

Abstract
Asparagine Synthetase Deficiency (ASD) is a recently described inborn error of metabolism caused by bi-allelic pathogenic variants in the asparagine synthetase (ASNS) gene. ASD typically presents congenitally with microcephaly and severe, often medically refractory, epilepsy. Development is generally severely affected at birth. Tone is abnormal with axial hypotonia and progressive appendicular spasticity. Hyperekplexia has been reported. Neuroimaging typically demonstrates gyral simplification, abnormal myelination, and progressive cerebral atrophy. The present report describes two siblings from consanguineous parents with a homozygous Arg49Gln variant associated with a milder form of ASD that is characterized by later onset of symptoms. Both siblings had a period of normal development before onset of seizures, and development regression. Primary fibroblast studies of the siblings and their parents document that homozygosity for Arg49Gln blocks cell growth in the absence of extracellular asparagine. Functional studies with these cells suggest no impact of the Arg49Gln variant on basal ASNS mRNA or protein levels, nor on regulation of the gene itself. Molecular modelling of the ASNS protein structure indicates that the Arg49Gln variant lies near the substrate binding site for glutamine. Collectively, the results suggest that the Arg49Gln variant affects the enzymatic function of ASNS. The clinical, cellular, and molecular observations from these siblings expand the known phenotypic spectrum of ASD.
Keywords: amino acids, metabolism, inborn errors, neurotransmitters, brain dysfunction
1 INTRODUCTION
1.1 ASNS protein structure and enzyme activity
Asparagine synthetase (ASNS) catalyzes the synthesis of asparagine and glutamate from the substrates aspartate and glutamine [1, 2]. The common name of the protein highlights its role in asparagine synthesis, but whether or not the enzyme activity impacts the cellular levels of one or more of the other three reactants has not been investigated. Particularly for neural tissue, the possible influence of ASNS activity on glutamate and aspartate levels must be a key point of interest. ASNS expression among tissues varies considerably, with much greater expression in the pancreas than most other tissues analysed [3]. However, ASNS abundance in brain is similar to many other tissues in the body (http://www.proteinatlas.org/ENSG00000070669-ASNS/tissue). The human ASNS gene is located at chromosome 7 region 7q21.3 [4, 5] and is 35 kb long with 13 exons [6]. ASNS expression is highly regulated in response to a wide variety of cell stresses, primarily by increased transcription [2]. Central among the genomic elements that control ASNS transcription is the C/EBP-ATF response element (CARE) within the promoter [7]. Protein limitation or an imbalanced dietary amino acid composition activate the ASNS gene through the GCN2-eIF2-ATF4 pathway culminating with ATF4 binding to the CARE. Endoplasmic reticulum stress also increases ASNS transcription through the PERK-eIF2-ATF4 arm of the unfolded protein response (UPR) [8].
The human ASNS enzyme is a 65 kDa protein that has two primary domains, termed the N- and C- terminal domains (Fig. 1A). As there is no crystal structure of human ASNS reported in the protein data base (PDB), an in silico model based on the crystal structure of E. coli asparagine synthetase B (PDB: 1CT), which shares 40% sequence homology with human ASNS, is often used to highlight the main secondary structural features of the enzyme [9]. The N-terminal domain is composed of an antiparallel β-sheet core and contains the glutamine binding pocket. Glutamine interacts with residues Arg49, Asn77, Glu97, and Asp98 through hydrogen bonding (3.0, 3.0, 3.1, and 2.6 Å, respectively) (Fig. 1B). In contrast, the C-terminal domain consists of mainly alpha helices and binds ATP through hydrogen bonds with residues Leu256, Ile288, Asp295, Ser363, Gly364, Glu365, and Asp401 (3.2, 3.1, 3.2, 3.0, 2.8, 3.3, and 3.4 Å, respectively) (Fig. 1C). With the exception of Ile288, which is a valine in E. coli and Drosophila melanogaster (UniProt ID: P22106 and Q7KTW9, respectively), the ASNS residues in the glutamine and ATP binding pockets are conserved between species, emphasizing their importance in the function of the enzyme.
Fig. 1.

Model of human ASNS protein structure. A human ASNS protein model was generated utilizing the crystal structure of E. coli asparagine synthetase B (PDB: 1CT9) as a template and then adapted to contain variants observed in patients with ASD, as described in the Methods section. (Panel A) N-terminal (light grey) and C-terminal (dark grey) domains with the substrate glutamine (purple) and the product AMP (orange) shown as sticks. (Panel B) Glutamine binding pocket illustrating glutamine hydrogen bonds. (Panel C) ATP/AMP binding pocket with AMP binding shown. Hydrogen bonds are represented as black dashes and the distance of each is shown in angstroms (Å).
1.2 Mutations within the gene encoding ASNS cause Asparagine Synthetase Deficiency (ASD)
Asparagine Synthetase Deficiency (ASD) was first described in 2013 on the basis of four families with homozygosity or compound heterozygosity for missense mutations in ASNS [10]. The affected individuals shared the features of severe encephalopathy, congenital microcephaly, brain atrophy, early onset seizures, axial hypotonia, and severe appendicular spasticity [10]. These features can be considered characteristic of the syndrome and are shared by the subsequently reported patients. The vast majority of patients with ASD have missense mutations.
1.3 Functional cellular consequences of a novel variant in the ASNS enzyme
The clinical and functional effects of a homozygous ASNS variant, Arg49Gln, a novel variant shared by two siblings, are described within the current report. Functional studies on fibroblasts from the two affected siblings and their parents revealed that fibroblasts from the parents exhibited typical basal levels of ASNS mRNA or protein, and their ASNS genes each responded appropriately to transcriptional induction by amino acid deprivation. Fibroblasts from one of the two affected siblings expressed higher basal ASNS levels than the other three family members tested, but this difference was not reflected in a change in proliferation rate. Relative to either parent, proliferation of cells from both affected siblings was severely suppressed in the absence of asparagine. Collectively, the results indicate that the Arg49Gln variant blocks cell growth in the absence of sufficient extracellular asparagine, which suggests that Arg49Gln affects the enzymatic function of ASNS. Modelling the location of the variant within the ASNS protein indicates that the affected residue may disrupt protein structure in the substrate binding site for glutamine because of a loss of hydrogen bonding.
2 MATERIALS AND METHODS
2.1 Exome sequencing and targeted mutation analysis
Using genomic DNA, the Agilent Clinical Research Exome kit was used to target the exonic regions and flanking splice junctions of the genome. These regions were sequenced simultaneously by massive parallel (NextGen) sequencing on an Illumina HiSeq 2000 sequencing system with 100 bp paired-end reads. For targeted mutation analysis, the relevant portion of the gene of interest was PCR amplified and capillary sequencing was performed. For both exome sequencing and mutation analysis, bi-directional sequence was assembled, aligned to reference gene sequences based on human genome build GRCh37/UCSC hg19, and analyzed for sequence variants using a custom-developed analysis tool (Xome Analyzer). Capillary sequencing or another appropriate method was used to confirm all potentially pathogenic variants identified in each sample. Sequence alterations were reported according to the Human Genome Variation Society (HGVS) nomenclature guidelines.
2.2 Protein modeling
Given their 40% sequence identity, the human ASNS (Uniprot ID: P08243) protein model was generated in SWISS-MODEL using the coordinates of the crystal structure of E. coli asparagine synthetase B (PDB: 1CT9) as a template [11–13]. The human ASNS sequence was adapted to contain the Arg49Gln variant observed in patients with ASD. The molecular graphics programs Coot [14] and PyMOL (PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC) were then used to superimpose the human wild type and mutated ASNS structures to analyze the effects of the Arg49Gln variant on protein structure and predict the potential impact on enzyme activity and stability.
2.3 Fibroblast cell line generation and culturing
Fibroblast cell lines were generated from the two affected siblings and both parents. These primary fibroblasts were cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 mM glutamine, 1X nonessential amino acids, 10% fetal bovine serum, and ABAM (streptomycin, penicillin G, and amphotericin B). Fibroblasts were seeded at a density of 1.5 × 105 per well in a 6-well plate, with three wells per condition and time point for RNA isolation. For protein determination, cells were plated at 5 × 105 per 60 mm dish and for cell growth studies, cells were plated at 5 × 104 in 35 mm dishes with 3 dishes per condition. To achieve a nutritional “basal state”, the culture medium was changed to fresh medium 12–14 h before experimental treatment.
2.4 RNA isolation and quantitative RT-PCR
RNA was collected using TRIzol Reagent (Invitrogen) per the manufacturer’s protocol. A 1 μg sample of total RNA was converted to cDNA using the qScript cDNA synthesis kit (Quanta Biosciences). To measure ASNS or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA, real-time quantitative polymerase chain reaction (RT-PCR) was performed using SYBR Green (Life Technologies) and a CFX-Connect Real-Time System (Bio-Rad) following a protocol described previously [15]. Steady state mRNA was measured using the following primers: ASNS; forward 5′-GGCGCTGAAAGAAGCCCAAGT-3′ and reverse 5′-TGTCTTCCATGCCAATTGCA-3′ and GAPDH; forward 5′-TTGGTATCGTGGAAGGACTC-3′ and reverse 5′-ACAGTCTTCTGGTGTGCAGT-3′.
2.5 Protein isolation and immunoblotting
Fibroblasts were washed with PBS and lysed using 300 μL of RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% sodium deoxycholate, 0.1% SDS, and 1% triton X-100) supplemented with Pierce Protease and Phosphatase Inhibitor Mini Tablets (ThermoFisher Scientific). Immunoblotting was performed as described previously [7]. Monoclonal anti-ASNS primary antibody [16] and anti-mouse-HRP secondary antibody (Santa Cruz Biotechnology) were used. Bound antibody was detected using Pierce ECL Western Blotting substrate (ThermoFisher Scientific).
2.6 Cell growth and ASNase challenge
Fibroblasts were plated at a density of 50,000 cells per 35 mm dish. After 24 h, the cells were incubated in control DMEM medium or DMEM containing 0.1 U/ml E. coli L-asparaginase (ASNase) for an additional 24, 48, or 72 h, with fresh enzyme provided each 24 h. At the end of the incubation, the cells were collected by treatment with 0.25% trypsin. The number of cells per dish was established by counting using a hemocytometer. Each sample was counted twice.
3 RESULTS
3.1 Case reports
Proband-1
At the time of this report, proband-1 is a 7-year old Emirati male with intractable epilepsy, microcephaly, and severe global developmental delay. His parents are first cousins, and he has one affected sibling and six unaffected siblings. Proband-1 was born at term following a pregnancy complicated by a maternal febrile upper respiratory infection requiring hospitalization. His birth weight was 3.4 kg and head circumference was 34 cm (10th to 25th percentiles). He had an uneventful neonatal course and normal development in early infancy. There were no medical concerns until his first seizure at six mo of age. He was started on phenobarbital, but continued to have up to five seizures a day. Seizures were right hemi-clonic or generalized tonic clonic, with no clear trigger. Given the lack of response to phenobarbital, he was switched to valproic acid and was seizure free for 3 mo. When seizures recurred, he was subsequently trialed on one or a combination of carbamazepine, topiramate, levetiracetam, clonazapem, clobazam and lamotrigine. At presentation to our center at the age of 5 years, he was having generalized tonic seizures with some asymmetry, at times followed by a clonic phase. Seizure frequency improved on restarting valproic acid, from multiple seizures a week to one seizure every 1–2 weeks. His seizure duration was typically longer than 5 min, frequently requiring rectal diazepam rescue. He later developed a possible new seizure type with staring and loss of tone, but this was not captured on electroencephalogram (EEG) to be confirmed. Notable EEG observations include continuous generalized slowing with lack of posterior dominant rhythm and anterior-posterior gradient, lack of normal sleep architecture, multifocal epileptiform discharges that became more abundant with time, and on one occasion a photoparoxysmal response.
The growth parameters of proband-1 started to gradually plateau at 25 mo of age, at which time his head circumference was less than the 3rd percentile. Now, at age seven years, his height is at the 1st–3rd percentile, weight is less than 1st percentile, and head circumference is less than 1st percentile (Z score −3.5). His developmental trajectory, while initially normal, began to plateau around the onset of his seizures at six mo with speech regression at one year of age after he lost the few words he had learned. He had gross motor delays; he sat independently after two years of age, pulled to stand at 2.5 years, and began walking when he was almost three years. He remained non-verbal and did not use any signs. He had no eye contact and poor social interaction. He experienced periods of regression over his course with worsening head control, gait, and vocalization, which he partially recovered over time. A formal neuropsychological evaluation was consistent with severe intellectual disability, lack of basic communication and life-care skills, and self-harming behaviours. Magnetic resonance imaging demonstrated possible cerebral atrophy and delayed myelination at 3 years of age. Magnetic resonance spectroscopy demonstrated a lactate peak.
At present, he has marked global developmental delay and behavioural dysregulation, including hyperactivity and self-harming behaviours. He has poor fine motor skills, although he is not overtly ataxic. He cannot utilize a pincer grasp. His gait remains unsteady and wide-based, with more recent regression and imbalance. He has oropharyngeal dysphagia and is gastric tube dependent. He is not overtly dysmorphic. He was found to have a type 2 laryngeal cleft, which was surgically repaired. He also has mild astigmatism, intermittent exotropia (status-post repair), and reduced visual acuity in the range of 20/130, attributed to cortical visual impairment.
Proband-1 had previous extensive genetic testing. Chromosomal microarray showed no genomic imbalance, but did demonstrate regions of homozygosity encompassing 6% of the genome consistent with known history of consanguinity. Previously performed plasma amino acids were normal. We arranged for repeat cerebral spinal fluid (CSF) metabolic studies and discovered that the CSF asparagine level was borderline-reduced at 3.8 μmol/l (reference range 3.8–7.9 μmol/l). Fasting plasma amino acids performed concurrently with the lumbar puncture demonstrated an asparagine level of 38 μmol/l (reference range 17–88 μmol/l).
Proband-2
Proband-2, the sister of Proband-1, presented for evaluation at 2.5 years of age due to moderate global developmental delay, hypotonia, and epilepsy. She was three kg at birth following a full term, uncomplicated pregnancy. She underwent an EEG at two days of life, because of the known history of epilepsy in her brother, and this EEG was normal by report. At 6 mo of age her head circumference was just under the 3rd percentile. She had her first witnessed seizure at 6 mo, described as full body shaking and facial cyanosis, similar to her brother. There was no clear tonic component to that seizure. She was started on levetiracetam and had a one-year seizure-free period, with a breakthrough seizure followed by another seizure-free period of nearly one year. At 2.5 years of age, generalized tonic clonic seizures recurred. Occasionally there was eye deviation to the right with more pronounced right-sided clonic activity. Despite the addition of zonisamide and later lamotrigine, she had daily seizures with eye deviation and extremity twitching, with around 10 seizures per month characterized as tonic clonic, frequently requiring diazepam rescue. With a recent increase in lamotrigine, her frequency improved to 1–2 seizures per week. Her EEG at three years of age showed continuous generalized slowing with lack of posterior dominant rhythm and absent anterior-posterior gradient and sleep architecture, with multifocal and generalized spikes. She did not undergo photic stimulation and no seizures were captured.
As with proband-1, there were no concerns about her development until the seizure onset after which she noticed to have a plateau, followed by global delays. In terms of gross motor skills, she sat at one year of age and started walking at age two. She developed good eye control, and used some non-verbal cues to communicate. Her initial development was delayed; she sat at one year of age and started walking at age two. A battery of developmental testing at age three, on Scales of Independent Behaviour-Revised, found that her development was equivalent to a 1-year old. Bayley Scales of Infant and Toddler Development demonstrated a cognitive composite score of 55 (0.1%, age equivalent of 13 mo), language composite score of 59 (0.3%, age equivalent of 12–17 mo), and a motor composite score of 55 (0.1%, age equivalent levels of 13–18 mo). She had not exhibited regression, but had very slow progression of developmental milestones. On physical exam at age four years, her head circumference was at the 3rd percentile, height was at the 77th percentile, and weight was at the 58th percentile. She was non-dysmorphic. She had mild axial and limb hypotonia, with brisk deep tendon reflexes in the lower extremities. Her gait was unsteady and wide-based. Dermatologic observations include ichthyosis and chronic paronychia.
Proband-2 had limited genetic and metabolic testing, but normal electrolytes and liver function was observed. Urine organic acids showed a trace increase in lactate and pyruvate. Plasma amino acids were normal, with an asparagine level of 93 μmol/l. She did not undergo CSF amino acid testing. Chromosomal microarray showed several large regions of homozygosity, encompassing greater than 11% of the genome. Magnetic resonance imagining showed marginally prominent ventricles and basal cisterns in the brain.
3.2 Exome sequencing and mutation analysis
For proband-1, exome sequencing was initially done through a clinical diagnostic laboratory. It demonstrated a homozygous variant c.146G>A (p.Arg49Gln) in the ASNS gene (transcript NM_183356; chromosome 7:97498323). Targeted variant analysis revealed that Proband-2 was homozygous for the same variant as her brother. Both patients were enrolled in The Manton Center for Orphan Disease Research, Gene Discovery Core under informed consent governed by the Institutional Review Board of Boston Children’s Hospital. Research laboratory-based sequencing was done to confirm the results and test other family members. Sequencing for the ASNS gene was done for the five of the six unaffected siblings of the probands and none were homozygous for the suspected pathogenic variant in ASNS, but rather heterozygous carriers or non-carriers. The parents were confirmed to be heterozygous carriers. Consistent with a rare, recessive mutation, only one allele is present in the ExAC database (http://exac.broadinstitute.org/variant/7-97498323-C-T) for a frequency of of 8.24e-6 and two alleles are reported in the gnomAD database (http://gnomad.broadinstitute.org/variant/7-97498323-C-T) for a frequency of 7.21e-6. The variant was deemed likely pathogenic by various in silico methods including Polyphen-2 (genetics.bwh.harvard.edu/pph2), SIFT (sift.jcvi.org), PROVEAN (provean.jcvi.org) and MutationTaster (mutationtaster.org).
3.3 Modelling of the Arg49Gln ASNS variant
A model of human ASNS incorporating the ASD-associated variant Arg49Gln was produced using the E. coli ASNS-B crystal structure as a template. In the wild type ASNS structure, residue Arg49 is expected to be stabilized through hydrogen bonds with proximal residues Val53, Asp54, and Pro55 (2.9, 2.7, and 2.8 Å, respectively) (Fig. 2A). The Arg49Gln variant, effectively shortening the side chain, results in the loss of these hydrogen bonds and the Gln49 mutant residue is only stabilized by a single hydrogen bond with the carbonyl oxygen of Gly58 (3.2 Å) (Fig. 2B, 2C). This loss of interactions has the potential to destabilize the loop between the two predominant β-sheets of the N-terminal domain. It is particularly important to note that the Arg49 residue is located in the glutamine binding pocket (Figs. 1B, 3A). Although the Gln49 variant residue may maintain a weak hydrogen bond with the carbonyl oxygen of the glutamine substrate (3.8 Å), the disease-associated variant is predicted to decrease the binding affinity for glutamine, potentially reducing catalytic activity (Fig. 3B, 3C).
Fig. 2.

Proposed role of residue 49 in human ASNS. (Panel A) Hydrogen bonding between wild type R49 and proximal residues. (Panel B) Predicted hydrogen bond between Q49 mutant and G58. (Panel C) Overl ay of wild type R49 and mutant Q49 residues. Hydrogen bonds are represented as black dashes and the distance of each is shown in angstroms (Å).
Fig. 3.

Glutamine binding site within the human ASNS model. (Panel A) Glutamine binding pocket in the N-terminal domain of wild type ASNS showing hydrogen bonds. The glutamine substrate is shown in purple. (Panel B) Predicted alteration in glutamine binding for the Arg49Gln mutant of ASNS. (Panel C) Overlay of wild type R49 and mutant Q49 residues in glutamine binding pocket. Hydrogen bonds are represented as black dashes and the distance of each is shown in angstroms (Å).
3.4 ASNS mRNA, as well as protein expression and stability is unaffected by the Arg49Gln variant
The ASNS gene is subject to transcriptional up-regulation by amino acid limitation of cells that triggers an adaptive program called the amino acid response (AAR) [2, 17]. Deprivation of the intracellular content for any amino acid will activate the AAR pathway, and if the ASNS gene is functional, the AAR will increase the steady state ASNS mRNA content. To test for the responsiveness of the ASNS gene in the fibroblasts of the affected children and their parents, cells were incubated in medium lacking the essential amino acid histidine for up to 24 h (Fig. 4). ASNS mRNA was increased within each cell line with qualitatively similar time kinetics. The fold increase was similar for three of the four individuals, with Proband-1 being about half of that observed for the other three. Upon analysis of the data, this difference may be the result of the basal level of ASNS mRNA being about 4-fold higher in Proband-1 (basal real-time PCR crossing point cycle values: Proband-1 = 24, Proband-2 = 26, mother = 26, father = 26). Given that both siblings harbor the same mutation, it is not likely that this difference for Proband-1 represents a mutation-associated change in gene transcription or mRNA stability. It is more likely that this expression level is a function of this particular cell line isolate. Overall, the results indicate that regulation of the ASNS gene and mRNA stability are unaffected by the genomic c. 146G>A mutation. The higher basal mRNA level of Proband-1 was also reflected in higher basal protein content (Fig. 4B). However, the expected increase in ASNS protein abundance following activation of the AAR for 24 h was similar for all family members tested. Thus, the Arg49Gln variant within the protein did not appear to significantly affect synthesis or stability. Collectively, the mRNA and protein data support the interpretation that the mutation does not alter the regulated expression or absolute abundance of ASNS mRNA or protein.
Fig. 4.

The Arg49Gln variant does not alter ASNS gene expression, nor does it change ASNS mRNA or protein metabolism. Skin fibroblasts from the parents and both probands were maintained in culture and then incubated in control DMEM medium or DMEM lacking the single amino acid histidine. After incubation for the indicated period of time, total RNA and protein was isolated. Real-time quantitative PCR was performed to measure steady state mRNA levels for ASNS and GAPDH (Panel A). The relative abundance of mRNA compared to time zero is plotted. The data are the averages ± standard deviations of three determinations within an experiment. Each experiment was repeated at least once to verify qualitative reproducibility. Where not visible, the standard deviation bars are contained with in the symbol. For protein analysis, immunoblots were performed using antibodies specific for ASNS and GAPDH, as described in the Methods section (Panel B). Each sample was tested at least twice to assess reproducibility of the results and a representative blot is shown.
3.5 Asparagine depletion increases ASNS expression in cells with the Arg49Gln variant
Activation of the AAR by depletion of the essential amino acid histidine indicated that transcriptional regulation of the ASNS gene was intact (Fig. 4). If the Arg49Gln variant causes a significant loss of ASNS activity, asparagine may become an essential amino acid for those cells. As in childhood acute lymphoblastic leukemia, in which the leukemic cells lack sufficient ASNS activity [18], loss of an extracellular asparagine source would trigger the AAR. To determine if asparagine depletion activated the ASNS gene, mRNA content was measured after treatment of the cells with 0.1 U/ml of the enzyme L-asparaginase (ASNase), which rapidly degrades extracellular asparagine, and subsequently, the intracellular level as well [19, 20]. Consistent with the hypothesis that cells with the Arg49Gln variant do not synthesize sufficient asparagine for cellular homeostasis, ASNase treatment resulted in an induction of ASNS mRNA levels in fibroblasts from either sibling (Fig. 5A). In contrast, the parental fibroblasts showed little or no change in ASNS mRNA following ASNase exposure. Measurement of ASNS protein abundance with or without ASNase treatment produced similar results in that ASNS protein content was increased by asparagine depletion in both affected siblings, whereas there was no induction of protein content in the cells from either parent (Fig. 5B). Collectively, the data document that Arg49Gln homozygous cells induce ASNS expression when challenged with asparagine limitation, whereas it appears that asparagine synthesis in heterozygous cells is sufficient even when the extracellular supply is limited.
Fig. 5.

Limiting extracellular asparagine causes up-regulation of the ASNS gene in the proband cells. Skin fibroblasts from the parents and both probands were maintained in culture and then incubated in control DMEM medium or DMEM containing 0.1 U/ml of ASNase to deplete extracellular asparagine. After incubation for the indicated period of time, total RNA and protein was isolated. Real-time quantitative PCR was performed to measure steady state mRNA levels for ASNS and GAPDH (Panel A). The data are the averages ± standard deviations of three determinations within an experiment. Each experiment was repeated at least once to verify qualitative reproducibility. Where not visible, the standard deviation bars are contained within the symbol. For protein analysis, immunoblots were performed using antibodies specific for ASNS and GAPDH, as described in the Methods section (Panel B). Each sample was tested at least twice to assess reproducibility of the results and a representative blot is shown.
3.6 Fibroblasts from the affected siblings require extracellular asparagine for proliferation
ASNase is used clinically to treat childhood acute lymphoblastic leukemia because the leukemic cells do not express sufficient ASNS protein [18, 21]. However, cells with sufficient functional ASNS activity will continue to grow despite the loss of an extracellular source of the amino acid. For example, it was shown previously that fibroblasts from a compound heterozygous ASD patient (Gly289Ala, Thr337Ile) were suppressed in their growth in response to asparagine depletion [22]. To test for the effect of the Arg49Gln variant on cell function, fibroblasts from all four family members were plated for 24 h and then transferred to culture medium depleted of asparagine by addition of 0.1 U/ml ASNase to the medium (Fig. 6). The results show that for the heterozygous parental cells the response to asparagine depletion of the medium was a modest reduction in growth. In contrast, the cells from both affected siblings exhibited little or no growth when challenged with asparagine limitation. For comparison, we have shown previously that unrelated, wild type human fibroblasts show little or no reduction in growth in medium containing 0.1 U/ml ASNase [22]. The present results clearly demonstrate that homozygotes expressing the Arg49Gln variant do not have sufficient ASNS activity to maintain cell proliferation.
Fig. 6.
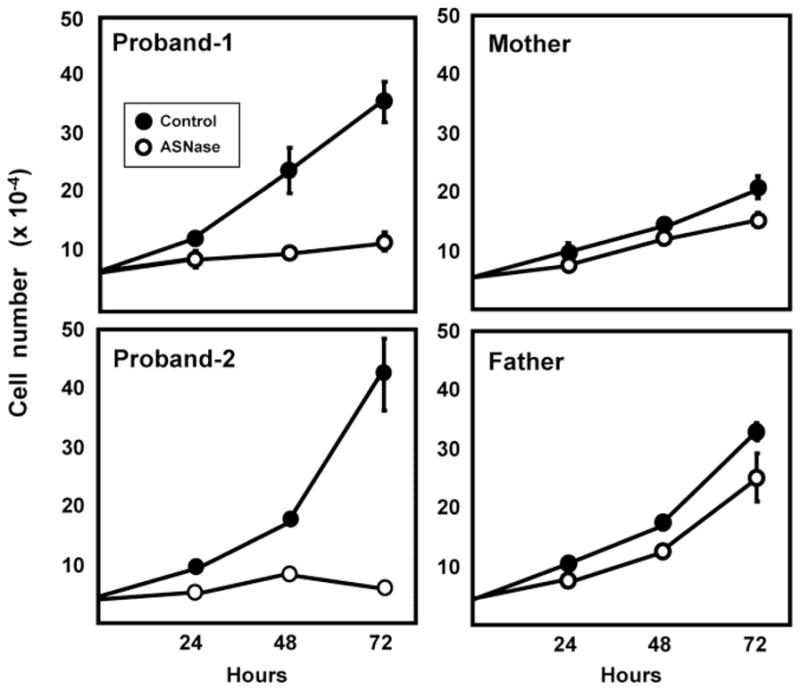
Growth of cells expressing the Arg49Gln ASNS variant is supressed. Fibroblasts from the parents and both probands were maintained in culture and then plated at 50,000 cells per 35 mm dish with three dishes per condition. After a 24 h adaption period, the cells were transferred to control DMEM medium or DMEM containing 0.1 U/ml of ASNase. The medium was replaced every 24 h. After incubation for the indicated period of time, cells were harvested and the cell number per dish counted as described in the Methods section. The cell number for each condition was determined twice and representative data are illustrated as the averages ± standard deviations for each condition and time point. Where not visible, the standard deviation bars are contained within the symbol.
4 DISCUSSION
4.1 Asparagine synthetase
The ASNS protein is composed of two domains that contribute to the overall reaction. The N-terminal domain generates glutamate and ammonia by hydrolysis of glutamine, while the C-terminal domain catalyzes an ATP-dependent activation of the aspartate side-chain carboxyl group and transfer of the ammonia to the aspartate to form asparagine and glutamate [1]. Human tissues express ASNS to varying degrees, but asparagine is considered a ‘non-essential’ amino acid as even in the absence of dietary intake sufficient amounts can be generated from the amino acid precursors glutamine and aspartate via ASNS. With the exception of the very high levels of ASNS expression in the pancreas [3], ASNS abundance in brain is similar to many other tissues in the body (http://www.proteinatlas.org/ENSG00000070669-ASNS/tissue). In young children, the CSF concentration of asparagine has been reported to be only 8–13% of the level in plasma [23, 24]. Thus, asparagine transport across the blood brain barrier becomes a consideration and may represent a limiting factor in the absence of sufficient brain ASNS enzymatic activity.
4.2 Asparagine Synthetase Deficiency
ASD is a recently described inborn error of metabolism, typically presenting congenitally with microcephaly and severe, often medically refractory epilepsy. In most of the previously reported cases, development has been severely impaired from birth. Tone is abnormal with axial hypotonia and progressive appendicular spasticity, and there may be hyperekplexia. Neuroimaging typically demonstrates gyral simplification, disturbed myelination, pontocerebellar hypoplasia, and progressive cerebral atrophy. Prior studies have identified 15 unique variants within ASNS, most of which arise from missense mutations [10, 22, 25–31]. Patients with missense mutations and those with one missense mutation and one frame-shift or deletion seem to share the classic characteristic features of ASD [27, 30]. Although patients with missense mutations vary from mild to severe in the ASD spectrum, the lone patient described with a homozygous nonsense mutation exhibited a severe phenotype, with microcephaly detected prenatally, and at birth the head circumference was at -3 standard deviations [27]. He had cerebral atrophy, simplified gyral pattern, and hypoplasia of cerebellum and pons. He died at 6 weeks from status epilepticus. One case of an attenuated form of this disorder with onset at 9 mo of life has been described previously [10]. In this report, we describe two additional cases of a hypomorphic form of this disorder with infantile onset. The probands in this study had later onset features and achieved more developmental progress than most cases previously reported.
The pathophysiology of ASD is not yet known. One hypothesis is that CNS asparagine deficiency in utero may limit neuronal proliferation resulting in congenital microcephaly and gyral simplification in severe cases. However, it is clear from our probands and the literature that there is also a progressive neurodegenerative component to this disorder that is at least partially independent of the neurocognitive effects of the epilepsy itself and treatment with anticonvulsants. Based on the fasting levels of CSF asparagine in proband-1, it is probable that the pathophysiology of this disorder extends beyond mere CNS asparagine deprivation. Although the diet can be supplemented with asparagine, it does not easily pass the blood brain barrier and a decrease in ASNS catalytic activity in the brain is presumed to cause the disease phenotype [10, 22, 25]. As mentioned, in healthy children the CSF asparagine concentration is only one tenth that of the plasma level, and a few patients with ASD have reduced CSF asparagine levels [10, 23]. It is more likely that disease in ASD results from a disruption of the delicate amino acid balance within the CNS. This interpretation is supported by the recent report of worsening seizures in a patient with ASD treated with dietary asparagine supplementation [32]. Elevating the circulating asparagine level may also perturb amino acid homeostasis in the brain, and many other tissues, as increased asparagine would compete for plasma membrane transport [33]. There have been few reports of biochemical analyses in ASD, including CSF and plasma asparagine levels. In two of the four cases that reported CSF asparagine values, they were undetectable [reviewed in 31]. In addition, 50% of patients previously described had low asparagine in blood. It is clear from previous studies and those described here that asparagine levels in CSF and blood are not reliable biomarkers for this disorder, in contrast to low respective fasted amino acid levels observed for serine and glutamine deficiency syndromes [34, 35]. However, asparagine values may still be meaningful if they are very low or undetectable.
4.3 Fibroblast growth in culture
Expression from the ASNS gene from either the heterozygotic parents or the Arg49Gln-containing siblings appeared to be unaffected by the mutation in that steady state mRNA and protein was increased following activation of the AAR by either incubation in histidine-deficient medium or asparagine depletion of the medium by ASNase treatment. These results indicate that transcription from the gene, translation from the mRNA as well as mRNA and protein turnover all appear to be largely unaffected by the Arg49Gln variant. However, the growth of fibroblasts from both siblings was significantly suppressed by culture in asparagine-depleted medium, suggesting that these cells do not contain sufficient ASNS enzymatic activity to maintain proliferation. In contrast, growth of cells from the heterozygotic parents was only mildly slowed. In a previous report, investigation of fibroblasts from parents of an ASD patient with compound heterozygosity (Gly289Ala, Thr337Ile) documented that cells from the father (Gly289Ala) exhibited little or no suppression of growth at 0.1 U/ml ASNase, and cells from the mother (Thr337Ile) were more sensitive to this level of asparagine depletion of the medium [22]. Both of those variants are predicted to be located near the ATP binding site of the enzyme.
4.4 Novel ASNS variant near the glutamine binding site
Although past investigations of ASD patients have revealed variants near the ATP binding site, the amino acid substitutions were predicted to affect ATP binding indirectly. This report describes the first discovery of a variant that is located within the substrate pocket for glutamine. Based on the structural model for human ASNS, the mutated residue, Arg49Gln variant is likely to directly alter glutamine binding through hydrogen bonding (Figs. 2C, 3C). Future analysis will be required to determine if there is a relationship between the Arg49Gln variant and the fact that the two probands described in the present study had later onset features and achieved more developmental progress than most cases previously reported.
5 CONCLUSIONS
This study illustrates an expanded phenotypic spectrum for ASD to include later infantile onset and greater developmental progress. Diagnosis for this inborn error of metabolism remains challenging and relies on molecular diagnosis, because a reliable biochemical diagnosis has not yet been identified. A clinical suspicion for this disorder may be based on phenotype, but with the level of current knowledge most cases will probably be diagnosed through exome sequencing. Diagnostic laboratories should include the ASNS gene in screening panels for patients with brain malformations, epilepsy, microcephaly and intellectual disability that reflect the entire phenotypic ASD spectrum. The pathophysiology for this disorder is still not known, but may be more complex than simple CNS asparagine deficiency. We hypothesize that ASD results in disease by dysregulating the delicate metabolic balance within the CNS. The in utero effects of ASD suggest that a possible disease modifying therapy for this disorder may be difficult.
Highlights.
Asparagine synthetase (ASNS) is an enzyme that catalyzes the interconversion of glutamine and aspartate to asparagine and glutamate.
Mutations in the human ASNS gene lead to an inborn error of metabolism termed Asparagine Synthetase Deficiency (ASD) that typically presents congenitally with microcephaly and severe, often medically refractory, epilepsy.
In this report two siblings are described who express a new ASNS variant, Arg49Gln, that is characterized by later onset of overt symptoms.
Homozygosity for Arg49Gln blocks cell growth in the absence of extracellular asparagine.
Molecular modeling of the ASNS protein structure indicates that the Arg49Gln variant lies near the substrate binding site for glutamine.
Acknowledgments
MSK is supported by a grant from the National Cancer Institute (CA-203565). CLL is supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under University of Florida Clinical and Translational Science Awards TL1TR001428 and UL1TR001427. We thank the Gene Discovery Core of The Manton Center for Orphan Disease Research for providing resources and support in patient consenting, sample collection, sequencing and sharing of information and samples.
Abbreviations
- ASD
Asparagine Synthetase Deficiency
- ASNase
Asparaginase
- ASNS
asparagine synthetase
- CARE
C/EBP-ATF response element
- CSF
cerebral spinal fluid
- EEG
electroencephalogram
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- PDB
protein data base
- SD
standard deviations
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Richards NG, Kilberg MS. Asparagine synthetase chemotherapy. Annu Rev Biochem. 2006;75:629–654. doi: 10.1146/annurev.biochem.75.103004.142520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balasubramanian MN, Butterworth EA, Kilberg MS. Asparagine Synthetase: Regulation by Cell Stress and Involvement in Tumor Biology American journal of physiology. Endocrinology and metabolism. 2013;304:E789–799. doi: 10.1152/ajpendo.00015.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milman HA, Cooney DA. The distribution of L-asparagine synthetase in the principal organs of several mammalian and avian species. Biochem J. 1974;142:27–35. doi: 10.1042/bj1420027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greco A, Gong SS, Ittmann M, Basilico C. Organization and expression of the cell cycle gene, ts 11, that encodes asparagine synthetase. Mol Cell Biology. 1989;9:2350–2359. doi: 10.1128/mcb.9.6.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heng HHQ, Shi XM, Scherer SW, Andrulis IL, Sui LCT. Refined localization of the asparagine synthetase gene ASNA) to chromosome 7, region q 21.3, and characterization of teh somatic cell hybrid line 4AF/106/KO15 Cytogenet. Cell Genet. 1994;66:135–138. doi: 10.1159/000133685. [DOI] [PubMed] [Google Scholar]
- 6.Zhang YP, Lambert MA, Cairney AEL, Wills D, Ray PN, Andrulis IL. Molecular structure of the human asparagine synthetase gene. Gene. 1989;4:259–265. doi: 10.1016/0888-7543(89)90329-7. [DOI] [PubMed] [Google Scholar]
- 7.Chen H, Pan YX, Dudenhausen EE, Kilberg MS. Amino acid deprivation induces the transcription rate of the human asparagine synthetase gene through a timed program of expression and promoter binding of nutrient-responsive bZIP transcription factors as well as localized histone acetylation. J Biol Chem. 2004;279:50829–50839. doi: 10.1074/jbc.M409173200. [DOI] [PubMed] [Google Scholar]
- 8.Barbosa-Tessmann IP, Chen C, Zhong C, Schuster SM, Nick HS, Kilberg MS. Activation of the unfolded protein response pathway induces human asparagine synthetase gene expression. J Biol Chem. 1999;274:31139–31144. doi: 10.1074/jbc.274.44.31139. [DOI] [PubMed] [Google Scholar]
- 9.Larsen TM, Boehlein SK, Schuster SM. Three-dimensional structure of Escherichia coli asparagine synthetase B: A short journey from substrate to product. Biochemistry (Mosc) 1999;38:16146–16157. doi: 10.1021/bi9915768. [DOI] [PubMed] [Google Scholar]
- 10.Ruzzo EK, Capo-Chichi JM, Ben-Zeev B, Chitayat D, Mao H, Pappas AL, Hitomi Y, Lu YF, Yao X, Hamdan FF, Pelak K, Reznik-Wolf H, Bar-Joseph I, Oz-Levi D, Lev D, Lerman-Sagie T, Leshinsky-Silver E, Anikster Y, Ben-Asher E, Olender T, Colleaux L, Decarie JC, Blaser S, Banwell B, Joshi RB, He XP, Patry L, Silver RJ, Dobrzeniecka S, Islam MS, Hasnat A, Samuels ME, Aryal DK, Rodriguiz RM, Jiang YH, Wetsel WC, McNamara JO, Rouleau GA, Silver DL, Lancet D, Pras E, Mitchell GA, Michaud JL, Goldstein DB. Deficiency of asparagine synthetase causes congenital microcephaly and a progressive form of encephalopathy. Neuron. 2013;80:429–441. doi: 10.1016/j.neuron.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 12.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 13.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 15.Shan J, Donelan W, Hayner JN, Zhang F, Dudenhausen EE, Kilberg MS. MAPK signaling triggers transcriptional induction of cFOS during amino acid limitation of HepG2 cells. Biochim Biophys Acta. 2015;1854:539–548. doi: 10.1016/j.bbamcr.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hutson RG, Kitoh T, Amador DAM, Cosic S, Schuster SM, Kilberg MS. Amino acid control of asparagine synthetase: relation to asparaginase resistance in human leukemia cells. Amer J Physiol. 1997;272:C1691–C1699. doi: 10.1152/ajpcell.1997.272.5.C1691. [DOI] [PubMed] [Google Scholar]
- 17.Kilberg MS, Balasubramanian M, Fu L, Shan J. The transcription factor network associated with the amino Acid response in Mammalian cells. Adv Nutr. 2012;3:295–306. doi: 10.3945/an.112.001891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aslanian AM, Fletcher BS, Kilberg MS. Asparagine synthetase expression alone is sufficient to induce L-asparaginase resistance in MOLT-4 human leukaemia cells. Biochem J. 2001;357:321–328. doi: 10.1042/0264-6021:3570321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Avramis VI. Asparaginases: biochemical pharmacology and modes of drug resistance. Anticancer Res. 2012;32:2423–2437. [PubMed] [Google Scholar]
- 20.Chan WK, Lorenzi PL, Anishkin A, Purwaha P, Rogers DM, Sukharev S, Rempe SB, Weinstein JN. The glutaminase activity of l-asparaginase is not required for anticancer activity against ASNS-negative cells. Blood. 2014;123:3596–3606. doi: 10.1182/blood-2013-10-535112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su N, Pan YX, Zhou M, Harvey RC, Hunger SP, Kilberg MS. Correlation between asparaginase sensitivity and asparagine synthetase protein content, but not mRNA, in acute lymphoblastic leukemia cell lines. Pediatr Blood Cancer. 2008;50:274–279. doi: 10.1002/pbc.21213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmer EE, Hayner J, Sachdev R, Cardamone M, Kandula T, Morris P, Dias KR, Tao J, Miller D, Zhu Y, Macintosh R, Dinger ME, Cowley MJ, Buckley MF, Roscioli T, Bye A, Kilberg MS, Kirk EP. Asparagine Synthetase Deficiency causes reduced proliferation of cells under conditions of limited asparagine. Mol Genet Metab. 2015;116:178–186. doi: 10.1016/j.ymgme.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scholl-Burgi S, Haberlandt E, Heinz-Erian P, Deisenhammer F, Albrecht U, Sigl SB, Rauchenzauner M, Ulmer H, Karall D. Amino acid cerebrospinal fluid/plasma ratios in children: influence of age, gender, and antiepileptic medication. Pediatrics. 2008;121:e920–926. doi: 10.1542/peds.2007-1631. [DOI] [PubMed] [Google Scholar]
- 24.Akiyama T, Kobayashi K, Higashikage A, Sato J, Yoshinaga H. CSF/plasma ratios of amino acids: reference data and transports in children. Brain Dev. 2014;36:3–9. doi: 10.1016/j.braindev.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 25.Alfadhel M, Alrifai MT, Trujillano D, Alshaalan H, Al Othaim A, Al Rasheed S, Assiri H, Alqahtani AA, Alaamery M, Rolfs A, Eyaid W. Asparagine Synthetase Deficiency: New Inborn Errors of Metabolism. JIMD Rep. 2015;22:11–16. doi: 10.1007/8904_2014_405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ben-Salem S, Gleeson JG, Al-Shamsi AM, Islam B, Hertecant J, Ali BR, Al-Gazali L. Asparagine synthetase deficiency detected by whole exome sequencing causes congenital microcephaly, epileptic encephalopathy and psychomotor delay. Metab Brain Dis. 2015;30:687–694. doi: 10.1007/s11011-014-9618-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seidahmed MZ, Salih MA, Abdulbasit OB, Samadi A, Al Hussien K, Miqdad AM, Biary MS, Alazami AM, Alorainy IA, Kabiraj MM, Shaheen R, Alkuraya FS. Hyperekplexia, microcephaly and simplified gyral pattern caused by novel ASNS mutations, case report. BMC Neurol. 2016;16:105. doi: 10.1186/s12883-016-0633-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gataullina S, Lauer-Zillhardt J, Kaminska A, Galmiche-Rolland L, Bahi-Buisson N, Pontoizeau C, Ottolenghi C, Dulac O, Fallet-Bianco C. Epileptic Phenotype of Two Siblings with Asparagine Synthesis Deficiency Mimics Neonatal Pyridoxine-Dependent Epilepsy. Neuropediatrics. 2016;47:399–403. doi: 10.1055/s-0036-1586222. [DOI] [PubMed] [Google Scholar]
- 29.Sun J, McGillivray AJ, Pinner J, Yan Z, Liu F, Bratkovic D, Thompson E, Wei X, Jiang H, Asan, Chopra M. Diaphragmatic Eventration in Sisters with Asparagine Synthetase Deficiency: A Novel Homozygous ASNS Mutation and Expanded Phenotype. JIMD Rep. 2017;34:1–9. doi: 10.1007/8904_2016_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamamoto T, Endo W, Ohnishi H, Kubota K, Kawamoto N, Inui T, Imamura A, Takanashi JI, Shiina M, Saitsu H, Ogata K, Matsumoto N, Haginoya K, Fukao T. The first report of Japanese patients with asparagine synthetase deficiency. Brain Dev. 2017;39:236–242. doi: 10.1016/j.braindev.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 31.Gupta N, Tewari VV, Kumar M, Langeh N, Gupta A, Mishra P, Kaur P, Ramprasad V, Murugan S, Kumar R, Jana M, Kabra M. Asparagine Synthetase deficiency-report of a novel mutation and review of literature. Metab Brain Dis. 2017 doi: 10.1007/s11011-017-0073-6. [DOI] [PubMed] [Google Scholar]
- 32.Alrifai MT, Alfadhel M. Worsening of Seizures After Asparagine Supplementation in a Child with Asparagine Synthetase Deficiency. Pediatr Neurol. 2016;58:98–100. doi: 10.1016/j.pediatrneurol.2016.01.024. [DOI] [PubMed] [Google Scholar]
- 33.Hawkins RA, O’Kane RL, Simpson IA, Vina JR. Structure of the blood-brain barrier and its role in the transport of amino acids. J Nutr. 2006;136:218S–226S. doi: 10.1093/jn/136.1.218S. [DOI] [PubMed] [Google Scholar]
- 34.Spodenkiewicz M, Diez-Fernandez C, Rufenacht V, Gemperle-Britschgi C, Haberle J. Minireview on Glutamine Synthetase Deficiency, an Ultra-Rare Inborn Error of Amino Acid Biosynthesis Biology (Basel) 2016;5:40–56. doi: 10.3390/biology5040040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Koning TJ. Amino acid synthesis deficiencies. J Inherit Metab Dis. 2017;40:609–620. doi: 10.1007/s10545-017-0063-1. [DOI] [PMC free article] [PubMed] [Google Scholar]