

Abstract
Sei-1 is a potential oncogene that plays an important role in promoting genomic instability. Double minute chromosomes (DMs) are hallmarks of gene amplification and contribute to tumorigenesis. Defects in the DNA double-strand break (DSB) repairing pathways can lead to gene amplification. To date, the mechanisms governing the formation of DMs induced by Sei-1 are not fully understood. We established DMs induced by Sei-1 in the NIH-3T3 cell line. RNA-sequencing was used to identify key characteristics of differentially expressed genes. Metaphase spreads were used to calculate DM numbers. Immunofluorescence was employed to detect γH2AX foci. Western blot and Akt pathway inhibition experiments were performed to reveal the role of the PI3K/Akt/BRCA1-Abraxas pathway in Sei-1-induced DMs. Luciferase reporter assay was employed to explore the regulatory mechanisms between Sei-1 and BRCA1. DM formation was associated with a deficiency in DSB repair. Based on this finding, activation of the PI3K/Akt/BRCA1-Abraxas pathway was found to increase the DM population with passage in vivo, and inhibition resulted in a reduction of DMs. Apart from this, it was shown for the first time that Sei-1 could directly regulate the expression of BRCA1. Our results suggest that the PI3K/Akt/BRCA1-Abraxas pathway is responsible for the formation of DMs induced by Sei-1.
Introduction
Sei-1 (also known as TRIP-Br1 and SERTAD1) is a member of the TRIP-Br family, which acts as a nuclear factor that regulates the cell cycle by interacting with cyclin-dependent kinase 4 (CDK4) and E2F/DP-11. As an identified oncogene, Sei-1 is located in a frequent amplification region, 19q13.1, in ovarian cancer2,3 and is associated with mechanisms that lead to chromosomal instability, such as increasing the number of chromosomes, and double minute chromosomes (DMs) and micronuclei formation4,5. In addition, Sei-1 also inhibits apoptosis by stabilizing the X-linked inhibitor of apoptosis protein6 and by promoting metastasis through AKT or ILK modulation in HER2/neu-suppressed or expressing cancer cells7. However, few reports about the mechanisms that govern the formation of DMs and Sei-1 have been published so far, and as a result, further exploration in this field is needed.
DMs are paired, circular, acentric, and self-replicating DNA and are the cytogenetic hallmark of extrachromosomal gene amplification8,9. DMs can be detected in many tumors, such as acute myeloid leukemia10, ovarian carcinoma11,12, glioblastoma multiforme13,14, and colonic carcinoma15,16. Moreover, genes located on DMs can play crucial roles in the development of cancer, such as DHFR amplification in methotrexate resistance17 and MYC promotion of proliferation and invasion18. Therefore, the study of the DM formation is important for exploring the mechanisms underlying the development of tumors.
DNA double-strand breaks (DSBs) are a form of DNA damage that can lead to genomic instability or apoptosis if not appropriately repaired19,20. When a DSB occurs, it can initiate two main repair mechanisms, homologous recombination (HR) and classical non-homologous end joining. In addition, alternative end joining and single-strand annealing have been found to take part in DSB repair under certain conditions20,21. Breast cancer type 1 susceptibility protein (BRCA1) is recruited to the sites of DSBs with the help of the BRCA1-Abraxas (BRCA1-A) complex to effectively repair DNA. The BRCA1-A complex is composed of BRCA1 and Abraxas (also named FAM175A, CCDC98, and Abra1) as well as other components, including Rap80, BRCC36, BRE, and NBA122,23. Abraxas possesses the domains necessary to interact with the BRCT domains of BRCA1 and other components, which reveals the role of central adaptor protein24. The BRCA1-A complex plays important roles in maintaining genomic integrity during the DNA damage response.
Previous studies have shown that Sei-1 not only transforms the NIH-3T3 passage in vivo model to possess characteristics of cancerous formations but it also increases the incidence of genomic instability3,4. Moreover, DMs can appear during the passages in vivo in this model, which has aroused interest in exploring how these DMs are produced during this process. Although we previously reported the role of the met pathway in the promotion of the formation of DMs induced by Sei-1 in NIH-3T3 murine fibroblasts5, deep-seated and unknown mechanisms are also involved. Therefore, we selected cells from different passages in vivo with high and low DM populations for RNA-sequencing (RNA-seq) to explain the process governing the formation of DMs.
Results
Sei-1 promotes DM formation with increasing passage number in vivo
To investigate the relationship between Sei-1 and the formation of DMs, we established the Sei-1-transformed NIH-3T3 model as previously described5. Consistent with previous results, the Sei-1-transfected group appeared to generate xenografts while the vector group failed. The primary cell culture from the original generation in vivo was named CPX1, and the sixth generation in vivo passage was named CPX6. The mean number of DMs for Vec, the control group transfected with null-vector-NIH-3T3 cells, and CPX1 and CPX6 was measured as ~0, 10, and 50, respectively (Fig. 1a, b). Next, quantitative real-time PCR (qPCR) was used to quantify Sei-1 mRNA expression and showed that relative mRNA expression increased with the increasing number of DMs (Fig. 1c). In addition, the Sei-1 protein expression also increased (Fig. 1d, e). These results indicate that Sei-1 acts as a promoter of DM formation in vivo.
Fig. 1. Sei-1 overexpression promotes the formation of DMs during in vivo passages.

. a Metaphase spread images of Vec, CPX1, and CPX6 samples. Red arrows represent the DMs. b Quantification of DMs in Vec, CPX1, and CPX6 samples; n = 100 from three independent experiments for each group. c Quantitative real-time PCR results of Sei-1 relative mRNA expression, n = 3. d Western blot results show increases in Sei-1 expression from the Vec, CPX1, and CPX6 samples during in vivo passages. e Quantification of the protein levels in d, n = 3. *P < 0.05; **P < 0.01; ***P < 0.001
DM generation is related to the occurrence of DSBs
The gene amplification process is associated with DSBs. Furthermore, DMs, which serve as one form of gene amplification, might be strongly related to DSBs. To explore this concept, γH2AX3,25,26 was selected as a marker to verify the existence of DSBs in this model. Immunofluorescence (IF) showed that the number of γH2AX foci increased significantly as the population of DMs increased (Figs. 1a, b and 2a, b). Western blot analysis also indicated that γH2AX protein levels correlated with the IF results (Fig. 2c, d). These results suggest that DM formation is inseparable from DSBs.
Fig. 2. DSB occurrence is related to DM formation.

. a IF shows increases of γH2AX foci in the Vec, CPX1, and CPX6 samples. Yellow: γH2AX; blue: DAPI; magnification ×1000. b The percentage of γH2AX foci in A, n = 30. c Western blot shows increases of γH2AX. d Quantification of the protein levels in c, n = 3. *P < 0.05
BRCA1 was selected as a pivotal gene from the RNA-seq data
The DMs were counted in all of the primary cells, including CPX1, CPX4, CPX5, and CPX6, from each in vivo passage. These data suggest that the DM population increased gradually as the number of in vivo passages increased (Fig. 3a). CPX1 (C1), which had the lowest count, and CPX6 (C2), which had the highest count, were chosen for the RNA-seq experiments. In order to screen the differentially expressed genes (DEGs), the expression of all genes (P < 0.05) were analyzed and shown in supplementary figure 1A, and the detail information of RNA-seq data was supplied in the SRA database (SRP130842). Then, the enrichment analysis based on the DEGs showed that the DEGs were significantly associated with changes of DNA conformation, DNA metabolism, and chromosome-related regulation in Gene Ontology terms and PI3K/Akt signaling pathway was significantly enriched based on Kyoto Encyclopedia of Genes and Genomes database (Supplementary Figure 1B). In our previous study, we found that met could activate the PI3K/Akt signaling pathway and was exactly enriched in the abovementioned pathway in RNA-seq data (data not shown)5. All of these suggested that DEGs associated with changes of DNA structures and PI3K/Akt pathway could play essential roles in the process of DM formation. Therefore, many DEGs that were involved in DSB repair (Table 1) were filtered according to the analysis of RNA-seq data and the detection of DSBs. Almost all genes were downregulated except for ATM, while BRCA1, Rad51, and Chek1 reached significance (P < 0.05). The qPCR was used to verify the accuracy of the RNA-seq data. All genes listed in the table were downregulated and only BRCA1, Rad51, and Chek1 reached the same degree of significance as the original data (Fig. 3b). Hence, BRCA1, Rad51, and Chek1 protein expression were subsequently measured. BRCA1 was the only protein to show results that were consistent with the RNA-seq data, including the expression tendency of protein and statistical significance (Figs. 3c, d). These results indicate that there are defects in the DSB-associated repair pathway and BRCA1 is a key gene in this process. As a result, BRCA1 was the focus of subsequent experiments.
Fig. 3. BRCA1 is a key DEG.

. a The increase in the number of DMs in CPX1, CPX4, CPX5, and CPX6 for each group. n = 100 from three independent studies. b Quantitative real-time PCR results of relative mRNA expression of the genes in Table 1, n = 3. c Western blot results show decreases of Rad51, BRCA1, and Chk1 levels. d Quantification of the protein levels in c, n = 3. *P < 0.05; **P < 0.01; ***P < 0.001
Table 1.
DSB-associated gene expression from RNA-seq data
| Gene | Read count | p value | q value | Log2.Fold_change | |
|---|---|---|---|---|---|
| C1 | C2 | ||||
| BRCA1 | 56.15029303 | 27.91804213 | 0.00076613 | 0.010053 | −1.0081 |
| Chek1 | 24.362124 | 9.603806492 | 0.0062685 | 0.057512 | −1.343 |
| Rad51 | 57.55918695 | 22.03664125 | 1.75E−05 | 0.00037981 | −1.3851 |
| ATM | 32.46326403 | 46.08338154 | 0.19742 | 0.67448 | 0.50544 |
| ATR | 37.86402405 | 28.14138646 | 0.1525 | 0.5796 | −0.42813 |
| BRCA2 | 17.75793376 | 10.90664846 | 0.15062 | 0.57492 | −0.70326 |
| Chek2 | 11.47661504 | 8.375412638 | 0.40965 | 0.96019 | −0.45447 |
Note: C1, CPX1; C2, CPX6; q value, the lower value, the more difference between C1 and C2; log 2.Fold_change, C2/C1; A significant difference is defined as P < 0.05
DSB double-strand break, RNA-seq RNA-sequencing
The PI3K/Akt/BRCA1-A complex pathway is activated when the DM population increases
BRCA1 is involved in many cellular processes including cell cycle checkpoints27,28 and DNA damage repair28,29. Furthermore, BRCA1 deficiency in breast epithelial cells could disable HR mechanisms for DNA damage, resulting in genomic instability and increased breast cancer risk. BRCA1-deficient mouse embryonic stem cells showed disabled DSB repair via HR29. Therefore, the role of BRCA1 in maintaining genomic integrity occupies an important position for DSBs. Previous studies demonstrated that BRCA1 is negatively regulated by the Akt signaling pathway30,31. Based on this finding and the enrichment analysis of RNA-seq data, we questioned whether the generation of DMs in this model was related to the activation of the Akt signaling pathway and loss of BRCA1 function. Thus, we measured the proteins involved in the Akt pathway and BRCA1 with immunoblot. We found a prominent increase in PI3K, phospho-Akt (Ser473), and phospho-Akt (Thr308), while there was no change in total Akt, and a significant reduction in BRCA1 (Fig. 4). BRCA1 has been shown to possess BRCT domains that interact with the pSPxF motif of Abraxas (A), Bach1/FancJ (B), and CtIP (C) to form three kinds of complexes in different phospho-dependent manners23,24. The BRCA1-A complex plays essential roles in maintaining genomic stability. Deficiencies in either BRCA132,33 or Abraxas22 increased tumor incidence in mouse models. A decreased protein level of Abraxas was detected (Fig. 4), which is consistent with changes in BRCA1 expression. PTEN acts as a negative regulator for the PI3K/Akt pathway34, which suggests that it may regulate the PI3K/Akt pathway in the process of the formation of DMs. No changes in PTEN expression were observed (Fig. 4). Since Sei-1 is also a transcription factor, it would be interesting to explore its relationship with the pivotal BRCA1. Luciferase reporter assay showed that the luciferase activity was significantly upregulated in Sei-1-co-transfected and pGL3-BRCA1 promoter-co-transfected group in 293T cells (Supplementary figure 2). These findings suggest that the PI3K/Akt/BRCA1-A complex signaling pathway is activated as the DM population increases and PTEN has no influence on DM formation (Figs. 1a, b and 4). This also indicates that Sei-1 can also directly regulate the expression of BRCA1.
Fig. 4. The PI3K/Akt/BRCA1-A complex pathway is activated as the population of DM increases.

. a Western blot results show increases in the PI3K/Akt pathway without the influence of PTEN as well as the reduction of BRCA1-A complex from Vec, CPX1, and CPX6 samples as DMs increase. b Quantification of the protein levels in a, n = 3. *P < 0.05; **P < 0.01; ***P < 0.001
DM population is reduced through the inhibition of the PI3K/Akt/BRCA1-A complex pathway
To explore the main function of the PI3K/Akt/BRCA1-A complex pathway in the production of DMs in Sei-1-transformed NIH-3T3 cells, MK2206 (an allosteric Akt inhibitor)35,36 was employed to inhibit the Akt signaling pathway. Western blot analysis showed decreasing levels of phospho-Akt (Ser473) and phospho-Akt (Thr308) in addition to significant decreases in total Akt. BRCA1 and Abraxas showed dramatic increases in expression after 10 µM drug treatment for 48 h (Fig. 5a, b). PI3K was not detected because MK2206 only inhibits a downstream molecule of the PI3K/Akt pathway. Furthermore, metaphase spreads were prepared to quantify the number of DMs at 0 and 48 h after 10 µM drug. CPX6 was chosen for counting DMs due to insufficient numbers of CPX1 samples. This revealed that the mean number of DMs decreased from ~50 to 20 after undergoing drug treatment (Fig. 5c, d). Collectively, these results demonstrate that the PI3K/Akt/BRCA1-A complex signaling pathway plays a vital role in the Sei-1-induced DM formation.
Fig. 5. Inhibition of the PI3K/Akt/BRCA1-A complex pathway can result in the loss of DMs.

. a, b Western blot results indicate effective inhibition of the Akt pathway with the MK2206 inhibitor at 10 µM and increases of the downstream BRCA1-A complex in CPX1 or CPX6. Quantification of the protein levels, n = 3. c Metaphase spread images of CPX6 after inhibition of the Akt pathway. Red arrows indicate DMs. d Quantification of DMs in C, n = 100 from three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001
Discussion
RNA-seq, which is a deep-sequencing technology, is used to map and quantify transcriptomes and has revealed new transcriptomic insights into gene and exon boundaries, transcript complexity, novel transcription mechanisms, and DEG identification37,38. In the present study, we aimed to sift through the DEGs related to DM formation, and in doing so identified BRCA1 as an essential gene. This was done using RNA-seq, which differs from the previous study we performed5. In addition, we found that most genes associated with the DSB repair pathway were downregulated, which indicated that a defect existed in repairing DSBs. Gene amplification increases when the factors involved in the occurrence of DSBs, such as IR, DNA synthesis inhibitors, and restriction endonucleases, are added. Defects in either HR-relevant genes such as ATM39, Rad51D40, and Rad5441 or in NHEJ interrelated DNA-PKcs42 can enhance the predisposition of gene amplification mechanisms that include homogeneous staining regions and DMs. Consequently, these phenomena hint that Sei-1 may act as a factor that promotes DSBs and that there is a relationship between the production of DSBs and DMs in the Sei-1-transformed NIH-3T3 model.
The PI3K/Akt pathway participates in many aspects of tumor biology, including cell proliferation, migration, invasion, metastasis, and survival. Activation of Akt, with the loss of PTEN, can impair Chk1 through ubiquitination, phosphorylation, and reduction of nuclear localization to mediate genomic instability43, but its relationship with the generation of DMs is still unknown. We found that the activation of PI3K/Akt promoted the appearance of DMs in a PTEN-independent manner. Likewise, not only can loss of BRCA1 lead to aneuploidy44 but it also contributes to activating the PI3K/Akt signaling pathway[30]. The BRCA1-A complex is important for genomic stability. However, the detailed mechanisms underlying DMs formation for BRCA1 or the BRCA1-A complex deficiency are not completely known. Our results uniquely support the claim that activating the PI3K/Akt pathway induced loss of function of the BRCA1-A complex to cause the production of DMs under the action of Sei-1 in NIH-3T3 cells, and Sei-1 could also target the promoter and regulate the expression of BRCA1 (Fig. 6). Therefore, the regulation of BRCA1 was realized in the abovementioned manner. Loss of function of BRCA1 or Abraxas can lead to tumorigenesis22,33. Therefore, tumor formation induced by Sei-1 is likely related to the low expression of BRCA1 and Abraxas. On the other hand, genomic instability is also responsible for promoting oncogenesis, which may induce tumor xenografts in Sei-1-transformed NIH-3T3 models.
Fig. 6. Schematic overview of the PI3K/Akt/BRCA1-A complex pathway in the Sei-1-transformed NIH-3T3 cell model.
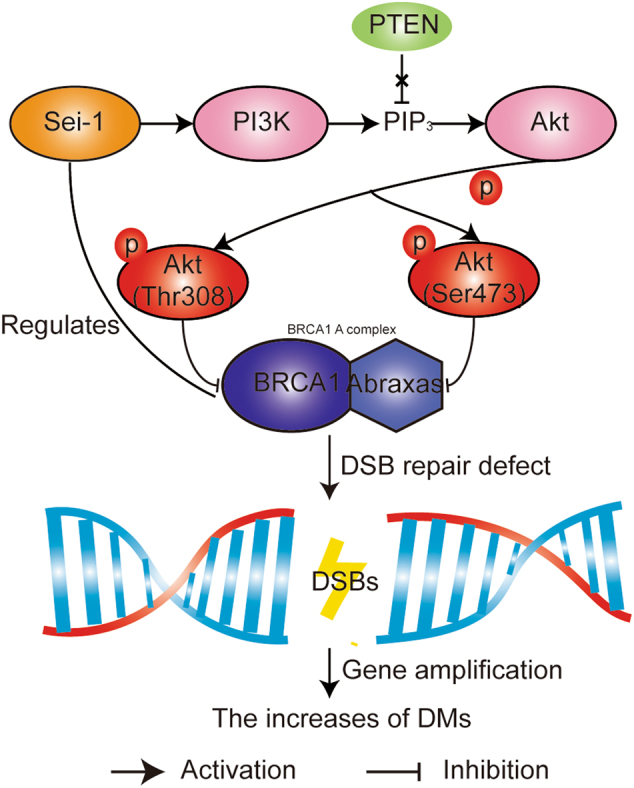
. Sei-1 activates the PI3K/Akt pathway independent of PTEN and inhibits the expression of the BRCA1-A complex to result in DSBs, leading to the formation of DMs in vivo. Sei-1 can also target the promoter and regulate the expression of BRCA1
Sei-1, a newly discovered oncogene, is connected to chromosome instability, particularly the formation of DMs, which was the focus of a previous study4. The DM population increases with increased passages in vivo but decreases in in vitro cultures due to micronuclei export5. Tumors likely adapt to the selective pressure created by the microenvironment through genes carried on DMs. Met, Tspan12, and Fam3c, which are located on DMs, facilitate cancer malignancies, and are correlated with poor prognosis45–47. Therefore, the Sei-1-transformed NIH-3T3 model mimics therapeutic approaches that reduce the population of DMs to treat DM-containing tumors. Here, the inhibition of the PI3K/Akt/BRCA1-A complex pathway may serve as an effective measurement to assess therapeutic treatments in Sei-1-associated and DM-containing tumors.
In this study, significantly increased expression of Sei-1 was found using qPCR and immunoblot analysis, which showed that Sei-1 acts as a promoter in accelerating the production of DMs in vivo. In summary, our results revealed a DSB repair defect and that the PI3K/Akt/BRCA1-A complex pathway plays a key role in the formation of DMs. It is necessary to do further studies on the regulatory mechanisms between Sei-1 and BRCA1. Moreover, DM production needs to be explored further by establishing Sei-1-relevant models to extend our understanding of DM evolution under the pressure of microenvironments.
Materials and methods
Cell line
NIH-3T3, a mouse embryonic fibroblast cell line, and 293T cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and authenticated by means of the STR (short tandem repeat) test (Microread, Beijing, China). The detailed procedures of cell culture, cell passage, and transfection were previously reported5.
Antibodies and reagents
The following antibodies were used for the experiments: anti-BRCA1 and PTEN antibodies (Santa Cruz Biotechnology, Dallas, TX, USA); anti-Rad51, Chk1, and Abraxas antibodies (Abcam, Cambridge, UK); anti-PI3K, pan-Akt, phospho-Akt (Ser473), phospho-Akt (Thr308), and anti-γH2AX antibodies (Cell Signaling Technology, Danvers, MA, USA); IRDye 800DX-conjugated affinity-purified anti-mouse IgG and IRDye 700DX-conjugated affinity-purified anti-rabbit IgG (Rockland Immunochemicals, Gilbertsville, PA, USA); and anti-mouse IgG/HRP and anti-rabbit IgG/HRP secondary antibodies (ZSGB-bio, Beijing, China). The MK2206 2HCl inhibitor (Selleckchem, Houston, TX, USA) was also used.
Metaphase spread preparation
Cells in metaphase reached 70–80% confluence and were treated with Colchicine (Sigma-Aldrich, St. Louis, MO, USA) for 1.5 h. The cells were trypsinized and then washed with phosphate-buffered saline (PBS) and centrifuged. The cell precipitate was re-suspended with 0.075 M KCl and incubated in a 37 °C water bath for 13 min. After undergoing fixation three times, the cell suspension was dropped on an ice-cold clean slide. The drops spread evenly and were stained with Gimsa (YuanMu, Shanghai, China). The pictures were then photographed using an Olympus BX41 microscope (Melville, NY, USA) equipped with a JVC TKC75U color video camera (JVC, Yokohama, Japan).
Semi-quantitative and quantitative real-time PCR
Cell precipitates were treated with Trizol (Invitrogen, Carlsbad, CA, USA) to extract the RNA. cDNA samples were obtained using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany). Semi-quantitative and quantitative real-time qPCR were performed as previously reported5,48.
Western blot
Cells were lysed with RIPA buffer and centrifuged at 12,000 × g at 4 °C for 30 min to extract the total protein. The protein samples were separated on a sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and transferred to a PVDF membrane. After blocking with 5% blocking buffer, the membranes were incubated with the appropriate antibodies and then scanned using the Odyssey Imaging system (Li-COR, Lincoln, NE, USA) or the Fluorchem R system (FluorChem R, Santa Clara, CA, USA). ImageJ software was used for band analysis.
Immunofluorescence
The cells covered 80–90% of the glass coverslips. The cells were washed three times with PBS and fixed in 4% paraformaldehyde (Boster, Wuhan, China). Then, the cells were permeabilized with 0.1% Triton X-100 and blocked in 4% bovine serum albumin. Images were obtained using a DM-RXA2 fluorescence microscope (Leica) after incubation with γH2AX antibody (Merck Millipore, Darmstadt, Germany), a fluorescence-conjugated secondary antibody, and DAPI (4′,6-diamidino-2-phenylindole) stain (Helixgen, Guangzhou, China).
RNA-sequencing
Primary cells were cultured from mouse xenografts as previously described5. The cells were sent to the Novogene Company (Beijing, China) for RNA-seq and analysis. We then independently screened, analyzed the data by R (Windows 64 bits, 3.4.2), and validated the DEGs.
Luciferase reporter assay
The construction of Sei-1 vectors (EX-A3731-Lv103) and pGL3-BRCA1-promoter luciferase reporter vectors were completed by GeneCopoeia company (Guangzhou, China) and Aizhe Biological Technology Co., Ltd (Guangzhou, China), respectively. The 293T cells were plated in 24-well plates at 50,000/well. The co-transfection of Sei-1 and luciferase reporter vectors were accomplished according to the instruction of HilyMax reagents (Dojindo, Shanghai, China). The detection of luciferase activity was based on the instruction of Luc-Pair Duo-Luciferase Assay Kits 2.0 (GeneCopoeia, Rockville, MD, USA) and completed by microplate reader (Molecular Devices, Shanghai, China).
Statistics
Two-tailed Student’s t test and one-way analysis of variance with the Student–Newman–Keuls test were employed for data analysis of DM counts, relative mRNA expression, luciferase reporter assay, and western blots. All data are presented as the mean ± S.D or mean ± S.E.M. Significant differences were defined as P < 0.05.
Electronic supplementary material
Figure legends of supplementary materials
Acknowledgements
This study was supported by Programme of The National Natural Science Foundation of China (Grant No. 81672572, to C.Z.; Grant No. 81272868, to C.Z.); The New Century Support Programme for the Excellent Scholar, Ministry of Education of China (Grant No. NCET-13-0758, to C.Z.); The Open Funds of State Key Laboratory of Oncology in South China (Grant No. HN2017-05, to C.Z.); and The International Science and Technology Cooperation Programme of China (Grant No. 2013DFA31610, to S.F.).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by R. Aqeilan
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41419-018-0362-y.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
R Aqeilan.
References
- 1.Watanabe-Fukunaga R, Iida S, Shimizu Y, Nagata S, Fukunaga R. SEI family of nuclear factors regulates p53-dependent transcriptional activation. Genes Cells. 2005;10:851–860. doi: 10.1111/j.1365-2443.2005.00881.x. [DOI] [PubMed] [Google Scholar]
- 2.Tang TC, et al. Identification of a candidate oncogene SEI-1 within a minimal amplified region at 19q13.1 in ovarian cancer cell lines. Cancer Res. 2002;62:7157–7161. [PubMed] [Google Scholar]
- 3.You J, et al. SEI1 induces genomic instability by inhibiting DNA damage response in ovarian cancer. Cancer Lett. 2017;385:271–279. doi: 10.1016/j.canlet.2016.09.032. [DOI] [PubMed] [Google Scholar]
- 4.Tang DJ, et al. Oncogenic transformation by SEI-1 is associated with chromosomal instability. Cancer Res. 2005;65:6504–6508. doi: 10.1158/0008-5472.CAN-05-0351. [DOI] [PubMed] [Google Scholar]
- 5.Bao Y, et al. Met promotes the formation of double minute chromosomes induced by Sei-1 in NIH-3T3 murine fibroblasts. Oncotarget. 2016;7:56664–56675. doi: 10.18632/oncotarget.10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hong SW, et al. p34SEI-1 inhibits apoptosis through the stabilization of the X-linked inhibitor of apoptosis protein: p34SEI-1 as a novel target for anti-breast cancer strategies. Cancer Res. 2009;69:741–746. doi: 10.1158/0008-5472.CAN-08-1189. [DOI] [PubMed] [Google Scholar]
- 7.Jung S, et al. Distinct regulatory effect of the p34SEI-1 oncoprotein on cancer metastasis in HER2/neu-positive and -negative cells. Int J. Oncol. 2014;45:189–196. doi: 10.3892/ijo.2014.2403. [DOI] [PubMed] [Google Scholar]
- 8.Shimizu N, Shingaki K, Kaneko-Sasaguri Y, Hashizume T, Kanda T. When, where and how the bridge breaks: anaphase bridge breakage plays a crucial role in gene amplification and HSR generation. Exp. Cell Res. 2005;302:233–243. doi: 10.1016/j.yexcr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Sun W, et al. Constitutive ERK1/2 activation contributes to production of double minute chromosomes in tumour cells. J. Pathol. 2015;235:14–24. doi: 10.1002/path.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto K, et al. Micronuclei-associated MYC amplification in the form of double minute chromosomes in acute myeloid leukemia. Am. J. Hematol. 2013;88:717–718. doi: 10.1002/ajh.23431. [DOI] [PubMed] [Google Scholar]
- 11.Guan XY, et al. Isolation of a novel candidate oncogene within a frequently amplified region at 3q26 in ovarian cancer. Cancer Res. 2001;61:3806–3809. [PubMed] [Google Scholar]
- 12.Olinici. CD. Double minute chromatin bodies in a case of ovarian ascitic carcinoma. Br. J. Cancer. 1971;25:350–353. doi: 10.1038/bjc.1971.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanborn JZ, et al. Double minute chromosomes in glioblastoma multiforme are revealed by precise reconstruction of oncogenic amplicons. Cancer Res. 2013;73:6036–6045. doi: 10.1158/0008-5472.CAN-13-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canute GW, et al. The hydroxyurea-induced loss of double-minute chromosomes containing amplified epidermal growth factor receptor genes reduces the tumorigenicity and growth of human glioblastoma multiforme. Neurosurgery. 1998;42:609–616. doi: 10.1097/00006123-199803000-00031. [DOI] [PubMed] [Google Scholar]
- 15.Dolf G, et al. Extrachromosomal amplification of the epidermal growth factor receptor gene in a human colon carcinoma cell line. Gene Chromosome Cancer. 1991;3:48–54. doi: 10.1002/gcc.2870030109. [DOI] [PubMed] [Google Scholar]
- 16.Lin CC, et al. Evolution of karyotypic abnormalities and C-MYC oncogene amplification in human colonic carcinoma cell lines. Chromosoma. 1985;92:11–15. doi: 10.1007/BF00327240. [DOI] [PubMed] [Google Scholar]
- 17.Meng X, et al. Novel role for non-homologous end joining in the formation of double minutes in methotrexate-resistant colon cancer cells. J. Med. Genet. 2015;52:135–144. doi: 10.1136/jmedgenet-2014-102703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji W, et al. Expulsion of micronuclei containing amplified genes contributes to a decrease in double minute chromosomes from malignant tumor cells. Int J. Cancer. 2014;134:1279–1288. doi: 10.1002/ijc.28467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 2016;37-38:51–64. doi: 10.1016/j.semcancer.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castillo A, et al. The BRCA1-interacting protein Abraxas is required for genomic stability and tumor suppression. Cell Rep. 2014;8:807–817. doi: 10.1016/j.celrep.2014.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu Q, et al. Structure of BRCA1-BRCT/Abraxas complex reveals phosphorylation-dependent BRCT dimerization at DNA damage sites. Mol. Cell. 2016;61:434–448. doi: 10.1016/j.molcel.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang. B. BRCA1 tumor suppressor network: focusing on its tail. Cell Biosci. 2012;2:6. doi: 10.1186/2045-3701-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paull TT, et al. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000;10:886–895. doi: 10.1016/S0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 26.Limoli CL, Giedzinski E, Bonner WM, Cleaver JE. UV-induced replication arrest in the xeroderma pigmentosum variant leads to DNA double-strand breaks, gamma-H2AX formation, and Mre11 relocalization. Proc. Natl. Acad. Sci. USA. 2002;99:233–238. doi: 10.1073/pnas.231611798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat. Genet. 2002;30:285–289. doi: 10.1038/ng837. [DOI] [PubMed] [Google Scholar]
- 28.Rosen EM. BRCA1 in the DNA damage response and at telomeres. Front Genet. 2013;4:85. doi: 10.3389/fgene.2013.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol. Cell. 1999;4:511–518. doi: 10.1016/S1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 30.Xiang T, et al. Negative Regulation of AKT Activation by BRCA1. Cancer Res. 2008;68:10040–10044. doi: 10.1158/0008-5472.CAN-08-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guirouilh-Barbat JK, Wilhelm T, Lopez BS. AKT1/BRCA1 in the control of homologous recombination and genetic stability: the missing link between hereditary and sporadic breast cancers. Oncotarget. 2010;1:691–699. doi: 10.18632/oncotarget.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thompson ME, Jensen RA, Obermiller PS, Page DL, Holt JT. Decreased expression of BRCA1 accelerates growth and is often present during sporadic breast cancer progression. Nat. Genet. 1995;9:444–450. doi: 10.1038/ng0495-444. [DOI] [PubMed] [Google Scholar]
- 33.McCoy ML, Mueller CR, Roskelley CD. The role of the breast cancer susceptibility gene 1 (BRCA1) in sporadic epithelial ovarian cancer. Reprod. Biol. Endocrinol. 2003;1:72. doi: 10.1186/1477-7827-1-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirai H, et al. MK2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010;9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, et al. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell. 2015;160:161–176. doi: 10.1016/j.cell.2014.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mondello C, et al. Gene amplification in fibroblasts from ataxia telangiectasia (AT) patients and in X-ray hypersensitive AT-like Chinese hamster mutants. Carcinogenesis. 2001;22:141–145. doi: 10.1093/carcin/22.1.141. [DOI] [PubMed] [Google Scholar]
- 40.Hinz JM, Yamada NA, Salazar EP, Tebbs RS, Thompson LH. Influence of double-strand-break repair pathways on radiosensitivity throughout the cell cycle in CHO cells. DNA Rep. 2005;4:782–792. doi: 10.1016/j.dnarep.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 41.Ruiz-Herrera A, et al. Gene amplification in human cells knocked down for RAD54. Genome Integr. 2011;2:5. doi: 10.1186/2041-9414-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salzano A, et al. Enhanced gene amplification in human cells knocked down for DNA-PKcs. DNA Repair. 2009;8:19–28. doi: 10.1016/j.dnarep.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 43.Puc J, et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell. 2005;7:193–204. doi: 10.1016/j.ccr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 44.Schlegel BP, Starita LM, Parvin JD. Overexpression of a protein fragment of RNA helicase A causes inhibition of endogenous BRCA1 function and defects in ploidy and cytokinesis in mammary epithelial cells. Oncogene. 2003;22:983–991. doi: 10.1038/sj.onc.1206195. [DOI] [PubMed] [Google Scholar]
- 45.Weidner KM, Sachs M, Birchmeier W. The Met receptor tyrosine kinase transduces motility, proliferation, and morphogenic signals of scatter factor/hepatocyte growth factor in epithelial cells. J. Cell Biol. 1993;121:145–154. doi: 10.1083/jcb.121.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knoblich K, et al. Tetraspanin TSPAN12 regulates tumor growth and metastasis and inhibits beta-catenin degradation. Cell Mol. Life Sci. 2014;71:1305–1314. doi: 10.1007/s00018-013-1444-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu YH, et al. Prognostic significance of FAM3C in esophageal squamous cell carcinoma. Diagn. Pathol. 2015;10:192. doi: 10.1186/s13000-015-0424-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu J, et al. De novo-generated small palindromes are characteristic of amplicon boundary junction of double minutes. Int J. Cancer. 2013;133:797–806. doi: 10.1002/ijc.28084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure legends of supplementary materials