,b,d). There was a significant effect of AAV that was dependent on genotype (three-way RM-ANOVA across all brain regions: genotype × virus, F(1,29) = 10.09, p = 0.0035). Because effects were also brain-region dependent (region × genotype, F(3,27) = 7.845, p = 0.0006), we performed separate analyses of each brain region. AAV-Grn significantly reduced lipofuscinosis in the CA3 and thalamus of Grn−/− mice (Fig. 3a,b,d). There were similar trends for reduction of lipofuscinosis in the mPFC (ANOVA: genotype × virus, F(1,30) = 3.902, p = 0.0575) and motor cortex (ANOVA: genotype × virus, F(1,31) = 3.43, p = 0.0736) of AAV-Grn-treated Grn−/− mice. These data show a therapeutic benefit of progranulin restoration to Grn−/− mice even after the onset of lipofuscinosis. The significant improvement in lipofuscinosis in the hippocampus and thalamus of AAV-Grn-treated Grn−/− mice, despite the very modest increase in progranulin in these brain regions (Fig. 1), indicates that the progranulin from small numbers of transduced cells is sufficient to produce beneficial effects. The fact that reduction of lipofuscinosis did not reach significance in the mPFC (p = 0.0575) or motor cortex (p = 0.0736) could be due to a floor effect because these regions have less lipofuscin accumulation than the hippocampus and thalamus.
As an additional measure of lipofuscinosis, we performed immunostaining for subunit C of mitochondrial ATP synthase (SCMAS), a protein component of lipofuscin (Hall et al., 1991; Kominami et al., 1992; Elleder et al., 1997) that accumulates in Grn−/− mice (Götzl et al., 2014; Zhou et al., 2017a). As expected, Grn−/− mice exhibited a striking increase in SCMAS immunoreactivity (Fig. 3c,e). AAV-Grn reduced SCMAS immunoreactivity in Grn−/− mice, providing further support for a reduction in lipofuscinosis in AAV-Grn-treated Grn−/− mice.
AAV-Grn improves microgliosis away from the injection site in the mPFC
In addition to lipofuscinosis, the pathology of Grn−/− mice is characterized by microgliosis (Ahmed et al., 2010; Wils et al., 2012; Filiano et al., 2013; Tanaka et al., 2014; Arrant et al., 2015; Petkau et al., 2016). To determine the effects of AAV-Grn on microgliosis, we measured CD68 immunoreactivity in AAV-GFP- and AAV-Grn-treated wild-type and Grn−/− mice. Global analysis across all brain regions indicated an effect of AAV-Grn that varied by brain region (RM-ANOVA region × genotype × virus, F(3,26) = 3.03, p = 0.0473; region × genotype, F(3,26) = 13.51, p < 0.0001). CD68 immunoreactivity was significantly reduced in motor cortex and CA3 of AAV-Grn-treated Grn−/− mice relative to their GFP-treated controls (Fig. 4a,b). As a further analysis, we measured the soma size of Iba1-positive cells. Grn−/− mice exhibit abnormal microglial morphology as they age, with Iba1-positive microglia exhibiting less ramification and larger soma size, indicative of activation (Ahmed et al., 2010; Karperien et al., 2013). Iba1-positive soma size was increased in Grn−/− mice in the VPM/VPL thalamus, consistent with a prior report (Fig. 4c,d) (Ahmed et al., 2010). AAV-Grn significantly reduced Iba1-positive soma size, partially correcting Iba1-positive cell morphology (Fig. 4c,d). Together, the CD68 and Iba1 analyses indicate that AAV-Grn reduced microgliosis in brain regions away from the injection site, including motor cortex, CA3, and VPM/VPL thalamus. This improvement in microgliosis in the absence of detectable increases in microglial progranulin (Fig. 2b) has several potential interpretations. Microgliosis could be reactive to neuronal dysfunction and indirectly improved by amelioration of neuronal dysfunction by neuronal progranulin restoration. Alternatively, AAV-expressed progranulin secreted from neurons could reduce microgliosis through effects on microglial cell surface receptors or could be taken up and very rapidly degraded by microglia.
AAV-Grn causes localized injection site inflammation in Grn−/− mice and a nonself reaction
In an unexpected contrast to regions away from the injection site, AAV-Grn strongly increased microglial markers near the injection site in the mPFC in Grn−/− mice, but not in wild-type mice. Both CD68 and Iba1 immunoreactivity were strongly increased in mPFC in AAV-Grn-treated Grn−/− mice, but not in AAV-Grn-treated wild-type mice (Fig. 5a–c). Iba1-positive cell size was not significantly affected by AAV-Grn in the mPFC of Grn−/− mice (data not shown), although there was a difference in staining pattern, with more diffuse Iba1 labeling and more Iba1-positive cells (Fig. 5a). To determine whether these increases in microglial markers reflected immune activation, we immunostained for MHCII, which is expressed by activated microglia during inflammation (Lynch, 2009; Franco and Fernández-Suárez, 2015). There was an increase in MHCII immunoreactivity in Grn−/− mice (RM-ANOVA effect of genotype, F(1,21) = 7.471, p = 0.0125), which was driven by very high expression of MHCII in the mPFC of AAV-Grn-treated Grn−/− mice, indicating local inflammation (Fig. 5a,d). In the motor cortex, CA3, and VPM/VPL thalamus of AAV-Grn-treated Grn−/− mice, MHCII staining was not significantly elevated upon statistical analysis. These data indicate a strong local inflammatory response to AAV-Grn in Grn−/− mice, but not in wild-type mice. Importantly, Grn−/− mice did not have a similar reaction to AAV-GFP, indicating that this local inflammation was not a general response to AAV injection.
In considering the opposing, region-specific effects of AAV-Grn on microgliosis in Grn−/− mice (improvement of microgliosis in multiple regions except at the mPFC injection site, where it was worse), we hypothesized that Grn−/− mice may have reacted to the exogenous progranulin as a nonself protein, given the complete absence of progranulin throughout their lifespan. In support of this possibility, wild-type mice (Fig. 5) and Grn+/− mice (data not shown) did not exhibit microgliosis or increased MHCII expression at the AAV-Grn injection site. In addition, antigen-presenting cells use MHC-II to present antigens to T cells, indicating a cell-mediated immune response to AAV-expressed progranulin in Grn−/− mice (Roche and Furuta, 2015). Furthermore, previous studies have shown that expressing foreign proteins with AAV vectors can induce nonself reactions that resemble the effects of AAV-Grn in Grn−/− mice when using AAV serotypes capable of transducing antigen-presenting cells (Ciesielska et al., 2013; Samaranch et al., 2014). We hypothesized that a similar mechanism could be at work in our study, in which Grn−/− mice have a large immune response to progranulin, a secreted protein, but a very small response to GFP, which, although it is a foreign protein, should remain primarily intracellular. Therefore, progranulin secreted from AAV-Grn-transduced neurons would be readily accessible to antigen-presenting cells, whereas GFP from AAV-GFP-transduced neurons would not.
To determine whether Grn−/− mice had a nonself reaction to progranulin and GFP, we tested for progranulin and GFP antibodies in the plasma of AAV-treated mice. We ran recombinant mouse progranulin or recombinant GFP from Aequorea victoria on SDS-PAGE and transferred the protein to PVDF membranes, and then probed the membranes with plasma isolated from individual mice. An anti-mouse secondary antibody was used to determine whether circulating antibodies were bound to the recombinant proteins. For progranulin, we tested plasma from wild-type mice treated with AAV-Grn and Grn−/− mice treated with AAV-GFP or AAV-Grn. Of these groups, only AAV-Grn-treated Grn−/− mice had circulating anti-mouse progranulin antibodies (Fig. 6a). Of the nine AAV-Grn-treated Grn−/− mice tested, eight were positive for circulating anti-mouse progranulin antibodies (Fig. 6b), suggesting that AAV-Grn induced a nonself reaction in these mice. This effect is probably unique to the Grn−/− mouse model and is unlikely to be an issue in patients with GRN mutations, who still express some progranulin and so are not at risk of nonself recognition.
In testing for GFP antibodies, we analyzed plasma from wild-type and Grn−/− mice treated with AAV-GFP, and from mice that received no AAV. Plasma from both wild-type and Grn−/− mice treated with AAV-GFP detected recombinant GFP, indicating the presence of anti-GFP antibodies (see Fig. 9c). The lack of genotype specificity is unsurprising as GFP is a foreign protein for both genotypes. However, the presence of detectable anti-GFP antibodies was somewhat surprising given the lack of a similar inflammatory response as observed in AAV-Grn-treated Grn−/− mice. Given our hypothesis that GFP should be less likely to generate an immune response than progranulin due to GFP's almost exclusively intracellular location, we hypothesized that AAV-GFP-treated wild-type and Grn−/− mice might have mounted a much weaker nonself response to GFP than AAV-Grn-treated Grn−/− mice generated to progranulin. To test this hypothesis, we analyzed anti-GFP and anti-progranulin antibody titers by progressively diluting the plasma from AAV-GFP-treated wild-type and Grn−/− mice and AAV-Grn-treated Grn−/− mice and determining the maximum dilution at which immunoreactivity was maintained. The anti-progranulin antibodies generated by AAV-Grn-treated Grn−/− mice were still detectable at plasma dilutions up to 1:2500, whereas the anti-GFP antibodies generated by AAV-GFP-treated wild-type and Grn−/− mice were detectable at plasma dilutions of 1:100–1:500 (Fig. 6c,d). These data indicate greater levels of anti-progranulin antibodies in AAV-Grn-treated Grn−/− mice than of anti-GFP antibodies in AAV-GFP-treated mice, which, along with the greater inflammation in AAV-Grn-treated Grn−/− mice, is consistent with a stronger nonself reaction to progranulin than to GFP.
To determine whether the mPFC inflammation induced by AAV-Grn in Grn−/− mice had negative functional effects, we tested the mice in two mPFC-dependent behavioral assays: spontaneous alternation in the Y-maze (Yang et al., 2014) and extinction of conditioned fear (Giustino and Maren, 2015). We observed no deficits in either Y-maze spontaneous alternation (Fig. 5e) or conditioned fear extinction (Fig. 5f) in AAV-Grn-treated Grn−/− mice relative to AAV-GFP-treated Grn−/− mice or uninjected Grn−/− mice. Therefore, the inflammation in the mPFC was not sufficient to produce detectable mPFC-dependent behavioral impairments in Grn−/− mice. These data are consistent with our prior study showing that AAV-Grn does not disrupt mPFC-dependent behaviors in wild-type or Grn+/− mice (Arrant et al., 2017).
Effects of AAV-Grn are independent of sortilin binding
Having observed that AAV-Grn reduced lipofuscinosis and microgliosis in Grn−/− mice (Figs. 3, 4), we began to investigate the mechanisms by which progranulin mediated these beneficial effects. First, we determined whether the beneficial effects of progranulin gene therapy with AAV-Grn depend on progranulin binding to sortilin, a major progranulin receptor (Hu et al., 2010). The C terminus of progranulin is critical for sortilin binding and C-terminal tags on progranulin have been reported to disrupt sortilin binding (Zheng et al., 2011). We therefore assessed whether our C-terminal myc-tagged progranulin binds sortilin. Consistent with prior reports, the C-terminal myc tag on our AAV-Grn construct almost completely eliminated sortilin binding (Fig. 7). Therefore, the improvement in pathology by AAV-Grn that we observed does not require sortilin binding.
Figure 7.

The C-terminal Myc tag on AAV-Grn disrupts sortilin binding. The C-terminal myc-tag on the AAV-Grn almost completely eliminated coimmunoprecipitation of progranulin with sortilin in HEK-293 cells cotransfected with the AAV-Grn construct and a human sortilin plasmid. In contrast, an identical AAV-Grn construct with an HA tag inserted at the N terminus of the protein after the signal peptide exhibited strong coimmunoprecipitation with sortilin. Coimmunoprecipitation data are representative of four experiments.
AAV-expressed progranulin improves lysosomal function
Lipofuscin consists of undegraded lysosomal storage material, so it is likely that the reduction in lipofuscinosis in AAV-Grn-treated Grn−/− mice was due to improved lysosomal function. To test this hypothesis, we assessed CatD levels and activity, as well as levels of the lysosomal membrane protein LAMP-1. CatD expression is increased in the brain of Grn−/− mice at ages ranging from 16–24 months and recent studies have shown that progranulin binds to CatD and may regulate its activity (Wils et al., 2012; Götzl et al., 2014; Beel et al., 2017; Valdez et al., 2017; Zhou et al., 2017b). This effect is age dependent because a recent study of 2-month-old Grn−/− mice did not observe elevated CatD levels in the brain (Zhou et al., 2017b).
First, we investigated the frontal cortex of 2- to 3-month-old and 8- to 10-month-old wild-type and Grn−/− mice (Fig. 8) and observed increased CatD activity and protein levels in 8- to 10-month-old (Fig. 8e–g), but not 2- to 3-month-old (Fig. 8a–c) Grn−/− mice, consistent with prior reports on CatD protein levels and activity (Götzl et al., 2014; Beel et al., 2017; Zhou et al., 2017b). Others have reported that Grn−/− mice or cells actually have lower CatD activity when normalized to levels of mature CatD protein, suggesting a functional impairment of CatD in Grn−/− mice (Beel et al., 2017; Valdez et al., 2017). We assessed this “relative activity” by dividing the fluorescence generated at the final time point of the CatD activity assay by the relative levels of mature CatD. We found reduced relative CatD activity in 8- to 10-month-old Grn−/− mice (Fig. 8h), but not in 2- to 3-month-old Grn−/− mice (Fig. 8d), indicating that the impairments in relative CatD activity are also age dependent. LAMP-1 levels followed a similar time course and were significantly increased in 8- to 10-month-old Grn−/− mice (Fig. 8f,g), but not 2- to 3-month-old Grn−/− mice (Fig. 8b,c). The age-dependent increase in CatD and LAMP-1 levels likely reflects a compensatory increase in lysosomal biogenesis induced in response to the lysosomal dysfunction associated with progranulin deficiency.
A notable feature of progranulin-immunoreactive neurons (Fig. 2a) in AAV-Grn-treated Grn−/− mice was the presence of very bright puncta within each cell that resembled vesicular immunolabeling. Endogenous progranulin strongly localizes to lysosomes, so we performed triple immunostaining for progranulin, NeuN, and LAMP-1, a lysosomal membrane protein, to determine whether these bright progranulin puncta represented lysosomal progranulin. We observed strong colocalization of progranulin and LAMP-1 (Fig. 9a), showing that AAV-expressed progranulin is transported to lysosomes in neurons, placing it in the proper cellular context to improve lipofuscinosis (Fig. 3).
We then tested whether AAV-Grn would normalize CatD activity and protein levels in Grn−/− mice. We investigated the ventral striatum of AAV-treated wild-type and Grn−/− mice because the PFC and other brain regions were processed with protease inhibitors for progranulin ELISA (Fig. 5). As in other regions, CatD activity was increased in the ventral striatum of AAV-GFP-treated Grn−/− mice (Fig. 9b). AAV-Grn significantly reduced CatD activity in Grn−/− mice to levels similar to wild-type mice. AAV-Grn also reduced CatD activity in wild-type mice. We further explored these changes by immunoblotting for CatD.
Immunoblotting for CatD revealed potentially different mechanisms behind the reduction in CatD activity in Grn−/− and wild-type mice. As expected, Grn−/− mice treated with AAV-GFP exhibited increased levels of pro-CatD and mature CatD relative to wild-type (Fig. 9c,d). AAV-Grn reduced pro-CatD levels in Grn−/− mice to levels not significantly higher than AAV-GFP-treated wild-type mice (Fig. 9c,d). A similar pattern was observed with levels of mature CatD, although the effect of AAV-Grn did not reach significance in Grn−/− mice (Fig. 9c,d). In contrast, AAV-Grn strongly reduced levels of mature CatD (decreased by ∼39%) in wild-type mice, whereas levels of pro-CatD did not differ from AAV-GFP-treated wild-type mice (Fig. 9c,d). To further test the association between CatD protein levels and activity in AAV-Grn-treated mice, we analyzed their correlation (Fig. 9e,f). We observed strong correlations between the fluorescence generated at the final time point of the CatD assay and both mature CatD and pro-CatD in AAV-Grn-treated wild-type and Grn−/− mice. The CatD immunoblotting data suggest that the reduction in CatD activity in Grn−/− mice is caused by a reduction in CatD expression without a change in CatD maturation, but in wild-type mice is caused by a reduction in CatD maturation without a change in CatD expression. This hypothesis was further supported by analysis of the ratio of mature/pro-CatD, which revealed a strong reduction in the mature/pro-CatD ratio in AAV-Grn-treated wild-type mice, but no significant change in AAV-Grn-treated Grn−/− mice (Fig. 9g).
We next analyzed the relative activity (fluorescence at 40 min time point divided by mature CatD protein levels) of CatD to better understand the effects of AAV-Grn on CatD activity. Despite a reduction in both raw activity and mature CatD levels, AAV-Grn-treated wild-type mice exhibited a ∼50% increase in relative CatD activity (Fig. 9h), indicating that progranulin may enhance the per-molecule activity of CatD in wild-type mice. In contrast, AAV-Grn-treated Grn−/− mice did not exhibit a significant increase in relative CatD activity (Fig. 9h) despite the normalization of raw CatD activity (Fig. 9b) and partial normalization in protein levels (Fig. 9c,d). These data again demonstrate that AAV-Grn has different effects on CatD in wild-type and Grn−/− mice and show a partial, although incomplete, normalization of CatD abnormalities in Grn−/− mice.
To further understand the lysosomal changes in AAV-Grn-treated Grn−/− mice, we measured LAMP-1 levels in AAV-treated mice as an additional measure of lysosomal dyshomeostasis. As expected, AAV-GFP-treated Grn−/− mice exhibited increased LAMP-1 levels relative to AAV-GFP-treated wild-type mice (Fig. 9c,d). AAV-Grn significantly reduced LAMP-1 levels in Grn−/− mice (Fig. 9c,d). Unlike CatD, LAMP-1 levels were not significantly altered by AAV-Grn in wild-type mice. These data suggest that AAV-Grn causes a general reduction in the elevated levels of lysosomal proteins in the brain of Grn−/− mice, whereas its effects in wild-type mice may be more specific for CatD and other progranulin-interacting proteins.
Discussion
This study shows that AAV-mediated restoration of progranulin to Grn−/− mice after the onset of pathology improves lipofuscinosis and microgliosis across widespread brain regions, providing in vivo preclinical data supporting progranulin gene therapy or related progranulin replacement approaches. Several of our findings are informative for the future design of such therapies, including the beneficial effects observed with even low levels of progranulin and selective targeting of neurons, and the fact that sortilin binding is not required. AAV-expressed progranulin was delivered to the lysosome and improved lysosomal abnormalities, including normalizing CatD activity and partially normalizing CatD and LAMP-1 protein levels. The findings also shed light on progranulin biology and pathogenic mechanisms in progranulin deficiency. We observed improvement in microgliosis, although progranulin was delivered selectively to neurons, suggesting that the microglial response in progranulin deficiency occurs at least in part in response to neuronal dysfunction.
Our data suggest that even low levels of progranulin may be beneficial under conditions of progranulin deficiency. We observed widespread reduction of lipofuscinosis and microgliosis 8–10 weeks after AAV administration to Grn−/− mice (Figs. 3, 4) despite very modest effects of AAV-Grn on progranulin levels outside of the mPFC (Fig. 1). Progranulin protein levels in AAV-Grn-treated Grn−/− mice were not significantly increased outside of the mPFC when measured by ELISA, although immunostaining showed small numbers of progranulin-immunoreactive cells throughout the brain. The beneficial effects of such small increases in progranulin may be due to the requirement for nearly complete progranulin deficiency to develop NCL, as has been observed with other lysosomal storage disorders (Sandhoff and Harzer, 2013; Schulze and Sandhoff, 2014).
The apparent nonself reaction of Grn−/− mice to AAV-Grn was an unexpected result, but likely has very limited clinical significance. A nonself reaction to progranulin is unlikely in FTD-GRN patients, who still have 25–30% of normal circulating progranulin levels (Finch et al., 2009). In support of this idea, we did not observe mPFC inflammation in a recent study after identical injections of AAV-Grn in Grn+/− mice (Arrant et al., 2017). In addition, wild-type mice treated with AAV-Grn, unlike those treated with AAV-GFP, did not develop antibodies to the virally expressed protein, showing that wild-type mice did not recognize AAV-expressed progranulin as a foreign protein. For NCL patients with near-complete progranulin deficiency, the possibility of a nonself reaction may depend on the patient's specific loss-of-function mutation (Smith et al., 2012; Almeida et al., 2016). Some GRN mutations do not cause complete loss of the protein, but allow generation of nonfunctional mutant progranulin proteins (Shankaran et al., 2008; Wang et al., 2010). Even low levels of endogenous progranulin protein could prevent a nonself reaction in patients, but the Grn−/− mouse line used in this study has deletion of the entire coding region of the Grn gene (Martens et al., 2012). This complete lack of progranulin may have led to progranulin's recognition as a nonself protein and is likely specific to this type of experimental model, thus limiting the clinical significance of this result.
The nonself reaction of Grn−/− mice to AAV-Grn also appears to have had limited confounding effects on our study. The inflammation associated with the reaction was largely limited to the mPFC because microgliosis was reduced in regions outside of the mPFC (Fig. 4). The nonself reaction at the mPFC injection site did not appear to have adverse functional effects because mPFC-dependent behaviors were not impaired in AAV-Grn-treated Grn−/− mice (Fig. 5e,f). In addition, it is unlikely that the microgliosis induced by the nonself reaction mediated the improvement in lipofuscinosis due to increased microglial clearance of lipofuscin because microgliosis was decreased in the two brain regions where lipofuscinosis was significantly improved: CA3 and VPM/VPL thalamus (Figs. 3, 4). Therefore, the nonself reaction to AAV-Grn does not appear to confound the measures of lipofuscinosis and lysosomal dysfunction in regions distal to the injection site. However, the nonself reaction of Grn−/− mice to AAV-Grn likely explains the reduced expression of progranulin in AAV-Grn-treated Grn−/− mice versus wild-type mice (Fig. 1c–e). Prior studies have shown that immune responses to AAV transgenes can suppress expression of the AAV transgene, although therapeutic benefits may still be observed (Yuasa et al., 2007; Mendell et al., 2010; Haurigot et al., 2013). In two studies, nonself reactions to AAV transgenes provoked a cell-mediated immune response in which effector T cells killed muscle cells expressing the AAV transgene, resulting in gradual loss of transgene expression (Yuasa et al., 2007; Mendell et al., 2010). This may also occur in the brain because expression of nonself proteins in rats induces an immune response with high MHCII expression and T-cell infiltration at the injection site, production of antibodies against the AAV transgene, and loss of neurons and transgene expression at the injection site (Ciesielska et al., 2013). A similar mechanism could be at work in AAV-Grn-treated Grn−/− mice given the massive increase in MHCII expression at the AAV-Grn injection site. If so, then longer exposure of Grn−/− mice to AAV-Grn could result in detectable adverse outcomes such as neuronal loss and functional deficits. However, this is likely of limited clinical relevance because most GRN mutation carriers are heterozygous for loss-of-function mutations.
Our findings shed light on interactions between neurons and microglia, the primary cell types expressing progranulin in the brain. The improvement in microgliosis in regions distal to the mPFC in the absence of detectable microglial progranulin demonstrates that increasing neuronal progranulin is sufficient to modulate microglial function. Our progranulin construct was secreted normally by cultured cells (data not shown) and progranulin secreted by neurons could in theory be taken up by microglia via multiple pathways (Hu et al., 2010; Zhou et al., 2015). The fact that we detected no microglial progranulin in AAV-Grn-treated Grn−/− mice suggests that this uptake may not be efficient in microglia in vivo. Alternatively, progranulin taken up by microglia may be cleaved into granulins in the lysosome (Holler et al., 2017), but we would still expect to detect low levels of intracellular progranulin and the antibody we used for immunostaining has been reported to detect granulins by Western blot (Zhou et al., 2017c). The improvement in microgliosis could therefore be an indirect result of improved neuronal function.
In contrast, selective depletion of neuronal progranulin is not sufficient to reproduce the microglial pathology of Grn−/− mice. Selective knock-out of neuronal progranulin using CaMKII-Cre or Nestin-Cre does not produce microgliosis (Arrant et al., 2017; Petkau et al., 2017). Normally, most brain progranulin is produced by neurons because Nestin-Cre neuronal progranulin knock-out mice exhibit 50–60% reduction of brain progranulin protein, whereas depletion of microglial progranulin with Cx3Cr1-CreERT2 produces ∼30% reduction of brain progranulin (Arrant et al., 2017; Krabbe et al., 2017; Petkau et al., 2017). The remaining microglia-derived progranulin can thus prevent microgliosis and lipofuscinosis in neuronal progranulin knock-out mice, although it is insufficient to prevent social behavior deficits such as those seen in Grn± mice (Arrant et al., 2017). Neuronal–microglial crosstalk will be an important area for future investigation of progranulin biology.
The improvement in lipofuscinosis and microgliosis in Grn−/− mice likely reflects some clearance of preexisting pathology. At the age of AAV administration (10–12 months) Grn−/− mice exhibit lipofuscinosis and microgliosis throughout the brain (Ahmed et al., 2010; Wils et al., 2012; Filiano et al., 2013). Although these pathologies continue to progress with age (Ahmed et al., 2010; Wils et al., 2012), the rate of progression is insufficient to explain the differences that we observed between AAV-GFP- and AAV-Grn-treated Grn−/− mice. We observed reductions of 40–50% for both lipofuscinosis (Fig. 1) and CD68 immunostaining (Fig. 2) over 8–10 weeks. These changes greatly exceed the magnitude of increases expected in that time frame in untreated Grn−/− mice if AAV-Grn simply arrested pathology because it takes ∼11 months for lipofuscinosis to double (from 6 months to 16–18 months) and ∼10 months for microgliosis to increase by ∼50% (from 12 months to 21–23 months) (Ahmed et al., 2010; Wils et al., 2012). AAV-GFP-treated Grn−/− mice had similar pathology as uninjected Grn−/− mice, so AAV-GFP did not artificially worsen the Grn−/− phenotype. It is therefore likely that the beneficial effects of AAV-Grn in Grn−/− mice reflect clearance of preexisting pathology.
The clearance of lipofuscin in Grn−/− mice is likely due to improved lysosomal function. Recent studies of lysosomal function in progranulin-insufficient models have consistently found abnormalities in CatD, with cell culture models revealing reduced CatD activity (Valdez et al., 2017; Ward et al., 2017) and Grn−/− mouse brain revealing an age-dependent increase in CatD activity and RNA and protein levels (Wils et al., 2012; Götzl et al., 2014; Beel et al., 2017; Zhou et al., 2017b). The age-dependent increase in CatD in mouse brain is similar to the increased levels of CatD in postmortem FTD-GRN brain (Götzl et al., 2014) and may be a compensatory response to underlying lysosomal dysfunction. Although we did not perform functional assays that would clarify the status of CatD activity in living mouse brain, the normalization of postmortem CatD activity, pro-CatD levels, and LAMP-1 levels indicate a shift toward more normal lysosomal homeostasis in AAV-Grn-treated Grn−/− mice.
The reduction of CatD maturation and total activity after AAV-Grn treatment in wild-type mice was an unexpected finding because several studies suggest that progranulin may enhance CatD activity (Beel et al., 2017; Valdez et al., 2017). However, our finding that AAV-Grn enhanced relative CatD activity (activity/mature CatD protein levels) in wild-type mice is consistent with these prior studies. One potential interpretation of these data is that the high levels of progranulin in AAV-Grn-treated wild-type mice enhance in vivo CatD activity, resulting in a homeostatic reduction in CatD maturation. The mechanism behind these effects remains unclear, but will be an interesting topic for future investigation.
Progranulin-boosting therapies are a rational approach to treating patients with GRN mutations, and this study provides important preclinical data relevant to several possible strategies. First, this study together with our previous study in Grn+/− mice show that an AAV gene therapy approach can improve pathology and behavioral abnormalities caused by progranulin deficiency (Arrant et al., 2017). AAV gene therapy was shown to be effective in several NCL models (Griffey et al., 2006; Passini et al., 2006; Cabrera-Salazar et al., 2007; Sondhi et al., 2007; Macauley et al., 2012; Hughes et al., 2014; Katz et al., 2015) and recent studies in nonhuman primates have raised the possibility of widespread cortical transduction with a single thalamic AAV injection (Yazdan-Shahmorad et al., 2018). Second, this study shows that progranulin's beneficial effects are sortilin-independent, which supports the strategy of blocking sortilin-dependent progranulin uptake to increase levels of extracellular progranulin (Hu et al., 2010; Lee et al., 2014). Progranulin's neurotrophic effects were shown to be sortilin-independent in primary cultured neurons (Gass et al., 2012) and our study provides the first in vivo evidence that progranulin's beneficial effects do not require sortilin.
Because of the clear loss-of-function mechanism, progranulin-deficient neurodegenerative diseases present an attractive opportunity for effective intervention. Our data support the concept of progranulin gene therapy or related progranulin-boosting approaches and inform the future design of such treatments.
Footnotes
This work was supported by the Consortium for FTD Research and the Bluefield Project to Cure FTD, a Ruth L. Kirschstein National Research Service Award Fellowship (F32NS090678), the National Institute of Neurological Disorders and Stroke–National Institutes of Health (NIH Grant R01NS075487, P30NS47466), the National Institute on Aging–NIH (K99AG056597), and the Civitan International Research Center. We thank James Black and Miriam Roberson for help with mouse breeding and colony management; Ashley Harms for helpful discussion and technical advice regarding MHCII immunostaining; Alexandra Nicholson for technical advice regarding progranulin/sortilin coimmunoprecipitation; Robert Farese, Jr., for providing Grn−/− mice; Joachim Herz for providing progranulin constructs; Yasuo Uchiyama for providing SCMAS antibody; and Talene Yacoubian and David Standaert for providing AAV expression constructs. Behavior experiments were performed in the Evelyn F. McKnight Brain Institute Behavior Core Facility.
The authors declare no competing financial interests.
References
- Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, Yu X, Wuertzer CA, Hou H, Chiba S, Yamanouchi K, Leissring M, Petrucelli L, Nishihara M, Hutton ML, McGowan E, Dickson DW, Lewis J (2010) Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol 177:311–324. 10.2353/ajpath.2010.090915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida MR, Macário MC, Ramos L, Baldeiras I, Ribeiro MH, Santana I (2016) Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol Aging 41:200.e1–200.e5. [DOI] [PubMed] [Google Scholar]
- Arrant AE, Patel AR, Roberson ED (2015) Effects of exercise on progranulin levels and gliosis in progranulin-insufficient mice. eNeuro 2: pii: 0061. 10.1523/ENEURO.0061-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrant AE, Filiano AJ, Unger DE, Young AH, Roberson ED (2017) Restoring neuronal progranulin reverses deficits in a mouse model of frontotemporal dementia. Brain 140:1447–1465. 10.1093/brain/awx060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, et al. (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919. 10.1038/nature05016 [DOI] [PubMed] [Google Scholar]
- Bateman A, Bennett HP (1998) Granulins: the structure and function of an emerging family of growth factors. J Endocrinol 158:145–151. 10.1677/joe.0.1580145 [DOI] [PubMed] [Google Scholar]
- Beel S, Moisse M, Damme M, De Muynck L, Robberecht W, Van Den Bosch L, Saftig P, Van Damme P (2017) Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum Mol Genet 26:2850–2863. 10.1093/hmg/ddx162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Salazar MA, Roskelley EM, Bu J, Hodges BL, Yew N, Dodge JC, Shihabuddin LS, Sohar I, Sleat DE, Scheule RK, Davidson BL, Cheng SH, Lobel P, Passini MA (2007) Timing of therapeutic intervention determines functional and survival outcomes in a mouse model of late infantile batten disease. Mol Ther 15:1782–1788. 10.1038/sj.mt.6300249 [DOI] [PubMed] [Google Scholar]
- Canafoglia L, Morbin M, Scaioli V, Pareyson D, D'Incerti L, Fugnanesi V, Tagliavini F, Berkovic SF, Franceschetti S (2014) Recurrent generalized seizures, visual loss, and palinopsia as phenotypic features of neuronal ceroid lipofuscinosis due to progranulin gene mutation. Epilepsia 55:e56–e59. 10.1111/epi.12632 [DOI] [PubMed] [Google Scholar]
- Capell A, Liebscher S, Fellerer K, Brouwers N, Willem M, Lammich S, Gijselinck I, Bittner T, Carlson AM, Sasse F, Kunze B, Steinmetz H, Jansen R, Dormann D, Sleegers K, Cruts M, Herms J, Van Broeckhoven C, Haass C (2011) Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J Neurosci 31:1885–1894. 10.1523/JNEUROSCI.5757-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik B, Sephton CF, Dewey CM, Xian X, Wei S, Yu K, Niu W, Coppola G, Coughlin SE, Lee SE, Dries DR, Almeida S, Geschwind DH, Gao FB, Miller BL, Farese RV Jr, Posner BA, Yu G, Herz J (2011) Suberoylanilide hydroxamic acid (vorinostat) up-regulates progranulin transcription: rational therapeutic approach to frontotemporal dementia. J Biol Chem 286:16101–16108. 10.1074/jbc.M110.193433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik B, Sephton CF, Kutluk Cenik B, Herz J, Yu G (2012) Progranulin: a proteolytically processed protein at the crossroads of inflammation and neurodegeneration. J Biol Chem 287:32298–32306. 10.1074/jbc.R112.399170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciesielska A, Hadaczek P, Mittermeyer G, Zhou S, Wright JF, Bankiewicz KS, Forsayeth J (2013) Cerebral infusion of AAV9 vector-encoding non-self proteins can elicit cell-mediated immune responses. Mol Ther 21:158–166. 10.1038/mt.2012.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, et al. (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924. 10.1038/nature05017 [DOI] [PubMed] [Google Scholar]
- Elleder M, Sokolová J, Hrebícek M (1997) Follow-up study of subunit c of mitochondrial ATP synthase (SCMAS) in batten disease and in unrelated lysosomal disorders. Acta Neuropathol 93:379–390. 10.1007/s004010050629 [DOI] [PubMed] [Google Scholar]
- Eriksen JL, Mackenzie IR (2008) Progranulin: normal function and role in neurodegeneration. J Neurochem 104:287–297. [DOI] [PubMed] [Google Scholar]
- Filiano AJ, Martens LH, Young AH, Warmus BA, Zhou P, Diaz-Ramirez G, Jiao J, Zhang Z, Huang EJ, Gao FB, Farese RV Jr, Roberson ED (2013) Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J Neurosci 33:5352–5361. 10.1523/JNEUROSCI.6103-11.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, Bisceglio G, Rovelet-Lecrux A, Boeve B, Petersen RC, Dickson DW, Younkin SG, Deramecourt V, Crook J, Graff-Radford NR, Rademakers R (2009) Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132:583–591. 10.1093/brain/awn352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Fernández-Suárez D (2015) Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol 131:65–86. 10.1016/j.pneurobio.2015.05.003 [DOI] [PubMed] [Google Scholar]
- Gass J, et al. (2006) Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 15:2988–3001. 10.1093/hmg/ddl241 [DOI] [PubMed] [Google Scholar]
- Gass J, Lee WC, Cook C, Finch N, Stetler C, Jansen-West K, Lewis J, Link CD, Rademakers R, Nykjær A, Petrucelli L (2012) Progranulin regulates neuronal outgrowth independent of sortilin. Mol Neurodegener 7:33. 10.1186/1750-1326-7-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustino TF, Maren S (2015) The role of the medial prefrontal cortex in the conditioning and extinction of fear. Front Behav Neurosci 9:298. 10.3389/fnbeh.2015.00298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, Janssens J, van der Zee J, Lang CM, Kremmer E, Martin JJ, Engelborghs S, Kretzschmar HA, Arzberger T, Van Broeckhoven C, Haass C, Capell A (2014) Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol 127:845–860. 10.1007/s00401-014-1262-6 [DOI] [PubMed] [Google Scholar]
- Griffey MA, Wozniak D, Wong M, Bible E, Johnson K, Rothman SM, Wentz AE, Cooper JD, Sands MS (2006) CNS-directed AAV2-mediated gene therapy ameliorates functional deficits in a murine model of infantile neuronal ceroid lipofuscinosis. Mol Ther 13:538–547. 10.1016/j.ymthe.2005.11.008 [DOI] [PubMed] [Google Scholar]
- Hall NA, Lake BD, Dewji NN, Patrick AD (1991) Lysosomal storage of subunit c of mitochondrial ATP synthase in Batten's disease (ceroid-lipofuscinosis). Biochem J 275:269–272. 10.1042/bj2750269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurigot V, Marco S, Ribera A, Garcia M, Ruzo A, Villacampa P, Ayuso E, Anor S, Andaluz A, Pineda M, Garcia-Fructuoso G, Molas M, Maggioni L, Munoz S, Motas S, Ruberte J, Mingozzi F, Pumarola M, Bosch F (2013) Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. J Clin Invest. 123:3254–3271. 10.1172/jci66778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler CJ, Taylor G, Deng Q, Kukar T (2017) Intracellular proteolysis of progranulin generates stable, lysosomal granulins that are haploinsufficient in patients with frontotemporal dementia caused by GRN mutations. eNeuro 4: pii: ENEURO.0100–17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM (2010) Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68:654–667. 10.1016/j.neuron.2010.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SM, Hope KM, Xu JB, Mitchell NL, Palmer DN (2014) Inhibition of storage pathology in prenatal CLN5-deficient sheep neural cultures by lentiviral gene therapy. Neurobiol Dis 62:543–550. 10.1016/j.nbd.2013.11.011 [DOI] [PubMed] [Google Scholar]
- Izquierdo A, Wellman CL, Holmes A (2006) Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci 26:5733–5738. 10.1523/JNEUROSCI.0474-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian J, Tian QY, Hettinghouse A, Zhao S, Liu H, Wei J, Grunig G, Zhang W, Setchell KDR, Sun Y, Overkleeft HS, Chan GL, Liu CJ (2016) Progranulin recruits HSP70 to beta-glucocerebrosidase and is therapeutic against gaucher disease. EBioMedicine 13:212–224. 10.1016/j.ebiom.2016.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karperien A, Ahammer H, Jelinek HF (2013) Quantitating the subtleties of microglial morphology with fractal analysis. Front Cell Neurosci 7:3. 10.3389/fncel.2013.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz ML, Tecedor L, Chen Y, Williamson BG, Lysenko E, Wininger FA, Young WM, Johnson GC, Whiting RE, Coates JR, Davidson BL (2015) AAV gene transfer delays disease onset in a TPP1-deficient canine model of the late infantile form of Batten disease. Sci Transl Med 7:313ra180. 10.1126/scitranslmed.aac6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Nakanishi H, Saftig P, Ezaki J, Isahara K, Ohsawa Y, Schulz-Schaeffer W, Watanabe T, Waguri S, Kametaka S, Shibata M, Yamamoto K, Kominami E, Peters C, von Figura K, Uchiyama Y (2000) Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci 20:6898–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kominami E, Ezaki J, Muno D, Ishido K, Ueno T, Wolfe LS (1992) Specific storage of subunit c of mitochondrial ATP synthase in lysosomes of neuronal ceroid lipofuscinosis (Batten's disease). J Biochem 111:278–282. 10.1093/oxfordjournals.jbchem.a123749 [DOI] [PubMed] [Google Scholar]
- Krabbe G, et al. (2017) Microglial NFkappaB-TNFalpha hyperactivation induces obsessive-compulsive behavior in mouse models of progranulin-deficient frontotemporal dementia. Proc Natl Acad Sci U S A 114:5029–5034. 10.1073/pnas.1700477114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Almeida S, Prudencio M, Caulfield TR, Zhang YJ, Tay WM, Bauer PO, Chew J, Sasaguri H, Jansen-West KR, Gendron TF, Stetler CT, Finch N, Mackenzie IR, Rademakers R, Gao FB, Petrucelli L (2014) Targeted manipulation of the sortilin-progranulin axis rescues progranulin haploinsufficiency. Hum Mol Genet 23:1467–1478. 10.1093/hmg/ddt534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Hall AM, Kelinske M, Roberson ED (2014) Seizure resistance without parkinsonism in aged mice after tau reduction. Neurobiol Aging 35:2617–2624. 10.1016/j.neurobiolaging.2014.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. (2009) The multifaceted profile of activated microglia. Mol Neurobiol 40:139–156. 10.1007/s12035-009-8077-9 [DOI] [PubMed] [Google Scholar]
- Macauley SL, Roberts MS, Wong AM, McSloy F, Reddy AS, Cooper JD, Sands MS (2012) Synergistic effects of central nervous system-directed gene therapy and bone marrow transplantation in the murine model of infantile neuronal ceroid lipofuscinosis. Ann Neurol 71:797–804. 10.1002/ana.23545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens LH, Zhang J, Barmada SJ, Zhou P, Kamiya S, Sun B, Min SW, Gan L, Finkbeiner S, Huang EJ, Farese RV Jr (2012) Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J Clin Invest 122:3955–3959. 10.1172/JCI63113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, Bowles D, Gray S, Li C, Galloway G, Malik V, Coley B, Clark KR, Li J, Xiao X, Samulski J, McPhee SW, Samulski RJ, Walker CM (2010) Dystrophin immunity in Duchenne's muscular dystrophy. N Engl J Med 363:1429–1437. 10.1056/NEJMoa1000228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AD, Nguyen TA, Martens LH, Mitic LL, Farese RV Jr (2013) Progranulin: at the interface of neurodegenerative and metabolic diseases. Trends in endocrinology and metabolism: TEM 24:597–606. 10.1016/j.tem.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L, Roberson ED (2011) Quantifying biomarkers of cognitive dysfunction and neuronal network hyperexcitability in mouse models of Alzheimer's disease: depletion of calcium-dependent proteins and inhibitory hippocampal remodeling. In: Alzheimer's disease and frontotemporal dementia: methods and protocols (Roberson ED, ed), pp 245–262. Totowa, NJ: Humana. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini MA, Dodge JC, Bu J, Yang W, Zhao Q, Sondhi D, Hackett NR, Kaminsky SM, Mao Q, Shihabuddin LS, Cheng SH, Sleat DE, Stewart GR, Davidson BL, Lobel P, Crystal RG (2006) Intracranial delivery of CLN2 reduces brain pathology in a mouse model of classical late infantile neuronal ceroid lipofuscinosis. J Neurosci 26:1334–1342. 10.1523/JNEUROSCI.2676-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkau TL, Hill A, Leavitt BR (2016) Core neuropathological abnormalities in progranulin-deficient mice are penetrant on multiple genetic backgrounds. Neuroscience 315:175–195. 10.1016/j.neuroscience.2015.12.006 [DOI] [PubMed] [Google Scholar]
- Petkau TL, Blanco J, Leavitt BR (2017) Conditional loss of progranulin in neurons is not sufficient to cause neuronal ceroid lipofuscinosis-like neuropathology in mice. Neurobiol Dis 106:14–22. 10.1016/j.nbd.2017.06.012 [DOI] [PubMed] [Google Scholar]
- Petoukhov E, Fernando S, Mills F, Shivji F, Hunter D, Krieger C, Silverman MA, Bamji SX (2013) Activity-dependent secretion of progranulin from synapses. J Cell Sci 126:5412–5421. 10.1242/jcs.132076 [DOI] [PubMed] [Google Scholar]
- Roche PA, Furuta K (2015) The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol 15:203–216. 10.1038/nri3818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CL, Baranowski DC, Chitramuthu BP, Malik S, Li Z, Cao M, Minotti S, Durham HD, Kay DG, Shaw CA, Bennett HP, Bateman A (2009) Progranulin is expressed within motor neurons and promotes neuronal cell survival. BMC Neurosci 10:130. 10.1186/1471-2202-10-130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranch L, Sebastian WS, Kells AP, Salegio EA, Heller G, Bringas JR, Pivirotto P, DeArmond S, Forsayeth J, Bankiewicz KS (2014) AAV9-mediated expression of a non-self protein in nonhuman primate central nervous system triggers widespread neuroinflammation driven by antigen-presenting cell transduction. Mol Ther 22:329–337. 10.1038/mt.2013.266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhoff K, Harzer K (2013) Gangliosides and gangliosidoses: principles of molecular and metabolic pathogenesis. J Neurosci 33:10195–10208. 10.1523/JNEUROSCI.0822-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze H, Sandhoff K (2014) Sphingolipids and lysosomal pathologies. Biochim Biophys Acta 1841:799–810. 10.1016/j.bbalip.2013.10.015 [DOI] [PubMed] [Google Scholar]
- Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, Haass C (2008) Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem 283:1744–1753. 10.1074/jbc.M705115200 [DOI] [PubMed] [Google Scholar]
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, Sims KB, Lewis J, Lin WL, Dickson DW, Dahl HH, Bahlo M, Berkovic SF (2012) Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet 90:1102–1107. 10.1016/j.ajhg.2012.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondhi D, Hackett NR, Peterson DA, Stratton J, Baad M, Travis KM, Wilson JM, Crystal RG (2007) Enhanced survival of the LINCL mouse following CLN2 gene transfer using the rh.10 rhesus macaque-derived adeno-associated virus vector. Mol Ther 15:481–491. 10.1038/sj.mt.6300049 [DOI] [PubMed] [Google Scholar]
- St Martin JL, Klucken J, Outeiro TF, Nguyen P, Keller-McGandy C, Cantuti-Castelvetri I, Grammatopoulos TN, Standaert DG, Hyman BT, McLean PJ (2007) Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J Neurochem 100:1449–1457. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Chambers JK, Matsuwaki T, Yamanouchi K, Nishihara M (2014) Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol Commun 2:78. 10.1186/s40478-014-0078-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez C, Wong YC, Schwake M, Bu G, Wszolek ZK, Krainc D (2017) Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum Mol Genet 26:4861–4872. 10.1093/hmg/ddx364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Van Hoecke A, Lambrechts D, Vanacker P, Bogaert E, van Swieten J, Carmeliet P, Van Den Bosch L, Robberecht W (2008) Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol 181:37–41. 10.1083/jcb.200712039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Van Damme P, Cruchaga C, Gitcho MA, Vidal JM, Seijo-Martínez M, Wang L, Wu JY, Robberecht W, Goate A (2010) Pathogenic cysteine mutations affect progranulin function and production of mature granulins. J Neurochem 112:1305–1315. 10.1111/j.1471-4159.2009.06546.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward ME, et al. (2017) Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci Transl Med 9: pii: eaah5642. 10.1126/scitranslmed.aah5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, Pereson S, Janssens J, Capell A, Van Dam D, Cuijt I, Joris G, De Deyn PP, Haass C, Van Broeckhoven C, Kumar-Singh S (2012) Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J Pathol 228:67–76. 10.1002/path.4043 [DOI] [PubMed] [Google Scholar]
- Yang ST, Shi Y, Wang Q, Peng JY, Li BM (2014) Neuronal representation of working memory in the medial prefrontal cortex of rats. Mol Brain 7:61. 10.1186/s13041-014-0061-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda Y, Kageyama T, Akamine A, Shibata M, Kominami E, Uchiyama Y, Yamamoto K (1999) Characterization of new fluorogenic substrates for the rapid and sensitive assay of cathepsin E and cathepsin D. J Biochem 125:1137–1143. 10.1093/oxfordjournals.jbchem.a022396 [DOI] [PubMed] [Google Scholar]
- Yazdan-Shahmorad A, Tian N, Kharazia V, Samaranch L, Kells A, Bringas J, He J, Bankiewicz K, Sabes PN (2018) Widespread optogenetic expression in macaque cortex obtained with MR-guided, convection enhanced delivery (CED) of AAV vector to the thalamus. J Neurosci Methods 293:347–358. 10.1016/j.jneumeth.2017.10.009 [DOI] [PubMed] [Google Scholar]
- Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A (2010) Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med 207:117–128. 10.1084/jem.20091568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa K, Yoshimura M, Urasawa N, Ohshima S, Howell JM, Nakamura A, Hijikata T, Miyagoe-Suzuki Y, Takeda S (2007) Injection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene products. Gene Ther 14:1249–1260. 10.1038/sj.gt.3302984 [DOI] [PubMed] [Google Scholar]
- Zheng Y, Brady OA, Meng PS, Mao Y, Hu F (2011) C-terminus of progranulin interacts with the beta-propeller region of sortilin to regulate progranulin trafficking. PLoS One 6:e21023. 10.1371/journal.pone.0021023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Sun L, Bastos de Oliveira F, Qi X, Brown WJ, Smolka MB, Sun Y, Hu F (2015) Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin. J Cell Biol 210:991–1002. 10.1083/jcb.201502029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Sun L, Brady OA, Murphy KA, Hu F (2017a) Elevated TMEM106B levels exaggerate lipofuscin accumulation and lysosomal dysfunction in aged mice with progranulin deficiency. Acta Neuropathol Commun 5:9. 10.1186/s40478-017-0412-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Paushter DH, Feng T, Pardon CM, Mendoza CS, Hu F (2017b) Regulation of cathepsin D activity by the FTLD protein progranulin. Acta Neuropathol 134:151–153. 10.1007/s00401-017-1719-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Paushter DH, Feng T, Sun L, Reinheckel T, Hu F (2017c) Lysosomal processing of progranulin. Mol Neurodegener 12:62. 10.1186/s13024-017-0205-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, Ding A (2002) Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell 111:867–878. 10.1016/S0092-8674(02)01141-8 [DOI] [PubMed] [Google Scholar]