

Abstract
Deregulation of the cyclin‐dependent kinases (CDKs) has been implicated in the pathogenesis of multiple cancer types. Consequently, CDKs have garnered intense interest as therapeutic targets for the treatment of cancer. We describe herein the molecular and cellular effects of CCT068127, a novel inhibitor of CDK2 and CDK9. Optimized from the purine template of seliciclib, CCT068127 exhibits greater potency and selectivity against purified CDK2 and CDK9 and superior antiproliferative activity against human colon cancer and melanoma cell lines. X‐ray crystallography studies reveal that hydrogen bonding with the DFG motif of CDK2 is the likely mechanism of greater enzymatic potency. Commensurate with inhibition of CDK activity, CCT068127 treatment results in decreased retinoblastoma protein (RB) phosphorylation, reduced phosphorylation of RNA polymerase II, and induction of cell cycle arrest and apoptosis. The transcriptional signature of CCT068127 shows greatest similarity to other small‐molecule CDK and also HDAC inhibitors. CCT068127 caused a dramatic loss in expression of DUSP6 phosphatase, alongside elevated ERK phosphorylation and activation of MAPK pathway target genes. MCL1 protein levels are rapidly decreased by CCT068127 treatment and this associates with synergistic antiproliferative activity after combined treatment with CCT068127 and ABT263, a BCL2 family inhibitor. These findings support the rational combination of this series of CDK2/9 inhibitors and BCL2 family inhibitors for the treatment of human cancer.
Keywords: ABT263, CCT068127, CDK, MCL1, seliciclib
Abbreviations
- BrdU
bromodeoxyuridine
- CDK
cyclin‐dependent kinase inhibitor
- CDKI
cyclin‐dependent kinase inhibitor
- CI
combination index
- GI50
50% growth inhibition
- HDAC
histone deacetylase
- IAP
inhibitor of apoptosis
- MAPK
mitogen‐activated protein kinase
- RT‐qPCR
real‐time quantitative polymerase chain reaction
- SDS/PAGE
sodium dodecyl sulfate/polyacrylamide gel electrophoresis
- siRNA
short interfering ribonucleic acid
1. Introduction
Loss of cell cycle control is a hallmark of human cancer, and this can be achieved by overexpression of the CDK partner cyclins or by decreased expression or mutation of CDK inhibitors (CDKIs) from the INK4 and CIP/KIP families (Malumbres and Barbacid, 2009). For example, amplification of the 19q12 locus has been reported in up to 15% of estrogen receptor (ER)‐negative breast cancers, typically resulting in amplification of CCNE1 (cyclin E1) (Adélaïde et al., 2007; Natrajan et al., 2012). The G1‐to‐S‐phase transition is regulated by CDK4/cyclin D and CDK6/cyclin D. CDK2/cyclin A, CDK2/cyclin E, and CDK1/cyclin A mediate progression through S phase to G2 and CDK1/cyclin B activity is involved in the initiation of mitosis (Malumbres and Barbacid, 2009). Importantly, CDK activities converge to phosphorylate the RB protein that functions to suppress the activity of E2F‐1, which in turn is required to stimulate the transcription of genes required for DNA synthesis. Current evidence supports a model whereby cumulative phosphorylation of RB displaces bound histone deacetylase (HDAC) and E2F‐1, relieving transcriptional repression and permitting gene transcription from E2F‐1‐dependent promoters (Zhang et al., 2000).
Targeting of CDK2/cyclin E activity by RNA interference or by small molecules is effective against human breast cancer cell lines exhibiting CCNE1 amplification (Natrajan et al., 2012). CDKs are not only involved in orchestrating the correct passage through the cell cycle, but also key mediators of transcriptional control, in part by the regulation of RNA polymerase II (Bregman et al., 2000). CDK7/cyclin H and CDK9/cyclin T phosphorylate RNA polymerase II on its C‐terminal, which is permissive for initiation and elongation of nascent mRNA transcripts. Interestingly, a number of transcripts involved in cell cycle control and apoptosis are highly responsive to alterations in transcriptional regulation, the mRNAs having short half‐lives, and are thus rapidly lost following inhibition of transcription (Lam et al., 2001). Hence, some CDK inhibitors are capable of suppressing the transcription of key genes such as MCL1, MYC, CCNB2 (cyclin B2), and several IAP family members (BIRC1, BIRC2, and BIRC3). Therefore, CDK inhibitors have the potential to perturb cell cycle progression through direct catalytic inhibition of CDK/cyclin complexes, loss of expression of the partner cyclins, and also sensitize cells to proapoptotic stimuli.
While it has been reported that CDKs can be dispensable for survival or cell cycle progression, it appears that this is due to a high degree of redundancy among the CDKs and their partner cyclins. For example, CDK2 knockout in mice proved to be nonlethal and RNAi‐mediated suppression of CDK2 did not affect cell proliferation (Ortega et al., 2003; Tetsu and McCormick, 2003). However, CDK1 is essential for survival in mice (Diril et al., 2012) and dual suppression of CDK1 and CDK2 presumably overcomes any redundancy in CDK2 function, resulting in cell cycle arrest and/or apoptosis (L'Italien et al., 2006). Combinatorial targeting of multiple CDKs therefore remains an attractive approach for cancer therapy (Whittaker et al., 2017).
We embarked upon a drug discovery program to develop CDK2 and 9 inhibitors that exhibited significantly improved pharmacological properties compared with the parental clinical drug seliciclib (CYC202, R‐roscovitine) (McClue et al., 2002; Raynaud et al., 2005; Whittaker et al., 2004). To this end, we identified CCT068127, a novel trisubstituted purine with improved potency, selectivity, metabolic stability, and antitumor activity compared with seliciclib (Wilson et al., 2011). Here, we describe the biochemical and cellular characterization of CCT068127 and identify synergistic drug combinations to further improve the efficacy of such CDK2/9 inhibitors for the treatment of colorectal cancer.
2. Materials and methods
2.1. Cell culture and reagents
All human cancer cell lines (HT29, colorectal adenocarcinoma; HCT116, colon carcinoma; COLO205, colon adenocarcinoma; RKO, colon carcinoma; SKMEL28, malignant melanoma; WM266.4, malignant melanoma) were obtained from the American Type Culture Collection and grown in the recommended culture medium, supplemented with 10% FBS, at 37 °C and an atmosphere of 5% CO2. Cell lines were passaged for less than 6 months upon receipt. CCT068127 was synthesized as described (Wilson et al., 2011) and solubilized in dimethyl sulfoxide to a stock concentration of 10 mmol·L−1. The BCL2 family inhibitor ABT263 was purchased from Selleck Chemicals (Tse et al., 2008). The ERK inhibitor VTX‐11e was purchased from Tocris (Aronov et al., 2009).
2.2. In vitro biochemical kinase assays
Profiling of CCT068127 against a panel of ~ 30 recombinant human kinases was performed as previously described (McIntyre et al., 2010; Wang et al., 2004; Wilson et al., 2011).
2.3. Enzyme expression and purification
The gene encoding full‐length human CDK2 was cloned into the pGEX‐6P‐1 and transformed into BL21(DE3)pLysS cells (Promega, Southampton, UK). Cultures were grown for 2–3 h at 37 °C, and then, the temperature was decreased to 18 °C prior to induction with 0.25 mmol·L−1 IPTG at OD600 = 0.6. The cultures were allowed to grow for an additional 20–24 h at 18 °C and were harvested by centrifugation. All purification steps were performed by FPLC at 4 °C. Harvested cells were resuspended in 50 mmol·L−1 HEPES buffer (pH 7.5) containing 150 mmol·L−1 NaCl, 2 mmol·L−1 DTT, and 0.5 mg·mL−1 lysozyme at 4 °C for 1 h. After sonication and centrifugation (1 h at 29 000 g), the supernatant was purified by immobilized glutathione Sepharose chromatography (GE LifeSciences, Little Chalfont, UK). Following incubation of peak fractions with 3C protease (20 : 1) at 4 °C, the cleaved GST‐tag was separated by size exclusion chromatography using a Superdex 75 (26/60) column and eluted with 50 mmol·L−1 HEPES buffer (pH 7.4) containing 150 mmol·L−1 NaCl and 1 mmol·L−1 DTT. Purified CDK2 was buffer‐exchanged into 100 mmol·L−1 Na/K phosphate buffer (pH 7.4) containing 1 mmol·L−1 DTT and concentrated to 10 mg·mL−1 for crystallization.
2.4. X‐ray crystallography
Crystallization was performed at 20 °C using the sitting drop vapor diffusion method. Crystals of human CDK2 were grown from 0.1 m HEPES pH 7.5 and 10% PEG3350 over a reservoir of 50 mmol·L−1 HEPES pH7.5 and 50 mmol·L−1 Na/K phosphate buffer (pH 7.4). Crystals were harvested in cryoprotectant containing 1 mmol·L−1 CCT068127 (50 mmol·L−1 HEPES pH 7.5, 50 mmol·L−1 Na/K phosphate buffer (pH 7.4), 15% PEG3350, 25% (v/v) ethylene glycol, 1% DMSO, and 1 mmol·L−1 CCT068127) 24 h prior to data collection. X‐ray diffraction data were recorded at Diamond Light Source (Harwell, Oxfordshire, UK). Data processing was carried out using XDS, POINTLESS/AIMLESS (Evans, 2011), and other programs of the CCP4i suite (Collaborative Computational Project, 1994) run through the CCP4i2 GUI. The structure was solved by molecular replacement using PHASER (McCoy et al., 2007) and pdb 3QXP as a starting model. REFMAC (Murshudov et al., 1997) was employed for refinement, and model building was performed using COOT (Emsley et al., 2010). Figures were prepared using PYMOL (Schrödinger, LLC, Cambridge, UK). The coordinates of the CDK2‐CCT068127 crystal structure have been deposited in the Protein Data Bank with accession code 5MHQ.
2.5. Cell proliferation
The effect of CCT068127 and seliciclib on cancer cell proliferation was measured using the sulforhodamine B (SRB) assay as described previously (Whittaker et al., 2004). For long‐term colony formation assays, HT29 colon cancer cells were seeded into 12‐well plates and then treated with the inhibitors for 5 days. Compounds were then washed away and the cells were allowed to proliferate for a further 7 days. After this, cells were fixed with 4% formaldehyde for 30 min, and cellular protein and DNA were stained with 0.5% crystal violet solution for 30 min and washed in water to remove excess dye. The colony plates were imaged on a FluorChem E System (ProteinSimple, Oxford, UK). Bound dye was subsequently resolubilized in 10% acetic acid and absorbance measured at 595 nm to quantify cell number.
2.6. Western blotting
Cells were seeded at a density of approximately 4 × 105 cells/well in 6‐well plates and incubated overnight prior to drug treatment. They were then harvested in NP‐40 buffer as previously described (Whittaker et al., 2004) and lysates were normalized using the Bradford assay (Bio‐Rad, Watford, UK). Equal amounts of protein were resolved by SDS/PAGE using 4–12% bis‐tris gels (or 6% tris‐glycine gels to demonstrate a mobility shift in RB in Fig. 3D) and transferred to nitrocellulose membranes (Life Technologies, Paisley, UK). The antibodies used in this study are described in Table S1. Primary antibodies were labeled with fluorescently labeled IRDye secondary antibodies (Li‐Cor, Cambridge, UK), and proteins were visualized using an Odyssey scanner (Li‐Cor).
Figure 3.
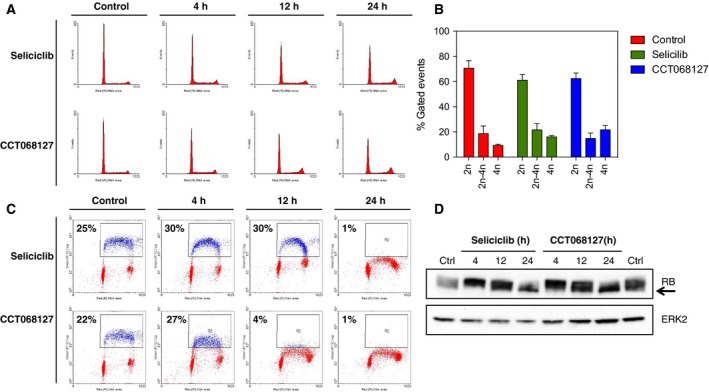
Induction of cell cycle arrest by CCT068127 and seliciclib. (A) HT29 human colon cancer cells were incubated with equiactive (3xGI 50, SRB assay) concentrations of CCT068127 or seliciclib for 4‐24 h. Cell cycle distribution was assessed by propidium iodide staining and analysis by flow cytometry. (B) Quantification of cell cycle distribution profiles from (A). Data presented are the mean of three independent experiments ± SE. (C) HT29 cells were treated as in (A) and were pulse‐labeled with BrdU, then fixed, and stained with propidium iodide. BrdU incorporation was determined using a FITC‐conjugated antibody to BrdU and detected by flow cytometry. The percentage of BrdU positive cells is indicated. (D) HT29 cells were treated as in (A) and cell lysates were analyzed by western blotting for the indicated proteins. The arrow indicates hypophosphorylated RB.
2.7. Cell cycle effects
Cell cycle analysis was performed as previously described (Whittaker et al., 2004). Briefly, for cell cycle distribution profiles, cells were exposed to inhibitors for the indicated times, and then attached cells were harvested by trypsinization. Cells were washed in PBS and fixed in ice‐cold 70% ethanol. Cellular DNA was stained using propidium iodide and analyzed by flow cytometry. To determine the number of cells undergoing DNA synthesis, cells were pulsed with 10 μmol·L−1 bromodeoxyuridine (BrdU, Sigma, Gillingham, UK) for 30 minutes prior to harvesting. Cells were fixed in 70% ethanol and nuclei were isolated using 2 mol·L−1 HCl and 0.2 mg·mL−1 pepsin. Incorporated BrdU was detected using an anti‐BrdU antibody coupled to FITC (Molecular Probes, Paisley, UK), and cells were quantified by flow cytometry.
2.8. Gene expression analysis
Cells were exposed to the inhibitors for 24 h prior to RNA extraction with Trizol (Invitrogen, Paisley, UK) and subsequent mRNA purification by Oligotex (Qiagen, Manchester, UK). Eight microgram of mRNA was labeled with either Cy3 or Cy5 dCTP via RT‐PCR and the labeled probe was then purified and 2 μg of each sample was hybridized to a custom cDNA array of 5808 genes spotted onto poly‐lysine‐coated glass slides at 65 °C overnight. Slides were scanned using a GenePix 4000B (Axon Labs, Union City, CA, USA) and spots verified using genepix software. Data analysis was performed in GeneSpring (Silicon Genetics, Redwood City, CA, USA). Gene expression data were analyzed relative to the DMSO controls by comparative marker selection using a t‐test to rank expression changes in Gene‐E (http://www.broadinstitute.org/cancer/software/GENE-E/). A ≥ twofold change in expression was used to refine the selection criteria. Gene expression profiles were analyzed using Connectivity Map (Lamb et al., 2006) and STRING databases (Franceschini et al., 2013).
For RT‐qPCR assays, mRNA was isolated from cancer cells using RNeasy Mini kit (Qiagen) according to the manufacturer's recommendations. Extracted RNA was used to generate cDNA with the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Paisley, UK). Primer combinations for the respective genes were designed according to the Harvard Primer Bank (http://pga.mgh.harvard.edu/primerbank) and are listed in Table S2. Gene expression was analyzed using Power SYBR Green gene expression assays (Life Technologies). Quantitative PCR was carried out in triplicate on the ViiA 7 Real‐Time PCR System (Applied Biosystems).
2.9. siRNA transfection
HT29 cells were reverse‐transfected in 6‐well or 96‐well plates using 0.4% RNAiMAX (Life Technologies) transfection reagent and 25 nm of ON‐TARGETplus siRNA (Dharmacon, Cambridge, UK) in OptiMem. Cells were then cultured for 4 days and analyzed as required. A nontargeting siRNA (siCTRL) and four different siRNAs targeting CDK5 were used (siCTRL: D‐001810‐01‐05; siCDK5: J‐003239‐9, J‐003239‐10, J‐003239‐11, J‐003239‐12).
2.10. Statistical analysis
For statistical analysis of protein expression data, the mean of three independent repeats per condition was used for two‐tailed t‐tests to determine significance of change, relative to the appropriate control. This was deemed the most appropriate test as nonparametric tests such as the Mann–Whitney U‐test require greater sample numbers to enable significance testing, as discussed by Krzywinski and Altman (2014).
3. Results
3.1. CCT068127 is a potent and stable inhibitor of CDK2 and 9
We previously reported a medicinal chemistry campaign to generate more potent, selective, and metabolically stable analogs of the clinical cyclin‐dependent kinase inhibitor seliciclib (CYC202, R‐roscovitine) (Wilson et al., 2011). CCT068127 was identified as having significantly increased potency toward human CDK1/cyclin B (15‐fold), CDK2/cyclin E (22‐fold), CDK5 (15‐fold), and CDK9/cyclin T (11‐fold) kinase activities, when compared to the parental seliciclib (Table 1). Importantly, selectivity toward CDK2 and 9 versus CDK4 and CDK7 is enhanced compared to seliciclib, as the concentration required to inhibit kinase activity by 50% (IC50) remained relatively unchanged for CDK4/cyclin D and CDK7/cyclin H. Although the potency of CCT068127 for CDK1 is increased over seliciclib, the IC50 is still relatively high at 1.12 μm. Overall, CCT068127 showed greatest in vitro potency against CDK2, CDK5, and CDK9.
Table 1.
In vitro kinase inhibition by CCT068127 and seliciclib. Compounds were tested against human recombinant enzymes in vitro as previously described (McIntyre et al., 2010; Wang et al., 2004; Wilson et al., 2011) except for CDK5/p35, which was carried out using the Invitrogen Select Screen service. Results are the mean of three independent repeats ± SE except for CDK5/p35, which is the mean of two determinations. The following kinases had IC50 values greater than 10 μmol·L−1 for both compounds: ABL, AKT, CAMKII, CK2, ERK2, GSK3, PKA, PKC, PLK1, S6, SAP2KA, AURKA, FLT3, SRC, LCK, PDGFB, AURKB, VEGFR1, VEGFR2, and IKKA. Data for seliciclib were reported previously (Mariaule and Belmont, 2014; Wilson et al., 2011)
| Kinase assay | CCT068127 Mean IC50 (μmol·L−1) ± SE | Seliciclib IC50 (μmol·L−1) |
|---|---|---|
| CDK1/B | 1.1 ± 0.29 | 17 |
| CDK2/A | 0.11 ± 0.02 | ND |
| CDK2/E | 0.010 ± 0.001 | 0.22 |
| CDK4/D1 | 4.8 ± 0.6 | 27 |
| CDK5/p35 | 0.07 | 1.0 |
| CDK6/D3 | 6.2 ± 0.6 | ND |
| CDK7/H | 0.52 ± 0.09 | 0.52 |
| CDK9/T | 0.09 ± 0.03 | 0.80 |
CCT068127 displays notably improved cellular activity compared with seliciclib, with the average concentration to reduce cell proliferation by 50% (GI50) in a panel of human colon cancer and melanoma cell lines being 0.5 μmol·L−1 versus 12 μmol·L−1, respectively. This 20‐fold increase in cellular potency was consistent with the enhanced activity against CDK2, CDK5, and CDK9 (Table 2). In addition, we have previously reported that CCT068127 displays greater metabolic stability than seliciclib (Wilson et al., 2011).
Table 2.
Antiproliferative activity of CCT068127 and seliciclib in human colon cancer and melanoma cell lines. GI50 values for cell proliferation were determined in human colon cancer and melanoma cell lines following exposure for four population doublings using SRB analysis as the endpoint (Whittaker et al., 2004). Values reported are the mean of three independent repeats ± SE. ND, not determined
| Cell line | Tissue | CCT068127 GI50 (μmol·L−1) ± SE | Seliciclib GI50 (μmol·L−1) ± SE |
|---|---|---|---|
| HT29 | Colorectal adenocarcinoma | 0.85 ± 0.03 | 15 ± 3.4 |
| HCT116 | Colon carcinoma | 0.25 ± 0.03 | 6.4 ± 1.8 |
| COLO205 | Colon adenocarcinoma | 0.50 ± 0.09 | 11 ± 1.2 |
| RKO | Colon carcinoma | 0.66 ± 0.03 | 10 ± 0.26 |
| SKMEL28 | Malignant melanoma | 0.31 ± 0.07 | 8.5 ± 2.1 |
| WM266.4 | Malignant melanoma | 0.40 ± 0.06 | 18 ± 2.4 |
| Average | – | 0.5 | 12 |
3.2. Crystal structure of CCT068127 bound to CDK2
Based on structure–activity relationships for synthesized analogs (Wilson et al., 2011), we hypothesized that the more potent inhibition of CDK2 and CDK9 for CCT068127 in comparison with seliciclib (Table 1) is due to the presence of the pyridyl nitrogen and the methyl group of defined stereochemistry in the hydroxyalkyl side chain of CCT068127 (Fig. 1A). Our initial in silico docking suggested that, compared to seliciclib, the hydroxyl group of CCT068127 could form an additional hydrogen bond with Asp145 of the DFG motif in human CDK2 (data not shown).
Figure 1.
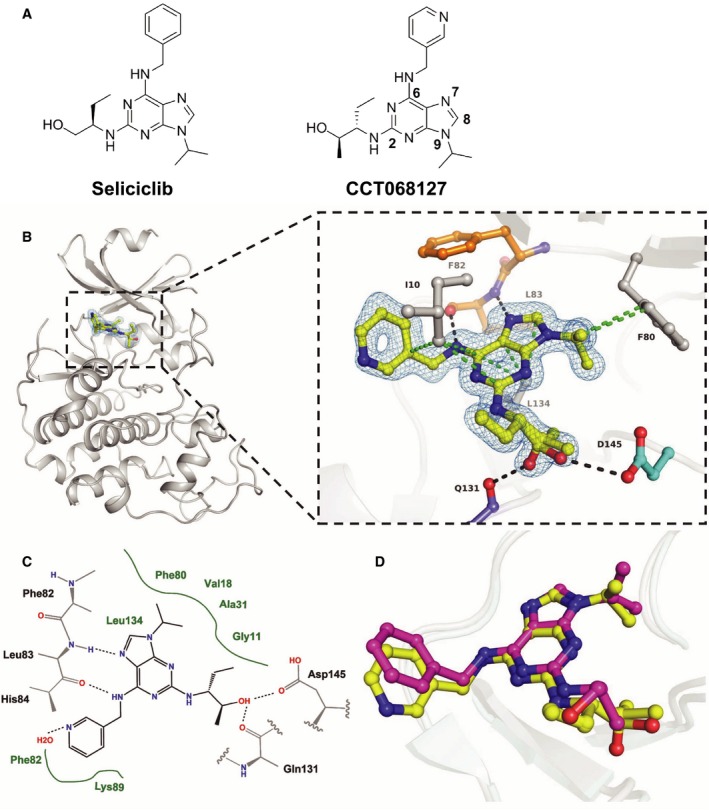
Enhanced potency of CCT068127 over seliciclib is achieved by additional ligand interactions with CDK2 and CDK9. (A) Chemical structures of seliciclib and CCT068127; numbering of the purine scaffold is indicated for CCT068127. (B) Secondary structure representation of human CDK2 in complex with CCT068127 determined by X‐ray crystallography at 1.3 Å resolution. The inset shows the binding interactions of CCT068127 within the ATP binding pocket. The hinge region is indicated in orange, Asp145 of the DFG motif in cyan, and CCT068127 in yellow. Displayed in blue is the 2Fo‐Fc electron density, contoured at 1σ around CCT068127. The data and refinement statistics are shown in the Supporting information (Table S3). The hydrogen bonding and van der Waals (hydrophobic) interactions are shown as black and green dotted lines, respectively. PDB: 5MHQ. (C) Schematic presentation of the binding interactions between CCT068127 and CDK2. (D) Alignment of CCT068127 (yellow) and seliciclib (magenta) (PDB: 3DDQ) bound to CDK2.
To determine the precise binding mode, we determined the X‐ray crystal structure of CCT068127 with CDK2 (Fig. 1B). The CCT068127–CDK2 complex formed crystals with space group P212121 that diffracted to 1.3Å resolution. The structure was refined to Rfactor and Rfree values of 19.6% and 23.4%, respectively (Table S3). This shows that CCT068127 acts as a type I kinase inhibitor of CDK2 and binds to the hinge region of the ATP binding pocket through two hydrogen bonds, formed by the purine N7 and the exocyclic nitrogen at C6 of the purine scaffold that interact with the main chain amine and carbonyl of Leu83, respectively (Fig. 1B,C). In addition, our high‐resolution electron density map of the inhibitor–CDK2 complex has allowed unambiguous assignment of the conformation of the hydroxymethyl moiety. As predicted by our modeling, the side chain hydroxyl forms an additional hydrogen bond with the carboxylic acid side chain of Asp145, part of the DFG motif. Electron density indicates two alternate conformations for the hydroxyl group, with the minor pose forming an interaction with the main chain carbonyl of Gln131. Also CCT068127 forms multiple van der Waals (hydrophobic) interactions with surrounding residues (3.4Å < d < 4.0Å). The purine core is sandwiched between Ile10 and Leu134, with the N9‐isopropyl moiety stacking up against the gatekeeper residue Phe80.
Superimposition of CDK2 structures bound to CCT068127 and seliciclib illustrates a key rotation of the C2 substituent that orients the pendant hydroxyl moiety toward Asp145. The additional hydrogen bound formed in this altered pose provides a plausible explanation for the increased potency CCT068127 relative to seliciclib (Fig. 1D). The pyridine moiety of CCT068127 is slightly shifted relative to the phenyl group of seliciclib (Fig. 1D) possibly resulting from the engagement of the pyridine nitrogen in water‐mediated hydrogen bonds with CDK2.
3.3. Inhibition of CDK2 and CDK9 activity correlates with decreased substrate phosphorylation in cells
To confirm that CCT068127 retains activity against CDKs in cells, the phosphorylation of selected CDK substrates was determined. We used a cell‐based assay for protein phosphatase 1 phosphorylation to measure CDK1 activity following compound treatment. PP1 phosphorylation at T320 shows inhibition with an IC50 of 6 μm for CCT068127 and 71 μm for seliciclib, 4–9 times greater than the respective GI50 values (Fig. S1) (Kwon et al., 1997). Of note, cyclin B1 expression was reduced only at higher concentrations, with an IC50 of 15 μm for CCT068127 and >100 μm for seliciclib, suggesting that loss of cyclin B1 contributes to, but is not primarily responsible for, the antiproliferative activity of these compounds. Phosphorylation of the RB protein by CDKs facilitates DNA synthesis and cell cycle progression through the displacement of HDACs and E2F family transcription factors from RB (Brehm et al., 1998). The phosphorylation status of RB was therefore assessed following a 24‐h exposure to CCT068127 or seliciclib in HT29, RKO, and COLO205 human colon cancer cells (Figs 2A and S2A). We found that a concentration of 3 μmol·L−1 CCT068127 or greater reduces phosphorylation of RB at S780, whereas 30 μmol·L−1 seliciclib is required to elicit the same effect, consistent with at least a 10‐fold greater potency of CCT068127 over seliciclib in the biochemical assays with recombinant proteins. Notably, the inhibition of RB phosphorylation was only partial (~50%) as determined by quantification of the blots and was less pronounced in the COLO205 cells, due to the loss of total protein (Figs 2B and S2B).
Figure 2.
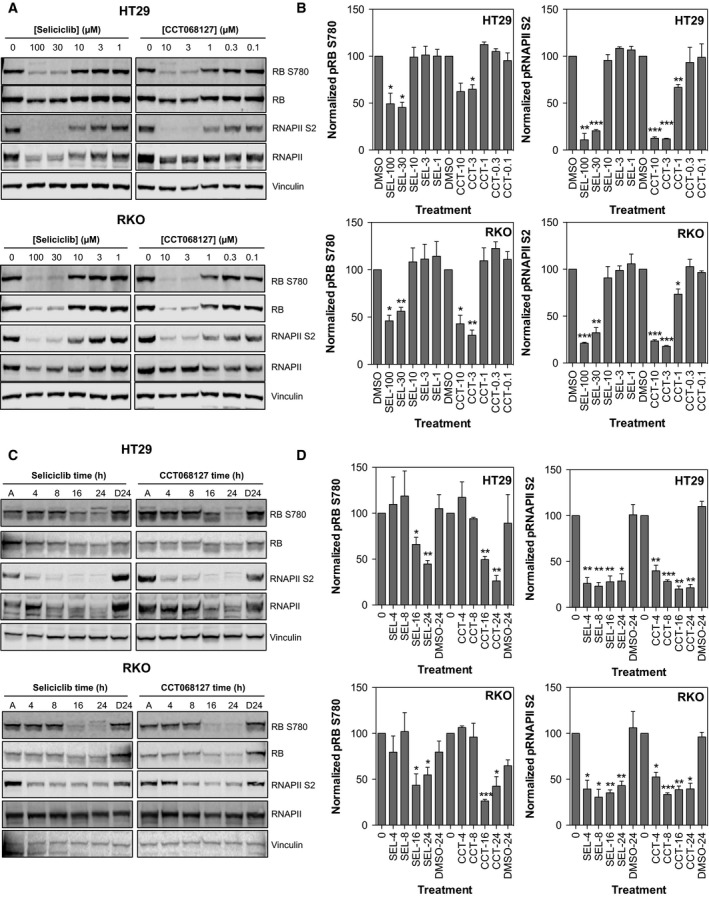
CCT068127 potently inhibits RB and RNA polymerase II phosphorylation in human cancer cells. (A) HT29 and RKO colon cancer cells were treated with increasing concentrations of CCT068127 or seliciclib for 24 h. Cell lysates were analyzed by western blotting for the phosphorylation of RB (a measure of CDK2 inhibition) and phosphorylation of RNA polymerase II (a measure of CDK9 inhibition). (B) Quantification of RB S780 and RNAPII S2 phosphorylation in (A), normalized to total RB or total RNAPII, respectively, and expressed as a percentage of the DMSO control (mean percentage of control values presented from three independent repeats ± SE, significant difference from control indicated by *P < 0.05, **P < 0.01, ***P < 0.001, two‐tailed t‐test). (C) HT29 and RKO cells were treated with equiactive (3xGI 50, SRB assay) concentrations of CCT068127 (2.55 μm for HT29 and 1.98 μm for RKO) or seliciclib (45 μm for HT29 and 30 μm for RKO) for the indicated times. A: asynchronously growing, untreated cells; D24: cells treated with DMSO for 24 h. Cell lysates were analyzed by western blotting for the indicated proteins. (D) Quantification of RB S780 and RNAPII S2 phosphorylation in (C), normalized to total RB or total RNAPII, respectively, and expressed as a percentage of the time 0 control (mean percentage of control values presented from three independent repeats ± SE, significant difference from control indicated by *P < 0.05, **P < 0.01, ***P < 0.001, two‐tailed t‐test).
In addition to effects on the cell cycle mediated through inhibition of RB phosphorylation, CDKs are known to regulate transcription through modification of RNA polymerase II, which is required for the generation of some transcripts arising from pol II‐dependent promoters (Oelgeschlager, 2002). Phosphorylation of RNA polymerase II at S5 and S2 is catalyzed by CDK7 and CDK9, respectively, although other CDKs have also been shown to target these sites (Jeronimo et al., 2016). Inhibition of these CDKs by compounds such as flavopiridol and seliciclib has been shown to have dramatic effects on the transcriptional machinery leading to a profound inhibition of transcription (Lam et al., 2001; Whittaker et al., 2004, 2007). RNA polymerase II phosphorylation was assessed at S2 following treatment with CCT068127 or seliciclib (Figs 2A and S2A). We found that inhibition of RNA polymerase II phosphorylation occurs following treatment with 3 μmol·L−1 or greater CCT068127. Seliciclib is less potent, again requiring approximately 30 μmol·L−1 to reduce RNA polymerase II phosphorylation to equivalent levels. This is likely in part due to the loss of total protein as previously observed (Whittaker et al., 2004). In contrast to RB, RNA polymerase II S2 phosphorylation was more completely inhibited (up to 80%) by both compounds (Figs 2B and S2B). Interestingly, we found that phosphorylation of S5 of RNA polymerase II was less sensitive to treatment and reflected loss of the total protein more than specific loss of phosphorylation (data not shown).
Time‐course experiments using a 3xGI50 concentration of both compounds were also performed in HT29 and RKO human colon cancer cells. Equiactive concentrations of each compound were used to enable a comparison of the molecular alterations induced by each compound at concentrations that have similar effects on cell proliferation/survival. We found that both compounds inhibit RB phosphorylation with similar kinetics from 16 to 24 h (Figs 2C,D and S2C,D). RNA polymerase II phosphorylation is inhibited more rapidly, with both compounds reducing phosphorylation at S2 from 4 to 24 h (Figs 2C,D, and S2C,D). As expected, the inhibition of RNA polymerase II phosphorylation is associated with decreased expression of the protein at later time points. These data demonstrate that loss of RNA polymerase II phosphorylation precedes loss of RB phosphorylation. To evaluate whether the expression of CDKs was being modulated by seliciclib and CCT068127, the levels of CDK1, 2, 5, and 9 were assessed following a 24‐h exposure to these compounds (Fig. S3A). The expression of these proteins was largely unaltered, with the exception of CDK9, which was decreased at 30–100 μm seliciclib and 3–10 μm CCT068127 in HT29 cells and at 100 μm seliciclib and 10 μm CCT068127 in RKO cells. The expression of cyclins E1, T1, B1, and D1 was also assessed and cyclin D1 levels were found to be decreased by CCT068127 and seliciclib (Fig. S3A); more modest decreases in cyclin T1 were observed with CCT068127. Notably, cyclin E1 expression was increased by seliciclib (10–30 μm) and by CCT068127 (0.3–1 μm) (Fig. S3A). This may reflect inhibition of CDK2, which is known to phosphorylate cyclin E1 and target it for degradation (Clurman et al., 1996), and has been reported as a marker of CDK2 inhibition (Sakurikar et al., 2016). We could not detect expression of the p35 regulatory subunit of CDK5, which is reportedly expressed primarily in postmitotic neurons, although a role in cancer is emerging (Shupp et al., 2017). To specifically address the effect of reduced CDK5 activity on cell proliferation, we used four different siRNAs to deplete HT29 cells of CDK5 by 80–90% (Fig. S4A). Under these conditions, we observed minor reductions of up to 10–20% in proliferation with two of the four siRNAs tested, with the remaining two siRNAs having no effect despite good knockdown of CDK5 expression (Fig. S4B). A time course of seliciclib and CCT068127 showed that the decrease in CDK9, cyclin T1, and cyclin D1 expression occurred at 16–24 h (Fig. S3B), whereas inhibition of RNA polymerase II phosphorylation was observed as early as 4 h (Fig. 2C,D). This is consistent with direct inhibition of CDK9 mediating the early effects of these compounds, although at later time points effects on transcription may also contribute to the loss of CDK activity, for example, by cyclin depletion.
3.4. Inhibition of cell cycle progression and DNA synthesis by CCT068127
To investigate the cell cycle effects of CCT068127, HT29 colon cancer cells were exposed to the respective 3xGI50 concentrations of seliciclib or CCT068127 for 4, 12, and 24 h and the effect on cell cycle distribution and DNA synthesis was assessed by incorporation of PI measured by flow cytometry (Fig. 3A). A modest reduction in the proportion of cells with 2n DNA content (~ 10%) and an increase in cells with 4n DNA content (~ 10%) are seen with both compounds (Fig. 3B). CCT068127 and seliciclib both blocked DNA synthesis, as determined by BrdU incorporation, although CCT068127 achieved this more rapidly than seliciclib and with 14‐fold greater potency (Fig. 3C). In both cases, the block in DNA synthesis is near complete at 24 h. Analysis of parallel treatments demonstrated a loss of RB phosphorylation as demonstrated by a mobility shift in the protein on SDS/PAGE (Fig. 3D).
3.5. Gene expression profiling of seliciclib analogs
In order to explore the potential similarities between seliciclib and CCT068127 in an unbiased manner, gene expression profiling was performed on HT29 human colon cancer cells treated with the respective 3xGI50 concentrations of the inhibitors for 24 h. As a control, cells were also treated with an inactive analog of seliciclib, CCT068152, and mRNA expression was measured. This compound displays no inhibitory activity against purified CDKs, has a GI50 > 50 μm on HT29 cells, and does not inhibit RB phosphorylation (Fig. S5). Comparison with this compound would potentially highlight any off‐target effects of the amino‐purine chemical backbone of the series. CCT068152 was used at an equimolar concentration when compared with seliciclib. Gene expression was determined using custom expression arrays (Whittaker et al., 2007). A table showing the transcriptional changes induced by each compound is presented in Table S4. When the gene expression changes induced by seliciclib and CCT068127 compound are plotted against each other, they show good concordance (Fig. 4A).
Figure 4.
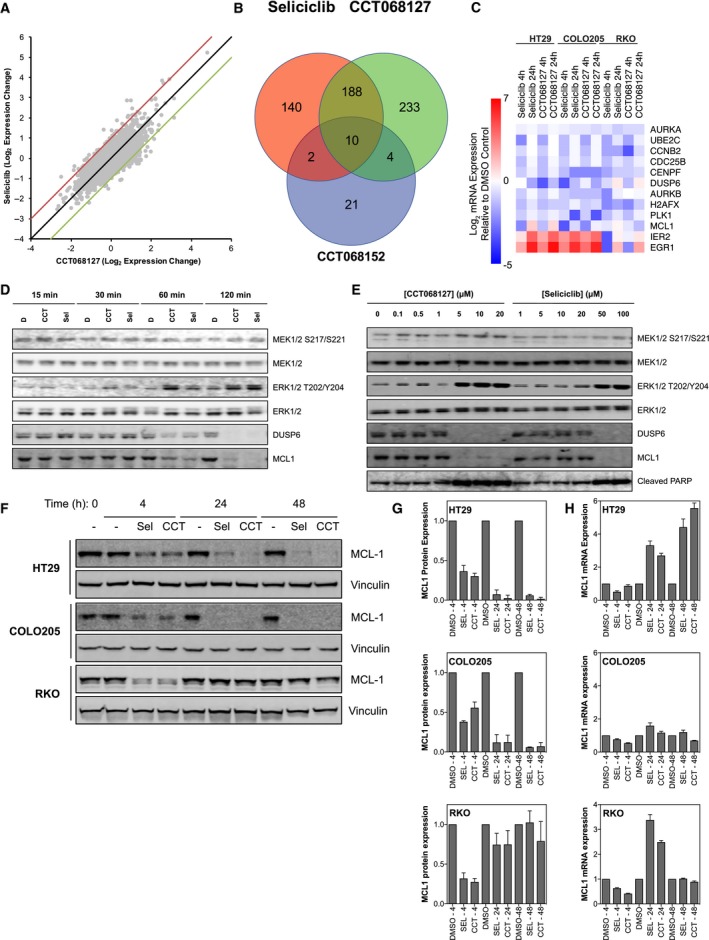
Gene expression analysis of colon cancer cells following treatment with CCT068127 and seliciclib. (A) HT29 cells were treated with equiactive concentrations (3xGI 50, SRB assay) of seliciclib (45 μm) and CCT068127 (2.55 μm) or 45 μm CCT068152. mRNA expression profiles were generated using a custom microarray enriched for 5808 cDNAs implicated in cancer. Genes that were differentially expressed between the CDK inhibitor‐ and the DMSO‐treated cells were determined using comparative marker selection (t‐test) and a ≥ twofold change in expression as a filter. The mRNA expression changes for seliciclib and CCT068127 were plotted against each other. (B) Venn diagram was constructed to show the number of genes in common between the two CDK inhibitors, and the inactive analog CCT068152, which displayed either increased or decreased expression following treatment. (C) mRNA expression changes following treatment of HT29, COLO205, or RKO human colon cancer cells with 3xGI 50 concentrations of CCT068127 or seliciclib for 4 and 24 h as determined by RT‐qPCR. (D) HT29 cells were treated with 5xGI 50 concentrations of CCT068127 (4.25 μm) or seliciclib (75 μm) for 15, 30, 60, or 120 minutes and cell lysates analyzed for the indicated proteins by western blotting. (E) HT29 cells were treated with a range of concentrations of CCT068127 or seliciclib for 24 h. Cells were lysed and extracts were analyzed by western blotting for the proteins listed. (F) HT29, COLO205, and RKO cells were treated with either DMSO or 3xGI 50 concentrations of CCT068127 or seliciclib for 4, 24, and 48 h and protein lysates analyzed by western blotting for the indicated proteins. Data are representative of three independent repeats. (G) The expression of MCL1 in (F) was quantified and normalized to vinculin expression. Data are the mean of three independent repeats ± SE. (H) Cells were treated as in (F) and the expression of MCL1 mRNA was quantified by qRT‐PCR, normalized to the housekeeping gene GUSB. The mean ± SE of triplicate measurements is presented.
We found that seliciclib and CCT068127 share a core set of 198 genes (of a total of 577 affected) that are either increased or decreased in expression following treatment (Fig. 4B). This is consistent with the view that CCT068127 retains some of the same characteristics as seliciclib, although other effects may contribute. Of these shared 198 genes, the expression of only 10 is also altered by the inactive analog CCT068152, suggesting that these are likely off‐target effects, whereas the others are on‐target. In fact, we found that the inactive analog changes the expression of only 37 genes, markedly fewer than either seliciclib or CCT068127. For further analysis, genes that were altered in expression by CCT068152 were excluded.
The mRNA expression changes induced by treatment of HT29 cells with either seliciclib or CCT068127 were used to define a signature for each compound by identifying genes altered in expression by ≥ twofold following treatment. We then analyzed these signatures for similarity to known drug‐induced gene expression changes using the Connectivity Map (Lamb et al., 2006). Strikingly, signatures for both compounds show a high level of correlation with those of other small‐molecule CDK inhibitors, namely 0175029‐0000 (Khan et al., 2012), alsterpaullone (Schultz et al., 1999), and GW8510 (Bramson et al., 2001), as well as HDAC inhibitors vorinostat/SAHA (Richon et al., 1998), trichostatin A (Yoshida et al., 1990), valproic acid (Gottlicher et al., 2001) (Tables S5 and S6).
We also utilized the STRING database (Franceschini et al., 2013) to look for functional associations between genes with altered expression in response to CDK inhibition (Fig. S6). A cluster of genes involved in G2/M cell cycle progression is immediately evident, including decreased expression of several mitosis‐related genes, including PLK1, CCNB2, AURKA, AURKB, CDC25B, UBE2C, and CENPF (Fig. S6). These changes could bring about a G2/M‐phase arrest, which would be consistent with the 4n DNA population reflecting an increased G2/M phase and the reduced transcriptional output induced by the CDK inhibitors as we have previously characterized for seliciclib (Whittaker et al., 2007). We confirmed these gene expression changes by RT‐qPCR (Fig. 4C). Alternatively, this could also reflect a population of G1‐phase cells with 4n DNA content, which may arise through mitotic slippage, due to premature loss of CDK1/cyclin B activity (Rieder and Maiato, 2004).
As part of the overall core transcriptional profile determined by microarray (see above), we identified transcription factors downstream of the MAPK pathway whose expression is increased in response to the CDK inhibitors; these changes were also confirmed by RT‐qPCR (Fig. 4C). For example, the expression of the transcription factor‐encoding genes EGR1 (coding for early growth response protein 1) and IER2 (encoding immediate early response 2) was induced (Boros et al., 2009; Hodge et al., 1998). In contrast, the expression of DUSP6—encoding dual‐specificity phosphatase 6 that acts as a negative regulator of ERK2 by decreasing phosphorylation (Muda et al., 1996)—is reduced. Consistent with this, we have previously observed phosphorylation/activation of extracellular signal‐regulated kinases (ERKs) in response to seliciclib in human colon cancer cell lines (Whittaker et al., 2004). Our observation here of increased expression of transcripts regulated by the MAPK pathway was of particular interest as this suggested that activation of the pathway has functional consequences. We confirmed that treatment of HT29 cells with CCT068127 or seliciclib for 15, 30, 60, and 120 minutes results in rapid phosphorylation of ERK1/2 and that this is seen in the absence of any dramatic changes in the phosphorylation state of the upstream kinases MEK1 and MEK2 (Fig. 4D). Therefore, we determined the expression at the protein level of the ERK2‐phosphatase DUSP6, which as noted above shows reduced mRNA expression. We found that DUSP6 protein levels reduce rapidly, within 1–2 h following treatment with either CCT068127 or seliciclib, commensurate with increased ERK1/2 phosphorylation (Fig. 4D).
We also noted that CDK inhibitor treatment alters transcription of the antiapoptotic gene MCL1, which belongs to the BCL2 family. Although we observed an increase in MCL1 mRNA at 24 h by microarray, RT‐qPCR analysis showed an initial decrease at 4 h, followed by a rebound to untreated levels or higher by 24 h (Fig. 4C). We found that MCL1 protein expression is decreased by both CCT068127 and seliciclib within 2 h of treatment (Fig. 4D) (Whittaker et al., 2007). Interestingly, unlike the mRNA, expression of MCL1 protein does not recover for 24 h with either seliciclib or CCT068127 (Fig. 4E). Sustained suppression of MCL1 protein expression by seliciclib and CCT068127 was confirmed in HT29 and COLO205 cells out to 48 h, whereas in RKO cells MCL1 protein was decreased at 4 h but recovered to near‐control levels at 24 and 48 h (Fig. 4F–H). qRT‐PCR of all of these cell lines treated as in Fig. 4F demonstrated that mRNA levels for MCL1 showed modest decreases at 4 h of treatment, followed by recovery to control levels or greater at 24 and 48 h. Notably, loss of MCL1 protein occurs at the same concentrations at which we also observe PARP cleavage—a marker of caspase activation and apoptosis (Fig. 4E). Hence, loss of MCL1 protein might present a deficit in the balance of antiapoptotic versus proapoptotic signals, resulting in the induction of apoptosis.
3.6. Combined inhibition of CDKs and the BCL2 family is synergistic
Our observation that CCT068127 treatment gives rise to enhanced MAPK signaling prompted us to hypothesize that activation of the MAPK pathway might provide a survival signal to the cells. We therefore treated HT29 human colon cancer cells with a selective inhibitor of ERK known as VTX‐11e (Aronov et al., 2009) in combination with CCT068127; we then determined the effect on cell proliferation and survival after 4 days and calculated synergy using a well‐established method (Chou and Talalay, 1984). Under these conditions, the ERK inhibitor plus CCT068127 gave combination index (CI) values of >1 and was classified as antagonistic (Fig. 5A).
Figure 5.
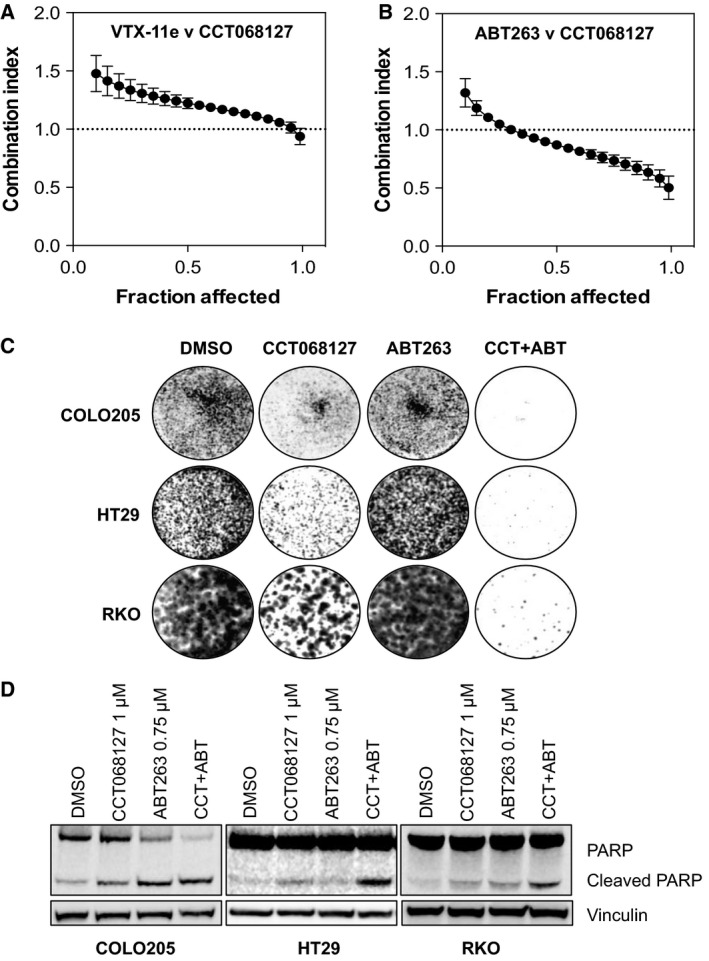
Targeting CDK2/9 and BCL2 family members synergistically inhibits proliferation and induces apoptosis. (A) HT29 human colon cancer cells were treated with increasing concentrations of CCT068127 in combination with a small‐molecule inhibitor of ERK, VTX‐11e. Cells were treated with both compounds in a 1.5‐fold dilution series starting at 5X the GI 50 (4.25 μm CCT068127 and 243 nm VTX‐11e) in a 1 : 1 ratio. Cell proliferation/survival was assessed by SRB assay, and synergy was calculated using the combination index (CI) methodology (Chou and Talalay, 1984). Data from four independent repeats were analyzed by calcusyn software, and the mean CI ± SE is plotted relative to the fraction of cells affected by the treatment. Values < 1 are indicative of synergy. (B) HT29 cells were treated as in (A) except the ERK inhibitor was replaced by the BCL2 family inhibitor ABT263. Cells were treated with both compounds in a 1.5‐fold dilution series starting at 5X the GI 50 (4.25 μm CCT068127 and 12.7 μm ABT263) in a 1 : 1 ratio. The mean combination index ± SE is plotted relative to the fraction of cells affected by the treatment. Values < 1 are indicative of synergy. (C) HT29, COLO205, and RKO human colon cancer cells were treated with 1 μm CCT068127 or 750 nm ABT263 alone, or in combination for 5 days after which the medium was replaced and cells were cultured in medium alone for a further 7 days. Cells were then fixed and stained with crystal violet. Data shown are representative of three independent experiments. (D) HT29, COLO205, and RKO cells were treated with 1 μm CCT068127 in the presence and absence of 750 nm ABT263 for 48 h. Cell lysates were analyzed by western blotting for the indicated proteins.
As we also observed a loss of the antiapoptotic MCL1 protein in response to CDK inhibitor treatment, we posited that this depletion of MCL1 might create a dependency upon other antiapoptotic proteins, such as additional members of the BCL2 family, and that a BCL2 family inhibitor might be synergistic with CCT068127. Hence, we investigated the response of HT29 cells to a 96‐h exposure to the combination of CCT068127 and ABT263, an inhibitor of BCL2 family proteins (Tse et al., 2008). Excitingly, we observed with a CI value ~0.5 or lower, indicating a synergistic antiproliferative effect with the combination of these two agents (Fig. 5B). We confirmed that the combination of these two agents results in an increased antiproliferative/cytotoxic effect as measured by colony formation assay in HT29, COLO205, and RKO cells (Fig. 5C). Colorectal cancer cells were exposed to 1 μm CCT068127, 750 nm ABT263, or a combination of both drugs for a period of 5 days. The drugs were then washed out and the cells grown in medium only for a further 7 days. We observed that the combination of CCT068127 and ABT263 results in a significantly greater suppression of measurable cell colony formation than when used as single agents (Fig. S7). Furthermore, exposure of HT29, COLO205, and RKO human colon cancer cells to both compounds in combination for 48 h results in a greater degree of PARP cleavage, indicative of apoptotic cell death (Fig. 5D).
4. Discussion
We set out to discover a CDK2/9 inhibitor with greater potency and metabolic stability compared with the parent compound seliciclib (Wilson et al., 2011). CCT068127 exhibits 15‐fold greater potency against CDK1/cyclin B, 14‐fold greater potency against CDK5/p35, 22‐fold greater potency against CDK2/cyclin E, and 11‐fold greater potency against CDK9/cyclin T. In contrast, potency toward CDK4 and CDK6 increased only by 6‐ to 8‐fold and that for CDK7/cyclin H potency remained the same; therefore, selectivity for CDK2 and 9 was increased over CDKs 4, 6, and 7. Despite increased potency against CDK1, a cell‐based assay for PP1 phosphorylation at T320 demonstrated that CDK1 was not inhibited at concentrations of CCT068127 (or seliciclib) where cellular effects on proliferation and signaling occurred. The basis for greater potency against CDK2 was suggested by the crystal structure of CCT068127 bound to CDK2, whereby the pendant C2 side chain hydroxyl moiety forms an additional hydrogen bond with Asp145 of the DFG motif. Improved enzyme potency translated into greater antiproliferative activity against human cancer cell lines with an average GI50 24‐fold lower than that observed with seliciclib. Importantly, there was a clear correlation between the suppression of key pharmacodynamic markers for CDK2 and CDK9 (phosphorylation of RB and RNA polymerase II, respectively) and the inhibition of cell proliferation. Notably, RNA polymerase II was inhibited more rapidly and near completely compared to RB by both seliciclib and CCT068127, suggesting that inhibition of CDKs targeting RNA polymerase II may be a primary driver of the cellular response to these agents. This is consistent with a recent study in which seliciclib was shown to bind to CDK7, 12, 9, and 2 in an affinity pull‐down assay (Delehouze et al., 2014). The consequence of CDK9 inhibition appears to be a gradual reduction in the expression of cyclin D1, cyclin T1, and CDK9 itself presumably via inhibition of transcriptional elongation by RNA polymerase II. Thus, these agents may primarily target CDKs that regulate transcription but, through cyclin or CDK depletion, result in inhibition of additional cell cycle CDKs as well. While in vitro biochemical data suggest that CCT068127 showed greatest potency for CDK2, CDK5, and CDK9, our cellular studies suggest that CDK9 may be a key target of this compound, supported by the key observations of the greater magnitude of inhibition of RNA polymerase II phosphorylation over RB, that the catalytic subunit of CDK5 was not detectable in the HT29 and RKO cell lines, and that siRNA‐mediated reduction in CDK5 expression did not have marked effects on HT29 cell proliferation. However, we cannot rule out the possibility that inhibition of these, and potentially other CDKs, may contribute to the mechanism of action of these compounds or additional effects in vivo.
In terms of cell cycle effects, CCT068127 caused the same arrest of cell cycle progression as seen with seliciclib, with a reduction in cells with 2n DNA content and an increase in cells with 4n DNA content, but was able to achieve this at a concentration 15‐fold lower than that required for seliciclib. Overall, CCT068127 retained the same cellular effects as the developmental clinical drug seliciclib but with significantly improved potency and, as we have reported previously, has improved metabolic stability and displays greater in vivo efficacy in a tumor xenograft model (Wilson et al., 2011).
We further investigated the mechanism of action of CCT068127 by comparing the transcriptional signature induced by the compound in the HT29 human colon cancer cell line to the expression signatures available in the Connectivity Map database. The signatures most highly correlated with that of CCT068127 are those for other CDK inhibitors and, in addition, HDAC inhibitors. The close correlation between these two compound classes may stem from their ability to broadly modulate transcription and that functional RB recruits histone deacetylase to suppress E2F‐regulated transcription (Brehm et al., 1998), which is enhanced by CDK inhibition. More global effects on transcription are also likely to be a result of decreased RNA polymerase II activity, through inhibition of CDKs including CDK9.
It is interesting to note that treatment with CCT068127 induces rapid activation of ERK1/2 and we were keen to determine how this was mediated and whether there was an associated dependency upon MAPK signaling for survival of the cells. As the phosphorylation state and expression of MEK1/2 appeared to be unaltered by CCT068127 treatment, our results suggested that upstream activation of the MAPK pathway was likely not mediating this effect. In fact, decreased protein expression of the ERK phosphatase DUSP6 occurs at the same time and concentrations of CCT068127 as the observed induction of ERK1/2 phosphorylation. It therefore seems likely that loss of DUSP6 expression permits increased ERK1/2 phosphorylation and activation of the pathway (Muda et al., 1996). Consistent with this, the mRNAs for EGR1 and IER2 were induced by both inhibitors, suggesting some capacity of ERK to positively modulate transcription. Given the positive regulation of cell proliferation and survival mediated by the MAPK pathway, we hypothesized that addition of an ERK inhibitor would block any proliferative or survival signaling through MAPK. Surprisingly, this was not the case and rather than showing synergy with CCT068127, the ERK inhibitor was antagonistic. ERK inhibition reportedly induces a G1 arrest (Morris et al., 2013) and therefore could potentially protect cells from CDKi‐mediated apoptosis, particularly if driven by deregulation of E2F during S phase (Qin et al., 1994; Wu and Levine, 1994).
The mRNA expression profiling data showed an increase in the transcript levels of the antiapoptotic gene MCL1 in the HT29 cell line. Previously, we had seen this with seliciclib treatment but the expression of MCL1 protein was rapidly decreased in response to CDK inhibition (Whittaker et al., 2007) as has been described for several other CDK9 inhibitors (Gregory et al., 2015; Lemke et al., 2014; Ma et al., 2003). Further RT‐qPCR analysis showed that MCL1 mRNA was initially inhibited in all three cancer cell lines at 4 h but was substantially increased in HT29 and RKO cells by both seliciclib and CCT068127 at 24–48 h. However, MCL1 protein levels were decreased by both compounds at 4–48 h in HT29 and COLO205 cells, whereas RKO cells showed a decrease at 4 h and recovery at 24–48 h. Given the effect of CDK2 and CDK9 inhibition on the cell cycle and global transcriptional regulation, presumably sustained MCL1 protein suppression at 24–48 h is due to either inhibition of translation of the mRNA or accelerated protein degradation (Choudhary et al., 2015). Notably, the RKO cells recover expression of MCL1 protein at 24–48 h so may regulate MCL1 protein expression differently to the HT29 and COLO205 cells. Irrespective of the precise mechanism, the loss of MCL1 protein presented a possible therapeutic opportunity to further target the antiapoptotic BCL2 family with a small‐molecule inhibitor and enhance the proapoptotic activity of CCT068127. Combined use of CCT068127 and ABT263, a BCL2 family inhibitor, led to synergistic antiproliferative activity and consistent with this, we saw elevated caspase activation indicative of apoptosis and reduced cell survival in colony formation assays. Excitingly, this presents a highly rational and mechanistically defined combinatorial approach to target cancer cell survival. Given that MCL1 is amplified in approximately 11% of cancers and overexpression of the protein is associated with chemoresistance (Beroukhim et al., 2010), the combination of a CDK2/9 inhibitor with a BCL2 family inhibitor presents an attractive therapeutic strategy for those cancers that may have an increased dependency on MCL1. To our knowledge, this is the first demonstration that combination of a CDK2/9 inhibitor and a BCL2 family inhibitor is beneficial in human colorectal cancer cell lines and may support widening the scope of indications in which these agents are tested. Overall, our data support the continued clinical development of this series of CDK2/9 inhibitors, from which CYC065 has now entered first‐in‐human studies (Cyclacel Ltd. press release October 22, 2015).
Conflict of interests
The Institute of Cancer Research has a commercial interest in the development of CDK inhibitors and operates a reward for discoverers scheme. PW has been or is a consultant to Novartis, Astex Pharmaceuticals, Chroma Therapeutics, Piramed Pharma, Nuevolution, and NexTech Ventures.
Author contributions
SRW, CB, CM, SWa, AS, EB, SS, RTP, and PAC carried out cell‐based studies including cell proliferation assays, RT‐qPCR, western blotting, microarray analysis, and colony formation assays. NB and JB conducted and interpreted in silico docking studies of CCT068127 and seliciclib. MPM and MN generated and analyzed the cocrystal structure of CCT068127 and CDK2/cyclin A. SWi and EMcD designed and synthesized CCT068127. WJ and PMF contributed to the biochemical characterization and development of CCT068127. SRW, PAC, MIW, JB, MDG, and PW were responsible for study conception, design, and data interpretation. SRW, MPM, MN, JB, and PW wrote the manuscript.
Supporting information
Fig. S1. Inhibition of CDK1 activity in HT29 colon cancer cells treated with CDK inhibitors.
Fig. S2. CCT068127 is a potent inhibitor of RNA polymerase II phosphorylation in COLO205 cells.
Fig. S3. CCT068127 treatment decreases the expression of cyclin T1, CDK9 and cyclin D1.
Fig. S4. CDK5 is not required for cell proliferation.
Fig. S5. CCT068152 is an inactive analogue of CCT068127.
Fig. S6. Gene expression profiling of seliciclib and CCT068127 identifies a cluster of genes involved in G2/M cell cycle control.
Fig. S7. HT29, COLO205 and RKO human colon cancer cells were treated with 1 μm CCT068127 or 750 nm ABT263 alone, or in combination for 5 days after which the medium was replaced and cells were cultured in medium alone for a further 7 days.
Table S1. Primary antibodies used, supplier, catalogue number, species and protein molecular weight.
Table S2. Primer sequences used for qRT‐PCR assays.
Table S3. Summary of Crystallographic Data Collection and Structure Refinement.
Table S4. mRNA expression data for HT29 cells treated with 3xGI50 concentrations of seliciclib, CCT068127 or CCT068152 for 24 h. Probe intensities were log2 normalised.
Table S5. CMAP correlations of gene expression changes induced by CCT068127.
Table S6. CMAP correlations of gene expression changes induced by seliciclib.
Acknowledgements
PW acknowledges program grant support from Cancer Research UK (CRUK grant numbers C309/A2187, C309/A8274, and C309/A11566) and support from The Institute of Cancer Research, London, and Cyclacel Ltd. He is also a Cancer Research UK Life Fellow (CRUK grant number C309/A8992), MN is supported by the MRC (grant number G0901526), and MM is supported by Cancer Research UK (grant number C2115/A21421). SRW was supported by the Sir Samuel Scott of Yews Trust. ICR authors acknowledge support from the CRUK Centre and NHS funding to the NIHR Biomedical Research Centre at The Royal Marsden and the ICR. CB, CM, EB, R te P, SS, NB, SW, PAC, MIW, EM, JB, and MDG acknowledge program grant support from Cancer Research UK (CRUK grant numbers CA309/A2187, C309/A8274, and C309/A11566). We thank Jenny Titley for assistance with flow cytometry.
Contributor Information
Steven R. Whittaker, Email: steven.whittaker@icr.ac.uk
Paul Workman, Email: paul.workman@icr.ac.uk.
References
- Adélaïde J, Finetti P, Bekhouche I, Repellini L, Geneix J, Sircoulomb F, Charafe‐Jauffret E, Cervera N, Desplans J, Parzy D et al (2007) Integrated profiling of basal and luminal breast cancers. Cancer Res 67, 11565–11575. [DOI] [PubMed] [Google Scholar]
- Aronov AM, Tang Q, Martinez‐Botella G, Bemis GW, Cao J, Chen G, Ewing NP, Ford PJ, Germann UA, Green J et al (2009) Structure‐guided design of potent and selective pyrimidylpyrrole inhibitors of extracellular signal‐regulated kinase (ERK) using conformational control. J Med Chem 52, 6362–6368. [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M et al (2010) The landscape of somatic copy‐number alteration across human cancers. Nature 463, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros J, Donaldson IJ, O'Donnell A, Odrowaz ZA, Zeef L, Lupien M, Meyer CA, Liu XS, Brown M and Sharrocks AD (2009) Elucidation of the ELK1 target gene network reveals a role in the coordinate regulation of core components of the gene regulation machinery. Genome Res 19, 1963–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramson HN, Corona J, Davis ST, Dickerson SH, Edelstein M, Frye SV, Gampe RT Jr, Harris PA, Hassell A, Holmes WD et al (2001) Oxindole‐based inhibitors of cyclin‐dependent kinase 2 (CDK2): design, synthesis, enzymatic activities, and X‐ray crystallographic analysis. J Med Chem 44, 4339–4358. [DOI] [PubMed] [Google Scholar]
- Bregman DB, Pestell RG and Kidd VJ (2000) Cell cycle regulation and RNA polymerase II. Front Biosci 5, D244–D257. [DOI] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ and Kouzarides T (1998) Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391, 597–601. [DOI] [PubMed] [Google Scholar]
- Chou TC and Talalay P (1984) Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances Enzyme Reg 22, 27–55. [DOI] [PubMed] [Google Scholar]
- Choudhary GS, Tat TT, Misra S, Hill BT, Smith MR, Almasan A and Mazumder S (2015) Cyclin E/Cdk2‐dependent phosphorylation of Mcl‐1 determines its stability and cellular sensitivity to BH3 mimetics. Oncotarget 6, 16912–16925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clurman BE, Sheaff RJ, Thress K, Groudine M and Roberts JM (1996) Turnover of cyclin E by the ubiquitin‐proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev 10, 1979–1990. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Delehouze C, Godl K, Loaec N, Bruyere C, Desban N, Oumata N, Galons H, Roumeliotis TI, Giannopoulou EG, Grenet J et al (2014) CDK/CK1 inhibitors roscovitine and CR8 downregulate amplified MYCN in neuroblastoma cells. Oncogene 33, 5675–5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, Tessarollo L and Kaldis P (2012) Cyclin‐dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re‐replication but not for liver regeneration. Proc Nat Acad Sci USA 109, 3826–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG and Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR (2011) An introduction to data reduction: space‐group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr 67, 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C et al (2013) STRING v9.1: protein‐protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41, D808–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG et al (2001) Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 20, 6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory GP, Hogg SJ, Kats LM, Vidacs E, Baker AJ, Gilan O, Lefebure M, Martin BP, Dawson MA, Johnstone RW et al (2015) CDK9 inhibition by dinaciclib potently suppresses Mcl‐1 to induce durable apoptotic responses in aggressive MYC‐driven B‐cell lymphoma in vivo. Leukemia 29, 1437–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge C, Liao J, Stofega M, Guan K, Carter‐Su C and Schwartz J (1998) Growth hormone stimulates phosphorylation and activation of elk‐1 and expression of c‐fos, egr‐1, and junB through activation of extracellular signal‐regulated kinases 1 and 2. J Biol Chem 273, 31327–31336. [DOI] [PubMed] [Google Scholar]
- Jeronimo C, Collin P and Robert F (2016) The RNA polymerase II CTD: The increasing complexity of a low‐complexity protein domain. J Mol Biol 428, 2607–2622. [DOI] [PubMed] [Google Scholar]
- Khan SA, Faisal A, Mpindi JP, Parkkinen JA, Kalliokoski T, Poso A, Kallioniemi OP, Wennerberg K and Kaski S (2012) Comprehensive data‐driven analysis of the impact of chemoinformatic structure on the genome‐wide biological response profiles of cancer cells to 1159 drugs. BMC Bioinformat 13, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M and Altman N (2014) Points of significance: nonparametric tests. Nat Methods 11, 467–468. [DOI] [PubMed] [Google Scholar]
- Kwon YG, Lee SY, Choi Y, Greengard P and Nairn AC (1997) Cell cycle‐dependent phosphorylation of mammalian protein phosphatase 1 by cdc2 kinase. Proc Nat Acad Sci USA 94, 2168–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam LT, Pickeral OK, Peng AC, Rosenwald A, Hurt EM, Giltnane JM, Averett LM, Zhao H, Davis RE, Sathyamoorthy M et al (2001) Genomic‐scale measurement of mRNA turnover and the mechanisms of action of the anti‐cancer drug flavopiridol. Genome Biol 2, RESEARCH0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN et al (2006) The Connectivity Map: using gene‐expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- Lemke J, von Karstedt S, Abd El Hay M, Conti A, Arce F, Montinaro A, Papenfuss K, El‐Bahrawy MA and Walczak H (2014) Selective CDK9 inhibition overcomes TRAIL resistance by concomitant suppression of cFlip and Mcl‐1. Cell Death Differ 21, 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L'Italien L, Tanudji M, Russell L and Schebye XM (2006) Unmasking the redundancy between Cdk1 and Cdk2 at G2 phase in human cancer cell lines. Cell Cycle 5, 984–993. [DOI] [PubMed] [Google Scholar]
- Ma Y, Cress WD and Haura EB (2003) Flavopiridol‐induced apoptosis is mediated through up‐regulation of E2F1 and repression of Mcl‐1. Mol Cancer Ther 2, 73–81. [PubMed] [Google Scholar]
- Malumbres M and Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9, 153–166. [DOI] [PubMed] [Google Scholar]
- Mariaule G and Belmont P (2014) Cyclin‐dependent kinase inhibitors as marketed anticancer drugs: where are we now? A short survey. Molecules 19, 14366–14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClue SJ, Blake D, Clarke R, Cowan A, Cummings L, Fischer PM, MacKenzie M, Melville J, Stewart K, Wang S et al (2002) In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R‐roscovitine). Int J Cancer 102, 463–468. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC and Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre NA, McInnes C, Griffiths G, Barnett AL, Kontopidis G, Slawin AM, Jackson W, Thomas M, Zheleva DI, Wang S et al (2010) Design, synthesis, and evaluation of 2‐methyl‐ and 2‐amino‐N‐aryl‐4,5‐dihydrothiazolo[4,5‐h]quinazolin‐8‐amines as ring‐constrained 2‐anilino‐4‐(thiazol‐5‐yl)pyrimidine cyclin‐dependent kinase inhibitors. J Med Chem 53, 2136–2145. [DOI] [PubMed] [Google Scholar]
- Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S et al (2013) Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov 3, 742–750. [DOI] [PubMed] [Google Scholar]
- Muda M, Theodosiou A, Rodrigues N, Boschert U, Camps M, Gillieron C, Davies K, Ashworth A and Arkinstall S (1996) The dual specificity phosphatases M3/6 and MKP‐3 are highly selective for inactivation of distinct mitogen‐activated protein kinases. J Biol Chem 271, 27205–27208. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA and Dodson EJ (1997) Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Natrajan R, Mackay A, Wilkerson PM, Lambros MB, Wetterskog D, Arnedos M, Shiu KK, Geyer FC, Langerod A, Kreike B et al (2012) Functional characterization of the 19q12 amplicon in grade III breast cancers. Breast Cancer Res 14, R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelgeschlager T (2002) Regulation of RNA polymerase II activity by CTD phosphorylation and cell cycle control. J Cell Physiol 190, 160–169. [DOI] [PubMed] [Google Scholar]
- Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M and Barbacid M (2003) Cyclin‐dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet 35, 25–31. [DOI] [PubMed] [Google Scholar]
- Qin XQ, Livingston DM, Kaelin WG Jr and Adams PD (1994) Deregulated transcription factor E2F‐1 expression leads to S‐phase entry and p53‐mediated apoptosis. Proc Nat Acad Sci USA 91, 10918–10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud FI, Whittaker SR, Fischer PM, McClue S, Walton MI, Barrie SE, Garrett MD, Rogers P, Clarke SJ, Kelland LR et al (2005) In vitro and in vivo pharmacokinetic‐pharmacodynamic relationships for the trisubstituted aminopurine cyclin‐dependent kinase inhibitors olomoucine, bohemine and CYC202. Clin Cancer Res 11, 4875–4887. [DOI] [PubMed] [Google Scholar]
- Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA and Marks PA (1998) A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Nat Acad Sci USA 95, 3003–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL and Maiato H (2004) Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell 7, 637–651. [DOI] [PubMed] [Google Scholar]
- Sakurikar N, Thompson R, Montano R and Eastman A (2016) A subset of cancer cell lines is acutely sensitive to the Chk1 inhibitor MK‐8776 as monotherapy due to CDK2 activation in S phase. Oncotarget 7, 1380–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz C, Link A, Leost M, Zaharevitz DW, Gussio R, Sausville EA, Meijer L and Kunick C (1999) Paullones, a series of cyclin‐dependent kinase inhibitors: synthesis, evaluation of CDK1/cyclin B inhibition, and in vitro antitumor activity. J Med Chem 42, 2909–2919. [DOI] [PubMed] [Google Scholar]
- Shupp A, Casimiro MC and Pestell RG (2017) Biological functions of CDK5 and potential CDK5 targeted clinical treatments. Oncotarget 8, 17373–17382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu O and McCormick F (2003) Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 3, 233–245. [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P et al (2008) ABT‐263: a potent and orally bioavailable Bcl‐2 family inhibitor. Cancer Res 68, 3421–3428. [DOI] [PubMed] [Google Scholar]
- Wang S, Meades C, Wood G, Osnowski A, Anderson S, Yuill R, Thomas M, Mezna M, Jackson W, Midgley C et al (2004) 2‐Anilino‐4‐(thiazol‐5‐yl)pyrimidine CDK inhibitors: synthesis, SAR analysis, X‐ray crystallography, and biological activity. J Med Chem 47, 1662–1675. [DOI] [PubMed] [Google Scholar]
- Whittaker SR, Mallinger A, Workman P, Clarke PA (2017) Inhibitors of cyclin‐dependent kinases as cancer therapeutics. Pharmacol Ther 173, 83–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker SR, Te Poele RH, Chan F, Linardopoulos S, Walton MI, Garrett MD and Workman P (2007) The cyclin‐dependent kinase inhibitor seliciclib (R‐roscovitine; CYC202) decreases the expression of mitotic control genes and prevents entry into mitosis. Cell Cycle 6, 3114–3131. [DOI] [PubMed] [Google Scholar]
- Whittaker SR, Walton MI, Garrett MD and Workman P (2004) The Cyclin‐dependent kinase inhibitor CYC202 (R‐roscovitine) inhibits retinoblastoma protein phosphorylation, causes loss of Cyclin D1, and activates the mitogen‐activated protein kinase pathway. Cancer Res 64, 262–272. [DOI] [PubMed] [Google Scholar]
- Wilson SC, Atrash B, Barlow C, Eccles S, Fischer PM, Hayes A, Kelland L, Jackson W, Jarman M, Mirza A et al (2011) Design, synthesis and biological evaluation of 6‐pyridylmethylaminopurines as CDK inhibitors. Bioorg Med Chem 19, 6949–6965. [DOI] [PubMed] [Google Scholar]
- Wu X and Levine AJ (1994) p53 and E2F‐1 cooperate to mediate apoptosis. Proc Natl Acad Sci USA 91, 3602–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Kijima M, Akita M and Beppu T (1990) Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem 265, 17174–17179. [PubMed] [Google Scholar]
- Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW and Dean DC (2000) Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC‐Rb‐hSWI/SNF and Rb‐hSWI/SNF. Cell 101, 79–89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Inhibition of CDK1 activity in HT29 colon cancer cells treated with CDK inhibitors.
Fig. S2. CCT068127 is a potent inhibitor of RNA polymerase II phosphorylation in COLO205 cells.
Fig. S3. CCT068127 treatment decreases the expression of cyclin T1, CDK9 and cyclin D1.
Fig. S4. CDK5 is not required for cell proliferation.
Fig. S5. CCT068152 is an inactive analogue of CCT068127.
Fig. S6. Gene expression profiling of seliciclib and CCT068127 identifies a cluster of genes involved in G2/M cell cycle control.
Fig. S7. HT29, COLO205 and RKO human colon cancer cells were treated with 1 μm CCT068127 or 750 nm ABT263 alone, or in combination for 5 days after which the medium was replaced and cells were cultured in medium alone for a further 7 days.
Table S1. Primary antibodies used, supplier, catalogue number, species and protein molecular weight.
Table S2. Primer sequences used for qRT‐PCR assays.
Table S3. Summary of Crystallographic Data Collection and Structure Refinement.
Table S4. mRNA expression data for HT29 cells treated with 3xGI50 concentrations of seliciclib, CCT068127 or CCT068152 for 24 h. Probe intensities were log2 normalised.
Table S5. CMAP correlations of gene expression changes induced by CCT068127.
Table S6. CMAP correlations of gene expression changes induced by seliciclib.