

Abstract
The exact regulation of the liver-secreted peptide hepcidin, the key regulator of systemic iron homeostasis, is still poorly understood. It is potently induced by iron, inflammation, cytokines or H2O2 but conflicting results have been reported on hypoxia. In our current study, we first show that pronounced (1%) and mild (5%) hypoxia strongly induces hepcidin in human Huh7 hepatoma and primary liver cells predominantly at the transcriptional level via STAT3 using two hypoxia systems (hypoxia chamber and enzymatic hypoxia by the GOX/CAT system). SiRNA silencing of JAK1, STAT3 and NOX4 diminished the hypoxia-mediated effect while a role of HIF1α could be clearly ruled out by the response to hypoxia-mimetics and competition experiments with a plasmid harboring the oxygen-dependent degradation domain of HIF1α. Specifically, hypoxia drastically enhances the H2O2-mediated induction of hepcidin strongly pointing towards an oxidase as powerful upstream control of hepcidin. We finally provide evidences for an efficient regulation of hepcidin expression by NADPH-dependent oxidase 4 (NOX4) in liver cells. In summary, our data demonstrate that hypoxia strongly potentiates the peroxide-mediated induction of hepcidin via STAT3 signaling pathway. Moreover, oxidases such as NOX4 or artificially overexpressed urate oxidase (UOX) can induce hepcidin. It remains to be studied whether the peroxide-STAT3-hepcidin axis simply acts to continuously compensate for oxygen fluctuations or is directly involved in iron sensing per se.
Abbreviations: β2mg, β2-microglobulin; BMP, Bone morphogenetic protein; CAT, Catalase; FPN1, ferroportin 1; GOX, Glucose Oxidase; HIF, Hypoxia inducible factor; IL-6, Interleukin 6; JAK, Janus kinase; NOX, NADPH oxidase; ODD, Oxygen-dependent degradation domain; PHD2, Prolyl hydroxylase domain protein 2; Prx2, Peroxiredoxin 2; SIH, Salicylaldehyde isonicotinoyl hydrazone; SOCS3, Suppressor of cytokine signaling 3; STAT3, Signal transducer and activator of transcription 3; UOX, Urate oxidase
Keywords: Hepcidin, Hypoxia, Hydrogen peroxide, NADPH oxidase 4 (NOX4), Oxidases, Iron metabolism
Graphical abstract
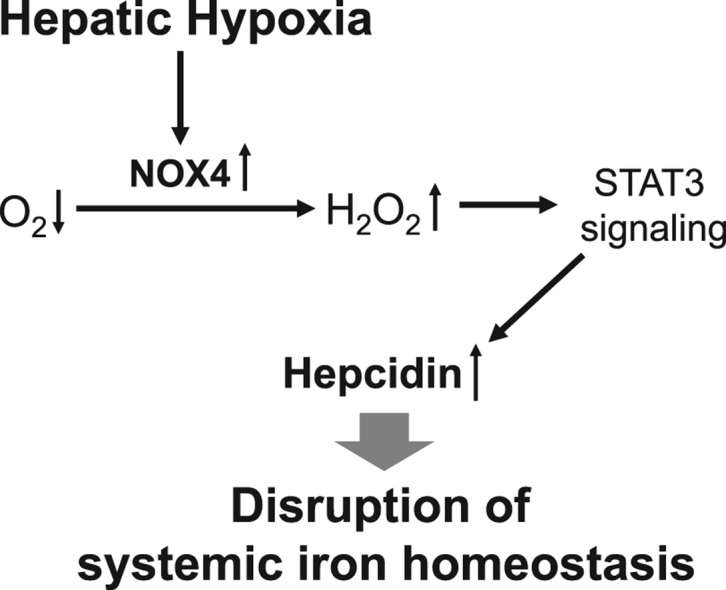
Highlights
-
•
Hypoxia strongly induces hepcidin via STAT3 signaling.
-
•
HIF1α is not involved in hepcidin regulation under hypoxia.
-
•
Hypoxia enhances hydrogen peroxide-mediated hepcidin induction.
-
•
Oxidases, such as NOX4 are powerful inducers of hepcidin.
1. Introduction
Hepcidin has emerged as central regulator of systemic iron homeostasis since its first identification as an antimicrobial peptide [1], [2]. The 25 amino acid peptide is primarily expressed in hepatocytes [3]. Deletion of hepcidin causes massive iron overload and many iron overload diseases are associated with suppressed hepcidin [4], [5]. In contrast, overexpression of hepcidin causes rapid decrease of serum iron ultimately leading to anemia [6] by binding to the iron exporter ferroportin (FPN1) finally leading to its internalization and subsequent proteasomal degradation [7], [8]. The impact on iron levels is conducted by the concerted blockage of FPN1 which is expressed on duodenal enterocytes (iron absorption), macrophages (iron recycling) and hepatocytes (iron storage).
Hepcidin is strongly induced by IL-6 and microbial molecules, such as lipopolysaccharide rapidly leading to the so called anemia of chronic disease [9]. The inflammation-mediated depletion of iron is thought to function as anti-bacterial defense mechanism. More recently, we could identify H2O2, a cellular key reactive oxygen species and important inflammatory cofactor, as potent inducer of hepcidin [10]. Interestingly, hepcidin shows a bivalent response to H2O2 depending on the peroxide level. Thus, H2O2 induces hepcidin in hepatocytes independent of IL-6 when exposed to very low and continuous non-toxic levels [10]. In contrast, artificial bolus treatment with high and toxic levels, drastically blocked hepcidin expression [11], [12] most likely due to unspecific inhibition of the mRNA transcription machinery [10].
So far, it remains poorly understood how exactly hepcidin is controlled by iron levels despite the discovery of various upstream regulators of hepcidin such as C/EBPα, BMP6, TFR1 and 2, Matriptase-2, IL-6, CREBH, CHOP, and recently PDGF-BB [13]. Controversial and partly conflicting findings have been especially reported on the role of hypoxia in regulating hepcidin. Thus, in vivo studies in rodent models have demonstrated that hypoxia leads to increased duodenal iron absorption and reduced hepcidin expression [12], [14], [15], [16], [17], suggesting a direct action of HIFs on hepcidin promoter via downregulation of CCAAT/enhancer binding protein alpha [18]. However, recent studies have refuted these conclusions showing that HIF-mediated suppression of hepcidin occurs indirectly through EPO-induced erythropoiesis [19], [20]. Interestingly, not all studies have demonstrated a decreased hepcidin expression in vitro. Thus, no diminished hepcidin levels or even an induction was seen in cultured HepG2 and Huh7 under hypoxic conditions [21], [22]. Although a direct role of HIF1α or HIF2α on hepcidin regulation has been suggested by several authors [18], [23], [24], [25], recent studies performed in hepatoma cell lines or in primary hepatocytes could not confirm former conclusions [12], [19], [21], [22].
A major challenge of in vivo studies on hypoxia is the very complex and difficult to interpret overlay by many adaptive responses of living organisms at various levels. For example, systemic hypoxia stimulates renal erythropoietin release which induces red blood cell formation and hence depletion of serum iron [19]. Moreover, hypoxia has important physiological adaptive effects such as an elevated cardiac output and a hyperventilation. In addition, hypoxic studies are sometimes performed using to high, aerobic levels of oxygen that are not mimicking physiological conditions found in specialized tissues. In fact, hepatocytes are usually surrounded by 4% and 8% in the pericentral and periportal area, respectively [26], [27], [28], [29], [30], [31]. In contrast, cultured cells are often exposed to artificially high oxygen levels of 21% instead of physiological low oxygen. Another pitfall with regard to ROS studies and hypoxia is the usage of artificially high peroxide bolus concentrations in the range up to 100 µM [32], [33].
Therefore, we here study in detail hepcidin expression under hypoxia (physiological oxygen levels found the liver). We primarily focus on the cellular level in hepatoma and primary liver cells to avoid systemic overlaying responses. In addition, we use both the hypoxia chamber and the recently established enzymatic hypoxia system, which allows the independent control of oxygen and peroxide levels [10], [34], [35]. Our data show a clear induction of hepcidin by physiological hypoxia from 1% to 7%. While a direct role of HIF1α is ruled out, promotor and signaling studies point towards the STAT3 signaling pathway. Moreover, we provide evidence for a drastically enhanced upregulation of hepcidin by peroxide under hypoxic conditions linking iron regulation directly to oxidases. Finally, our studies demonstrate that peroxide-generating oxidases such as NOX4 or UOX are able to efficiently control the expression of hepcidin via the STAT3 axis. Since hepcidin control is not only restricted to NOX4 but also other oxidases, our findings suggest that oxidase-mediated expression of hepcidin may be involved in iron sensing per se.
2. Material and methods
2.1. Cell culture and primary cells isolation
Huh7 cells from the Japanese Cancer Research Resources Bank (JCRB, Tokyo, Japan) were cultured under standard conditions using Dulbecco's modified Eagle medium (Sigma-Aldrich, Taufkirchen, Germany), 25 mM glucose and 10% fetal calf serum under 21% oxygen and 5% CO2 [10]. Primary human hepatocytes were isolated from healthy liver region of patients with liver metastasis undergoing partial liver resection using a collagenase-based protocol as published previously [36].
2.2. Chemicals and reagents
Actinomycin D, Rotenone and Cobalt (II) chloride were all purchased from Sigma-Aldrich (Taufkirchen, Germany). SIH was a kind gift of Dr. P. Ponka (McGill University, Montreal, Canada). Stock solutions prepared in DMSO were further diluted in culture medium with a final concentration of 0.1% DMSO.
2.3. Exposure of cells to H2O2 and/or hypoxia
Steady-state H2O2 (H2O2ss) treatment was performed using the glucose oxidase and catalase system (GOX/CAT system) as described previously [10], [34]. Briefly, glucose oxidase (GOX) and catalase (CAT) were purchased from Sigma-Aldrich (Taufkirchen, Germany) and the activities were determined using a sensitive chemiluminescence technique [33], [34], [37], [38]. During all experiments kGOX was kept at 4 × 10−8 M/s, while kCAT was adjusted to reach the final H2O2ss concentration of 2.5 µM. Since GOX metabolizes glucose and oxygen stochiometrically to H2O2 and δ-gluconolactone, attention was payed to decrease glucose during 24 h by no more than 3 mM. For hypoxia chamber treatments, cell culture plates were placed in a sealed chamber flushed with 5% O2, 5% CO2 and 90% N2 or with 1% O2, 5% CO2 and 94% N2. The hypoxia chamber was incubated at 37 °C for a maximal period of 24 h. In some experiments, the GOX/CAT system was used in combination with the hypoxia chamber to generate additional H2O2.
2.4. Hypoxia imaging
The cells were grown on chamber slides at 21, 5 or 1% O2. During the last 2 h, pimonidazole was added at a final concentration of 200 µM (Hypoxyprobe Plus Kit; HPI inc., Burlington, MA, USA). The slides were then washed with PBS and fixed in 100% methanol. Pimonidazole adducts were stained with the FITC-labeled mouse antibody according to the manufacturer's instructions. Cells were visualized using a 20 × objective on a Zeiss Axiovert 200M microscope (Zeiss, Göttingen, Germany) and the images were processed using Fiji ImageJ.
2.5. RNA isolation, cDNA synthesis and real time quantitative PCR analysis
RNA was isolated with Trifast (Peqlab biotechnology GmbH, Erlangen, Germany) according to the manufacturer specifications. Reverse transcription and the real time quantitative PCR reactions were performed as previously described [10]. Primers and probes were designed using the Probefinder software (Roche, Mannheim, Germany) and the sequences are provided in Supplemental Table S1.
2.6. Transfection experiments
Huh7 cells were transfected with an ODD containing plasmid construct, NOX4, UOX or hepcidin promoter plasmid constructs wild-type, truncated or mutated promoter constructs as previously described [39], [40], [41] using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). As previously described, the ODD containing plasmid (pCMV-ODD-GFP vector) was used for the competition experiments and the same vector without the ODD insert was used as control [31]. NOX4 cDNA was cloned into the KpnI and NotI sites of a pcDNA3 vector, resulting in antisense orientation in respect to the cytomegalovirus (CMV) promoter (Invitrogen, Carlsbad, CA). UOX cDNA was cloned into the EcoRI site of a pBluescriptSK vector. Hepcidin promoter constructs containing firefly luciferase and a control plasmid containing renilla luciferase were co-transfected at a ratio of 1:16 as published previously [10]. NOX4 and hepcidin promoter construct transfections were carried out for 48 h. For hypoxia studies, cells were then transferred to 5% O2 in a hypoxia chamber for additional 24 h.
2.7. RNA silencing
Huh7 cells were transfected using Lipofectamine 2000 and 10 nM siRNA. For the assessment of hepcidin promoter activity, the cells were co-transfected with hepcidin wild type promoter containing firefly luciferase construct, Renilla control plasmid and siRNA directed against individual molecules of the JAK-STAT3 signaling or universal negative siRNA as a control. 48 h after transfection, the cells were conditioned to 5% O2 in a hypoxia chamber for further 24 h. Validated siRNA for STAT3, IL-6 receptor, Jak1, Jak2 and NOX4 were purchased by Ambion (Foster City, CA, USA). Equimolar concentrations of universal negative siRNA (Sigma-Aldrich, Taufkirchen, Germany) were used as control.
2.8. Immunoblotting
Cells were washed in ice-cold 1xPBS and harvested in RIPA buffer plus 1 × Complete ® protease inhibitor with EDTA (Roche Applied Sciences, Penzberg, Germany) on ice. For Prx2 Western blotting, the cells were incubated with the thiol-blocking agent methanethiosulfonate (MMTS) at 80 mM prepared in ice-cold PBS for 10 min before harvesting, to avoid lysis-induced oxidation of Prx2 [35]. Prx2 samples were subjected to nonreducing SDS-PAGE and the Western Blotting was performed as described previously [10]. Equal protein loading was confirmed by protein staining with Ponceau-S solution as well as β-actin. Primary and secondary antibodies are listed in Supplemental Table S2. Immune-reactive bands were detected by chemiluminescence (Rotilumin, Carl Roth, Karlsruhe, Germany) and exposed to autoradiography film. Band intensities were quantified using ImageJ for further statistical analysis.
2.9. Statistical Analysis
All the data are expressed as mean ± SD. Significant differences (P < 0.05) between means of data sets were assessed by one-way ANOVA with Tukey's test or two-way ANOVA with Sidak's test using GraphPad Prism 6 software. Correlations with protein and mRNA expression data were assessed using Spearman's rank correlation coefficient (correlation coefficient r, r2, p). All p-values are two-sided. Differences were considered significant at p < 0.05. Statistical analysis was performed using PASW Statistics 18, version 18.0.0 (SPSS, Inc., Munich, Germany).
3. Results
3.1. Hypoxia strongly induces hepcidin in liver cells
In order to study the effect of hypoxia on hepcidin regulation, Huh7 cells were exposed for 24 h to 1%, 5% and 7% oxygen using the hypoxia chamber. As shown in Fig. 1A, hepcidin mRNA was significantly up-regulated in all hypoxic conditions. Pimonidazole was used to confirm cellular hypoxia under these conditions (Fig. 1B). These results could be also recapitulated in human primary liver cells exposed to 5% oxygen (Fig. 1C). We next compared the response of hepcidin to hypoxia either induced by the rapid enzymatic GOX/CAT system or the slower onset of hypoxia due to gas diffusion in the chamber. As shown in Fig. 1D both, GOX/CAT and hypoxia chamber showed a significant increase of hepcidin after 24 h. Notably, at this time point, HIF1α was only weakly induced with the GOX/CAT system due to PHD-mediated degradation mirroring the rapid onset of hypoxia by this system [31]. In contrast the hypoxia chamber showed a more profound increase of HIF1α as reported earlier [31]. In conclusion, both hypoxic systems induce hepcidin without a significant difference in liver cells and no suppression was observed even at lowest oxygen levels (1%).
Fig. 1.

Hypoxia induces hepcidin in liver cells. (A) Hypoxia significantly induces hepcidin mRNA in hepatocytes. Huh7 cells were exposed to 1% and 5% and 7% O2 over 24 h using a hypoxia chamber. (B) Confirmation of cellular hypoxia using pimonidazole adducts. Huh7 cells were conditioned to different oxygen levels over 24 h and fixed. Hypoxia was visualized using an antibody against pimonidazole (green). Scale bar = 10 µm. (C) Hypoxia also significantly induces Hepcidin mRNA in primary isolated human hepatocytes (*, P < 0.05). (D), Hepcidin is induced by two independent hypoxia systems. Rapid and slow onset hypoxia induces hepcidin after 24 h using the hypoxia chamber or the enzymatic GOX/CAT system (5% O2) in Huh7 cells. Changes in hepcidin mRNA levels and protein expression (HIF1α and PHD-2) are shown for a representative experiment. Hepcidin mRNA was quantified by quantitative real-time PCR and the results are represented as mean of hepcidin mRNA levels normalized to β2-microglobulin ± SD. Significant differences are marked by asterisks (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Representative data of at least three independent experiments are shown for protein data. n.s.: not significant.
3.2. STAT3 is required for hypoxia-dependent induction of hepcidin
Hypoxia mediated induction of hepcidin could be efficiently blocked in the presence of the transcription inhibitor actinomycin D (Fig. 2A). To further analyze the underlying transcriptional mechanisms, we performed reporter assays using full length wild type hepcidin promoter construct (WT 3 kb), three increasingly truncated wildtype (WT 1 kb, WT 385 bp and WT 165 bp) and four full length hepcidin promoter constructs with selective deletions of the STAT3-binding site (del-STAT3) or the BMP-responsive elements 1, 2, and 3 (BMP-RE1–3), respectively. As shown in Fig. 2B no change of promoter activity in response to hypoxia was observed when truncating the wildtype hepcidin promoter construct starting from the 5’-end. In contrast, the deletion of the STAT3-binding site completely blocked the hypoxia-dependent induction of hepcidin promoter activity. Selective deletions of BMP-RE1, -2 and -3 still allowed hypoxia-mediated hepcidin promoter activation. We then analyzed the JAK/STAT signaling cascade using siRNA-mediated silencing of IL-6R, JAK1, JAK2 and STAT3. Efficient silencing of the factors involved in JAK/STAT pathway is shown in Supplemental Fig. S3. Fig. 2D demonstrates that silencing of JAK1 and STAT3 suffices to block hypoxia-mediated hepcidin induction. As noted before, JAK1 is important for basal hepcidin expression under normoxia, therefore hepcidin is repressed in 21% oxygen after silencing of JAK1 [10]. In contrast, silencing of IL6-R and JAK2 only partly decreased the promoter responsiveness to hypoxia. Taken together, hypoxia induces hepcidin primarily at the transcriptional level via the STAT3 promotor domain. Moreover, and in addition to H2O2, JAK1 and STAT3 phosphorylation play a crucial role for the induction of hepcidin under hypoxia.
Fig. 2.

Hepcidin promoter activity is upregulated under hypoxia via the STAT3 signaling cascade. (A) The induction of hepcidin under hypoxia (5% O2) was efficiently blocked by the treatment with 0.1 µg/mL Actinomycin (**, P < 0.001). Results are presented as mean of hepcidin mRNA levels normalized to β2-microglobulin ± SD. (B) The deletion of the STAT3-binding site completely blocked the hypoxia-dependent induction of hepcidin promoter activity (*, P < 0.05). Huh7 cells were transfected with hepcidin promoter constructs including the wild type promoter (WT3kb), promoter regions with decreased length of the 5‘-flanking region (WT 1 kb, WT 855 bp and WT 165 bp) or the WT with specific deletions of transcription factor binding sites (STAT3-binding site (STAT3bs del) and the BMP-responsive elements 1–3 (bmp-RE1 to − 3 del)). The cells were transfected for 48 h and then conditioned to 5% O2 in a hypoxia chamber for 24 h. Cells transfected with the WT3kb construct showed a 2-fold increased hepcidin promoter activity relatively the control (21% O2). Renilla plasmid was used as control for expression and transfection. The results are expressed as fold induction ± SD of firefly/Renilla luciferase activity relatively to the 21% O2 control of each construct. (C) Although the silencing of IL6-R and JAK2 decreased the promoter responsiveness to hypoxia, only the silencing of JAK1 and STAT3 completely blocked the hypoxia effect significantly. The cells were co-transfected with hepcidin wild type promoter containing firefly luciferase construct, Renilla control plasmid and siRNA directed against individual molecules of the JAK-STAT3 signaling or universal negative siRNA as a control. 48 h after transfection, the cells were conditioned to 5% O2 in a hypoxia chamber for further 24 h. The results are presented as mean of the firefly/Renilla luciferase activity ratio ± SD. Significant differences are marked by asterisks (*, P < 0.05; **, P < 0.01; ***, P < 0.001). n.s.: not significant.
3.3. HIF1α is not directly involved in the hypoxia-mediated induction of hepcidin
Since HIF1α is one of the major hypoxia-responding transcription factors and conflicting data have been reported previously, we set up a series of experiments to test whether hepcidin induction was mediated by HIF1α. Data shown in Supplemental Fig. S1 suggested that HIF1α was not directly following or preceding hepcidin mRNA expression. Chemical HIF stabilizers, as CoCl2 or iron chelators could provide important information about the role of HIFs on hepcidin regulation. Notably, stabilization of HIF1α by exposure of Huh7 cells to CoCl2 or the membrane-permeable iron chelator SIH caused no significant changes on hepcidin mRNA levels while HIF1α was profoundly upregulated with PHD2 following the HIF1α response ( Fig. 3A). A strong stabilization of HIF2α was also detected after the treatment with CoCl2 without rendering hepcidin expression (data not shown) confirming that HIF2α does also not play a direct role in hepcidin regulation. To specifically test an involvement of HIF1α, we next performed competition experiments by overexpressing a plasmid harboring the HIF1-associated oxygen dependent degradation domain (ODD). This ODD domain confers hydroxylation by PHDs and specifically targets HIF1α for ubiquitination and proteasomal degradation [42], [43]. Furthermore, overexpression of the ODD containing plasmid has been previously shown to competitively block HIF1α degradation [31]. As shown in Fig. 3B, overexpression of the ODD-GFP construct indeed caused specific HIF1α accumulation. However, no hepcidin elevation was seen under these conditions. In our opinion, these data clearly exclude any direct role of HIF1α in the hypoxia-mediated upregulation of hepcidin.
Fig. 3.

HIF1α is not directly involved in the hypoxia-mediated induction of hepcidin. (A) Chemical stabilization of HIF1α by exposure of human hepatoma cells to CoCl2 or SIH caused no significant changes on hepcidin mRNA levels. The cells were treated for 24 h with CoCl2 (100 µM) or SIH (100 µM) and hepcidin mRNA was quantified by quantitative RT-PCR. (B) The presence of ODD-GFP constructs that specifically prevent HIF1α degradation decreased hypoxia-mediated induction of hepcidin. Huh7 cells were transfected for 6 h with an ODD-GFP plasmid or the plasmid only containing the GFP was a control and then conditioned to 5% O2 for further 16 h. Under 21% O2, the stabilization of HIF1α exerted no significant effect, however at 5% O2 the upregulation of hepcidin mRNA was blocked (**, P < 0.01). HIF1α and PHD-2 were analyzed by Western blot. Hepcidin mRNA was quantified by quantitative RT-PCR and the results are presented as mean of hepcidin mRNA levels normalized to β2-microglobulin ± SD. Representative data of at least three independent experiments are shown. n.s.: not significant.
3.4. Hypoxia enhances H2O2-mediated induction of hepcidin
We next analyzed how hypoxia would affect hepcidin expression in the presence of additional H2O2 which was recently identified as potent inducer of hepcidin via the STAT3 axis [10]. The GOX/CAT system was explored as it allows the independent control of oxygen and peroxide [34]. As shown in Fig. 4A, H2O2 was able to further induce hepcidin under hypoxia in a synergistic fashion. In fact, the peroxide-mediated response of hepcidin was even more pronounced under hypoxia. Here, H2O2 was generated by the GOX/CAT system in combination with the hypoxia chamber. In all cases, the presence of non-toxic steady-state H2O2 level was confirmed by Prx2 Western blotting (Fig. 4B). The expression of HIF1α, pSTAT3 and STAT3 was also assessed (Fig. 4A and C). Interestingly, Prx2 was more induced after exposure to H2O2 under hypoxic conditions. To get more insights into the underlying mechanisms, we performed kinetic studies of various peroxide and hypoxia sensitive signaling molecules with the two hypoxia systems. As shown in Supplemental Fig. S1, the kinetic response of hepcidin showed in both systems a more complex, bi-modal pattern with an initial 2–3 fold increase and a later induction from 2 to 10 times due to a negative loop feedback by SOCS3 as shown earlier [44], [45]. Supplemental Fig. S2 demonstrates kinetics of hepcidin and SOCS3 mRNA confirming the negative feedback role of the STAT3-inhibitor SOCS3 and explaining the bi-modal hepcidin response upon exposure to hypoxia. Notably, correlation analysis showed that hepcidin mRNA significantly correlated with pSTAT3 (r = 0.671, P < 0.05) but not with HIF1α or Prx2 protein expression (Table 1). In summary, H2O2 and hypoxia show a synergistic effect on hepcidin expression.
Fig. 4.

Hypoxia mediated hepcidin induction is further potentiated by the exposure to low ss H2O2levels. Hypoxia chamber and the GOX/CAT system were used to independently control low steady state H2O2 levels and to maintain low oxygen level (5% O2) in Huh7 cells. (A) Hypoxia in combination with low ss H2O2 caused a significant induction of hepcidin mRNA and increased HIF1α. (B and C) Densitometric analysis and representative Western blot of Prx2 and STAT3 protein expression under hypoxia and low ss H2O2. Hypoxia led to increased oxidized Prx2 and pSTAT3 level, which were further enhanced with additional low ss H2O2. Hepcidin mRNA was quantified by quantitative real-time PCR and the results are expressed as mean of hepcidin mRNA levels normalized to β2-microglobulin ± SD. Significant differences are marked by asterisks (**, P < 0.01; ***, P < 0.001). Representative data of at least three independent experiments are shown.
Table 1.
Correlation (Spearman Rho) of Hepcidin mRNA levels with Prx2, pSTAT3 and HIF1α expression from kinetic hypoxia experiments. Hypoxia (5% O2) was induced in Huh7 cells either by the hypoxia chamber or the enzymatic GOX/CAT system (see Suppl. Fig. S2).
| Parameter | Hepcidin mRNA | Pvalue |
|---|---|---|
| Prx2oxidized/ Prx2reduced | 0.000 | 1.000 |
| pSTAT3/STAT3 | 0.671* | 0.017 |
| HIF1α | −0.273 | 0.391 |
3.5. Efficient control of hepcidin by NOX4 in hepatocytes
The synergistic induction of hepcidin by hypoxia and H2O2 clearly suggested to us that oxidases could be potential upstream regulators of hepcidin since these enzymes are efficient peroxide sources by consuming and lowering oxygen levels. Since NOX are important peroxide sources in hepatocytes, we further investigated their involvement in regulating hepcidin. We first show in Fig. 5A that NOX4 was present in Huh7 cells and NOX4 mRNA as well as protein expression could be significantly induced by hypoxia (5% O2). Moreover, overexpression of NOX4 in Huh7 hepatoma cells caused significant upregulation of hepcidin (Fig. 5B), which was further enhanced under hypoxic conditions. Comparable results were obtained for Prx2 protein oxidation (Fig. 5C) used as a measure for endogenous H2O2, indicating that NOX4-generated H2O2 may be responsible for hepcidin upregulation. In addition, NOX4 overexpression also led to an increased pSTAT3 what was even more pronounced under hypoxia (Fig. 5D). Vice versa, siRNA mediated knockdown of NOX4 caused a significant decrease in hepcidin expression, nearly absent or diminished Prx2 protein oxidation under 5% O2 as well as reduced pSTAT3 expression levels if compared to the normoxic or hypoxic controls (Fig. 6A). Furthermore, siRNA mediated knockdown of STAT3 also reduced hepcidin mRNA expression back to baseline levels as found in normoxic control (Fig. 6B) but did not significantly change NOX4 mRNA levels (Supplemental Fig. S4). We finally tested whether other oxidases or cellular peroxide sources would also be able to regulate hepcidin expression. As shown in Supplemental Fig. S5 both, the peroxide release by complex I inhibition of the mitochondrial respiration by rotenone and overexpression of murine urate oxidase (UOX) strongly and significantly induced hepcidin. In conclusion, these data confirm NOX4 as potent upstream modulator of hepcidin in hepatocytes activating the STAT signaling cascade. Our data further suggest that also other oxidases are able to upregulate hepcidin and this effect is even more pronounced under physiologic low oxygen conditions (5% O2), offering a novel powerful mechanism of iron control.
Fig. 5.

NOX4-mediated release of intracellular H2O2induces hepcidin mRNA expression. (A) Hypoxia (5% O2) increases NOX4 mRNA levels. Huh7 hepatoma cells were conditioned to 5% O2 during 24 h and NOX4 mRNA was analyzed by quantitative RT-PCR. (B) Overexpression of NOX4 increases hepcidin mRNA levels and potentiates the hypoxia-mediated hepcidin induction. The cells were transfected with a NOX4 promoter construct for 48 h and then conditioned in a hypoxia chamber to a 5% O2 atmosphere for additional 24 h. Significant increases on hepcidin mRNA levels were observed after transfection with the NOX4 promoter construct. Hepcidin and NOX4 mRNA were quantified by quantitative RT-PCR and the results are represented as mean of hepcidin or NOX4 mRNA levels normalized to β2-microglobulin ± SD. Significant differences are marked by asterisks (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Representative data of at least three independent experiments are shown. (C and D) Densitometric analysis and representative Western blot of Prx2 and STAT3. Huh7 transfected with control or NOX4 plasmid were conditioned to 21 or 5% O2. The results are expressed as ratio of Prx2 oxidized (ox) to Prx2 reduced (red) or pSTAT3 to STAT3.
Fig. 6.
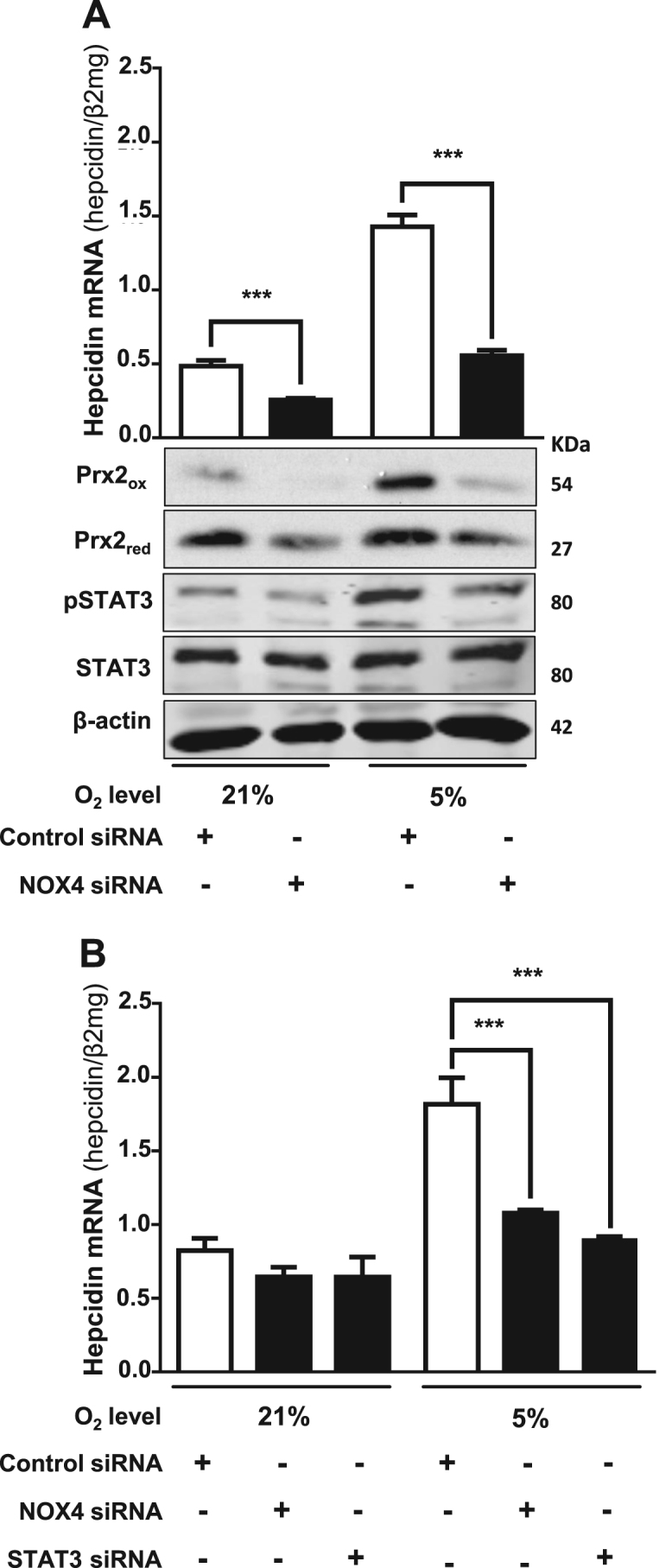
Silencing of NOX4 and STAT3 prevents hypoxia-mediated induction of hepcidin. (A) siRNA-mediated silencing of NOX4 decreased hepcidin mRNA expression and blocked the upregulation under hypoxia. Huh7 cells were transfected with siRNA against NOX4 or with universal negative siRNA control and conditioned to 21 or 5% O2 over 24 h. pSTAT3 and oxidized Prx2 expression were followed by Western blot. (B) Effect of NOX4 overexpression on hepcidin is attenuated by co-transfection with siRNA against NOX4 or STAT3. The cells were co-transfected with NOX4 plasmid and siRNA against NOX4, STAT3 or universal negative control. After transfection, Huh7 were exposed to 21 or 5% O2 over 24 h and hepcidin mRNA was quantified by quantitative RT-PCR. The results are expressed as mean of hepcidin mRNA levels normalized to β2-microglobulin ± SD. Significant differences are marked by asterisks (***, P < 0.001).
4. Discussion
We here show that hypoxia at normal liver tissue oxygen levels strongly induces hepcidin in liver cells predominantly at the transcriptional level via STAT3 and independent of HIF1α. We focused in our study on hepcidin mRNA expression levels since hepcidin regulation occurs primarily at the transcriptional level and data on protein expression and secreted peptide levels correspond well to mRNA data [46], [47]. Moreover, hypoxia drastically enhances the previously discovered H2O2-mediated induction of hepcidin mRNA. We next demonstrate that oxidases such as NOX4 in hepatocytes are potent inducers of hepcidin via the STAT3 signaling pathway and inhibition of NOX4 impeded the response. We finally demonstrate that also other oxidases such as UOX are able to upregulate hepcidin.
Despite an enormous progress in the understanding of the complex transcriptional regulation of hepcidin as systemic iron master switch, many aspects still remain unresolved including the response to iron per se and hypoxia [13]. Thus, conflicting data have been reported on the role of hypoxia on hepcidin. While hypoxia seems to suppress hepcidin in in vivo models mostly in rodents [14], [15], [16], [17], an upregulation was reported under hypoxic conditions in experiments with cultured cells [21], [22]. Moreover, controversies exist on the role of HIFs and an earlier reported identification of a potential hypoxia responsive element could not be confirmed so far [18], [19], [20]. New in vivo evidences have linked the suppression of hepcidin to HIF2α through systemic EPO-mediated increased erythropoiesis, however these studies are not able to describe the response of hepcidin to local liver hypoxia found in pathological inflammatory processes.
Using two strategies to generate hypoxia either enzymatically or by the hypoxia chamber, we clearly demonstrate a strong induction of hepcidin by mild or pronounced hypoxia both in hepatoma or primary liver cells, mimicking normal or pathological tissue oxygen levels. Similar to H2O2 [10], deletion of the STAT3 promotor site and siRNA silencing of JAK1 and STAT3 completely blocked hypoxia-mediated induction of hepcidin. In addition, we identify three major experimental arguments against an immanent role of HIF1α in hepatocytes. First, the kinetic response of hepcidin to hypoxia-mimetics such as cobalt chloride and iron chelators e.g. SIH clearly differed from HIF1α expression/stabilization which typically increases only transiently in response to oxygen lowering [31]. Second, overexpression of a plasmid containing the ODD highly specifically blocked HIF1α degradation through competitive inhibition but did not affect at all the hepcidin response. Third, in kinetic studies, hepcidin mRNA only correlated with the levels of STAT3 but not HIF1α or Prx2 which has been recently proposed to regulate hepcidin [48]. With regard to these findings and the complex interplay of oxygen and iron metabolism, we conclude that HIF1α is definitely not involved in hepcidin regulation and previous in vivo findings in rodent models may be due to an overlaid adaptive response at various levels including the blood building system. In addition, earlier in vitro reports could also not show changes of hepcidin regulation after deletion or overexpression of HIF1α, HIF2α or the dimer partner ARNT [12], [19], [21].
Another highly interesting finding of our work is that hypoxia drastically enhances the H2O2-mediated induction of hepcidin and, in fact, the hepcidin response mirrored the H2O2 signaling cascade including STAT3 [10]. Thus, hypoxia-mediated upregulation of hepcidin was blocked by the transcription inhibitor actinomycin D, which was almost exclusively restricted to the STAT3-binding site. In contrast, selective deletions of BMP-RE1, -2 and -3 still allowed hypoxia-mediated hepcidin promoter activation. Moreover, although the silencing of IL6-R and JAK2 decreased the promoter responsiveness to hypoxia, just the silencing of JAK1 and STAT3 diminished the hypoxia effect significantly. These findings strongly point towards oxidases as efficient upstream component of hepcidin regulation. Indeed, we provide evidence for an efficient control of hepcidin expression by liver expressed NADPH-dependent oxidases 4 (NOX4). NOX4 mRNA was significantly induced by hypoxia, which was accompanied by significant upregulation of hepcidin. NOX4 overexpression also induced hepcidin, which was further enhanced under hypoxic conditions. In contrast, siRNA mediated silencing of either NOX4 or STAT3 prevented hypoxia-induced hepcidin upregulation. In addition, silencing of NOX4 led to absent or diminished oxidized Prx2 protein level pointing towards reduced H2O2 level. Our findings indicate that NOX4 generated H2O2 may signal over JAK/STAT pathway thereby upregulating hepcidin. Since we are able to demonstrate hepcidin upregulation in response to other oxidases/cellular H2O2 sources such as the mitochondrial respiratory chain or artificially expressed urate oxidase (UOX) [49], [50], [51], various oxidases or a concerted integral action of oxidases may act as potential upstream inducers of hepcidin. Beside this, paracrine factors may also play a role to activate transcription factors like STAT3 under hypoxia. We believe that former studies with artificially high bolus H2O2 concentrations are a major reason for controversial findings on hepcidin expression [10]. Typically, 100 or even up to 200 µM H2O2 are applied [22] and shown to suppress hepcidin. We could show that these levels are rapidly eliminated within 30–60 min [33] and unspecifically inhibit mRNA expression and transcriptional machinery [10]. In contrast, we here used micromolar, or even in the hypoxia studies submicromolar concentrations of H2O2 mirroring physiologic conditions [10].
Taken together, our data suggest that oxidases induce hepatocyte hepcidin via STAT3. Notably, our findings on hypoxia are not in favor of a specific or classical hypoxia signaling cascade and it could resolve some of the confusions in the preexisting literature on hepcidin signaling and hypoxia [14], [15], [16], [17], [18], [21], [22]. According to our findings and the clear STAT3 dependence, it rather appears that the H2O2-STAT3 axis is the actual signaling cascade of hepcidin that is, of course, also responsive to hypoxia, as cellular H2O2 production will strongly depend on tissue oxygen levels. Thus, we believe that normal oxygen metabolism and oxygen fluctuations could strongly modulate the peroxide-hepcidin cascade. Since peroxide is continuously produced in cells either as metabolic side product of mitochondria or mandatory from peroxisomes [51] it could be responsible for a permanent basal expression of hepcidin. Although the kinetics in response to hypoxia and H2O2 unambiguously point towards STAT3 as major H2O2 sensing molecule upstream of hepcidin, several details still remain open. For instance, it is not clear why Prx2 expression levels do not directly correlate with hepcidin although it has been recently suggested to form a redox relay with STAT3 for H2O2 [52] and to be involved in hepcidin control [48]. It is suggested that Prx2 may act as H2O2 signal receptor due to its cysteine residues and forms a redox relay with other redox sensitive proteins such as the transcription factor STAT3 [52]. Since we could also see a more enhanced expression of Prx2 in response to H2O2 under hypoxia, these data could at least suggest a facilitated H2O2/STAT3 signaling by Prx2 and may lead to an induced transcriptional activity of the hamp promotor. In addition, increased H2O2 may alter redox signaling by enhancing tyrosine or serine phosphorylation by inactivating protein phosphatases while activating protein kinases (e.g. Src kinase or EGF receptor) [53].
Finally, it would be very attractive to postulate specific cellular H2O2 sources as mandatory upstream control of hepcidin. One such source could be the hepatocyte expressed NOX4. It is constitutively active and, in contrast to all other NOX enzymes, does not require upstream activators, either calcium or organizer/activator subunits (p47(phox), NOXO1/p67(phox), and NOXA1) [54]. NOX4 also reportedly releases hydrogen peroxide in contrast to NOX1-NOX3 and NOX5, which release superoxide. Interestingly, NOX4 has an unusually high Km for oxygen of ca. 18%, similar to the values of known oxygen-sensing enzymes, compared with a Km of 2–3% for NOX2, the major phagocyte NADPH oxidase [54]. This allows NOX4 to generate peroxide as a function of oxygen concentration throughout a physiological range of pO2 values and, thus, to respond rapidly to changes in oxygen. Based on these enzyme characteristics, NOX4 has been recently suggested to act as an oxygen sensor [55]. However and not in contradiction to these conclusions, it could also act to adapt to tissue oxygen fluctuations. This could explain why NOX4 is induced in response to hypoxia and confirms earlier reports on enhanced ROS production under hypoxia [22]. Consequently, the NOX4-STAT3-hepcidin axis could help to continuously compensate for oxygen fluctuations and stay tuned for additional interfering signals.
We are aware of the limitation that a direct in vivo proof of the role of STAT3 and NOX4 would be highly desirable to confirm this novel concept on oxidase-mediated hepcidin regulation. However, as mentioned above, classical approaches such as gene deletion are not feasible since e.g. STAT3-/- KO mice die early during embryogenesis [56] and NOX4-/- KO mice show no phenotype and reach normal life span [57], [58]. These findings may not speak against a role of NOX4 in iron homeostasis since its function may be replaced by many other oxidases.
In summary, we here demonstrate that hypoxia strongly enhances the peroxide-mediated induction of hepcidin via STAT3. Moreover, our data demonstrate that oxidases such as NOX4 are powerful inducers of hepcidin via synchronic modulation of both oxygen and peroxide levels and the STAT3 signaling pathway. Thus, oxidase-mediated hepcidin signaling may not only provide an important regulatory link to inflammation but could also be responsible for basal cellular hepcidin expression. It remains to be clarified in future studies whether the peroxide-STAT3-hepcidin axis simply acts to continuously compensate for oxygen fluctuations or is directly involved in iron sensing per se.
Acknowledgement
This work was supported by the Dietmar Hopp Foundation to SM and funded by a grant of the DFG to VR and SM (RA 2677/1-1). We would further like to thank Dr. P. Ponka (McGill University, Montreal, Canada) for providing us with the membrane permeable iron chelator SIH, Dr. G. D’Angelo (Institut de Pharmacologie Moleculaire et Cellulaire, CNRS, Valbonne, France), Dr. M. Muckenthaler (University of Heidelberg, Germany) and Dr. J.K. Reddy (Northwestern University Medical School, Chicago, USA) for the ODD, hepcidin promotor and UOX cDNA plasmids, respectively.
Acknowledgments
Conflict of interest
The authors have nothing to disclose.
Author contributions
IS and VR designed the experiments, analyzed the data and wrote the manuscript. IS and TP performed the experiments. VR, GM and HKS critically revised the manuscript. SM conceived the study, designed experiments, analyzed data and wrote the manuscript.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.02.005.
Appendix A. Supplementary material
Supplementary material
References
- 1.Krause A., Neitz S., Magert H.J., Schulz A., Forssmann W.G., Schulz-Knappe P., Adermann K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480(2–3):147–150. doi: 10.1016/s0014-5793(00)01920-7. [DOI] [PubMed] [Google Scholar]
- 2.Park C.H., Valore E.V., Waring A.J., Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001;276(11):7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 3.Sow F.B., Florence W.C., Satoskar A.R., Schlesinger L.S., Zwilling B.S., Lafuse W.P. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J. Leukoc. Biol. 2007;82(4):934–945. doi: 10.1189/jlb.0407216. [DOI] [PubMed] [Google Scholar]
- 4.Nicolas G., Bennoun M., Devaux I., Beaumont C., Grandchamp B., Kahn A., Vaulont S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA. 2001;98(15):8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roetto A., Papanikolaou G., Politou M., Alberti F., Girelli D., Christakis J., Loukopoulos D., Camaschella C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 2003;33(1):21–22. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 6.Nicolas G., Bennoun M., Porteu A., Mativet S., Beaumont C., Grandchamp B., Sirito M., Sawadogo M., Kahn A., Vaulont S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc. Natl. Acad. Sci. USA. 2002;99(7):4596–4601. doi: 10.1073/pnas.072632499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nemeth E., Ganz T. The role of hepcidin in iron metabolism. Acta Haematol. 2009;122(2–3):78–86. doi: 10.1159/000243791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hentze M.W., Muckenthaler M.U., Galy B., Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell. 2010;142(1):24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 9.Weiss G., Goodnough L.T. Anemia of chronic disease. N. Engl. J. Med. 2005;352(10):1011–1023. doi: 10.1056/NEJMra041809. [DOI] [PubMed] [Google Scholar]
- 10.Millonig G., Ganzleben I., Peccerella T., Casanovas G., Brodziak-Jarosz L., Breitkopf-Heinlein K., Dick T.P., Seitz H.K., Muckenthaler M.U., Mueller S. Sustained submicromolar H2O2 levels induce hepcidin via signal transducer and activator of transcription 3 (STAT3) J. Biol. Chem. 2012;287(44):37472–37482. doi: 10.1074/jbc.M112.358911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miura K., Taura K., Kodama Y., Schnabl B., Brenner D.A. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology. 2008;48(5):1420–1429. doi: 10.1002/hep.22486. [DOI] [PubMed] [Google Scholar]
- 12.Choi S.O., Cho Y.S., Kim H.L., Park J.W. ROS mediate the hypoxic repression of the hepcidin gene by inhibiting C/EBPalpha and STAT-3. Biochem. Biophys. Res. Commun. 2007;356(1):312–317. doi: 10.1016/j.bbrc.2007.02.137. [DOI] [PubMed] [Google Scholar]
- 13.Muckenthaler M.U., Rivella S., Hentze M.W., Galy B., Red A. Carpet for Iron Metabolism. Cell. 2017;168(3):344–361. doi: 10.1016/j.cell.2016.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leung P.S., Srai S.K., Mascarenhas M., Churchill L.J., Debnam E.S. Increased duodenal iron uptake and transfer in a rat model of chronic hypoxia is accompanied by reduced hepcidin expression. Gut. 2005;54(10):1391–1395. doi: 10.1136/gut.2004.062083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mastrogiannaki M., Matak P., Keith B., Simon M.C., Vaulont S., Peyssonnaux C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J. Clin. Invest. 2009;119(5):1159–1166. doi: 10.1172/JCI38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicolas G., Chauvet C., Viatte L., Danan J.L., Bigard X., Devaux I., Beaumont C., Kahn A., Vaulont S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Invest. 2002;110(7):1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Riordan D.K., Debnam E.S., Sharp P.A., Simpson R.J., Taylor E.M., Srai S.K. Mechanisms involved in increased iron uptake across rat duodenal brush-border membrane during hypoxia. J. Physiol. 1997;500(Pt 2):379–384. doi: 10.1113/jphysiol.1997.sp022028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peyssonnaux C., Zinkernagel A.S., Schuepbach R.A., Rankin E., Vaulont S., Haase V.H., Nizet V., Johnson R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) J. Clin. Invest. 2007;117(7):1926–1932. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mastrogiannaki M., Matak P., Mathieu J.R., Delga S., Mayeux P., Vaulont S., Peyssonnaux C. Hepatic hypoxia-inducible factor-2 down-regulates hepcidin expression in mice through an erythropoietin-mediated increase in erythropoiesis. Haematologica. 2012;97(6):827–834. doi: 10.3324/haematol.2011.056119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Q., Davidoff O., Niss K., Haase V.H. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J. Clin. Invest. 2012;122(12):4635–4644. doi: 10.1172/JCI63924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Volke M., Gale D.P., Maegdefrau U., Schley G., Klanke B., Bosserhoff A.K., Maxwell P.H., Eckardt K.U., Warnecke C. Evidence for a lack of a direct transcriptional suppression of the iron regulatory peptide hepcidin by hypoxia-inducible factors. PLoS One. 2009;4(11):e7875. doi: 10.1371/journal.pone.0007875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaston T.B., Matak P., Pourvali K., Srai S.K., McKie A.T., Sharp P.A. Hypoxia inhibits hepcidin expression in HuH7 hepatoma cells via decreased SMAD4 signaling. Am. J. Physiol. Cell Physiol. 2011;300(4):C888–C895. doi: 10.1152/ajpcell.00121.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lakhal S., Schodel J., Townsend A.R., Pugh C.W., Ratcliffe P.J., Mole D.R. Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxia-inducible factors: new link between hypoxia signaling and iron homeostasis. J. Biol. Chem. 2011;286(6):4090–4097. doi: 10.1074/jbc.M110.173096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson E.R., Taylor M., Xue X., Martin A., Moons D.S., Omary M.B., Shah Y.M. The hypoxia-inducible factor-C/EBPalpha axis controls ethanol-mediated hepcidin repression. Mol. Cell Biol. 2012;32(19):4068–4077. doi: 10.1128/MCB.00723-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silvestri L., Pagani A., Camaschella C. Furin-mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis. Blood. 2008;111(2):924–931. doi: 10.1182/blood-2007-07-100677. [DOI] [PubMed] [Google Scholar]
- 26.Jungermann K., Kietzmann T. Oxygen: modulator of metabolic zonation and disease of the liver. Hepatology. 2000;31(2):255–260. doi: 10.1002/hep.510310201. [DOI] [PubMed] [Google Scholar]
- 27.Stroka D.M., Burkhardt T., Desbaillets I., Wenger R.H., Neil D.A.H., Bauer C., Gassmann M., Candinas D. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. 2001;15(13):2445–2453. doi: 10.1096/fj.01-0125com. [DOI] [PubMed] [Google Scholar]
- 28.Sitkovsky M., Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat. Rev. Immunol. 2005;5(9):712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- 29.Carlsson P.O., Palm F., Andersson A., Liss P. Markedly decreased oxygen tension in transplanted rat pancreatic islets irrespective of the implantation site. Diabetes. 2001;50(3):489–495. doi: 10.2337/diabetes.50.3.489. [DOI] [PubMed] [Google Scholar]
- 30.Richardson R.S., Noyszewski E.A., Kendrick K.F., Leigh J.S., Wagner P.D. Myoglobin O2 desaturation during exercise. Evidence of limited O2 transport. J. Clin. Invest. 1995;96(4):1916–1926. doi: 10.1172/JCI118237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Millonig G., Hegedusch S., Becker L., Seitz H.K., Schuppan D., Mueller S. Hypoxia-inducible factor 1 alpha under rapid enzymatic hypoxia: cells sense decrements of oxygen but not hypoxia per se. Free Radic. Biol. Med. 2009;46(2):182–191. doi: 10.1016/j.freeradbiomed.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 32.Forman H.J. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic. Biol. Med. 2007;42(7):926–932. doi: 10.1016/j.freeradbiomed.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mueller S. Sensitive and nonenzymatic measurement of hydrogen peroxide in biological systems. Free Radic. Biol. Med. 2000;29(5):410–415. doi: 10.1016/s0891-5849(00)00261-6. [DOI] [PubMed] [Google Scholar]
- 34.Mueller S., Millonig G., Waite G.N. The GOX/CAT system: a novel enzymatic method to independently control hydrogen peroxide and hypoxia in cell culture. Adv. Med. Sci. 2009;54(2):121–135. doi: 10.2478/v10039-009-0042-3. [DOI] [PubMed] [Google Scholar]
- 35.Sobotta M.C., Barata A.G., Schmidt U., Mueller S., Millonig G., Dick T.P. Exposing cells to H2O2: a quantitative comparison between continuous low-dose and one-time high-dose treatments. Free Radic. Biol. Med. 2013;60:325–335. doi: 10.1016/j.freeradbiomed.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 36.Ganten T.M., Koschny R., Sykora J., Schulze-Bergkamen H., Buchler P., Haas T.L., Schader M.B., Untergasser A., Stremmel W., Walczak H. Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs. Clin. Cancer Res. 2006;12(8):2640–2646. doi: 10.1158/1078-0432.CCR-05-2635. [DOI] [PubMed] [Google Scholar]
- 37.Andriopoulos B., Hegedusch S., Mangin J., Riedel H.D., Hebling U., Wang J., Pantopoulos K., Mueller S. Sustained hydrogen peroxide induces iron uptake by transferrin receptor-1 independent of the iron regulatory protein/iron-responsive element network. J. Biol. Chem. 2007;282(28):20301–20308. doi: 10.1074/jbc.M702463200. [DOI] [PubMed] [Google Scholar]
- 38.Mueller S., Riedel H.D., Stremmel W. Determination of catalase activity at physiological hydrogen peroxide concentrations. Anal. Biochem. 1997;245(1):55–60. doi: 10.1006/abio.1996.9939. [DOI] [PubMed] [Google Scholar]
- 39.Verga Falzacappa M.V., Vujic Spasic M., Kessler R., Stolte J., Hentze M.W., Muckenthaler M.U. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood. 2007;109(1):353–358. doi: 10.1182/blood-2006-07-033969. [DOI] [PubMed] [Google Scholar]
- 40.Casanovas G., Mleczko-Sanecka K., Altamura S., Hentze M.W., Muckenthaler M.U. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J. Mol. Med. 2009;87(5):471–480. doi: 10.1007/s00109-009-0447-2. [DOI] [PubMed] [Google Scholar]
- 41.Verga Falzacappa M.V., Casanovas G., Hentze M.W., Muckenthaler M.U. A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. J. Mol. Med. 2008;86(5):531–540. doi: 10.1007/s00109-008-0313-7. [DOI] [PubMed] [Google Scholar]
- 42.D'Angelo G., Duplan E., Boyer N., Vigne P., Frelin C. Hypoxia up-regulates prolyl hydroxylase activity: a feedback mechanism that limits HIF-1 responses during reoxygenation. J. Biol. Chem. 2003;278(40):38183–38187. doi: 10.1074/jbc.M302244200. [DOI] [PubMed] [Google Scholar]
- 43.Schofield C.J., Ratcliffe P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004;5(5):343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 44.Pietrangelo A., Dierssen U., Valli L., Garuti C., Rump A., Corradini E., Ernst M., Klein C., Trautwein C. STAT3 is required for IL-6-gp130-dependent activation of hepcidin in vivo. Gastroenterology. 2007;132(1):294–300. doi: 10.1053/j.gastro.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 45.Williams J.J., Munro K.M., Palmer T.M. Role of ubiquitylation in controlling suppressor of cytokine signalling 3 (SOCS3) function and expression. Cells. 2014;3(2):546–562. doi: 10.3390/cells3020546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Domenico I., Ward D.M., Kaplan J. Hepcidin regulation: ironing out the details. J. Clin. Invest. 2007;117(7):1755–1758. doi: 10.1172/JCI32701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ganz T. Hepcidin and iron regulation, 10 years later. Blood. 2011;117(17):4425–4433. doi: 10.1182/blood-2011-01-258467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matte A., De Falco L., Federti E., Cozzi A., Iolascon A., Levi S., Mohandas N., Zamo A., Bruno M., Lebouef C., Janin A., Siciliano A., Ganz T., Federico G., Carlomagno F., Mueller S., Silva I., Carbone C., Melisi D., Kim D.W., Choi S.Y., Franceschi L. De. Peroxiredoxin-2: a novel regulator of iron homeostasis in ineffective erythropoiesis. Antioxid. Redox Signal. 2017 doi: 10.1089/ars.2017.7051. [DOI] [PubMed] [Google Scholar]
- 49.Boveris A., Oshino R., Erecinska M., Chance B. Reduction of mitochondrial components by durohydroquinone. Biochim. Biophys. Acta. 1971;245(1):1–16. doi: 10.1016/0005-2728(71)90002-8. [DOI] [PubMed] [Google Scholar]
- 50.Yeldandi A.V., Chu R., Reddy S.K., Pan J., Usuda N., Lin Y., Rao M.S., Reddy J.K. Functional expression and peroxisomal targeting of rat urate oxidase in monkey kidney cells. Gene Expr. 1995;5(2):125–132. [PubMed] [Google Scholar]
- 51.Fritz R., Bol J., Hebling U., Angermuller S., Volkl A., Fahimi H.D., Mueller S. Compartment-dependent management of H(2)O(2) by peroxisomes. Free Radic. Biol. Med. 2007;42(7):1119–1129. doi: 10.1016/j.freeradbiomed.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 52.Sobotta M.C., Liou W., Stocker S., Talwar D., Oehler M., Ruppert T., Scharf A.N., Dick T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015;11(1):64–70. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- 53.Rhee S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312(5782):1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 54.Nisimoto Y., Diebold B.A., Cosentino-Gomes D., Lambeth J.D. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry. 2014;53(31):5111–5120. doi: 10.1021/bi500331y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nisimoto Y., Jackson H.M., Ogawa H., Kawahara T., Lambeth J.D. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry. 2010;49(11):2433–2442. doi: 10.1021/bi9022285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene. 2000;19(21):2607–2611. doi: 10.1038/sj.onc.1203478. [DOI] [PubMed] [Google Scholar]
- 57.Rezende F., Schurmann C., Schutz S., Harenkamp S., Herrmann E., Seimetz M., Weissmann N., Schroder K. Knock out of the NADPH oxidase Nox4 has no impact on life span in mice. Redox Biol. 2017;11:312–314. doi: 10.1016/j.redox.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rezende F., Lowe O., Helfinger V., Prior K.K., Walter M., Zukunft S., Fleming I., Weissmann N., Brandes R.P., Schroder K. Unchanged NADPH oxidase activity in nox1-nox2-nox4 triple knockout mice: what do NADPH-stimulated chemiluminescence assays really detect? Antioxid. Redox Signal. 2016;24(7):392–399. doi: 10.1089/ars.2015.6314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material