

Abstract
Hypomorphic mutations in the genes encoding the MRE11/RAD50/NBS1 (MRN) DNA repair complex lead to cancer-prone syndromes. MRN binds DNA double strand breaks where it functions in repair and triggers cell cycle checkpoints via activation of the ataxia-telangiectasia mutated (ATM) kinase. To gain understanding of MRN in cancer, we engineered mice with B lymphocytes lacking MRN, or harboring MRN in which MRE11 lacks nuclease activities. Both forms of MRN deficiency led to hallmarks of cancer, including oncogenic translocations involving c-Myc and the immunoglobulin locus. These pre-neoplastic B lymphocytes did not progress to detectable B lineage lymphoma, even in the absence of p53. Moreover, Mre11 deficiencies prevented tumorigenesis in a mouse model strongly predisposed to spontaneous B cell lymphomas. Our findings indicate that MRN cannot be considered a standard tumor suppressor and instead imply that nuclease activities of MRE11 are required for oncogenesis. Inhibition of MRE11 nuclease activity increased DNA damage and selectively induced apoptosis in cells overexpressing oncogenes, suggesting MRE11 serves an important role in countering oncogene-induced replication stress. Thus, MRE11 may offer a target for cancer therapeutic development. More broadly, our work supports the idea that subtle enhancements of endogenous genome instability can exceed the tolerance of cancer cells and be exploited for therapeutic ends.
Keywords: Cancer, Lymphoma, Genome Stability, DNA Repair, Cancer Therapy
INTRODUCTION
Inherited cancer syndromes cause approximately 5-10% of cancers. Many of these diseases stem from mutations in genes involved in cell cycle checkpoint control and DNA repair. DNA double-strand breaks (DSBs) are highly toxic chromosomal lesions, estimated to arise nearly fifty times per genome per cell cycle resulting from stalled or collapsed replication forks (1). Failings of cellular responses to DSBs cause diseases with diverse sequelae, including cancer predisposition (2). For example, Li-Fraumeni syndrome (LFS) patients are susceptible to early onset of a broad range of tumors including sarcomas, leukemias, adrenal cortical carcinomas, and other cancers (3). LFS is caused by germline mutations in the TP53 gene, which plays crucial roles in cell cycle regulation upon DNA damage. Similarly, inherited deficiencies in ATM, the main kinase responsible for activation of DNA damage checkpoints, lead to ataxia-telangiectasia (A-T) featuring lymphoma and other cancers (4).
ATM is a serine/threonine kinase that is activated in response to DNA DSBs and initiates a signaling cascade that elicits apoptosis, cell cycle checkpoints and/or DNA repair. The recruitment and activation of ATM depends primarily on the MRE11/RAD50/NBS1 (MRN) complex, which rapidly binds and stabilizes one or both DNA ends (5,6). Depending on the nature of the break, the nucleolytic activities of MRE11 remove covalently bound moieties and initiate DNA resection (7–9). The functional relationship between MRN and ATM is underscored by identification of mutations in members of the complex in inherited genetic syndromes that resemble A-T. Partial loss-of-function mutations in MRE11 are responsible for ataxia-telangiectasia like disorder (ATLD), an inherited genome instability disorder. ATLD is characterized by cerebellar ataxia and variable predisposition to cancer (10,11). Hypomorphic mutations in NBS1 lead to Nijmegen breakage syndrome (NBS) and mutations in RAD50 lead to NBS-like disorder. Individuals with NBS exhibit severe predisposition to cancer, immunodeficiency, and microcephaly (12,13).
The cancer prone phenotypes associated with MRN syndromes suggest the complex functions as a tumor suppressor. Affected individuals harbor either homozygous hypomorphic mutations or compound heterozygous alleles. For example, most NBS patients are homozygous for the founder 657del allele, which is carried by approximately 0.6% of the Slavic population of Central Europe (14). Two Japanese brothers with ATLD died of pulmonary adenocarcinoma at ages 9 and 14, and were shown to harbor two distinct MRE11 mutant alleles (one from each parent) (11). Studies of engineered mouse models harboring NBS or ATLD disease alleles have reinforced the notion that MRN acts as a tumor suppressor. Like their human counterparts, the mice are commonly cancer prone (15–18). However, studies of murine models have also hinted that the biology of MRN is more complicated. Complete knockout of any murine MRN component results in early embryonic lethality (19–21). This is likely the same for humans, as no known disease alleles are null. Thus, human disease alleles must preserve some function(s) of the MRN complex, i.e., function(s) that are essential for mammalian development.
We previously generated a germline mouse allele in which Mre11 can be conditionally deleted, leading to deficiency of the entire MRN complex (19). In addition, we have targeted a single amino acid change in the endogenous Mre11 locus that expresses a mutant MRE11 (Mre11H129N) deficient in DNA nuclease activities, while preserving normal protein levels and integrity of the complex (19). Studies of mice and cells with combinations of these alleles demonstrated that nuclease activities of MRE11 are essential for embryonic development and are involved in DSB repair (19,22). However, nuclease activities are dispensable for activation of ATM, thus providing a clear separation of function between involvement in DNA repair and control of cell cycle responses.
In this study we have employed our engineered Mre11 alleles in combination with p53 deficiency to further understand roles for MRN in cancer predisposition. Mice were generated in which mature B lymphocytes were rendered deficient for the MRN complex, or maintain the complex with MRE11 deficient in nuclease activities. Surprisingly, despite the presence of early molecular events in lymphomagenesis, the MRE11 deficiencies did not predispose to detectable B cell malignancy. When present in a genetic background highly prone to early B lineage lymphomas, MRE11 deficiency prevented these tumors. Thus, we conclude that MRN cannot be considered a classic tumor suppressor. Rather, certain functions of the complex, such as DNA nuclease activity, may be required for progression to cancer.
MATERIALS AND METHODS
Mouse models
Mre11cond/+, Mre11cond/−, and Mre11cond/H129N mice with CD19-Cre expression were previously described (22). Trp53tm1Tyj mice were obtained from The Jackson Laboratory. Gene targeted Artemis null mice were previously described (23). Mice were housed in a specific pathogen free facility. All experiments complied with regulations and ethics guidelines of the National Institute of Health and were approved by the Institutional Animal Care and Use Committee of the University of Michigan (PRO00006263). Class switch experiments and translocation analyses were performed on 6-12 week old mice.
IgH:MYC Translocation PCR analyses
B cells were isolated from mice and cultured as previously described (22). Cultures were infected with AID-expressing lentivurus after 24 hours as previously described. DNA was prepared 48 hours after infection using Qiagen DNeasy kit. IgH:Myc translocations were detected using a nested PCR reaction using Expand Long Template PCR System (Roche) as described previously (24).
Two-color FISH
Two-color FISH analyses on metaphase spreads from stimulated B cells was performed as described (22) using BAC 199 (199M11, Invitrogen) and BAC 207 (RP22-207123, BAC PAC Resources), and a BAC harboring murine MYC genomic sequence (generous gift from Dr. Wesley Dunnick). FISH images were acquired on an Olympus BX61 microscope using a 60× objective, DAPI, FITC, and Texas Red filters, a CCD camera, and FISHview software (Applied Spectral Imaging).
Derivation and Transformation of MEFs
Primary MEFs of less than 4 passages were immortalized with SV40 large T antigen (19) or MYC using pLenti-Myc T58A (generous gift from Dr. David Lombard). Primary MEFs were derived in our laboratories and confirmed by PCR based genotyping, sequencing, and western blot analyses. Transformed MEFs derived from these lines were routinely confirmed by PCR and Western blot analyses. Experiments were performed on cells ranging from 5 to 10 passages after immortalization. Lines and media tested negative for Mycoplasma using MycoAlert Mycoplasma detection kit (Lonza). Additional details can be found in Supplementary Methods.
Translocation PCR
DR-GFP:Mmp24 translocations were identified by a nested PCR reaction using Expand Long Template PCR System (Roche). See Supplemental Methods for detailed PCR conditions and primer sequences.
Growth curves
MYC and T-antigen transformed Mre11cond/− and Mre11cond/+ MEFs were plated (5 × 105 cells) in 10cm dishes and cultured overnight. Cells were infected twice with either adeno-cre or adeno-empty (University of Michigan vector core) at an MOI of 500:1. Four days after the second infection, cells were plated at a low density (1 × 105 cells/10cm dish) and live cells were counted every 2 days using a hemacytometer.
Mre11 inhibitor treatment of MYC overexpressing cells
Authenticated U2OS cells were originally obtained from the ATCC and tested for mycoplasma using the MycoAlert Mycoplasma detection kit (Lonza) and a PCR detection method in 2015. U2OS-pBABE and U2OS-cMycER cell lines were generated by infecting the human U2OS cell line with the retroviral vector pBABE-PURO or pBabepuro-MYC-ER (Addgene plasmid #19128) expressing human c-MYC cDNA fused to the hormone-binding domain of the estrogen receptor (ER), then selected in puromycin (2.5mg/ml). For cellular survival assays, U2OS-pBABE and U2OS-cMycER cells were plated in triplicate in 96-well plates 24 hours before treatment with 200nM 4-OHT (Sigma). After 72 h, cells were treated with mirin (Cayman Chemical) or DMSO for 24 h and survival was determined by fixing and staining cells with crystal violet then measuring absorbance of solubilized dye at 595nm. Cells were passaged between three and five times after thawing for each experiment. Survival (%) was calculated relative to vehicle-treated controls. Three or more independent experiments were performed.
β-gal senescence assay
MYC and SV40 large T-antigen transformed, as well as primary, MEFs were cultured and stained according to the manufacturers instructions (Senescence β-Galactosidase Staining Kit - Cell Signaling). See Supplementary Methods for additional details.
Apoptosis assay
U2OS-pBABE and U2OS-cMycER cells were plated in 6-well plates 24 hours before treatment with 200nm 4-OHT (72 h) then treated with mirin for 24 h. After 48 hr, cells were harvested and stained with FITC Annexin V antibody (BD Pharmingen 556419) and propidium iodide then analyzed by flow cytometry (BD Accuri C6 Flow Cytometer). Data were analyzed using FlowJo software.
Metaphase spread analyses
Mre11cond/− and Artemis−/−Mre11cond/− MEFs were infected with adeno-empty or adeno-cre viruses at an MOI of 500. Cells were grown for 3 d, split, doubly infected then incubated with colcemid (KaryoMAX) for 8 h. DAPI stained chromosomes were imaged on an Olympus BX61 microscope using 60× objective (SKYview software; Applied Spectral Imaging). Chromosomal anomalies were scored in a blinded manner from at least 3 independent experiments.
Western Blotting
Western blots of lysates from MRE11 wild-type and deficient MEFs and tumor cells were performed as previously described (25) using antibodies against (#2360S), MRE11 (#4895S), and GAPDH (#2118L) (Cell Signaling).
Immunofluorescence Microscopy
U2OS-pBABE and U2OS-cMycER cells were plated on coverslips (4×104) in a 12-well dish (24h) then treated with 200nM 4-OHT for 72 h. Fixed cells were stained with primary antibodies (45 min) then secondary antibodies, Alexa Fluor 488 or 594 (45 min). Foci were visualized using Olympus BX61 microscope using 100× objective. Additional details are in Supplemental Materials.
RESULTS
Establishing Mre11 deficiencies in the murine B lymphocyte lineage
The existence of programmed DNA rearrangements in B lymphocytes makes this lineage ideal to study roles of errant DSB repair in cancer initiation and progression. Such mistakes in repair are directly responsible for oncogenic translocations involving the immunoglobulin heavy chain (IgH) locus and oncogenes such as c-MYC (MYC), BCL2 and BCL6(26). Similarly in mice, B lineage tumors harbor IgH:Myc translocations. Therefore, we generated mice that lack MRE11 entirely, or express an Mre11 nuclease-deficient allele, in the B lymphocyte lineage to assess the impact on the initiation or progression of lymphomagenesis (19,22).
To bypass embryonic lethality conferred by absence of MRE11 (Mre11−/−) or defective MRE11 nuclease activity (Mre11H129N/−), we utilized a conditional allele (Mre11cond) that can be inactivated by cre-mediated recombination of two LoxP sites flanking a conserved exon within the endogenous locus (Figure 1A) (19). Through breeding, we generated Mre11+/cond, Mre11−/cond, and Mre11H129N/cond mice, each harboring one allele of CD19-cre which expresses cre recombinase in bone marrow B lymphocyte progenitors (22,27). Cre expression can be detected in pro-B cells in bone marrow, prior to class switch recombination (CSR), which takes place in mature B cells in peripheral lymphoid organs (27). This breeding scheme results in mice with B lymphocytes of the following genotypes: Mre11+/−, Mre11−/−, and Mre11H129N/−. We previously reported that mice lacking MRE11 entirely, or expressing nuclease deficient MRE11H129N, in B cells produce approximately normal numbers of lymphocytes in bone marrow and spleen, demonstrating that Mre11 is not required for survival of mature B cells that emanate from bone marrow and populate peripheral lymph organs. However, splenic B cells exhibited a significant decrease in CSR owing to defective repair of programmed DNA DSBs (22). In the current study we bred mice with the Mre11 genotypes described above with germline p53 knockout mice (p53−/−) (28). We observed that p53 deficiency does not rescue the class switching defect caused by the MRE11 deficiencies (Figure 1B, Supplementary Fig. 1).
Figure 1. Isotype switching and end joining remain defective in MRE11 deficient B cells lacking p53.
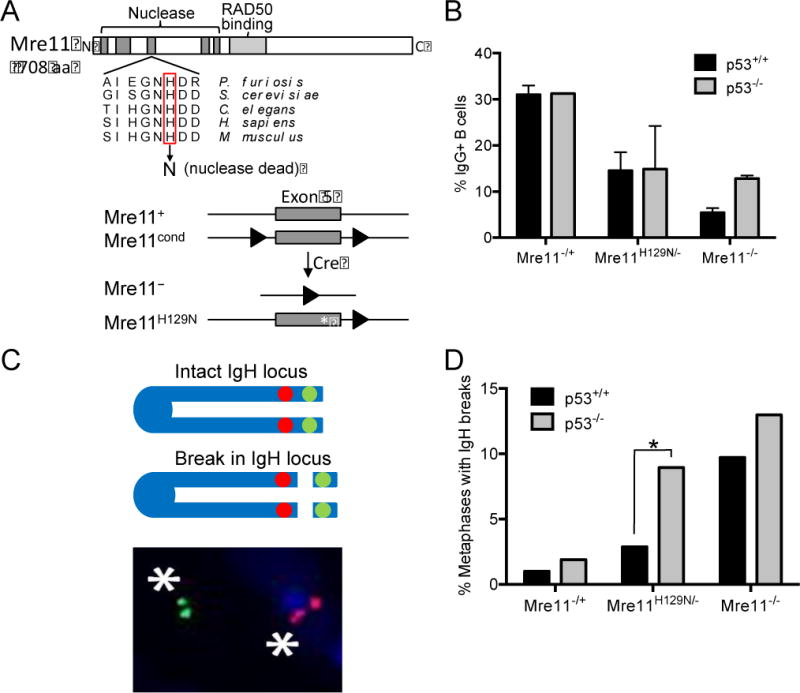
A) Mammalian MRE11 domains and location of the invariant histidine required for nuclease activity (top). The four murine germline Mre11 alleles used in this study and described previously (bottom)(19). Red rectangle, histidine in exon 5 (grey box) essential for nuclease activities; line, introns; triangles, LoxP sites; asterisk, H129N nuclease dead mutation. (B) Isotype class switching in stimulated primary splenic B cells. Quantitation of the percentage of IgG1+ B cells of the indicated genotypes. Mean ± standard error of 3 independent experiments. Primary splenic B cells were cultured then stimulated with α-CD40/IL-4 to induce class switching. CSR to IgG1 was analyzed by flow cytometry with α-B220 and α-IgG1 antibodies. (C) Two-color FISH analyses of breaks within the IgH locus. Asterisks, fluorescent signals using BACs located 3′ (red) and 5′ (green) of the IgH locus that have separated due to chromosome breakage. (D) Quantitation of the percentage of metaphases containing IgH breaks in stimulated B cells of the indicated genotypes. Data represent analyses from 3 independent experiments. >100 metaphases per genotype. *, p≤0.05 (two tailed T test).
Previously, decreased levels of isotype switching in Mre11−/− B cells coincided with increased accumulation of breaks in the IgH locus in mouse chromosome 12 (22). To determine if this aberrant outcome is impacted by p53 loss, two-color fluorescence in situ hybridization (FISH) was employed utilizing bacterial artificial chromosome (BAC) probes which flank the IgH locus. Breaks in the locus are identified as separated probes in metaphase spreads after induction of CSR in splenic B lymphocytes (Figure 1C). Loss of p53 did not significantly impact the number of chromosome breaks accumulated in Mre11+/− and Mre11−/− B cells as compared with p53 wild-type Mre11+/− and Mre11−/− B cells (Figure 1D). Interestingly, p53−/−Mre11H129N/− B cells displayed a significant increase in DNA breaks as compared to Mre11H129N/− B cells with wild-type p53 (Figure 1D). This difference is likely explained by the previous observation that cells expressing MRE11H129N maintain intact ATM dependent checkpoints, which include p53-dependent cellular responses (19). Removing p53, a substrate of ATM (29), from nuclease deficient cells likely permits B cells harboring breaks to survive and progress through the cell cycle allowing increased visualization of metaphases with broken chromosomes. The accumulation of chromosome breaks combined with defective cell cycle checkpoints provides ample substrates for aberrant repair capable of generating oncogenic translocations and consequent lymphoid neoplasia.
Lifespans and tumorigenesis in mice with combinations of p53 and B lymphocyte Mre11 deficiencies
Mice with B cell genotypes Mre11+/− (control), Mre11−/−, and Mre11H129N/−, with and without p53, were aged to evaluate survival and tumor development. p53 wild-type mice with B cell specific MRE11 deficiencies were observed to have a normal life span with a median survival of 27.5 months (compared to 29 months for the controls) (Figure 2A). Mice with p53−/−Mre11−/− and p53−/−Mre11H129N/− B cell genotypes became moribund at about 16-18 weeks of age (Figure 2B). However, the control p53−/−Mre11+/− mice also became moribund around 16-18 weeks of age (Figure 2B). Upon necropsy, the majority of p53−/− mice presented with an enlarged thymus that was negative for the B cell marker B220. Further flow cytometric analyses of these tumors indicated the masses were thymic lymphomas of T cell origin, a common outcome of murine p53 deficiency (Figure 2C) (30,31). A smaller percentage of mice succumbed to non-lymphoid tumors (Figure 2C). Therefore, it appears that B lymphocyte specific deficiency of MRE11, or MRE11 nuclease activity, does not impact the survival of mice. Furthermore, these studies imply that there is no predisposition to B cell malignancy despite the presence of genome instability and defective p53 dependent cell cycle checkpoints.
Figure 2. Survival and tumorigenesis in mice with combined p53 and B cell Mre11 deficiencies.
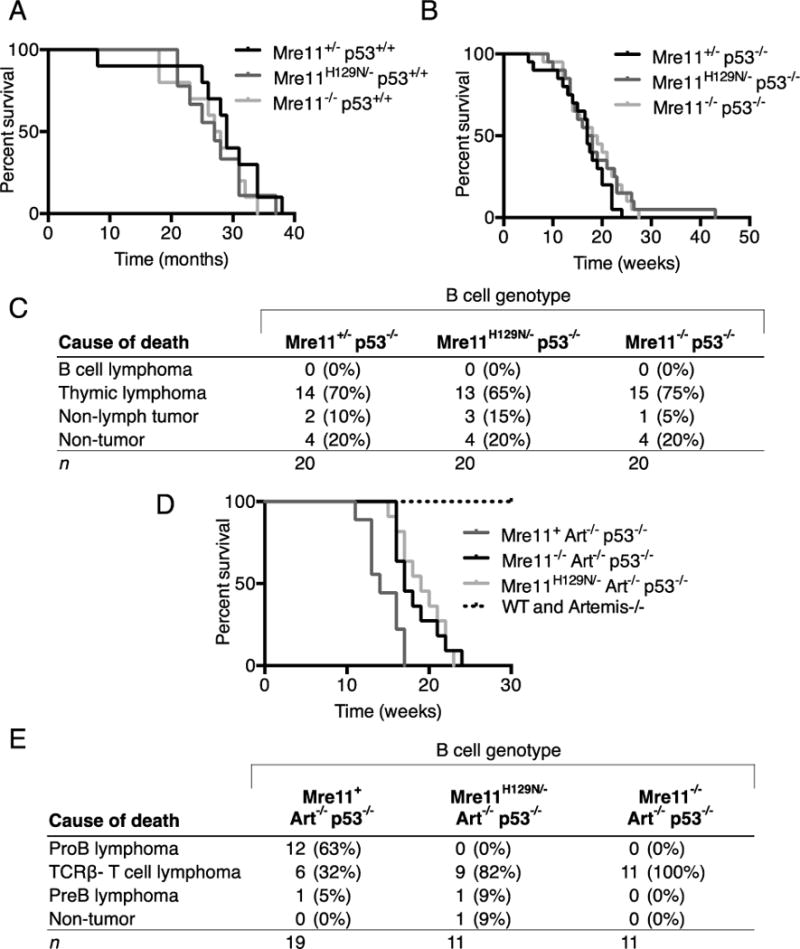
(A) Kaplan-Meier plots representing the survival percentages of mice with B cell genotypes of Mre11−/− (median survival, 27.5 months, p=0.72) and Mre11H129N/− (median survival, 29 months, p=0.50) in a p53 wild-type background. p values from Mantel-Cox, log rank test. (B) Plots as in (A) of mice with p53−/−Mre11−/− and p53−/−Mre11H129N/− B cell genotypes and p53−/−Mre11+/− controls with median survival of 17.5wks (p=0.36), 18.5wks (p=0.25). (C) Causes of death for mice of the indicated B cell genotypes. No mice developed B cell lymphoma. (D) Mre11 B-cell specific mutation suppresses pro-B lymphoma in an Artemis/p53 double null background. Kaplan-Meier survival curves representing the percent survival of mice versus age in weeks. Wild-type (n=20), Artemis−/− (n=9), Artemis−/−p53−/− (n=9), and mice with Artemis−/−p53−/− Mre11−/− (n=11), and Artemis−/−p53−/− Mre11H129N/− (n=11) B cell genotypes were observed for a period of 30 weeks. Artemis−/−p53−/−Mre11cond/− and Artemis−/−p53−/− Mre11cond/H129N mice harboring the CD19-cre transgene survive longer than Artemis−/−p53−/− mice (p≤0.01, Mantel-Cox, log rank test). (E) Table of causes of death and tumor types arising in Artemis/p53/Mre11 mutant mice. No pro-B lymphomas were observed in Artemis−/−p53−/− mice with germline Mre11cond/− or Mre11cond/H129N alleles and CD19-cre (p≤0.0001, Fisher’s exact, two tailed test). Data for Artemis−/−p53−/− mice from this study and reference 35.
MRE11 deficiency suppresses progenitor B cell lymphomagenesis
To further assess the roles of MRE11 in tumorigenesis, we examined the impact of Mre11 deficiencies in a mouse model that is strongly predisposed to B lineage lymphomas (32). The ARTEMIS DNA nuclease is required for processing DNA ends generated during V(D)J recombination, a lymphocyte specific DNA rearrangement that assembles variable region genes from component V, D and J exons (33). V(D)J recombination is required for lymphocyte development; thus, mutations in ARTEMIS (DCLRE1C) cause primary immunodeficiency disorders of varying severity, and a subset of patients harboring hypomorphic mutations exhibit cancer predisposition (34). Previous studies demonstrated that mice harboring a gene targeted null mutation in Artemis, when combined with p53 mutation, succumb to lymphoid tumors (32). The predominant tumor type observed is early onset, aggressive pro-B lymphoma harboring clonal translocations involving the IgH and Myc or N-myc genomic loci. The translocated loci are co-amplified, thereby leading to increased oncogene expression. Here, we took advantage of the Artemis−/−p53−/− mouse model of spontaneous pro-B lymphomagenesis to determine the impact of Mre11 mutation on tumor predisposition.
We introduced the targeted Mre11 mutant alleles into the Artemis/p53 double null background through mouse breedings and obtained Artemis−/− p53−/−Mre11−/cond and Artemis−/−p53−/− Mre11H129N/cond mice harboring the CD19-cre transgene. We observed that the Artemis−/−p53−/− mice expressing wild-type Mre11 (Mre11+/−) in B cells exhibited a median survival of 14 weeks (Figure 2D). In comparison, mice with B cell genotypes of Artemis−/− p53−/−Mre11−/− and Artemis−/−p53−/−Mre11H129N/− exhibited median survival times of 17 and 19 weeks, respectively. Thus, B cell specific loss of MRE11 or MRE11 nuclease activity did not markedly alter survival of these tumor prone mice.
The majority of Artemis/p53 double null mice succumbed to progenitor B lymphoma, and smaller subsets to thymic lymphoma of progenitor T cell origin or other tumor types, consistent with previous studies (32,35) (Figure 2E). Analyses of the pro-B tumors confirmed that they harbored clonal IgH rearrangements and amplification of the IgH and Myc or N-myc loci (Supplementary Fig. 2). We observed that nearly all of the mice analyzed harboring B lineage MRE11 deficiencies also succumbed to tumors. One Artemis−/− p53−/− Mre11H129N/− mouse became moribund and died with no obvious cause of death upon analysis. Remarkably, no pro-B-cell lymphomas arose in Artemis−/−p53−/− mice with either the Mre11−/− (n=11) or Mre11−/H129N (n=11, p≤0.005) B cell genotypes (Figure 2E). The mice in these cohorts primarily developed thymic lymphomas, and in one Artemis−/−p53−/−Mre11−/H129N case, a pre-B cell tumor. Importantly, this tumor as well as all of the other lymphoid tumors analyzed retained the Mre11 conditional allele and expressed MRE11 protein (Supplementary Fig. 3). These results indicate that Mre11 mutation suppresses pro-B cell lymphomas in a context where they normally arise in the majority of mice by 8–10 weeks of age.
Chromosome translocations in Mre11/p53 deficient B cells
Our observations that B cell specific loss of MRE11 or inactivation of MRE11 nuclease activity did not potentiate lymphomagenesis, and indeed, suppressed pro-B lymphoma in ARTEMIS/p53 deficiency suggest that MRE11 may be required for tumorigenesis. Programmed DSBs in the IgH locus are known intermediates in the formation of oncogenic translocations (26). Many deficiencies in DSB repair enhance the frequency of translocation formation by permitting mis-repair of breaks by alternative mechanisms. However, because the MRN complex has been implicated as functioning in all known DSB repair pathways (22,36–38), it is possible that translocations cannot be generated in the absence of MRE11.
To address this important question, the two-color FISH assay described above was employed to identify translocations involving IgH. Translocations were identified as separated centromere proximal and distal IgH probe signals, each located on different chromosomes (Figure 3A). We found that translocations were readily detectable in metaphases from Mre11−/− and Mre11H129N/− stimulated B cells (all were p53−/−) (Figure 3B). We also determined if specific IgH:Myc translocations occur in Mre11/p53 deficient B cells using a PCR based assay (24). This assay utilizes primers that amplify translocations occurring between the region of DNA spanning JH4 and Cμ in IgH (chromosome 12) and exon 1 of Myc (chromosome 15) (Figure 3C). Indeed, IgH:Myc translocations were detected in p53−/−Mre11−/− and p53−/−Mre11H129N/− B cells, as well as p53−/−Mre11+/− B cells (Figure 3D) and confirmed by sequencing (Supplementary Fig. 4A–C). Thus, p53−/−Mre11−/− and p53−/−Mre11H129N/− B cells retain the capacity to generate oncogenic chromosomal translocations.
Figure 3. Detection of translocations in Mre11-deficient cells.
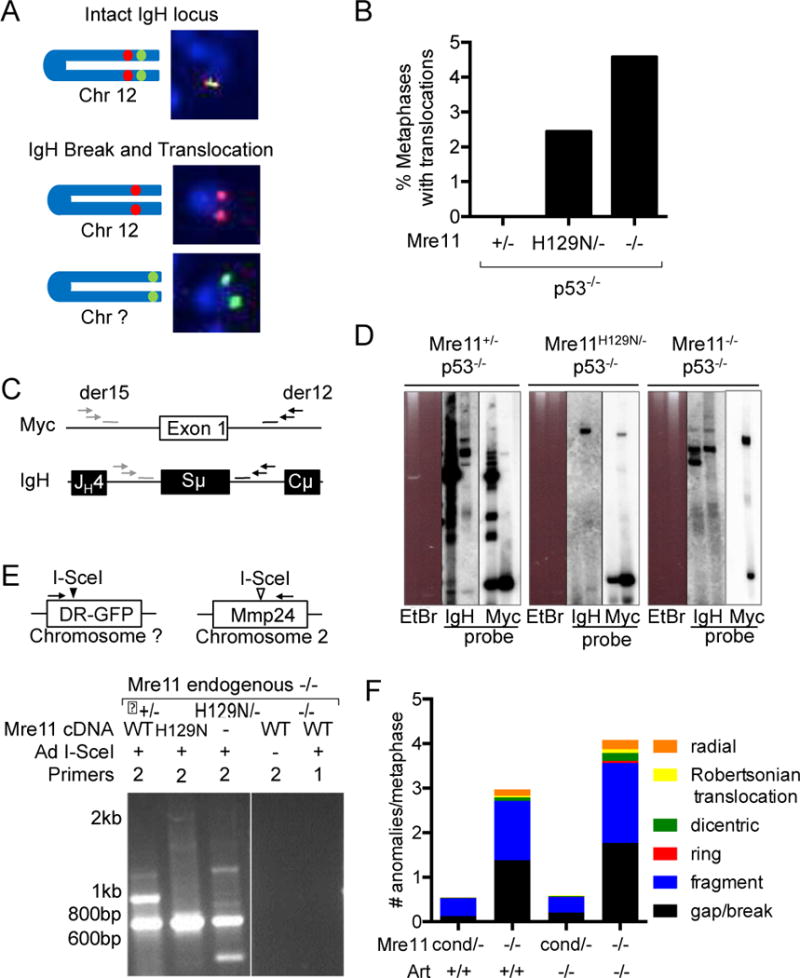
(A) Schematic of 2-color FISH IgH translocation assay. Translocations were scored when the telomeric (green) and centromeric (red) IgH probes were separated and each were localized to different chromosomes. (B) Quantitation of translocations in Mre11+/−, Mre11−/− and Mre11H129N/− B cells in a p53−/− background. Data are from 3 independent experiments, >100 metaphases/sample. (C) Schematic of IgH:Myc translocation PCR. Nested primers (arrows) flanking exon 1 of Myc and Sμ of the IgH locus detect translocations between the two loci. Probes, bars. (D) B cells of the indicated Mre11 genotypes were stimulated to undergo CSR in culture. Nested PCR products were analyzed by Southern blotting with chr. 12 (IgH) and chr. 15 (Myc) probes. (E) Top, Schematic of PCR based detection of I-SceI endonuclease induced translocations. Nested PCR primers detect translocations between the randomly integrated I-SceI site (black arrowhead) and endogenous, cryptic I-SceI site within the Mmp24 locus (open arrowhead) on chr. 2. Bottom, EtBr stained agarose gels of nested PCR products from cells expressing Mre11WT, Mre11H129N or no Mre11 (−). Addition of Ad I-SceI and nested (2) or first round only (1) PCR primers, as indicated. (F) Spontaneous chromosomal anomalies in Mre11−/− and Mre11−/−Art−/− MEFs. Metaphases were stained with DAPI and scored in a blinded manner for chromosomal anomalies. Bar graphs, number of anomalies per metaphase; type of anomaly, as indicated in the color scheme. Data are from 3 independent experiments.
Chromosome translocations in MRE11 and ARTEMIS/MRE11 deficient fibroblasts
We next examined the broader roles of the MRN complex in generating chromosomal translocations by examining sites outside of the rearranging IgH locus. We stably integrated a plasmid (DR-GFP) harboring an 18 base pair target site for the rare cutting endonuclease I-SceI in Mre11−/cond MEFs (Figure 3E). The conditional Mre11 allele was deleted by an adenovirus expressing cre to generate Mre11−/− cells (19). Study of nuclease deficient MRE11 was accomplished by stable expression of Mre11H129N cDNA to ensure comparison of an isogenic background. I-SceI can cleave the mouse genome at cryptic sites that diverge 1-5 nucleotides from the consensus sequence (39). Errant repair between the I-SceI induced DSB in the integrated substrate and cryptic site results in a translocation between the two loci, which can be detected by PCR amplification. After introduction of I-SceI via adenovirus, we detected translocations between the stably integrated I-SceI substrate and the endogenous Mmp24 gene on chromosome 2, which contains a cryptic cut site (39) in control (Mre11+/−) cells (Figure 3E). PCR products were cloned and sequenced to confirm the presence of translocations (Supplementary Fig. 5A,B). Notably, in Mre11−/− and Mre11−/− expressing Mre11H129N cDNA we also detected PCR products that were confirmed to be translocations (Figure 3E, Supplementary Fig. 5A,B and not shown). The sequenced translocation junctions from all three genotypes contained blunt ended joins, reflecting ligation by classical non homologous end joining (C-NHEJ), and microhomology mediated joins, reflecting alternative end joining mechanisms (A-NHEJ) (Supplementary Fig. 5A,B) (26). Thus, as previously observed at the IgH locus in stimulated B cells (22), the junctional sequences indicate Mre11 deficiency does not preferentially abrogate one end joining pathway. Collectively, these results demonstrate that translocations can be generated in both lymphocytes and fibroblasts that lack MRE11 entirely, or selectively lack MRE11 nuclease activity.
We next examined the spontaneous chromosomal anomalies that arise in Mre11−/− and Artemis−/−Mre11−/− MEFs for the presence of fusions and translocations. Upon adeno-cre deletion of Mre11, we observed a marked increase in chromosomal anomalies in Mre11−/− MEFs in comparison to control cells (Figure 3F), which is consistent with previous studies (19). We found that combined mutation of Artemis and Mre11 resulted in a moderate increase in chromosomal anomalies above that of Mre11 deficiency alone (Figure 3F, P<0.0001, chi-squared, two-tailed). It is noteworthy that in Mre11 deficiency alone or in combination with Artemis deficiency, elevated levels of chromosome and chromatid fusions, including dicentrics, ring chromosomes and Robertsonian translocations were identified (Table 1). These observations provide further evidence that chromosomal translocations can be generated in the absence of MRE11, not only in B cells, but also in non-lymphoid cell types at loci outside of the rearranging IgH locus, and even in cells lacking the ARTEMIS DSB repair nuclease.
Table 1.
Spontaneous chromosomal anomalies in Mre11/Artemis deficient MEFS
| Geneotype | Mre11cond/− | Mre11−/− | Mre11cond/− Art−/− | Mre11−/−Art−/− |
|---|---|---|---|---|
| Total Metaphases | 140 | 141 | 166 | 151 |
| Gaps/Breaks | 18 (0.13) | 195 (1.38) | 34 (0.20) | 67 (1.77) |
| Fragments | 56 (0.40) | 187 (1.33) | 58 (0.35) | 271 (1.79) |
| Ring | 0 (0) | 0 (0) | 1 (0.01) | 7 (0.05) |
| Dicentric | 1 (0.01) | 12 (0.09) | 3 (0.02) | 27 (0.18) |
| Radial | 1 (0.01) | 21 (0.15) | 1 (0.01) | 32 (0.21) |
| Robertsonian translocations | 0 (0) | 4 (0.03) | 1 (0.01) | 12 (0.08) |
|
| ||||
| Total anomalies | 76 | 419 | 98 | 616 |
| Anomalies per metaphase | 0.54 | 2.97 | 0.59 | 4.08 |
Impact of oncogene overexpression and Mre11 deficiency on cellular survival
Our findings led us to hypothesize that, rather than facilitating the generation of oncogenic chromosomal translocations, the MRN complex may promote survival of cells harboring oncogenic events. To this end we used retrovirus to express oncogenic Myc in primary Mre11cond/− MEFs. MEFs receiving MYC expressing virus were successfully immortalized, whereas parallel cultures infected with empty virus (adeno-empty) died within days (data not shown). We observed that in MYC immortalized Mre11−/cond MEFs, loss of Mre11 (Mre11−/−) resulted in significantly impaired growth (Figure 4A). For comparison, we also immortalized MEFs with SV40 large T antigen and observed that cells lacking MRE11 again exhibited a decreased proliferation rate (Figure 4A). The decrease in proliferative capacity of immortalized MRE11 deficient cells could potentially be due to increased senescence. Primary Mre11−/− MEFs senesce much more rapidly in comparison to wild-type, raising the possibility that oncogene immortalized Mre11−/− cells retain certain properties of primary cells (19). Additionally, overexpression of oncogenes has been reported to elicit senescence in human cells and mouse models, preventing progression of premalignant lesions in a mechanism known as oncogene-induced senescence (OIS)(40). To address the possibility that Mre11 deficient cells are more susceptible to OIS, we measured senescence in the Mre11 mutant and control MEFs using the senescence associated β–galactosidase detection assay (41). These studies confirmed the dramatic increase in senescence following Mre11 deletion in primary MEFs, but demonstrated no increase in the percentage of senescent cells immortalized with Myc or SV40 T-antigen (Figure 4B). Thus, OIS does not appear to be exacerbated in oncogene expressing, Mre11 deficient MEFs.
Figure 4. Mre11 dependent proliferation and a DNA damage response in oncogene overexpressing cells.
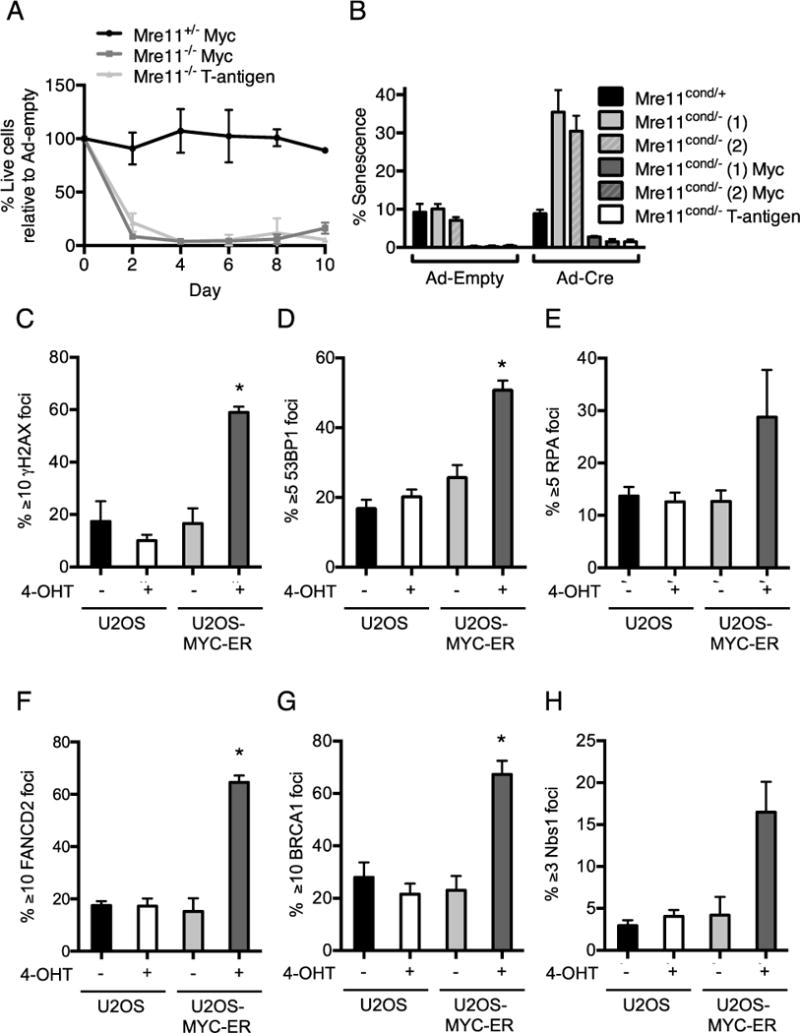
(A) Mre11cond/+ and Mre11cond/− MEFs expressing MYC or large T-antigen were treated with adeno-cre to delete the conditional Mre11 allele. Survival (%) of Mre11+/− and Mre11−/− cells relative to adeno-empty treated control cultures is plotted as a function of days. Mean ± SEM of 3 independent experiments. (B) Primary MEFs, or those expressing MYC or large T-antigen were treated with adeno-cre or adeno-empty as controls, and the percentage of cells that had undergone senescence was measured using a β-galactosidase colorimetric assay. Mean ± SEM of 3 independent experiments. 2 independently derived MEF lines were used, as indicated in parentheses. (C) Examination of DNA repair foci upon MYC overexpression in human cells. U2OS-pBABE and U2OS-MYC-ER cells were treated with DMSO or 200nM 4-OHT to induce MYC translocation to the nucleus. Localization of DNA repair proteins at sites of DNA damage was detected by immunofluorescence microscopy. Percentage of nuclei withγH2AX (C), 53BP1 (D), RPA (E), FANCD2 (F), BRCA1 (G), and NBS1 (H) foci is plotted. Mean ± SEM for 3 or more independent experiments. *, p≤0.05; paired t-test.
Myc induces a DNA damage response involving the MRN complex
Many activated oncogenes drive inappropriate entry into and progression through S phase of the cell cycle (42). This can lead to replication stress in the form of collapsed replication forks that result in generation of DSB repair intermediates. The presence of DSBs activates DNA damage responses that can be detected in some precancerous lesions and tumors (42). The damage response reflects an attempt to maintain genome stability, but has the unintended consequence of preserving the viability of cancerous cells. We therefore examined DNA damage responses triggered by MYC overexpression.
To induce MYC nuclear function, we generated human U2OS cell lines stably expressing the human MYC cDNA fused to the hormone-binding domain of the estrogen receptor (MYC-ER) (43). Upon 4-hydroxytamoxifen (4-OHT) treatment, MYC translocates to the nucleus where it is active and leads to increased accumulation of cells in S-phase, consistent with the known functions of MYC that cause accelerated entry into S phase (Supplementary Fig. 6A–D) (44). We examined the localization of DNA repair proteins to sites of damage, which can be detected as punctate subnuclear foci by immunofluorescence microscopy. Key DNA repair proteins, including phosphorylated H2AX (γH2AX), p53 binding protein 1 (53BP1), single strand DNA binding protein (RPA), Fanconi anemia group D2 protein (FANCD2), and breast cancer 1, early onset (BRCA1), localize to stalled replication forks and facilitate their restart and/or repair (45). We observed that MYC overexpression induced increased foci formation of γH2AX, 53BP1, RPA, FANCD2 and BRCA1 compared to controls (Figure 4C–G, Supplementary Fig. 7). Importantly, we also observed increased localization of the NBS1 component of MRN to foci in response to oncogene overexpression (Figure. 4H, Supplementary Fig. 7). These findings indicate that MYC overexpression induces genomic damage that triggers recruitment of the MRN complex.
Inactivation of MRE11 nuclease activity with a small molecule inhibitor preferentially decreases survival in cells overexpressing oncogenes
Our findings indicate that loss of MRE11 results in decreased proliferative capacity of cells overexpressing Myc. To selectively examine the importance of MRE11 nuclease activity in in this context, we used mirin, a well characterized small molecule inhibitor, to inhibit MRE11 exonuclease activity (7,46). We observed that mirin treatment of Mre11cond/− or Art−/−Mre11cond/− SV40 T antigen immortalized MEFs overexpressing Myc or the related N-myc oncogene led to a significant decrease in cellular survival compared to non-overexpressing controls (Figure 5A).
Figure 5. Mre11 deletion or inhibition in oncogene overexpressing cells reduces cellular survival.
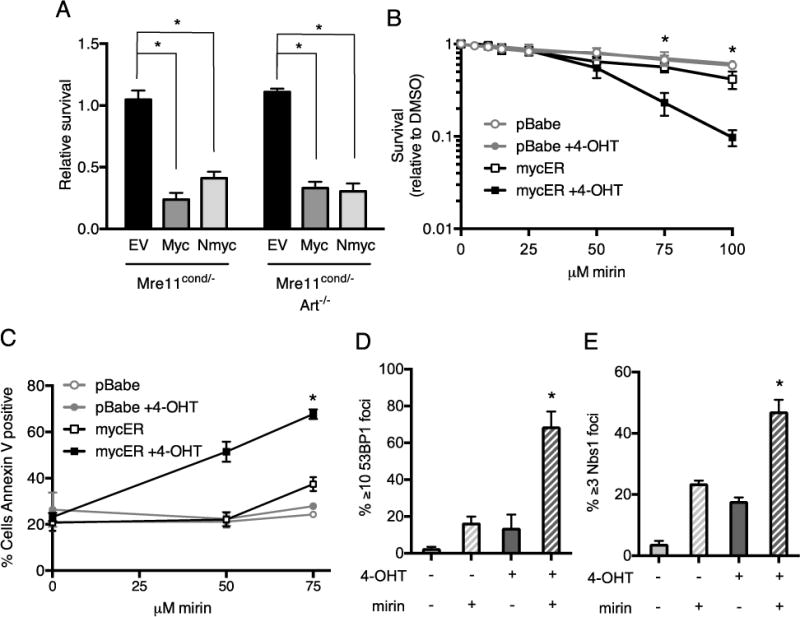
(A) Inhibition of MRE11 nuclease activity by the small molecule inhibitor, mirin, reduces survival of cells overexpressing oncogenes. Mre11cond/− and Mre11cond/−Artemis−/− MEFs were transduced with control retrovirus (empty vector, EV) or retroviruses expressing MYC or N-MYC, then treated with mirin or vehicle for 24h. Live cells were counted and survival (mirin/DMSO) relative to untransduced cells is plotted. Mean ± SEM of 3 or more independent experiments. *, p≤0.05. (B) MYC overexpressing human U2OS cells exhibit decreased survival upon treatment with mirin. U2OS-pBABE and U2OS-MYC-ER cells were treated with DMSO or 4-OHT for 72h, then with increasing concentrations of mirin for 24h. Cellular survival after 48h was determined relative to vehicle controls. Graph represents Mean ± SEM of 3 independent experiments. *p≤0.05. (C) Cells were treated as in B, and the percentages of annexin V positive cells were determined by flow cytometry. (D-E) U2OS-cMycER cells were treated as in C. Localization of 53BP1 (D) and NBS1 (E) to DNA repair foci was examined by immunofluorescence microscopy. Percentage of nuclei with indicated DNA repair foci is plotted. Mean + SEM of 3 independent experiments. *, p<0.05; paired t-test.
We next assessed the impact of inhibition of MRE11 nuclease activity by mirin on survival of the human U2OS cell line overexpressing MYC. We found that upon 4-OHT treatment, exposure to mirin resulted in markedly reduced survival at concentrations of the inhibitor that had minimal impact on survival of control cells (Figure 5B). To determine the cause of reduced viability of cells overexpressing MYC, we examined the extent of apoptosis induced by mirin. We observed that the percentage of cells undergoing apoptosis as measured by annexin V staining was significantly increased in mirin treated cells overexpressing MYC in a dose-dependent manner (Figure 5C). In contrast, at similar doses of the MRE11 inhibitor, control cells were largely unaffected (Figure 5C). These results indicate that the nuclease activity of MRE11 is important for survival of cells overexpressing oncogenes, and a small molecule inhibitor of MRE11 can induce selective killing of MYC expressing cells.
We hypothesized that inhibition of MRE11 dependent cellular responses to oncogene induced replication stress significantly increases accumulation of unrepaired genomic damage that ultimately causes cell death. To test this hypothesis, we examined the impact of MRE11 inhibition on key DNA damage responses in cells overexpressing MYC. We observed that exposure of cells to both 4-OHT and mirin markedly increased localization of the MRN subunit, NBS1, and 53BP1 to DNA damage foci, compared to either treatment alone (Figure 5D,E). The levels of DNA damage foci in 4-OHT and mirin treated cells increased between 8 and 24 hours (Figure 5D,E and Supplementary Fig. 8A–C), consistent with the notion that S phase specific chromosomal damage requiring MRE11 nucleolytic activity accumulates over time. These findings demonstrate that inhibition of MRE11 nuclease activity exacerbates accumulation of DNA damage in cells overexpressing MYC.
DISCUSSION
We sought to further understand roles of MRN as a tumor suppressor. To this end we generated mice with MRE11 deficiency in the B lymphocyte lineage. Splenic B cells demonstrated hallmarks of cancer such as genome instability and the formation of oncogenic IgH:Myc translocations. Despite these observations and the defective ATM-dependent checkpoint responses inherent to MRN loss, there was a striking absence of B lymphocyte malignancy, even in the background of p53 deficiency. This implies that partial loss of the MRN complex in inherited patient alleles generates a milieu favorable to cancer development, likely by retaining some function(s) of the complex that are necessary for tumor formation. The nuclease activities of MRE11 appear to be one such function. In support of this notion, we found that loss of the MRN complex or MRE11 nuclease activity prevents pro-B lymphomagenesis in the Artemis−/−p53−/− tumor prone mouse model.
The lack of tumors is even more surprising when compared to deficiencies of other factors involved in DSB repair. When XRCC4 was absent in mouse mature B lymphocytes, persistent chromosome breaks in IgH were observed at about the same frequency we observed with MRE11 deficiency (47). In this case however, mature B lineage lymphomas did form when XRCC4 deficiency was combined with germline p53 deficiency (48). Moreover, mouse models harboring targeted germline null mutations in Lig4, XRCC4 or Artemis, combined with p53 deficiency were predominantly predisposed to pro-B cell lymphomas characterized by translocation and co-amplification of the IgH and Myc loci (and IgH:N-myc translocations in the case of Artemis deficiency) (32,49,50). This is in stark contrast to inactivation of the MRN complex or MRE11 nuclease activities, where we found a conspicuous absence of these tumors. We cannot entirely exclude the possibility that the p53 deficient mice succumb to thymic lymphomas before B cell lymphomas can be detected. However, tissues from Mre11/p53 mutant mice at ages beyond development of tumors in the XRCC4/p53 deficient mice showed no evidence of malignancy (not shown).
One hypothesis for the absence of B cell tumors in Mre11 deficiency is that oncogenes, like MYC, play a role in tumor development in conjunction with MRE11. Previous studies have shown that overexpression of MYC deregulates S-phase, which increases replication stress and generates significant genomic instability (42). Second, MRE11 and its nuclease activity are required for cellular survival in the face of replication stress (19,51,52). Thus, it is possible that overexpression of oncogenes coupled with defective cellular responses to replication stress is incompatible with the cellular proliferation and survival required for tumor growth (53). We now demonstrate that cells overexpressing oncogenes exhibit enhanced DNA damage responses, decreased survival and increased apoptosis upon inactivation of MRE11.
Collectively, these findings suggest that while inherited hypomorphic mutations of MRN components lead to cancer predisposition, complete loss of MRN may not be compatible with malignancy. Thus, the hypomorphic mutations of MRN must retain specific functions of the complex that support cancer development. That the MRE11 nuclease activities represent such a function is supported by a lack of inherited mutations predicted to abrogate nuclease activities (19,25,54). For example, in humans and mice the C-terminally truncated Mre11ATLD1 allele can predispose to breast cancer (16,55). This mutation likely maintains sufficient nuclease activity for tumorigenesis given that it supports whole organism viability, and the N-terminal nuclease domain maintains its activity and structure even in the absence of a C terminus (15,25,54).
Our studies raise the specter that pharmacologic inhibition of MRE11 nuclease activity could represent a promising new avenue in cancer therapeutics. Indeed, consistent with this notion, we demonstrate that inhibition of MRE11 nuclease activity by the small molecule inhibitor, mirin, selectively decreases survival of oncogene expressing human cells. Specifically targeting the nuclease activities of MRE11 would disable the ability of tumor cells to endure oncogene induced replication stress, while maintaining responses controlled by the ATM kinase (19). This approach warrants further study, especially in light of the development of the next generation of selective MRE11 nuclease inhibitors (7). More broadly, this work supports the emerging notion that subtle enhancements of endogenous genome instability that initially fosters tumor development can be used as anti-tumor therapy (53).
Supplementary Material
Acknowledgments
We thank Dr. W. Dunnick (U. of Michigan) for helpful advice and discussions, Dr. P. Ng (McMaster University) for AdNGUS24i I-SceI adenovirus, Dr. M. Jasin (Memorial Sloan Kettering Cancer Center) for the DR-GFP reporter, and H. Moale and M. Zych for technical assistance with animal husbandry and immunofluorescence microscopy.
Financial Support: Support for this work was provided by US National Institutes of Health R01 AI063058 to J.M. Sekiguchi, R01 HL079118 to D.O. Ferguson, a Leukemia and Lymphoma Society Scholar Grant to D.O. Ferguson and the University of Michigan Cancer Center Support Grant 5-P30-CA46592 which provided partial support for use of the UM DNA Sequencing and Genome Analysis and Vector cores. Trainee support provided by T32 AI007413 to E. Spehalski and M.J. Morgan, T32 CA009676 to K.M. Capper, T32 GM007544 and F31 CA163530 to C.J. Smith.
Footnotes
Conflicts of Interest Statement. The authors declare that no conflicts of interest exist.
References
- 1.Aguilera A, Garcia-Muse T. Causes of genome instability. Annu Rev Genet. 2013;47:1–32. doi: 10.1146/annurev-genet-111212-133232. [DOI] [PubMed] [Google Scholar]
- 2.McKinnon PJ, Caldecott KW. DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet. 2007;8:37–55. doi: 10.1146/annurev.genom.7.080505.115648. [DOI] [PubMed] [Google Scholar]
- 3.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–69. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 5.Paull TT. Mechanisms of ATM Activation. Annu Rev Biochem. 2015;84:711–38. doi: 10.1146/annurev-biochem-060614-034335. [DOI] [PubMed] [Google Scholar]
- 6.Hartlerode AJ, Morgan MJ, Wu Y, Buis J, Ferguson DO. Recruitment and activation of the ATM kinase in the absence of DNA-damage sensors. Nat Struct Mol Biol. 2015;22:736–43. doi: 10.1038/nsmb.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deshpande RA, Lee JH, Arora S, Paull TT. Nbs1 Converts the Human Mre11/Rad50 Nuclease Complex into an Endo/Exonuclease Machine Specific for Protein-DNA Adducts. Mol Cell. 2016;64:593–606. doi: 10.1016/j.molcel.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 9.Hoa NN, Shimizu T, Zhou ZW, Wang ZQ, Deshpande RA, Paull TT, et al. Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol Cell. 2016;64:580–92. doi: 10.1016/j.molcel.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 10.Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–87. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 11.Uchisaka N, Takahashi N, Sato M, Kikuchi A, Mochizuki S, Imai K, et al. Two brothers with ataxia-telangiectasia-like disorder with lung adenocarcinoma. J Pediatr. 2009;155:435–8. doi: 10.1016/j.jpeds.2009.02.037. [DOI] [PubMed] [Google Scholar]
- 12.Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR, 3rd, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–86. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 13.Waltes R, Kalb R, Gatei M, Kijas AW, Stumm M, Sobeck A, et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am J Hum Genet. 2009;84:605–16. doi: 10.1016/j.ajhg.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Digweed M, Sperling K. Nijmegen breakage syndrome: clinical manifestation of defective response to DNA double-strand breaks. DNA Repair (Amst) 2004;3:1207–17. doi: 10.1016/j.dnarep.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Theunissen JW, Kaplan MI, Hunt PA, Williams BR, Ferguson DO, Alt FW, et al. Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol Cell. 2003;12:1511–23. doi: 10.1016/s1097-2765(03)00455-6. [DOI] [PubMed] [Google Scholar]
- 16.Gupta GP, Vanness K, Barlas A, Manova-Todorova KO, Wen YH, Petrini JH. The mre11 complex suppresses oncogene-driven breast tumorigenesis and metastasis. Mol Cell. 2013;52:353–65. doi: 10.1016/j.molcel.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams BR, Mirzoeva OK, Morgan WF, Lin J, Dunnick W, Petrini JH. A murine model of Nijmegen breakage syndrome. Curr Biol. 2002;12:648–53. doi: 10.1016/s0960-9822(02)00763-7. [DOI] [PubMed] [Google Scholar]
- 18.Kang J, Ferguson D, Song H, Bassing C, Eckersdorff M, Alt FW, et al. Functional interaction of H2AX, NBS1, and p53 in ATM-dependent DNA damage responses and tumor suppression. Mol Cell Biol. 2005;25:661–70. doi: 10.1128/MCB.25.2.661-670.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buis J, Wu Y, Deng Y, Leddon J, Westfield G, Eckersdorff M, et al. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135:85–96. doi: 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo G, Yao MS, Bender CF, Mills M, Bladl AR, Bradley A, et al. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci U S A. 1999;96:7376–81. doi: 10.1073/pnas.96.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu J, Petersen S, Tessarollo L, Nussenzweig A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr Biol. 2001;11:105–9. doi: 10.1016/s0960-9822(01)00019-7. [DOI] [PubMed] [Google Scholar]
- 22.Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, et al. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol. 2009;16:808–13. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rooney S, Sekiguchi J, Zhu C, Cheng HL, Manis J, Whitlow S, et al. Leaky scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Mol Cell. 2002;10:1379–90. doi: 10.1016/s1097-2765(02)00755-4. [DOI] [PubMed] [Google Scholar]
- 24.Kovalchuk AL, Muller JR, Janz S. Deletional remodeling of c-myc-deregulating chromosomal translocations. Oncogene. 1997;15:2369–77. doi: 10.1038/sj.onc.1201409. [DOI] [PubMed] [Google Scholar]
- 25.Regal JA, Festerling TA, Buis JM, Ferguson DO. Disease-associated MRE11 mutants impact ATM/ATR DNA damage signaling by distinct mechanisms. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boboila C, Alt FW, Schwer B. Chapter One - Classical and Alternative End-Joining Pathways for Repair of Lymphocyte-Specific and General DNA Double-Strand Breaks. In: Frederick WA, editor. Advances in Immunology. Academic Press; 2012. pp. 1–49. [DOI] [PubMed] [Google Scholar]
- 27.Rickert R, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucl Acids Res. 1997;25:1317–8. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 29.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–9. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 30.Giblin W, Chatterji M, Westfield G, Masud T, Theisen B, Cheng HL, et al. Leaky severe combined immunodeficiency and aberrant DNA rearrangements due to a hypomorphic RAG1 mutation. Blood. 2009;113:2965–75. doi: 10.1182/blood-2008-07-165167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 32.Rooney S, Sekiguchi J, Whitlow S, Eckersdorff M, Manis JP, Lee C, et al. Artemis and p53 cooperate to suppress oncogenic N-myc amplification in progenitor B cells. Proc Natl Acad Sci U S A. 2004;101:2410–5. doi: 10.1073/pnas.0308757101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 34.Woodbine L, Gennery AR, Jeggo PA. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst) 2014;16:84–96. doi: 10.1016/j.dnarep.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 35.Huang Y, Giblin W, Kubec M, Westfield G, St Charles J, Chadde L, et al. Impact of a hypomorphic Artemis disease allele on lymphocyte development, DNA end processing, and genome stability. J Exp Med. 2009;206:893–908. doi: 10.1084/jem.20082396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol. 2009;16:814–8. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol. 2009;16:819–24. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- 38.Deng Y, Guo X, Ferguson DO, Chang S. Multiple roles for MRE11 at uncapped telomeres. Nature. 2009;460:914–8. doi: 10.1038/nature08196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiarle R, Zhang Y, Frock Richard L, Lewis Susanna M, Molinie B, Ho Y-J, et al. Genome-wide Translocation Sequencing Reveals Mechanisms of Chromosome Breaks and Rearrangements in B Cells. Cell. 2011;147:107–19. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Courtois-Cox S, Jones SL, Cichowski K. Many roads lead to oncogene-induced senescence. Oncogene. 2008;27:2801–9. doi: 10.1038/sj.onc.1210950. [DOI] [PubMed] [Google Scholar]
- 41.Itahana K, Campisi J, Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007;371:21–31. doi: 10.1007/978-1-59745-361-5_3. [DOI] [PubMed] [Google Scholar]
- 42.Taylor EM, Lindsay HD. DNA replication stress and cancer: cause or cure? Future Oncol. 2016;12:221–37. doi: 10.2217/fon.15.292. [DOI] [PubMed] [Google Scholar]
- 43.Eilers M, Picard D, Yamamoto KR, Bishop JM. Chimaeras of myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature. 1989;340:66–8. doi: 10.1038/340066a0. [DOI] [PubMed] [Google Scholar]
- 44.Dominguez-Sola D, Gautier J. MYC and the control of DNA replication. Cold Spring Harb Perspect Med. 2014;4 doi: 10.1101/cshperspect.a014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011;25:1320–7. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dupre A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol. 2008;4:119–25. doi: 10.1038/nchembio.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–82. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 48.Wang JH, Alt FW, Gostissa M, Datta A, Murphy M, Alimzhanov MB, et al. Oncogenic transformation in the absence of Xrcc4 targets peripheral B cells that have undergone editing and switching. J Exp Med. 2008;205:3079–90. doi: 10.1084/jem.20082271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5:993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- 50.Gao Y, Ferguson DO, Xie W, Manis J, Sekiguchi J, Frank KM, et al. Interplay of p53 and DNA-repair protein XRCC4 in tumourigenesis, genomic stability and development. Nature. 2000;404:897–900. doi: 10.1038/35009138. [DOI] [PubMed] [Google Scholar]
- 51.Costanzo V, Robertson K, Bibikova M, Kim E, Grieco D, Gottesman M, et al. Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol Cell. 2001;8:137–47. doi: 10.1016/s1097-2765(01)00294-5. [DOI] [PubMed] [Google Scholar]
- 52.Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108:183–93. doi: 10.1016/s0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 53.Andor N, Maley CC, Ji HP. Genomic Instability in Cancer: Teetering on the Limit of Tolerance. Cancer Res. 2017;77:2179–85. doi: 10.1158/0008-5472.CAN-16-1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bartkova J, Tommiska J, Oplustilova L, Aaltonen K, Tamminen A, Heikkinen T, et al. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol Oncol. 2008;2:296–316. doi: 10.1016/j.molonc.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.