

Abstract
Targeting of drug carriers to endocytic cell-receptors facilitates intracellular drug delivery. Carrier size and number of targeting moieties (valency) influence cell binding and uptake. However, how these parameters influence receptor-mediated cell-signaling (the link between binding and uptake) remains uncharacterized. We studied this using polymer carriers of different sizes and valencies, targeted to endothelial intercellular adhesion molecule-1 (ICAM-1), a marker overexpressed in many pathologies. Unexpectedly, induction of cell-signals (ceramide and PKC enrichment and activation) and uptake, were independent of: carrier avidity, total number of carriers bound per cell, cumulative cell-surface area occupied by carriers, number of targeting antibodies at the carrier-cell contact, and cumulative receptor engagement by all bound carriers. Instead, “valency density” (number of antibodies per carrier surface area) ruled signaling, and carrier size independently influenced uptake. These results are key to understanding the interplay between carrier design parameters and receptor-mediated signaling conducive to endocytosis, paramount for intracellular drug delivery.
Keywords: Targeted drug carrier, carrier size and valency, carrier-induced signaling, endocytic uptake, ICAM-1, endothelial cell
TOC Image
Introduction
Targeting of drug carriers to cell-surface receptors associated with endocytic transport is a strategy broadly used to facilitate intracellular drug delivery.1, 2 Many parameters rule the efficiency of such endocytic uptake into cells. These parameters relate to both the biological regulation of the cell type and pathway targeted, and the physicochemical properties of the carrier used.1, 2 Among other design factors, the size of carriers greatly impacts uptake by cells,2–6 in addition to other aspects such as circulation, biodistribution, clearance, etc.7–13 For instance, professional phagocytic cells (such as macrophages) can internalize either pristine or IgG-opsonized particles across a wide range of sizes, from the submicrometer to the supramicrometer scale.14, 15 In contrast, in non-phagocytic cells (the most common targets for drug delivery) larger carriers typically associate with lower uptake efficiency when compared to smaller counterparts.5, 16–20
This is likely due to restrictions imposed by the biophysical properties of cell membranes and endocytic machinery involved.1, 2, 21–23 For example, opposite to phagocytosis, clathrin- and caveolae-mediated pathways are generally restricted by the size of membrane vesicles that form via these routes, which are in the range of ∼100–200 nm and ∼50–80 nm in diameter, respectively.1, 2 Consequently, carriers targeted to cell receptors of these routes suffer from analogous restrictions. As an example, 60 nm diameter liposomes targeted to the transferrin receptor show superior cell uptake compared to 120 nm counterparts, and anti-transferrin receptor antibody is internalized more readily than 250 nm polymer particles coated with this antibody.24, 25 Similarly, naked ligands targeted to caveolar markers (e.g., PV1, ganglioside GM1, aminopeptidase P) internalize better than larger carrier-coupled formulations.10, 26, 27 Even in cells that internalize carriers within a broad size range, the uptake mechanism may vary depending on the carrier size. For instance, internalization of submicrometer vs. supramicrometer IgG-opsonized particles by macrophages occurs via clathrin pits vs. phagocytosis, respectively.14 The shape, flexibility, and auxiliary cargoes associated with the carrier also influence uptake differently depending on the carrier size.3, 4, 12, 15, 23, 28 Therefore, the size of carriers is a parameter of recognized key relevance for intracellular drug delivery.
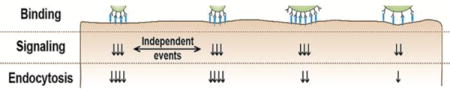
For drug carriers that present targeting moieties, the type and length of linkers coupling these moieties to the carrier surface, the number in which they are displayed (valency) on the carrier surface, the combination of targeting moieties, etc., are important parameters modulating binding to cell receptors, which may affect the mechanism and/or efficacy of endocytosis.13, 29–35 Often, a higher number of targeting moieties on the carrier surface results in an increased avidity toward cells, but exceeding a threshold may cause binding to decay due to steric hindrances.13, 36, 37 This relationship has been mostly explored in terms of carrier binding. However, how changes in the size and valency of multivalent carriers impact cell-signaling events that occur subsequent to binding and precede uptake is a phenomenon not understood. It is also unclear whether such signaling and the subsequent endocytosis depend on the total number of carriers bound on a cell, or whether each carrier engages in an independent signaling and uptake event. Whether carrier size impacts signaling and uptake independently from carrier valency is also unknown. Understanding these aspects will shine light over the regulation of receptor-mediated signaling, the link between carrier binding and endocytosis, which represents the focus of this work.
Intercellular adhesion molecule-1 (ICAM-1), a glycoprotein expressed on endothelial and other cell types,38, 39 is a good candidate to examine these questions, since the pathway of internalization of ICAM-1-targeted carriers (called cell adhesion molecule (CAM)-mediated endocytosis40) is relatively well understood and amenable for uptake of carriers within a wide size range, as demonstrated both in cell culture and in vivo.41, 42 Antibody- or peptide-coated carriers targeted to ICAM-1 induce signaling through the sodium/proton exchanger 1 (NHE1) and the enzyme acid sphingomyelinase,41, 43 which hydrolyzes sphingomyelin to ceramide at the cell-surface, thereby modifying the lipid composition and biophysical properties of the plasma membrane.41 Protein kinase C (PKC) has also been reported to contribute to this signaling, which along with the ceramide signal culminates in rearrangement of actin into stress fibers and carrier uptake.23, 39, 40, 42 Highlighting the translational relevance of this example, ICAM-1 targeting has been explored for imaging and drug delivery in the context of lung disease, cardiovascular conditions, inflammation, metabolic disease, cancer, autoimmune conditions, genetic syndromes, etc.13, 44–52 In particular, ICAM-1-targeted delivery to the vascular endothelium seems a viable option for treatment of endothelial conditions using carriers of different geometries and valencies.11, 42, 53 This is because this cell lining is in direct contact with the circulation and not constrained by the same size limitations of the enhanced permeability and retention (EPR) effect associated to other applications, such as treatment of primary tumors.54
Therefore, using the example of ICAM-1 targeting, we have examined the role of carrier size and valency on the receptor-mediated signaling process that links binding to endocytosis of targeted drug carriers. Our results reveal a complex interplay between these factors, advancing our understanding of these fundamental processes and guiding future applications.
Materials and Methods
Antibodies and reagents
Mouse anti-human ICAM-1 clone R6.5 (anti-ICAM) was from ATCC (Manassas, VA). Mouse IgM anti-ceramide was from Sigma-Aldrich (Saint Louis, MO). Rabbit anti-human PKC (H-300), rabbit anti-human GAPDH, and FITC-labeled goat anti-mouse IgM were from Santa Cruz Biotechnology (Dallas, TX). Rabbit anti-phosphorylated human PKCα (phospho T638) was from Abcam (Cambridge, United Kingdom). HRP-linked anti-rabbit IgG was from GE Healthcare (Pittsburgh, PA). Mouse IgG and fluorescently-labeled secondary antibodies were from Jackson Immunoresearch (West Grove, PA). Polystyrene beads were from Polysciences (Warrington, PA). 125Iodine (125I) was from Perkin-Elmer (Waltham, MA) and Pierce iodination tubes were from Thermo Scientific (Rockford, IL). Precast 4–15% polyacrylamide gels were from Biorad (Hercules, CA) and PVDF membranes were from Pall Life Sciences (Port Washington, NY). Unless noted, all other reagents were from Sigma-Aldrich.
Cell culture
Human umbilical vein endothelial cells (HUVECs) from Lonza (Walkersville, MD) were cultured in M-199 medium (Invitrogen, Carlsbad, CA) supplemented with 15% fetal bovine serum, 15 μg/mL endothelial cell growth supplement, 2 mM L-glutamine, 100 μg/mL heparin, 100 U/mL penicillin, and 100 μg/mL streptomycin. For experiments, cells were seeded on 12-mm2 1%-gelatin-coated glass coverslips and grown to confluence at 37°C, 5% CO2, and 95% relative humidity. Cells were treated for 16 h with 10 ng/mL tumor necrosis factor α (TNFα; BD Biosciences, San Jose, CA) to cause cell activation, as observed in many diseases.55
Preparation and characterization of targeted carriers
Model polymer carriers were prepared by coating standard polystyrene beads by surface adsorption with anti-ICAM IgG (anti-ICAM) vs. non-specific IgG (IgG), or mixtures containing different anti-ICAM-to-IgG ratios, as previously described.23, 30 Briefly, ∼5 μM antibody was incubated for 1 h at room temperature with a particle concentration equivalent to ∼7×106 - 3×108 μm2 of particle surface area/μL to allow surface adsorption, followed by removal of non-coated antibody by centrifugation at 13.8 g for 3 min. Coated carriers were resuspended at ∼1×106 to 2×107 μm2 of carrier surface area/μL in phosphate-buffered saline containing 1% bovine serum albumin and sonicated to eliminate potential aggregates. The size of coated carriers after preparation, or after 30 min incubation at 37°C in saline or 20% serum was determined by dynamic light scattering (Malvern Zetasizer Nano-ZS90, Worcestershire, UK) or optical microscopy (Olympus IX81, Olympus, Inc., Center Valley, PA). Alternatively, 125I-anti-ICAM or 125I-IgG were employed to determine the final coating by measuring the 125I content in a gamma counter (2470 Wizard2, Perkin Elmer; Waltham, MA) and calculating the number of antibodies per particle, based on the particle concentration provided by the vendor and the 125I-antibody specific activity (cpm/mass), as described.23, 55 The characterization of these formulations is provided in Tables 1, 2, and Figure S1. For all carrier binding and uptake experiments, coated carriers were diluted such that every formulation added to cells contained a total of ∼2×105 μm2 of carrier surface area/μL and, thus, cells were always exposed to the same carrier surface area regardless of carrier size and valency (except for those treated with 4×103 μm2 of carrier surface/μL, when specified).
Table 1.
Characterization of targeted vs. non-targeted carrier formulations
| Size (μm) | Coating | Binding avidity | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| Formulation | Mean | SEM | a Antibody molecules/μm2 | a Antibody molecules/carrier | Bmax (carriers/cell) | Kd (pM) |
| Anti-ICAM | ||||||
|
| ||||||
| Submicrometer | ||||||
| b v1 | 0.273 | 0.006 | 7,765 ± 273b | 242.1 ± 8.6 | 5,437.1 | 130.05 |
| Micrometer | ||||||
| v1 | 1.15 | 0.02 | 6,452 ± 243 | 20,268 ± 764 | ND | ND |
| v2 | 0.93 | 0.01 | 12,963 ± 513 | 40,725 ± 1,610 | 174.5 | 1.41 |
| b v3 | 1.09 | 0.02 | 30,564 ± 1,961b | 96,019 ± 6,161 | 637.4 | 0.87 |
|
| ||||||
| Supramicrometer | ||||||
| b v3 | 4.41 | 0.05 | 34,507 ± 447b | 2,195,270 ± 28,459 | 254.4 | 0.09 |
|
| ||||||
| Mouse IgG | ||||||
|
| ||||||
| Submicrometer | ||||||
| v1 | 0.259 | 0.015 | 5,431 ± 562 | 170.6 ± 17.6 | ND | ND |
|
| ||||||
| Micrometer | ||||||
| v2 | 0.96 | 0.02 | 15,451 ± 2,835 | 48,542 ± 8,907 | ND | ND |
|
| ||||||
| Supramicrometer | ||||||
| v3 | 4.37 | 0.06 | 37,294 ± 8,681 | 2,372,580 ± 552,233 | ND | ND |
some anti-ICAM carriers contain IgG to vary targeting valency while keeping constant the total number of antibodies on the coat, where anti-ICAM counterpart is shown;
carriers with only anti-ICAM (no IgG on the coat; v = valency; ND = not determined).
Table 2.
Characterization of additional submicrometer carriers for ceramide enrichment experiments
| Size (μm) | Coating | |||
|---|---|---|---|---|
|
|
||||
| Formulation | Mean | SEM | a Antibody molecules/μm2 | a Antibody molecules/carrier |
| Submicrometer | ||||
| b Anti-ICAM | 0.206 | 0.005 | 11,518 ± 294b | 361.8 ± 9.20 |
| Anti-ICAM | 0.197 | 0.003 | 5,685 ± 95 | 178.6 ± 2.99 |
| Anti-ICAM | 0.193 | 0.002 | 3,501 ± 25 | 109.99 ± 0.80 |
| Anti-ICAM | 0.195 | 0.005 | 1,581 ± 23 | 49.67 ± 0.74 |
| Anti-ICAM | 0.164 | 0.006 | 849 ± 15 | 26.66 ± 0.48 |
some anti-ICAM carriers contain IgG to vary targeting valency while keeping constant the total number of antibodies on the coat, where anti-ICAM counterpart is shown;
carriers with only anti-ICAM (no IgG on the coat; v = valency).
Avidity of targeted carriers
To test avidity toward cells without confounding effects of concomitant carrier uptake, activated HUVECs were fixed with 2% paraformaldehyde. Cells were then incubated for 1 or 3 h at room temperature with anti-ICAM carriers of different size (250 nm, 1 μm, or 4.5 μm) and targeting-coat densities (from 7,700 to 34,000 anti-ICAM molecules/μm2 carrier surface) at different concentrations (from 0.5 fM to 1.08 nM), followed by washing of non-bound carriers and analysis by optical microscopy. Although micrometer and, mainly, supramicrometer carriers may not be clinically significant given their size, they represent valuable research models to study the link between carrier size and endocytic cell-signaling. Submicrometer carriers were green Fluoresbrite®, having pH-stable fluorescence, and were visualized by fluorescence microscopy, while larger counterparts were visible by phase-contrast. Images were obtained using an Olympus IX81 microscope (Olympus, Inc., Center Valley, PA), 40× or 60× oil immersion objectives (UPlanApo, Olympus, Inc., Center Valley, PA), ORCA-ER camera (Hamamatsu Corporation, Bridgewater, NJ), and SlideBook™ 4.2 software (Intelligent Imaging Innovations, Denver, CO). The number of submicrometer carriers per cell was counted with an algorithm previously described40 using Image-Pro 6.3 (Media Cybernetics, Bethesda, MD), while larger carriers were manually counted.23 The algorithm used for submicrometer carriers provides the total count of carrier fluorescence bound to a cell, in pixels2. This number was divided by the pixels2 occupied by a single carrier to calculate the total number of carriers bound. Using these data, ligand-binding saturation regression was performed with SigmaPlot 11.0 (Systat Software Inc., San Jose, CA) to obtain the maximal binding (Bmax) and dissociation constant (Kd) of the different formulations. All curves obtained had a regression coefficient of R2 > 0.9.
Specific binding and endocytosis of targeted carriers
Anti-ICAM carriers vs. control IgG carriers of different sizes (250 nm, 1 μm, or 4.5 μm in diameter) and coat densities (from 7,700 to 34,000 antibody molecules/μm2 carrier surface) were incubated for 30 min, 3 h, or 24 h with activated HUVECs. Binding was first assessed at room temperature using fixed cells, to avoid confounding effects that may arise from concomitant cell uptake, and then confirmed using live cells. Endocytosis requires active metabolism; hence, it was only examined in live cells at 37°C. Green Fluoresbrite® submicrometer carriers were used for visualization by fluorescence microscopy, while larger counterparts were visible by phase-contrast. Microscopy was used to determine the number of carriers bound per cell, as described above. For uptake studies, cells were fixed after washing off non-bound carriers, followed by incubation with Texas Red-labeled goat anti-mouse IgG. This secondary antibody can only access anti-ICAM (or IgG) on the coat of carriers located on the cell-surface, not carriers inside cells. This allows differentiation between carriers bound on the cell-surface, which appear either yellow (red + green) if submicrometer type or red if larger, and internalized carriers that lack red staining.55 This differentiation can then be used to calculate the percentage of uptake with respect to the total amount of carriers associated with cells, as described before.55
Ceramide enrichment at sites of cell-binding of targeted carriers
Anti-ICAM carriers of different sizes (250 nm, 1 μm, or 4.5 μm in diameter) and coat densities (from 800 to 34,000 antibody molecules/μm2 carrier surface) were incubated 15 or 30 min at 37˚C with activated HUVECs. Non-bound carriers were washed off and cells were fixed with 2% paraformaldehyde and permeabilized with 0.2% Triton X-100. Ceramide, a signaling molecule associated with the ICAM-1 uptake pathway, was then immunostained in green and visualized using fluorescence microscopy. Image-Pro 6.3 was used to determine ceramide fluorescence at sites of carrier binding, as described previously.23, 41 Briefly, ceramide fluorescence intensity at the plane of a carrier “equator” was determined around the carrier circumference (membrane engulfment area) and outside the region of carrier binding (background), from which the ceramide enrichment associated with carrier binding was determined (engulfment – background).
PKC signaling upon cell-binding of targeted carriers
Anti-ICAM carriers of different sizes (250 nm or 1 μm in diameter) and coat densities (7,700 to 13,000 antibody molecules/μm2 carrier surface) were incubated for 10 or 30 min at 37˚C with activated HUVECs. Non-bound carriers were washed off and cells were fixed with 2% paraformaldehyde and permeabilized with 0.2% Triton X-100. In parallel experiments, either total PKC or activated (phosphorylated)-PKCα (pPKCα) were immunostained in red and visualized using fluorescence microscopy, from which total PKC and pPKCα enrichment at sites of carrier-cell binding was calculated as described above for ceramide enrichment. In addition, pPKCα present in the lysate of cells incubated in the absence vs. presence of anti-ICAM carriers (250 nm or 1 μm in diameter; 9,800 to 30,000 antibody molecules/μm2 carrier surface) was separated by SDS-PAGE. This was followed by Western blot to immunodetect and normalize the level of pPKCα to that of the housekeeping protein GAPDH. For both fluorescence and Western blot assays testing PKC signaling, two different times (10 min and 30 min) were analyzed and averaged to estimate signal enrichment. This is because PKC is involved in both endocytosis upon carrier binding to ICAM-1 and also subsequent intracellular trafficking,40, 42 events which occur sequentially. Since the uptake and intracellular trafficking kinetics of submicrometer carriers is slightly different from that of micrometer carriers, averaging the signal at two different time points minimizes potential confounding effects of a dual (uptake and trafficking) signal.
Calculations
The total surface area occupied by all carriers bound on a cell (herein called the “surface area occupied per cell”) was calculated by multiplying the total number of carriers bound per cell by the top-down surface area of a carrier (i.e., the area of a circle of the same diameter as the carrier). The “density of ICAM-1 engagement” was calculated as the density of anti-ICAM coat on each carrier (the number of anti-ICAM antibodies per carrier surface area) multiplied by a factor of 2, since anti-ICAM is bivalent and each antibody molecule can theoretically engage two ICAM-1 molecules on a cell. “ICAM-l engagement per carrier” was calculated by multiplying the “density of ICAM-1 engagement” by the “effective cell-surface area occupied per carrier”, where the latter parameter was the theoretical area of a carrier in contact with the cell membrane. This surface area was estimated by assuming that for each carrier size, a dome of height 9.35 nm (half the length of the extracellular domain of ICAM-1) is in contact with the cell membrane. Previous studies have shown that carriers accommodate into the cell membrane in a manner that depends on half the length of the bound receptor.56 “ICAM-1 engagement per cell” was calculated by multiplying “ICAM-1 engagement per carrier” by the total number of carriers bound on a cell. These data were used to perform linear regressions in SigmaPlot 11.0.
Statistics
Experiments encompass a total sample size of n ≥3. Data were calculated as mean ± standard error of the mean (SEM). For multiple comparisons, statistical significance was determined using One-way ANOVA followed by Tukey’s test (p<0.05). For two-way comparisons, statistical significance was determined using Student’s t-test with a threshold of p<0.05.
Results and Discussion
Characterization of anti-ICAM carriers with different sizes and valencies
Endocytosis of receptor-targeted drug carriers may depend upon the induced cell-signaling, which could be ruled by carrier size, targeting avidity, total number of carriers bound per cell, etc. We prepared carriers with varying sizes and targeting antibody-coating densities or valencies (called “v” in Table 1) to study these aspects. Model carriers consisted of non-biodegradable polystyrene particles coated by surface absorption with targeting (anti-ICAM) or non-specific (IgG) antibodies. This model was selected to avoid carrier degradation that may confound results on cellular binding and uptake (our focus). Adsorption of antibodies on the surface of particles favors outward display of variable regions at the used concentrations.57 A random orientation is also possible, which provides a similar coat batch-to-batch and is no different from random chemical conjugation of antibodies where the linkage may occur at any of the available antibody residues. Validating this model, these formulations are relatively stable (lack of: aggregation, antibody detachment, and albumin coating, shown previously45, 46 and verified in Figure S1) and render binding, endocytosis, intracellular trafficking, and in vivo biodistribution comparable to biodegradable poly(lactic-co-glycolic acid) carriers.47, 58 Three carrier sizes were used, representing submicrometer (∼250 nm), micrometer (∼1 μm), and supramicrometer counterparts (∼4.5 μm). These sizes were selected because carriers 250 nm in diameter are still close to the limit of uptake by classical pathways (e.g., clathrin-coated pits), 1 μm is still somewhat amenable for a cell to internalize a carrier, and 4.5 μm is far from the typical range permissible in a non-immune cell.1, 2 Although nanoparticles below 250 nm diameter are more suitable in a medical setting, they are not individually distinguishable by classical optical means used to evaluate signaling and uptake, and microparticles represent a better research tool to examine the aspects at hand without varying carrier shape or chemistry. Micro-sized carriers, such as filomicelles, rods, etc., have been shown to be suitable for drug delivery.10, 11 Importantly, in order to achieve carrier formulations with different sizes but similar targeting valency, anti-ICAM and control IgG were mixed at different ratios on the coating mixture (see Materials and Methods), so that only the targeting valency on the carrier surface would vary while the total amount of antibodies (anti-ICAM + IgG) and the resulting modification to the carrier size would remain similar, as optimized previously,23, 30 and shown here in Table 1.
First, as expected, covering the surface of carriers with saturating levels of anti-ICAM alone rendered different targeting valencies for each size group (Table 1), e.g., 240 vs. 96,000 vs. 2,300,000 anti-ICAM molecules/carrier for 250 nm vs. 1 μm (v3) vs. 4.5 μm carriers, respectively. This correlated well with increasing avidities of larger carriers toward activated endothelial cells: their respective dissociation constants (Kds) were 130 vs. 0.87 vs. 0.09 pM (Table 1 and Figure 1). Second, increasing the levels of anti-ICAM in anti-ICAM + IgG coating mixtures, examined for 1 μm carriers, resulted in increasing targeting valencies without affecting final carrier size: 20,300 (v1) vs. 40,700 (v2) vs. 96,000 (v3) anti-ICAM molecules/carrier, which accordingly rendered increasing avidities toward cells: e.g., Kds were 1.41 pM for v2 vs. 0.87 pM for v3 (Table 1 and Figure 1). Third, increasing carrier size while maintaining constant the number of antibody molecules per μm2 of carrier surface area, also resulted in increased avidity toward cells: e.g., 0.87 pM for 1 μm carriers (v3) vs. 0.09 pM for 4.5 μm carriers (v3), both bearing 30,500–34,500 anti-ICAM/μm2 (Table 1 and Figure 1). Finally, comparison of anti-ICAM carriers to respective control IgG carriers of similar sizes (250 nm, 1 μm, and 4.5 μm) and valencies (v1, v2, and v3) showed good specificity of binding toward endothelial cells: 95 vs. 3 carriers/cell, 20 vs. 0.01 carriers/cell, and 10 vs. 0.1 carriers/cell for 250 nm, 1 μm, and 4.5 μm carriers, respectively (30 min; Figure S2). Such high specificity over IgG carriers verifies that firm binding to cells (what we track), withstanding the washing process, is driven by ICAM-1 targeting and not by the Fc portion of antibodies (since Fc is also present in IgG) or sedimentation of micro-sized carriers. Hence, these carriers represent valid models for the subsequent experiments.
Figure 1.
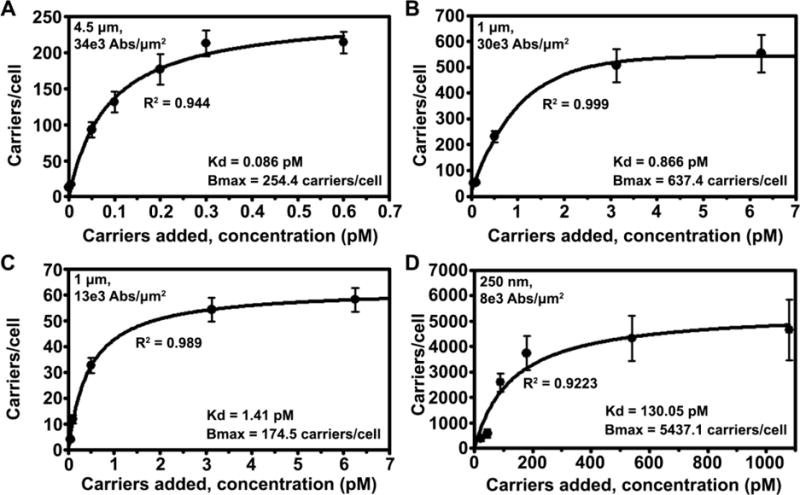
Avidity of anti-ICAM carriers toward endothelial cells. Fixed activated HUVECs were incubated for 1 h or 3 h at room temperature with various concentrations (0.5 fM to 1.08 nM) of anti-ICAM carriers with diameter: (A) 4.5 μm (34,000 anti-ICAM/μm2 carrier surface), (B) 1 μm (30,000 anti-ICAM/μm2), (C) 1 μm (13,000 anti-ICAM/μm2), or (D) 250 nm (7,700 anti-ICAM/μm2). Binding was quantified from microscopy (see Materials and Methods). Data are mean ± SEM, for which regression curves were fitted in order to determine the dissociation constant (Kd) and maximal binding at saturation (Bmax). The regression coefficient (R2) is shown.
Endothelial binding of anti-ICAM carriers with different sizes and valencies
Using carriers of different sizes and saturating levels of anti-ICAM alone (b in Table 1), we first examined binding separately from uptake, for which fixed cells were used. The total number of carriers bound per cell increased with time for all carrier sizes but, at any given time, binding decreased with increasing carrier size: e.g., 95 and 307 carriers/cell at 30 min and 3 h for 250 nm carriers, compared to 9 and 23 carriers/cell at these times for 4.5 μm carriers (Figure 2A–B). A similar trend was observed for the Bmax for these carriers (Table 1). This is expected since a larger carrier occupies a greater cell-surface area vs. a smaller carrier and, hence, more carriers of a small size can be packed within the same cell-surface area. However, an estimation of the net cell-surface area occupied by all carriers bound on a cell at saturation (Figure 2C) revealed that, given sufficient time, this parameter was similar for all three carrier sizes (360 μm2 at 24 h). Yet, this maximal value was reached faster for larger carriers (3 h for 4.5 μm carriers). Since cells had been incubated with solutions containing the same total carrier surface area regardless of size, and sedimentation was determined not to influence the specificity of firm binding after washings, this indicates that carriers of larger sizes exhibited faster binding, which is in agreement with their observed avidity Kds (Figure 1 and Table 1).
Figure 2.
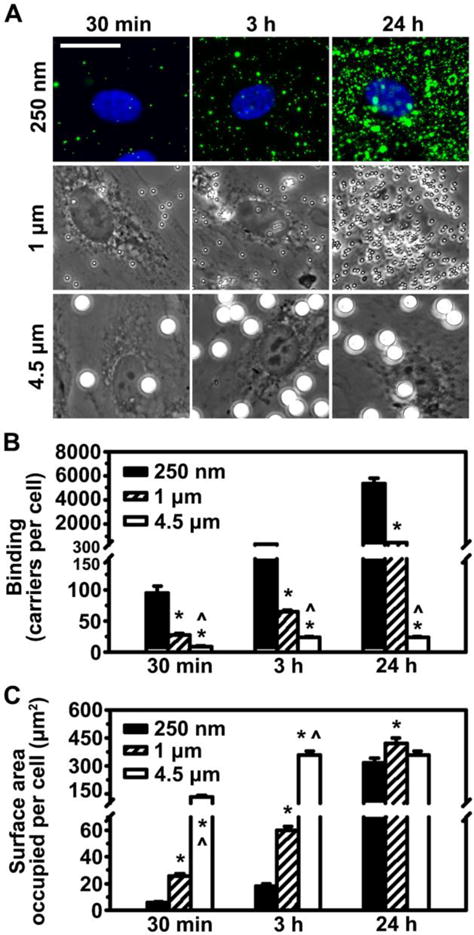
Binding of anti-ICAM carriers to endothelial cells. (A) Fluorescence (top) or phase-contrast (middle, bottom) microscopy showing binding of 250 nm, 1 μm or 4.5 μm anti-ICAM carriers bearing saturating anti-ICAM density on the carrier coat (b in Table 1) after 30 min, 3 h or 24 h incubation at room temperature with fixed activated HUVECs. Submicrometer carriers contain a green fluorophore. Scale bar = 20 μm. (B) Microscopy quantification of total carriers bound per cell. (C) Estimation of the cell-surface area occupied by all carriers bound on a cell (see Materials and Methods). Mean ± SEM. *Compares 1 μm or 4.5 μm to 250 nm; ^compares 4.5 μm to 1 μm (p<0.05 by Tukey’s test after One-way ANOVA).
An estimation of the total number of ICAM-1 molecules that are engaged on a cell by all carriers bound to that cell (Figure S3) showed that, theoretically, by 24 h binding of all three carrier sizes may have surpassed the level of ICAM-1 surface expression (reported to be between 105-106 molecules on these cells).39, 58 This occurred for all carrier sizes at saturating antibody-coat, implying that these carrier valencies were not limiting. Assuming an expression level of 105–106 ICAM-1 molecules/cell,39, 58 and an average cell-surface of 3,000 μm2 (measured by microscopy), a uniform distribution of ICAM-1 across the plasma membrane would render 35–350 ICAM-1 molecules/μm2, while the lower carrier valency used was 7,700 antibodies/μm2 (Table 1). Hence, we assume that for every carrier there was engagement of as many ICAM-1 molecules as sterically possible. This agrees with previous reports showing that binding positively depends on carrier valency, but the optimal valency to design a carrier depends on the level of expression of the receptor by the target cell.13, 36, 53, 59 In turn, at binding saturation, the cumulative cell-surface area occupied by all carriers bound on a cell was equal regardless of the carrier size (Figure 2C). Since this occupancy was significantly below the entire cell-surface area (400–500 μm2 occupied vs. 3,000 μm2 available), these point to ICAM-1 expression being the limiting factor. If this was not the case, one would expect that smaller carriers would pack more efficiently and would reach higher surface occupancy levels vs. larger counterparts.
These findings were then validated in live cells, only at early time points (up to 3 h; Figure S4) in order to minimize variation of binding resulting from concomitant endocytosis. The pattern of increasing number of carriers with time and lower number of carriers observed with higher carrier sizes remained. The total amount of submicrometer carriers associated per cell reached higher levels in live vs. fixed cells (e.g., 3-fold and 1.7-fold higher at 30 min and 3 h, respectively), while this difference was less apparent for micrometer and supramicrometer carriers (0.97-fold and 1.7-fold enhancement at 30 min; no variation between live and fixed cells at 3 h). This suggests that accumulation of submicrometer carriers in live cells vs. fixed cells may be more efficient. It also indicates that active responses of the targeted cell, e.g., dynamic mobilization of receptors on the membrane, concomitant uptake and possibly receptor recycling, may influence the net binding of carriers, as inferred in other studies.13, 36, 58 Hence, the kinetics of these biological responses (not only carrier design parameters) contributes to binding; this may appear counterintuitive since binding precedes uptake, but is logical taking into account that both processes occur concomitantly. For instance, one carrier may approach the cell while another carrier is being internalized and may remove a number of receptors from the surface, or recycling from a previous uptake event may increase the level of receptor available for binding. To the complexity of these intertwined events one must add additional factors of the biological environment, e.g., in the case of endothelial targeting, the presence of the blood flow may drag larger carriers from their cell interaction, etc. Hence, comparison of static studies to experimental and theoretical models introducing flow dynamics is highly relevant.7, 11, 13, 31, 53, 59, 60 Nevertheless, these results hold significance for tissues where flow is less apparent, and in vascular capillaries and postcapillary venules with almost-static conditions.61
Endothelial uptake of anti-ICAM-1 carriers with different sizes and valencies
Endothelial uptake of anti-ICAM carriers was examined next (Figure 3A). Uptake could not be traced for control IgG carriers given their low binding (Figure S2). For anti-ICAM carriers, the rate of uptake at an early time-point (30 min), expressed as the percentage of carriers internalized out of the total number of carriers associated with a cell, depended on the carrier size (Figure 3B), as inferred in the previous section. At 30 min, uptake of submicrometer and micrometer carriers was more efficient than that of supramicrometer ones (66%, 71%, and 32% uptake, respectively). This result supports the notion that uptake of larger size carriers would require more substantial plasmalemma deformations.3, 5 However, given enough time (3 h), the uptake was close to completion regardless of carrier size (90% uptake; Figure 3B), in agreement with reports showing the broad range of carrier sizes that can be internalized via the CAM pathway.24, 41, 42, 62
Figure 3.
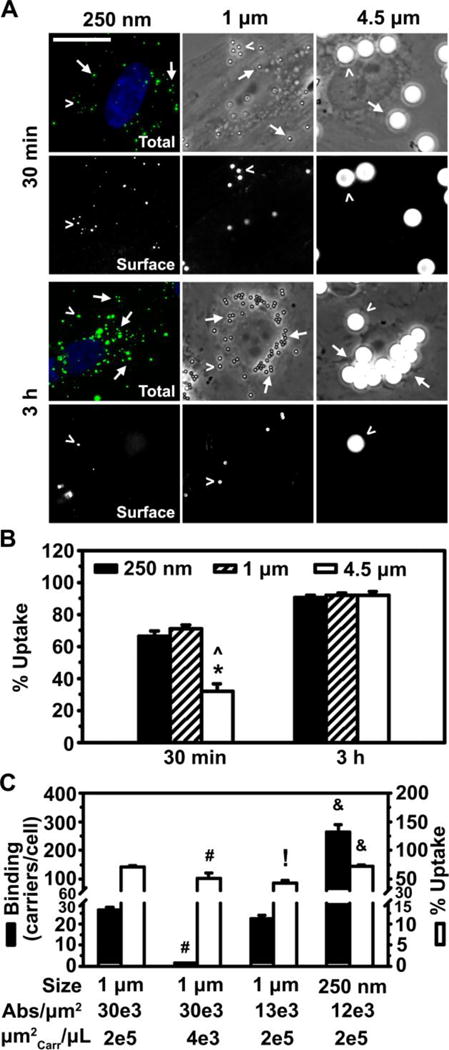
Uptake of anti-ICAM carriers by endothelial cells. (A) Endocytosis of 250 nm, 1 μm or 4.5 μm anti-ICAM carriers bearing saturating anti-ICAM density on the carrier coat (b in Table 1), after 30 min or 3 h incubation at 37˚C with activated HUVECs. For each time-point, top panels show total carriers, and bottom panels show surface-bound carriers. Submicrometer carriers contain a green fluorophore. In all cases, cell-surface located carriers are immunostained in red (see Materials and Methods). Arrows indicate internalized carriers and arrowheads mark surface-bound carriers. Scale bar = 20 μm. (B) Quantification of uptake, expressed as the percent of carriers inside cells from the total number of cell-associated carriers. Mean ± SEM. *Compares 1 μm or 4.5 μm to 250 nm; ^compares 4.5 μm to 1 μm (p<0.05 by Tukey’s test after One-way ANOVA). (C) Binding and uptake of carriers incubated at 37˚C with activated HUVECs for 30 min, according to size (250 nm vs. 1 μm), antibody (Ab) coating density per carrier surface area (12,000 to 30,000 Abs/μm2), and carrier surface area (μm2Carr) per μL of cell medium (4,000 or 200,000 μm2Carr/ μL). Mean ± SEM. #Compares carriers of same size and valency but different concentrations (two left sets of bars); !compares carriers of same size and concentration but different valencies (first-left and third sets of bars); &compares carriers of same valency and concentration but different sizes (two last-right sets of bars) (p<0.05 by Student’s t-test).
To examine whether carrier size alone impacts uptake or this is due to other parameters of carrier design, internalization was compared among carriers of similar size but different anti-ICAM valency, and carriers of similar targeting valency but different sizes (Figure 3C). First, microcarriers with similar size and targeting valency (1 μm diameter; 30,000 Abs/μm2 carrier surface; Table 1) were incubated with cells at different concentrations: 76,000 vs. 1,500 carriers/μL (equivalent to 200,000 vs. 4,000 μm2 of carrier surface/μL). As expected, this rendered different levels of binding to cells (27 vs. 1.5 carriers/cell within 30 min; Figure 3C) and different absolute levels of uptake (19 vs. 0.7 carriers internalized/cell; not shown). However, the rate of internalization was considerable in both cases and not significantly different: 71 ± 2% and 60 ± 11% of all cell-associated carriers within 30 min (Figure 3C). This indicates that uptake of each one carrier is an individual event, independent of the presence of other carriers bound to that cell.
Next, the size and concentration of anti-ICAM carriers were kept constant (1 μm diameter; 76,000 carriers/μL, equivalent to 200,000 μm2 of carrier surface/μL) but the density of anti-ICAM on the coat was varied: 30,000 vs. 13,000 anti-ICAM molecules/μm2 carrier surface. Although this decreased carrier avidity (Kd varied from 0.87 to 1.41 pM; Table 1), cell binding was not significantly decreased at the time point tested (30 min): 27 to 23 carriers/cell (Figure 3C). However, uptake was reduced from 71% to 44% uptake (Figure 3C). This verifies that the total number of carriers bound on a cell does not impact the rate of uptake of each individual carrier. This also demonstrates that for each individual carrier, uptake is ruled by the density of the targeting antibody on the carrier coat, which determines the density of engagement of the receptor on the cell-surface. Finally, comparison of carriers of different sizes (250 nm vs. 1 μm) but similar anti-ICAM density per carrier surface area (12,000 to 13,000 anti-ICAM molecules/μm2 carrier surface) and concentration (equivalent to 200,000 μm2 of carrier surface/μL) showed that smaller carriers were internalized more efficiently: 76% vs. 44% uptake (Figure 3C). These results suggest that uptake is ruled by a complex interplay between the density of engagement of the receptor and carrier size.
Ceramide signaling induced by anti-ICAM carriers of different sizes and valencies
The data obtained suggest that uptake of ICAM-1-targeted carriers is ruled by an interplay between the density of engagement of the receptor and the carrier size. While carrier size may influence the ability of the plasmalemma to deform and engulf a carrier, the density of engagement of the receptor (which relates to the targeting antibody density on the carrier) may rule the cell-signaling contributing to uptake. In the case of ICAM-1, enrichment of ceramide at regions of the plasmalemma where anti-ICAM carriers bind is a necessary signal toward uptake, and mimicking this signaling can also be used as a strategy to improve uptake via other routes.23, 41 This is because ceramide associates with biophysical changes in the plasmalemma and cytoskeletal reorganization, both of which facilitate carrier engulfment and uptake.23, 41 Hence, association of ceramide with carrier size and valency was studied next. First, ceramide immunostaining (Figure 4A) showed a clear difference between ceramide levels in regions of carrier binding and engulfment around the carrier perimeter vs. background levels at the surrounding regions (this difference is herein called “enrichment”; Figure 4B). For all carrier sizes, carrier-cell binding areas were enriched in ceramide, yet enrichment was lower for submicrometer carriers vs. micrometer or supramicrometer ones (Figure 4C). This finding is interesting given the previously noted result that larger carriers showed initially lower levels of internalization. Such lower internalization could suggest that supramicrometer carriers are less efficient inducing cell-signaling conducive to uptake, or that sufficient signaling needs to build up over time to enable uptake of such large carriers. The fact that ceramide enrichment (a key signal mediating plasmalemma deformability and actin reorganization via the CAM pathway)44 was greater for large carriers ruled out the first possibility: both supramicrometer and micrometer carriers induced similar levels of specific ceramide enrichment at binding sites, and this was higher compared to submicrometer counterparts (Figure 4B). Therefore, the second explanation is more plausible, which agrees with a greater requirement for remodeling of the cell to enable uptake of such large carriers.6, 15, 23, 41
Figure 4.
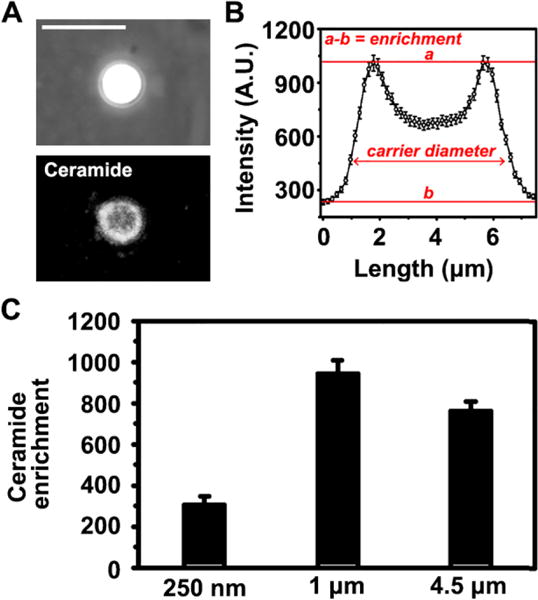
Ceramide enrichment at sites of binding of anti-ICAM carriers to endothelial cells. (A) Example of a 4.5 μm anti-ICAM carrier (phase-contract, upper panel) co-localizing with ceramide (immunofluorescence, bottom panel) at the plasma membrane of activated HUVECs after 30 min incubation. Scale bar = 10 μm. (B) Quantification of ceramide enrichment by fluorescence microscopy, where a = ceramide mean fluorescence intensity at the carrier periphery (engulfment peaks), b = ceramide mean fluorescence intensity at the neighboring cell-surface region (base of the curve), and hence a-b = “ceramide enrichment” at carrier engulfment sites. The x axis shows the length of the cell-surface area measured, encompassing the averages of 4.5 μm diameter carriers and 1.5 μm surrounding regions. (C) Ceramide enrichment at sites of binding of 250 nm, 1 μm or 4.5 μm anti-ICAM carriers. Mean ± SEM.
To examine whether carrier size alone impacts this specific signaling or this is due to other parameters of carrier design, a comparison in ceramide enrichment was made between carriers of similar size but different targeting valencies, and carriers of similar targeting valency but different sizes (Figure 5A). First, microcarriers with similar size and anti-ICAM density (1 μm and 30,000 Abs/μm2 carrier surface) were incubated with cells at different concentrations: 76,000 vs. 1,500 carriers/μL (equivalent to 200,000 vs. 4,000 μm2 of carrier surface/μL). This rendered different levels of carrier binding to cells (20-fold difference at 30 min), yet the ceramide enrichment induced by each individual carrier was comparable (1.2-fold difference; Figure 5A), just like in the case of uptake (Figure 3C). This indicates that along with uptake, the signal is individually relayed by each carrier bound to ICAM-1, independently of how many carriers are bound on a cell. Next, the size and concentration of anti-ICAM carriers were kept constant (1 μm diameter; 76,000 carriers/μL, equivalent to 200,000 μm2 of carrier surface/μL) but the density of anti-ICAM on the coat was varied: 30,000 vs. 13,000 anti-ICAM molecules/μm2. The ceramide enrichment was 2-fold decreased for carriers with lower anti-ICAM density (Figure 5A), despite the fact that binding was similar (1.1-fold difference). This indicates that the density of engagement of each individual carrier, not the total number of carriers bound on a cell, rules this signaling, just as observed for uptake (Figure 3C). Furthermore, carriers of different sizes (250 nm vs. 1 μm) but similar anti-ICAM density (12,000 to 13,000 anti-ICAM/μm2 carrier surface) and concentration used for cell binding (equivalent to 200,000 μm2 of carrier surface/μL) were compared. While smaller carriers bound at a greater extent (12-fold difference at 30 min), ceramide enrichment only varied by 1.2-fold for these carriers (Figure 5A). Therefore, it is the “density of engagement” of each individual carrier (the receptor engagement per surface area on the cell), not the absolute number of receptors engaged by a carrier or the cumulative binding of all carriers that regulates signaling conducive to endocytosis, and this process is regulated independently for each individual carrier.
Figure 5.
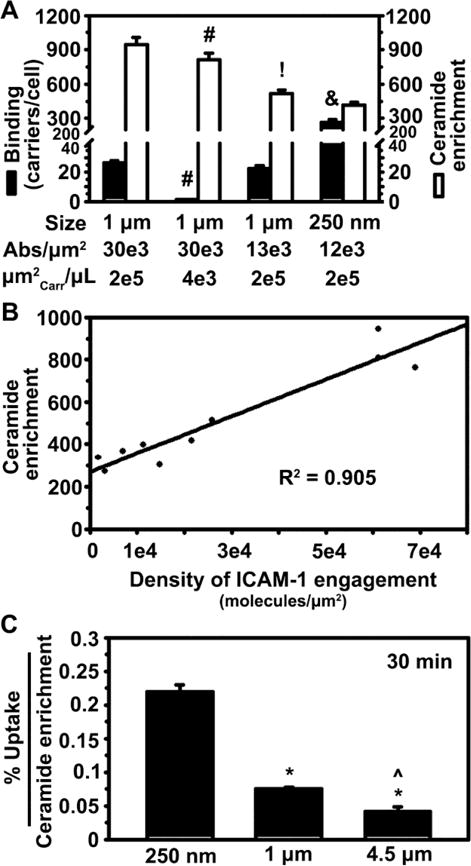
Role of carrier size, coating density, and concentration on ceramide enrichment induced by anti-ICAM carrier binding to endothelial cells. (A) Binding and ceramide enrichment of anti-ICAM carriers incubated with activated HUVECs at 37°C for 15 or 30 min, according to carrier size (250 nm vs. 1 μm), antibody (Ab) coating per carrier surface area (12,000 to 30,000 Abs/μm2), and carrier surface area (μm2Carr) per μL of cell medium (4,000 or 200,000 μm2 carrier surface/μL). Mean ± SEM. #Compares carriers of same size and valency but different concentrations (two left sets of bars); !compares carriers of same size and concentration but different valencies (first-left and third sets of bars); &compares carriers of same valency but different sizes (two last-right sets of bars) (p<0.05 by Student’s t-test). (B) Relationship between ceramide enrichment vs. the density of ICAM-1 engagement on the cell-surface by each carrier formulation, encompassing all anti-ICAM carriers shown in Tables 1 and 2. See Materials and Methods for details on the calculations. The line represents a linear regression, for which R2 = regression coefficient. (C) Uptake of anti-ICAM carriers (b in Table 1), normalized to ceramide enrichment. *Compares 1 μm or 4.5 μm to 250 nm; ^compares 4.5 μm to 1 μm (p<0.05 by Tukey’s test after One-way ANOVA).
In accord with this, regression analysis of all anti-ICAM formulations tested indicated an apparent direct linear correlation between ceramide enrichment and targeting antibody density on the carrier surface (R2=0.905; Figure 5B). This included the original formulations shown in Figure 5A, as well as supramicrometer carriers shown in Figure 4 and new submicrometer carriers with additional variations of the anti-ICAM valency, whose characterization is detailed in Table 2. There was no linear correlation when comparing ceramide enrichment to: the total cell-surface area that all bound carriers occupied on a cell (R2=0.069; Figure S5A), the cell-surface area occupied by each single carrier (R2=0.366; Figure S5B), the estimated total ICAM-1 engaged by all carriers bound on a cell (R2=0.055; Figure S5C), or the estimated ICAM-1 engagement by each single carrier (R2=0.368; Figure S5D).
Interestingly, the ratio between the uptake efficiency and the ceramide enrichment observed for these carriers decreased with increasing carrier size, and this difference was more dramatic between submicrometer carriers vs. larger counterparts (Figure 5C). This agrees with the fact that larger carriers would have a greater need for ceramide enrichment, plasmalemma deformability and/or cytoskeletal rearrangements in order to achieve uptake. This correlates well with our previous study indicating that coupling of sphingomyelinases on the surface of carriers targeted to non-CAM receptors (e.g., mannose-6-phosphate receptor or transferrin receptor) also enables ceramide generation and favors uptake of micrometer and supramicrometer carriers, with larger carriers depending more upon a higher dose of the ceramide-generating enzyme.23
Role of valency density of anti-ICAM carriers on PKC signaling
To further validate that valency density rules signaling upon carrier binding, we examined PKC recruitment and activation at anti-ICAM binding sites. PKC is another signaling mediator of the CAM pathway, which contributes to cytoskeletal rearrangements40, 41 and associates with ceramide production.63 We first used an immunofluorescence approach similar to the one described above to evaluate ceramide enrichment. Using an antibody capable of detecting all PKC isoforms regardless of their activation status, we found that total PKC enrichment at carrier binding sites depended on antibody valency, but not carrier size (Figure 6A; black bars). For instance, carriers with similar anti-ICAM coat density (12,000 and 13,000 Abs/μm2) but very different sizes (250 nm vs. 1 μm) rendered similar total PKC enrichment (1.05-fold difference; two left black bars in Figure 6A). Instead, for carriers with the same size (250 nm) lowering the valency from 12,000 to 7,700 Abs/μm2 decreased total PKC enrichment by 1.5-fold (two right black bars in Figure 6A). This pattern persisted when we evaluated a more specific marker of PKC signaling: active (phosphorylated)-PKCα (pPKCα), the main form previously related to ICAM-1 signaling.39 As in the case of total PKC, very different sizes (1 μm vs. 250 nm) but similar anti-ICAM valency (13,000 and 12,000 Abs/μm2) generated similar pPKCα enrichment (1.1-fold difference; two left white bars in Figure 6A), while carriers with the same size (250 nm) but different valency (12,000 vs. 7,700 Abs/μm2) rendered different enrichment (1.5-fold difference; two right white bars in Figure 6A). Western blot analysis of pPKCα, to confirm this result by a different method, suggested a similar trend when comparing carriers of the same size (1 μm) and different valency (30,000 vs. 13,000 Abs/μm2), which rendered different signal (1.24-fold difference; two left bars in Figure 6B). Carriers of very different size (250 nm vs. 1 μm) and more similar valency (9,800 vs. 13,000 Abs/μm2) rendered a similar signal (1.08-fold difference; two right bars in Figure 6B). This is despite Western blot not being ideal for this type of analysis, since it does not allow us to distinguish surface-bound from internalized particles (in contract to fluorescence visualization), and the goal is to relate binding valency to signals preceding endocytosis. Hence, the fact that Western blot results paired relatively well with fluorescence imaging strengthens our conclusion and suggests that, perhaps, intracellular trafficking following endocytosis is regulated by valency density in a similar manner.
Figure 6.
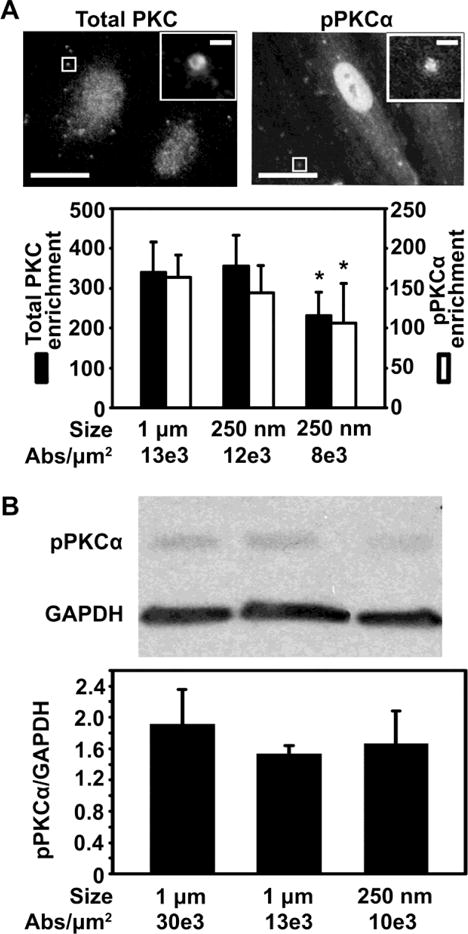
Role of carrier coating density on PKC signaling induced by binding of anti-ICAM carriers to endothelial cells. (A) Fluorescence microscopy (top panel, with magnified insets showing 1 μm anti-ICAM carriers) and image quantification (bottom panel) of the enrichment of either total PKC or pPKCα upon incubation of anti-ICAM carriers with activated HUVECs at 37°C (enrichment at 10 min and 30 was averaged). Different carrier sizes (250 nm vs. 1 μm) and antibody (Ab) coating densities (7,700 to 13,000 Abs/μm2) are shown. Scale bar = 10 μm (full) or 1 μm (inset). (B) Western blot (top panel) and densitometric quantification (bottom panel) showing pPKCα normalized to GAPDH levels, in cells incubated with anti-ICAM carriers (250 nm or 1 μm in diameter; 9,800 to 30,000 Abs/μm2) as in (A). Mean ± SEM. *Compares carriers of same size but different valency; #compares carriers of same valency but different sizes; (p<0.05 by Student’s t-test).
Conclusions
Understanding how design parameters of targeted drug carriers influence interaction with cells and the subsequent signaling leading to endocytosis is key to inform optimization of drug delivery systems.1, 2 Both the size and targeting valency of drug carriers impact cell binding and endocytic uptake.3–8, 12, 36, 64 However, how these parameters influence receptor-mediated signaling linking binding with internalization is rather obscure. For instance: Is the uptake efficiency correlative with the signaling level? Is this regulated similarly for carriers of different sizes and valencies? Are signaling and uptake events induced independently for each individual carrier or do they depend on cumulative carrier binding to cells? Is the number of receptors engaged by a carrier the factor ruling signaling and uptake, or does this depend on the density of engagement per surface area? Using model submicrometer, micrometer, and supramicrometer carriers targeted to ICAM-1 at varying valencies, we have examined these questions. Our results revealed an intricate interplay between these parameters, where the density of the targeting antibody per carrier surface area (which would determine the density of the receptor cluster on the cell-surface) is the main parameter controlling the signaling conducive to endocytosis (Figure 7). Larger carriers posed a requirement for a stronger signaling, since carrier size impacts the ability of the plasmalemma to efficiently engulf a carrier. It is likely that for endocytic pathways lacking signals that enable sufficient plasmalemma deformability (e.g., ceramide enrichment shown here for the CAM pathway), size will become restrictive regardless of the targeting density. Each carrier bound on a cell induced signaling based on its antibody-coat density (the density of the receptor cluster, not the absolute number of receptors engaged or the size of the cluster), and rendered endocytosis based on its size and enough build-up of the inducing signal. Each signaling-uptake event was independent for each single carrier bound on a cell, where the total number of carriers bound to the cell, the total surface area occupied by those carriers, and the total number of cell receptors engaged did not exert a major influence. It will be important to examine this regulation for other receptors and pathways in order to extract generic conclusions, and to evaluate how other carrier design parameters (e.g., smaller sizes, different shape, curvature, carrier type, surface chemistry, etc.) and biological environments (different cell types, presence of flow, pathological alterations, etc.) impact these outcomes.3–5, 7, 8, 12. Nevertheless, other natural routes associate to signaling common to the CAM pathway and it is expected that the findings obtained here would broadly apply to the use of said routes for drug delivery applications. For instance, ceramide and/or PKC are key for the formation of lipid domains, vesiculation, cytoskeletal rearrangements, and formation of membrane protrusions observed for phagocytic and macropinocytic pathways, intracellular invasion by certain viral and bacterial pathogens, leukocyte endothelial adhesion and extravasation, and certain events of transcytosis across cellular barriers.65–70 Therefore, these results have shed light on key factors modulating the efficacy of uptake of targeted drug carriers, which shall inform future design of vehicles for intracellular delivery.
Figure 7. Visual summary of findings.
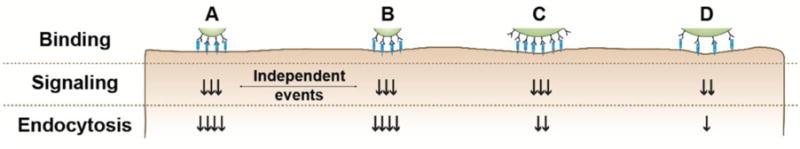
A and B: two carriers of the same formulation bind on a cell, yet the induction of signaling and uptake are independent events for each one carrier. B and C: the carrier size (hence, the surface area occupied on a cell) and the total receptor engagement do not affect signaling, but rather the same signal arises if the antibody density on the carrier surface (hence, the density of receptor engagement) is the same. Yet, for a similar signal, smaller carriers are endocytosed more efficiently. C and D: for carriers of similar size, the density of antibody on the carrier coat (hence, the density of receptor engagement) is the parameter that rules signaling (as shown in B-C), where lower signaling leads to lower uptake efficiency.
Supplementary Material
Acknowledgments
The authors thank Ronaldo Moscoso and Jordi Long for their contribution to Figure 6B. This work was supported by funds awarded to S.M. by the National Institutes of Health (R01-HL98416) and the National Science Foundation (CBET-1402756), as well as a Research Supplement to Promote Diversity in Health-Related Research to D.S. (R01 HL098416-05S1), and by the National Science Foundation graduate research fellowship (DGE-0750616), University of Maryland Flagship fellowship, and National Institutes of Health F31 fellowship (F31HL128121) to R.L.M.
Footnotes
Supporting Information Available
Stability of anti-ICAM carriers; Specificity of endothelial binding of anti-ICAM carriers; estimated ICAM-1 engagement by anti-ICAM carriers; anti-ICAM carrier association to live endothelial cells; relationship between ceramide enrichment at sites of anti-ICAM carrier binding and other targeting parameters. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Duncan R, Richardson SC. Mol Pharm. 2012;9:2380–2402. doi: 10.1021/mp300293n. [DOI] [PubMed] [Google Scholar]
- 2.Muro S. J Control Release. 2012;164:125–137. doi: 10.1016/j.jconrel.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitragotri S. Pharm Res. 2009;26:232–234. doi: 10.1007/s11095-008-9740-y. [DOI] [PubMed] [Google Scholar]
- 4.Simone EA, Dziubla TD, Muzykantov VR. Expert Opin Drug Deliv. 2008;5:1283–1300. doi: 10.1517/17425240802567846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM. Proc Natl Acad Sci USA. 2008;105:11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herd H, Daum N, Jones AT, Huwer H, Ghandehari H, Lehr CM. ACS Nano. 2013;7:1961–1973. doi: 10.1021/nn304439f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doshi N, Mitragotri S. PLoS ONE. 2010;5:e10051. doi: 10.1371/journal.pone.0010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charoenphol P, Mocherla S, Bouis D, Namdee K, Pinsky DJ, Eniola-Adefeso O. Atherosclerosis. 2011;217:364–370. doi: 10.1016/j.atherosclerosis.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 9.Geng Y, Dalhaimer P, Cai S, Tsai R, Tweari M, Minko T, Discher DE. Nat Nanotech. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shuvaev VV, Ilies MA, Simone E, Zaitsev S, Kim Y, Cai S, Mahmud A, Dziubla T, Muro S, Discher DE, Muzykantov VR. ACS Nano. 2011;5:6991–6999. doi: 10.1021/nn2015453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolhar P, Anselmo AC, Gupta V, Pant K, Prabhakarpandian B, Ruoslahti E, Mitragotri S. Proc Natl Acad Sci USA. 2013;110:10753–10758. doi: 10.1073/pnas.1308345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merkel TJ, Chen K, Jones SW, Pandya AA, Tian S, Napier ME, Zamboni WE, DeSimone JM. J Control Release. 2012;162:37–44. doi: 10.1016/j.jconrel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gunawan RC, Auguste DT. Mol Pharm. 2010;7:1569–1575. doi: 10.1021/mp9003095. [DOI] [PubMed] [Google Scholar]
- 14.Koval M, Preiter K, Adles C, Stahl PD, Steinberg TH. Exp Cell Res. 1998;242:265–273. doi: 10.1006/excr.1998.4110. [DOI] [PubMed] [Google Scholar]
- 15.Champion JA, Mitragotri S. Proc Natl Acad Sci USA. 2006;103:4930–4934. doi: 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brewer JM, Tetley L, Richmond J, Liew FY, Alexander J. J Immunol. 1998;161:4000–4007. [PubMed] [Google Scholar]
- 17.Chithrani BD, Chan WCW. Nano Lett. 2007;7:1542–1550. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
- 18.Chithrani BD, Ghazani AA, Chan WCW. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 19.Prabha S, Zhou WZ, Panyam J, Labhasetwar V. Int J Pharm. 2002;244:105–115. doi: 10.1016/s0378-5173(02)00315-0. [DOI] [PubMed] [Google Scholar]
- 20.Win KY, Feng SS. Biomaterials. 2005;26:2713–2722. doi: 10.1016/j.biomaterials.2004.07.050. [DOI] [PubMed] [Google Scholar]
- 21.Zhang GD, Yang Z, Lu W, Zhang R, Huang Q, Tian M, Li L, Liang D, Li C. Biomaterials. 2009;30:1928–1936. doi: 10.1016/j.biomaterials.2008.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ansar M, Serrano D, Papademetriou I, Bhowmick TK, Muro S. ACS Nano. 2013;7:10597–10611. doi: 10.1021/nn404719c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papademetriou J, Garnacho C, Serrano D, Bhowmick T, Schuchman EH, Muro S. J Inherit Metab Dis. 2013;36:467–477. doi: 10.1007/s10545-012-9534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hatakeyama H, Akita H, Maruyama K, Suhara T, Harashima H. Int J Pharm. 2004;281:25–33. doi: 10.1016/j.ijpharm.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 26.Oh P, Borgstrom P, Witkiewicz H, Li Y, Borgtröm BJ, Chrastina A, Iwata K, Zinn KR, Baldwin R, Testa JE, Schnitzer JE. Nat Biotechnol. 2007;25:327–337. doi: 10.1038/nbt1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frey A, Giannasca KT, Weltzin R, Giannasca PJ, Reggio H, Lencer WI, Neutra MR. J Exp Med. 1996;184:1045–1059. doi: 10.1084/jem.184.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolhar P, Mitragotri S. Adv Funct Mater. 2012;22:3759–3764. [Google Scholar]
- 29.Papademetriou IT, Garnacho C, Schuchman EH, Muro S. Biomaterials. 2013;34:3459–3466. doi: 10.1016/j.biomaterials.2013.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papademetriou J, Tsinas Z, Hsu J, Muro S. J Control Release. 2014;188:87–98. doi: 10.1016/j.jconrel.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eniola AO, Hammer DA. Biomaterials. 2005;26:7136–7144. doi: 10.1016/j.biomaterials.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Saul JM, Annapragada AV, Bellamkonda RV. J Control Release. 2006;114:277–287. doi: 10.1016/j.jconrel.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 33.Wang BB, Galliford CV, Low PS. Nanomedicine. 2014;9:313–330. doi: 10.2217/nnm.13.175. [DOI] [PubMed] [Google Scholar]
- 34.Stefanick JF, Ashley JD, Kiziltepe T, Bilgicer B. ACS Nano. 2013;7:2935–2947. doi: 10.1021/nn305663e. [DOI] [PubMed] [Google Scholar]
- 35.Kawano K, Maitani Y. J Drug Deliv. 2011;2011:160967. doi: 10.1155/2011/160967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng ZL, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Science. 2012;338:903–910. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghaffarian R, Muro S. Mol Pharm. 2014;11:4350–4362. doi: 10.1021/mp500409y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rothlein R, Springer TA. J Exp Med. 1986;163:1132–1149. doi: 10.1084/jem.163.5.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serrano D, Muro S. In: Mechanobiology of the Endothelium. Aranda-Espinoza H, editor. CRC Press; Boca Raton, FL: 2015. pp. 185–226. [Google Scholar]
- 40.Muro S, Wiewrodt R, Thomas A, Koniaris L, Albelda SM, Muzykantov VR, Koval MJ. Cell Sci. 2003;116:1599–1609. doi: 10.1242/jcs.00367. [DOI] [PubMed] [Google Scholar]
- 41.Serrano D, Bhowmick T, Chadha R, Garnacho C, Muro S. Arterioscler Thromb Vasc Biol. 2012;32:1178–1185. doi: 10.1161/ATVBAHA.111.244186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muro S, Garnacho C, Champion JA, Leferovich J, Gajewski C, Schuchman EH, Mitragotri S, Muzykantov VR. Mol Ther. 2008;16:1450–1458. doi: 10.1038/mt.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muro S, Mateescu M, Gajewski C, Robinson M, Muzykantov VR, Koval M. Am J Physiol Lung Cell Mol Physiol. 2006;290:L809–817. doi: 10.1152/ajplung.00311.2005. [DOI] [PubMed] [Google Scholar]
- 44.Murciano JC, Muro S, Koniaris L, Christofidou-Solomidou M, Harshaw DW, Albelda SM, Granger DN, Cines DB, Myzukantov VR. Blood. 2003;101:3977–3984. doi: 10.1182/blood-2002-09-2853. [DOI] [PubMed] [Google Scholar]
- 45.Hsu J, Serrano D, Bhowmick T, Kumar K, Shen Y, Kuo YC, Garnacho C, Muro S. J Control Release. 2011;149:323–331. doi: 10.1016/j.jconrel.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsu J, Northrup L, Bhowmick T, Muro S. Nanomedicine. 2012;8:731–739. doi: 10.1016/j.nano.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garnacho C, Dhami R, Simone E, Dziubla T, Leferovich J, Shuchman EH, Muzykantov V, Muro S. J Pharmacol Exp Ther. 2008;325:400–408. doi: 10.1124/jpet.107.133298. [DOI] [PubMed] [Google Scholar]
- 48.Hamilton AJ, Huang SL, Warnick D, Rabbat M, Kane B, Nagaraj A, Klegerman M, McPherson DD. J Am Coll Cardiol. 2004;43:453–460. doi: 10.1016/j.jacc.2003.07.048. [DOI] [PubMed] [Google Scholar]
- 49.Weller GE, Villanueva FS, Tom EM, Wagner WR. Biotechnol Bioeng. 2005;92:780–788. doi: 10.1002/bit.20625. [DOI] [PubMed] [Google Scholar]
- 50.Choi KS, Kim SH, Cai QY, Kim SY, Kim HO, Lee HJ, Kim EA, Yoon SE, Yun KJ, Yoon KH. Mol Imaging. 2007;6:75–84. [PubMed] [Google Scholar]
- 51.Zhang N, Chittasupho C, Duangrat C, Siahaan TJ, Berkland C. Bioconjug Chem. 2008;19:145–152. doi: 10.1021/bc700227z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park S, Kang S, Veach AJ, Vedvyas Y, Zarnegar R, Kim JY, Jin MM. Biomaterials. 2010;31:7766–7775. doi: 10.1016/j.biomaterials.2010.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calderon AJ, Bhowmick T, Leferovich J, Burman B, Pichette B, Muzykantov V, Eckmann DM, Muro S. J Control Release. 2011;150:37–44. doi: 10.1016/j.jconrel.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petros RA, DeSimone JM. Nat Rev Drug Discov. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 55.Muro S, Muzykantov VR, Murciano J. In: Methods in molecular biology Bioconjugation protocols. Niemeyer CM, editor. Humana Press; Totowa, NJ: 2004. pp. 21–36. [DOI] [PubMed] [Google Scholar]
- 56.Liu J, Weller GER, Zern B, Ayyaswamy PS, Eckmann DM, Muzykantov VR, Radhakrishnan R. Proc Natl Acad Sci USA. 2010;107:16530–16535. doi: 10.1073/pnas.1006611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wiseman ME, Frank CW. Langmuir. 2012;28:1765–1774. doi: 10.1021/la203095p. [DOI] [PubMed] [Google Scholar]
- 58.Muro S, Dziubla T, Qiu W, Leferovich J, Cui X, Berk E, Muzykantov VR. J Pharmacol Exp Ther. 2006;317:1161–1169. doi: 10.1124/jpet.105.098970. [DOI] [PubMed] [Google Scholar]
- 59.Calderon AJ, Muzykantov V, Muro S, Eckmann DM. Biorheology. 2009;46:323–341. doi: 10.3233/BIR-2009-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bhowmick T, Berk E, Cui X, Muzykantov VR, Muro S. J Control Release. 2012;157:485–492. doi: 10.1016/j.jconrel.2011.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perry MA, Granger DN. J Clin Invest. 1991;87:1798–1804. doi: 10.1172/JCI115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garnacho C, Albelda SM, Muzykantov VR, Muro S. J Control Release. 2008;130:226–233. doi: 10.1016/j.jconrel.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zeidan YH, Hannun YA. J Biol Chem. 2007;282:11549–11561. doi: 10.1074/jbc.M609424200. [DOI] [PubMed] [Google Scholar]
- 64.Rudnick SI, Adams GP. Cancer Biother Radiopharm. 2009;24:155–161. doi: 10.1089/cbr.2009.0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Canals D, Jenkins RW, Roddy P, Hernandez-Corbacho MJ, Obeid LM, Hannun YA. J Biol Chem. 2010;285:32476–32485. doi: 10.1074/jbc.M110.141028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cardone MH, Smith BL, Song W, Mochly-Rosen D, Mostov KE. J Cell Biol. 1994;124:717–727. doi: 10.1083/jcb.124.5.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheeseman KL, Ueyama T, Michaud TM, Kashiwagi K, Wang D, Flax LA, Shirai Y, Loegering DJ, Saito N, Lennartz MR. Mol Biol Cell. 2006;17:799–813. doi: 10.1091/mbc.E04-12-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grassme H, Becker KA, Zhang Y, Gulbins E. J Biol Chem. 2008;389:1371–1379. doi: 10.1515/BC.2008.162. [DOI] [PubMed] [Google Scholar]
- 69.Holopainen JM, Subramanian M, Kinnunen PK. Biochemistry. 1998;37:17562–17570. doi: 10.1021/bi980915e. [DOI] [PubMed] [Google Scholar]
- 70.Lopes Pinheiro MA, Kroon J, Hoogenboezem M, Geerts D, van Het Hof B, van der Pol SM, van Buul JD, de Vries HE. J Immunol. 2016;196:72–79. doi: 10.4049/jimmunol.1500702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.