

Abstract
Current tuberculosis (TB) drug development efforts are not sufficient to end the global TB epidemic. Recent efforts have focused on the development of whole-cell screening assays because biochemical, target-based inhibitor screens during the last two decades have not delivered new TB drugs. Mycobacterium tuberculosis (Mtb), the causative agent of TB, encounters diverse microenvironments and can be found in a variety of metabolic states in the human host. Due to the complexity and heterogeneity of Mtb infection, no single model can fully recapitulate the in vivo conditions in which Mtb is found in TB patients, and there is no single “standard” screening condition to generate hit compounds for TB drug development. However, current screening assays have become more sophisticated as researchers attempt to mirror the complexity of TB disease in the laboratory. In this review, we describe efforts using surrogates and engineered strains of Mtb to focus screens on specific targets. We explain model culture systems ranging from carbon starvation to hypoxia, and combinations thereof, designed to represent the microenvironment which Mtb encounters in the human body. We outline ongoing efforts to model Mtb infection in the lung granuloma. We assess these different models, their ability to generate hit compounds, and needs for further TB drug development, to provide direction for future TB drug discovery.
1. Introduction
1.1. Why We Need more TB Therapies
Tuberculosis (TB) is the number one cause of human death from infectious disease in the world. It is estimated that one-third of the world’s population is infected with Mycobacterium tuberculosis (Mtb), the pathogen that causes TB disease.1 Only 10% will develop active disease in their lifetime.2 Although, it is currently not possible to completely accurately predict who will develop the disease, diabetes mellitus has been identified as a significant comorbidity. The risk of developing active TB disease rises to 30% in diabetics.3
Typically, 80–90% of patients with drug-sensitive TB are cured after receiving 6–8 months of intensive antibiotic treatment. However, the side effects from the cocktails of antibiotic therapies used have led to adherence issues and high levels of drug-resistant Mtb strains in the infected population. Treatment of drug-resistant or multi drug-resistant TB (MDR-TB: resistant to isoniazid and rifampin with or without resistance to other first-line TB drugs) is more complex and takes up to two years of combination chemotherapy.4
The prevalence of MDR-TB is sufficiently high that 3.9%5 of first time TB patients are infected with drug-resistant Mtb strains. Thus, more effective treatments to eliminate this reservoir of future disease are required. Treatment of the high levels of drug-resistant Mtb infection, which include rifampin-resistant (Rif-TB), MDR-TB, and extensively resistant TB (XDR-TB: resistant to multiple first-line and second line TB drugs) requires new drugs with new mechanisms of action to limit crossover resistance. Development of new therapies is further complicated because TB drugs are given in combination to prevent development of resistance. The combinations may be antagonistic or have incompatible metabolic profiles that limit their use. The discovery of anti-TB drugs with lower toxicities and shorter treatment times will help to reduce further growth of drug-resistant TB in the population.
1.2. Postgenomic Era Approaches to Developing New TB Therapeutics
With the availability of the complete TB genome in 1998,6 drug development efforts focused on designing and selecting inhibitors of important enzymatic targets that were largely selected based on gene essentiality. These protein targets were purified and crystallized and binding sites identified. Then, inhibitors were designed to block enzyme activity. However, these efforts have not yielded new drugs to date.
Target-based enzymatic assays test in vitro inhibition of isolated target proteins and are generally binding affinity driven based on the assumption that high binding affinities will provide high specificities for the targets. However, these screening assays neglect essential factors such as cell wall permeability, metabolic stability, and drug target vulnerability. These properties can be difficult to design into a compound structure while maintaining inhibition potency.
After a decade’s worth of attempts at biochemical screening against enzymatic Mtb targets, both the pharmaceutical industry and academic laboratories have shifted their focus to whole cell-based phenotypic screens for TB drug discovery. The impetus for this approach arises partially from the knowledge that all current TB drugs were discovered in whole-cell screens for inhibition of Mtb growth or growth of an Mtb surrogate.
However, whole cell-based phenotypic screens using either Mtb or Mtb surrogates in in vitro liquid media conditions have raised questions about their physiological relevance. Targets inhibited in liquid culture may not be essential in vivo. Thus, compounds and targets may be selected that are only active under in vitro conditions.7
Therefore, different culture models fashioned on our current understanding of in vivo mechanisms of Mtb infection and survival have been developed. The hope is that mimicking the environment in which Mtb resides as a pathogen will lead to improved drug discovery. The optimal drugs will combat the existing resistance problems, shorten the current chemotherapy, and improve efficacy of sterilization.
Here, we summarize the major types of whole cell-based phenotypic screens for TB drug discovery that have been reported in the period January 2007 to September 2017. Prior reviews have summarized earlier work.8−12 Our focus is on the systems used in compound screening to model TB disease. We do not review the diversity of libraries or types of chemical entities used in this effort, although we do call out compound classes that have been identified in the literature.
First, we provide background on the in vivo physiology of Mtb infection. Second, we summarize the types of Mtb antibiotics currently available and their mechanisms of action. Third, we provide a broad overview of recent and current drug discovery approaches for finding new chemotherapeutic leads for treating Mtb infection. Last, we provide an analysis of future directions based on the apparent pros and cons of current methods.
2. Physiology of Mtb Infection in the Lung
The process of lung infection by Mtb has been extensively described in earlier reviews.13−15 Our understanding is based primarily on infection of the mouse, although the mouse model of infection is imperfect, as Mtb is supremely adapted to persist in its human host and the human immune system is not identical to that of the mouse.16−18 Some additional analyses have been performed in nonhuman primates,19 as well as other model organisms.20
In brief, an aerosol droplet containing Mtb is expelled through the cough of a person with an active TB infection, and the droplet is inhaled by the next person in the chain of infection. The bacilli then travel through the respiratory system into the lung. Upon reaching the alveoli, Mtb is phagocytosed by macrophages, as well as monocytes and dendritic cells.4 The infected macrophage invades the epithelial layer to reach the lung interstitium and the site of infection is established.21 An aerosol droplet containing one Mtb bacillus is sufficient for transmitting Mtb infection from one person to another.15
At this stage of infection, Mtb bacilli still replicate exponentially in macrophages until the infected macrophage is recognized by T-cells.22 T-cells then activate the macrophage by the secretion of interferon-γ (INF-γ) and tumor necrosis factor-α (TNF-α) in an attempt to bring bacillary replication under control.23 The infection of the macrophage induces the innate inflammatory response and the infected alveolar macrophages signal to recruit mononuclear cells from the surrounding blood vessels to form the initial building blocks of the granuloma.15 A localized chemokine gradient is created which results in waves of immune cell recruitment to the site of infection which is remodeled into a granuloma (Figure 1).
Figure 1.
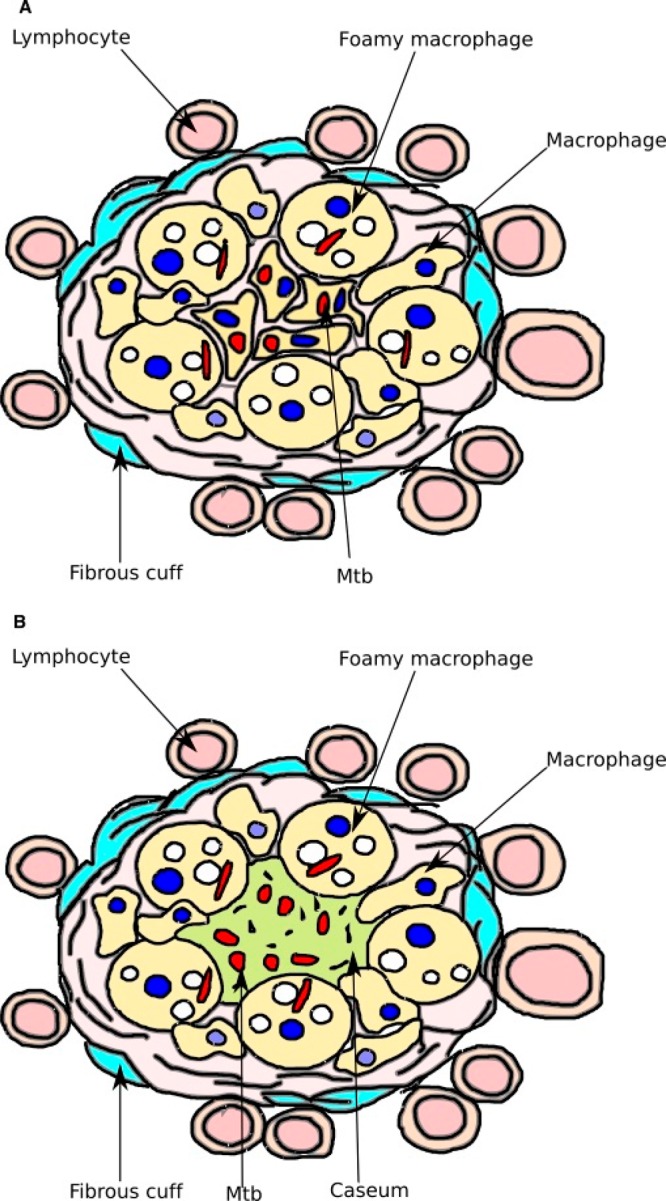
Diagrams of characteristic granulomas. (A) Cellular granuloma. Macrophages infected with Mtb (red) are at the center. Lipid bodies (white) are within foamy macrophages. (B) Necrotic granuloma. Macrophages have died and released Mtb into the necrotic center, which is hypoxic and filled with lipid caseum.
Granuloma formation marks the hallmark of Mtb infection. In the beginning state of granuloma formation, the tubercle is highly vascularized and the immune cells are actively recruited to the infection site due to the chemokines produced by infected macrophages. Cell recruitment is accompanied by the differentiation of macrophages into foamy macrophages, multinucleated giant cells, and epithelioid cells, as well as other mononuclear phagocytes.21,22 Once the immune response is triggered, lymphocytes and dendritic cells are recruited to the infection sites.
As Mtb multiplies and inflammation develops, the structure of the granuloma matures. With the formation of a fibrous cuff, the blood vessels within the structure start to diminish. The loss of vascularization leads to necrosis and a hypoxic environment, with the accumulation of caseum in the granuloma core.13,15,24 In this solid structure, the number of bacilli remains constant and at least some of the mycobacteria enter a nonreplicating persistent state.13,22 Many of the mycobacteria are extracellular and reside in the caseum (Figure 1). The killing of Mtb by the macrophage is limited at this stage due to the inhibition of superoxide and nitric oxide production in the hypoxic environment.25 In addition, during this persistent stage, Mtb arrests phagolysosome biogenesis to avoid the antimycobacterial intracellular environment. A single individual TB patient has granulomas in different states; their development is not synchronized.26 Eventually, the granuloma structure collapses and aerosolized infectious bacilli are released into the air through coughing.27
Therefore, when developing new TB drugs, the large array of microenvironments in which Mtb survives in the host should be considered. Traditionally, prospective compounds are first screened against actively replicating Mtb in culture which may not be the best model of Mtb environment in vivo. The complex microenvironments encountered by Mtb in vivo have been modeled in various ways as described in sections 7 and 8.
3. Current and Future TB Drug Regimens
The current drug regimen for drug sensitive TB treatment was established in the 1980s (Table 1).28 The regimen comprises four drugs, isoniazid (INH), rifampicin (RIF), ethambutol (EMB), and pyrazinamide (PZA; Figure 2), for 6 months of treatment. The primary targets of TB drugs are cell wall biosynthesis, DNA replication, RNA transcription, or protein synthesis. Newer TB drugs and TB drugs in development also target energy metabolism.
Table 1. Current TB Drugs Approved by the FDA/EMA.
| primary protein target | mechanism of action | pro-drug | |
|---|---|---|---|
| First-Line TB Drugs, Primarily Used for Drug-Sensitive TB Treatment | |||
| isoniazid INH, H | enoyl acyl-carrier protein reductase InhA | inhibits mycolic acid synthesis | activated by catalase-peroxidase-peroxynitritase (KatG) to form an adduct with NAD+/NADP+ |
| rifampicin RIF, R | RNA polymerase β subunit RpoB | inhibits RNA synthesis/transcription | N/Aa |
| pyrazinamide PZA, Z | pleiotropic | possible targets include FAS I, QAPRTase, RpsA, PanD, Rv2783 | converted by pyrazinamidase (PZase) to pyrazinoic acid |
| ethambutol EMB, E | arabinosyl transferase EmbB | inhibits arabinogalactan synthesis | N/A |
| Second-Line TB Drugs, Primarily Used for Drug-Resistant TB Treatment | |||
| streptomycin | 16S rRNA subunit | inhibits protein synthesis | N/A |
| kanamycin amikacin capreomycin | 30S rRNA subunit | inhibit protein synthesis | N/A |
| ofloxacin levofloxacin moxifloxacin | DNA gyrase and topoisomerase | inhibit DNA synthesis | N/A |
| p-amino salicylic acid | dihydrofolate reductase | inhibits DNA precursor synthesis | converted by dihydropteroate synthase (DHPS) and dihydrofolate synthase (DHFS) to an antifolate metabolite |
| ethionamide prothionamide | enoyl acyl-carrier protein reductase InhA | inhibit mycolic acid biosynthesis | activated by a monooxygenase (EthA) to form an adduct with NAD+/NADP+ |
| cycloserine | alanine racemase and d-alanine:d-alanine ligase | inhibits cell wall biosynthesis | N/A |
| bedaquiline | ATP synthase subunit ε | inhibits ATP production | N/A |
| delamanid pretomanid | not specific | generates NO and inhibits energy metabolism | activated by a nitroreductase (Ddn) |
N/A: not applicable.
Figure 2.

Chemical structures of front-line TB drugs.
Treatment of drug-resistant or multidrug resistant TB is much more complex. The treatment depends on the patient history and drug sensitivity screening, as well as the country-specific protocols that depend on drug pricing and availability. Multidrug resistant TB requires treatment for two years with a cocktail of at least five drugs (Figure 3).5 These second-line drugs (Table 1) tend to be more expensive and include bedaquiline, ethionamide, cycloserine, moxifloxacin, and streptomycin, as well as front-line drugs pyrazinamide and ethambutol. All of the multidrug resistant TB treatment regimens include daily injectables for up to 6 months, and some have toxicity issues including cardiotoxicity and ototoxicity.
Figure 3.

Chemical structures of second-line TB drugs.
Early bactericidal activity (EBA) is monitored during treatment of patients. This assay measures whether replicating, extracellular bacteria that can be accessed in sputum have been killed. In some cases, the bactericidal activity of a drug wanes after the first 14 days of treatment, suggesting that the Mtb microenvironment in the lung affects Mtb susceptibility to that drug. Ultimately, the sterilizing activity of a TB drug is assessed by whether a cure is maintained after completion of treatment. Thus, one reason for current multidrug regimens is to ensure sterilization of different Mtb populations.
3.1. Drug-Sensitive TB Treatment
The primary target of INH is enyol-ACP-reductase, InhA. INH activity requires bioactivation by a Mtb catalase, KatG, to form an isonicotinic acyl radical, which subsequently couples with NAD+/NADP+ to form an adduct.29,30 For drug-sensitive TB, INH has the highest early bactericidal activity (EBA). In the first 2 days of treatment, INH is able to kill >95% of bacilli found in the sputum, which are extracellular bacteria. However, after the first few days of treatment, with eradication of the actively replicating subpopulation, INH efficacy starts to diminish, and an INH-refractory population remains. This profile is consistent with a mechanism of action that targets inhibition of cell wall biosynthesis.31 The implication is that INH has limited efficacy for killing and sterilization of nonreplicating Mtb in the lung.
RIF is an inhibitor of RNA polymerase.32 Thus, its antimycobacterial activity is manifested through blocking of protein synthesis. In contrast to INH, RIF shows a relatively modest EBA.31,33 However, the bactericidal activity remains constant over the time course of pulmonary TB treatment. This profile results in excellent sterilization and implies that RIF also kills nonreplicating subpopulations of Mtb in the lung.
The mechanism of action of PZA is not completely defined.34−41 PZA is a prodrug which requires bioactivation to its pyrazinoic acid (POA) form. This hydrolytic bioactivation can be catalyzed by Mtb pyrazinamidase, PncA,42 or by host metabolism.43 There are multiple cellular targets of POA including fatty acid biosynthesis,37 NAD+ biosynthesis,38 RNA trans translation,35 membrane transport function,34 RNA and DNA metabolism,41 and CoA biosynthesis.39,40 PZA shows no EBA in the first 2 days of treatment, and its activity requires acidic conditions.31 However, it is still considered a sterilizing drug that shortens TB treatment.34
EMB inhibits the biosynthesis of arabinogalactan, a component of the bacterial cell wall.44 EMB is bacteriostatic and is not active against nonreplicating bacilli in a Wayne hypoxia culture model.45 EMB has a modestly high EBA which falls in between the profiles of INH and RIF. However, it has also been reported that EMB has poor or no sterilizing activity.31
All four drugs are typically used for the first two months of treatment, followed by four months of INH and RIF treatment. The extended duration of this regimen is to ensure relapse-free treatment. On the other hand, the extended treatment times required highlight the limitations of the current drugs, even in combination, to act as effective bactericidal agents and to sterilize Mtb infection completely.
3.2. Drug-Resistant TB Treatment
Ethionamide (and a related analog prothionamide) are derivatives of isonicotinic acid. The mechanism of action of ethionamide is similar to the mechanism of action of INH. Ethionamide is first activated by a Mtb monooxygenase EthA/EtaA46 that results in formation of an NAD+ adduct. The NAD+ adduct inhibits InhA, a enoyl-ACP reductase in the mycolic acid biosynthesis pathway. The expression level of ethA in vivo is highly correlated to the activation of ethionamide, and therefore drug potency. ethA expression is regulated by the repressor EthR. The efficacy of ethionamide can be boosted by inhibiting EthR binding to DNA, which increases EthA production levels.47,48 Thus, the potential for synergistic codrug action is high.
Cycloserine inhibits alanine racemase and d-alanine:d-alanine ligase in peptidoglycan biosynthesis.49 Cycloserine is bacteriostatic at the dose used in Mtb treatment.50 Moreover, cycloserine is highly toxic, especially to the central nervous system where it acts as an NMDA receptor agonist and induces psychosis.
Moxifloxacin is a fluoroquinolone antibiotic. Fluoroquinolones inhibit DNA gyrase and have broad spectrum antimicrobial activity. Moxifloxacin is the most effective fluoroquinolone against Mtb infection. It is bactericidal and its EBA profile is close to that of INH.51
Streptomycin is the first drug to be used for TB treatment. It inhibits protein synthesis by targeting the 16S rRNA subunit. Streptomycin shows low, dose-dependent early bactericidal activity.52 Streptomycin is bactericidal against replicating Mtb.53 However, it is less active against latent and intracellular bacilli.54 This drug is administered via intramuscular injection.
Clofazimine was originally developed for the treatment of leprosy, and it has been repurposed and evaluated for the treatment of MDR-TB and XDR-TB. The mechanism of clofazimine action is not completely elucidated. It appears to act as a pro-drug that is activated by NADH dehydrogenase (NADH-2) and competes with menaquinone for reduction by NADH dehydrogenase, and thereby releases reactive oxygen species upon nonenzymatic oxidation by O2.55,56 Clofazimine is bactericidal against replicating Mtb and nonreplicating bacilli in a Wayne hypoxia model.45
Linezolid is one of the oxazolidione antibiotics which was originally used for the treatment of Gram-positive bacterial infections and has been repurposed as an antimycobacterial agent for TB treatment. Linezolid targets 23S rRNA and inhibits protein synthesis.57 Linezolid shows bacteriostatic activity against Mtb including MDR-TB and XDR-TB strains in vitro.57
Bedaquiline is the first anti-TB drug approved by the FDA in nearly 50 years. It was identified and developed in the early 2000s and approved in 2013 for MDR-TB treatment. Bedaquiline targets the ε subunit of ATP synthase,58,59 thereby depleting the cellular ATP pool. It has also been shown that bedaquiline affects the proton gradient across the Mtb cell membrane acting as an uncoupler of the proton motive force.60 Bedaquiline is bactericidal against Mtb under both replicating condition and nonreplicating hypoxic conditions.61
Pretomanid and delamanid are nitroimidazole compounds. Nitroimidazoles are known for their activity against anaerobic microorganisms. Pretomanid was identified through a whole-cell screen of metronidazole derivatives and displays high bactericidal activity against both replicating and nonreplicating Mtb.62 The mechanisms of action of these two drugs are not fully understood. Both pretomanid and delamanid are activated by a nitroreductase and the corresponding des-nitroimidazole can generate reactive nitrogen species that kill bacilli.63,64 Transcription profiling revealed that pretomanid inhibits cell wall synthesis and interferes with cell respiration.65 Delamanid inhibits mycolic acid biosynthesis and exhibits potent activity against Mtb including MDR Mtb, and it also kills intracellular Mtb.66 In a 14-day assessment of EBA, delamanid shows a similar EBA profile to RIF,67 and delamanid has also been shown to enhance the treatment of MDR-TB.68 Delamanid was approved by the EMA in 2014.
3.3. Requirements for New TB Drugs
To address the unmet need for improved TB treatments, new drug candidates should be effective in sterilizing the diverse subpopulations of Mtb infection in the human. The candidate must have a novel mechanism of action to ensure activity against MDR- and XDR-TB disease. A favorable manageable metabolism profile and good oral availability are needed to reduce the potential for variability between patient populations. The pharmacokinetic and pharmacodynamic (PK/PD) profiles should be compatible with HIV treatment and diabetes mellitus management. Finally, any new treatment must achieve a stable and relapse-free cure.28,69
3.3.1. Pharmacokinetic and Pharmacodynamic Considerations for TB Drugs
Measurement of MIC and MBC, minimal inhibitory concentration and minimal bactericidal concentration, respectively, are important in evaluating the potential of a new compound to be effective in vivo. However, these measurements are made under constant drug concentration and evaluate growth or death after an extended incubation time in culture.
Pharmacokinetic (PK) and pharmacodynamic (PD) properties of a drug play an important role in determining in vivo efficacy. Furthermore, PK/PD properties such as compatibility with other TB drugs or medications for comorbid diseases, safety profile, oral bioavailability, and metabolic stability, determine the success of a new TB drug.
Oral administration is the route of choice for TB drug development. Therefore, oral bioavailability is crucial for a new TB drug. Aqueous solubility and gastrointestinal permeability are the two main factors affecting oral bioavailability. The general bioavailability of current TB drugs ranges from 40 to 90%,70 and new drugs must be similarly bioavailable.
To improve patient adherence, less frequent dosing is recommended. Once per day dosing is preferred. To dose less frequently, a relatively long in vivo half-life of a drug is needed. The half-lives of current TB drugs vary a lot between individuals, depending on their metabolism and comorbidities, and range from 1 to 14 h.70,71
An ideal TB drug should distribute to the lung, the primary infection site, and should be able to penetrate the granuloma to reach intracellular and extracellular bacilli in the hypoxic and possibly necrotic center. Ideally, the drug concentration in target tissue should be maintained at a concentration above the MIC between each dose. How this concentration is achieved depends on whether the drug binds to plasma protein, drug tissue distribution (volume of distribution), and plasma half-life of the drug. Drug lipophilicity is a major contributor to these properties. Decisions about dosing depend on PK/PD parameters and mechanism of action. The reader is referred to a TB-focused review on the topic for more details.71
In terms of drug safety, an ideal TB drug should have no acute or long-term toxicity. Because of the combination nature of TB therapy, an ideal drug should not show drug–drug interaction either chemically or biologically with other TB drugs in the regimen and other drugs for comorbid diseases.
3.3.2. HIV-Coinfection and TB Treatment
HIV-Mtb coinfection is the leading cause of death among HIV positive patients. In the treatment of HIV-Mtb coinfection, the coadministration of antiretroviral drug and anti-TB drugs is problematic due to drug–drug interactions.28 RIF induces the expression of CYP3A4 and p-glycoprotein, liver enzymes responsible for first pass metabolism of HIV-protease inhibitors.72,73 For HIV patients treated with an HIV-protease inhibitor-based regimen, RIF can increase the metabolism of these protease inhibitors and thus reduce the drug concentration below its effective range.74
3.3.3. Diabetes Mellitus and TB Treatment
Growing evidence has shown that there is an increasing rate of diabetes mellitus and TB co-occurrence.75,76 Diabetes puts TB infected individuals at higher risk of TB disease due to alteration of their immune system. In addition, TB infection might induce glucose intolerance and cause problems in glycemic control for diabetes patients.
Moreover, there are rising concerns about drug–drug interactions in the comanagement of diabetes and TB. Numerous studies have shown that with the induction of cytochrome P450 liver enzymes CYP2C8, CYP2C9, and CYP3A4 by RIF, antiobesity drugs such as glyburide, glipizide, rosiglitazone, nategkinide, and repaglinide undergo more rapid metabolism than dosing regimens anticipate. Thus, there is a high risk of causing hyperglycemia with frontline TB treatment because drug plasma concentrations of diabetes drugs drop rapidly and blood sugar levels rise uncontrollably.77−80
4. Target Identification
With the whole genome sequence of Mtb available,6 the capability to search through the entire Mtb genome for novel targets for antibiotic development became available.9 Novel targets and new chemical entities are expected to circumvent existing drug resistance and therefore enhance current treatments. An ideal target for antibiotic development should be essential in vivo, drug vulnerable, and druggable.
Genetic essentiality screens are a typical first step to establish which gene products might be targeted for TB chemotherapy. However, not all essential genes are equally vulnerable to drug action. Moreover, the target must be accessible to chemical inhibition. That is, the target should have the ability to bind a small molecule other than its substrate. Inhibition or activation of the protein’s function with an achievable concentration of the small molecule must lead to cell dysfunction, e.g., attenuated growth or cellular killing. In addition to being amenable to chemical inhibition, an inhibitor screen against the target should also yield drug-like compounds with specificity for affecting the function of the target in the absence of interference with any host orthologues.
4.1. Target Essentiality
Genetic and chemical approaches can be taken to validate the essentiality of a potential drug target.81−83 A gene knockout results in the complete absence of a specific cellular target product. Analysis of gene knockout phenotypes can give critical information about the potential targets’ essentiality.
Evaluation of gene knockout strain phenotypes can be made under various conditions both in vitro and in vivo to assess the gene’s essentiality during different stages of infection.84 Removal of a target that is essential for replication leads to attenuated growth. If a target is essential for survival, the depletion leads to cell death. A target can be essential for Mtb’s growth or survival under different environmental conditions and different replication states, and it is important to test knockout strain phenotypes that are relevant to infection.
One might expect that chemical inhibition of a genetically essential gene would lead to the same phenotype as seen in gene knockout strain. However, it turns out not to be that simple. Target vulnerability also plays an important role.
4.2. Target Vulnerability
The degree of inhibition required to affect cell viability and to be bactericidal determines the target’s vulnerability to being drugged. For an essential gene with high vulnerability, the drug need not occupy 100% of the protein target in the cell. An essential gene with low vulnerability is very difficult to drug. The high target engagement required to effect physiologic change may be very difficult to achieve. Therefore, methods to evaluate how much of the target protein must be deactivated in the cell have been developed to more accurately assess target vulnerability.
4.2.1. Gene Knockdown
Gene knockdown by reducing the expression level of the target protein can serve as a good tool to evaluate target vulnerability.85 Gene knockdown is an approach in which the expression of the target gene is modulated to different levels for assessment of target vulnerability. Knockdown can be achieved by engineering expression of antisense RNA to modulate mRNA levels or through a regulatable promoter that modulates transcription. In the latter system, the native promoter is replaced with a regulated promoter and the target gene is only transcribed in the presence or absence of the inducer.83 The transcription level can be titrated with inducer/repressor. A tetracycline-controlled promoter is most commonly used. Due to the bioavailability of anhydrotetracycline and other analogs, the regulated system can be used in animal models to study gene vulnerability and essentiality during infection.85
4.2.2. Controlled Protein Degradation
Other approaches such as inducible protein degradation utilizing the Clp protease system have been applied to determine target vulnerability.86 The Clp protease identifies the SsrA peptide sequence that is fused to the C-terminus of a protein and subsequently proteolyzes the entire protein. A synthetic C-terminal peptide was engineered that consists of the SsrA sequence fused with a protective peptide sequence that is recognized by HIV protease. Conditional induction of HIV protease expression leads to cleavage of the synthetic sequence, which in turn exposes the SsrA peptide on the C-terminus of the target protein. Exposure enables recognition of the targeted protein by Clp protease and the target’s subsequent degradation. Using this approach, the target vulnerability and sensitivity to depletion of existing antibiotic targets has been studied.86
Similarly, by utilizing a delivery protein from E. coli, SspB, and Clp protease, a dual-control gene switch was constructed in which a tetracycline inducer can simultaneously trigger transcriptional repression of the target gene and consequent degradation of the encoded protein.87 This approach has been utilized to identify and evaluate gene vulnerability in different models of metabolic state that mimic the microenvironmental niches encountered by Mtb in vivo. These models are described in further detail in section 8.
4.2.3. Cell Washout
Target vulnerability can be assessed using cell washout experiments for drugs with long residence times on their protein target.88 Drugs that have slow off-rates or which are covalent inhibitors will remain bound to the target for an extended period after drug has been removed from the system. In the case of a target with low vulnerability, washout of drug after treatment of cells will result in reversal of the drug’s effect. This may arise due to degradation of the drug-target complex in the cell or rapid synthesis of new target after removal of the drug. In the case of a highly vulnerable target, drug effects will be prolonged after washout of the drug. In the case of antibiotics, this is known as the postantibiotic effect.
4.2.4. Additional Considerations
Whether a target is vulnerable is also influenced by Mtb’s environmental conditions and replication state. Therefore, it is crucial to assess target vulnerability under different in vitro and in vivo conditions to evaluate the prospects for favorable chemical inhibition under varying in vivo conditions. Moreover, it has been reported that the expression level of some targets is much higher than needed for normal cell growth. As a result, if even 97% of the target is depleted or inhibited, little effect on cell growth is observed.86 High concentrations of drug are required to have efficacy with these types of targets.
5. Biochemical Assays to Identify Specific Hits against Protein Targets
Biochemical assays to drive antibiotic discovery against protein targets have been well utilized by big pharmaceutical companies and academic laboratories during the last two decades. Much preparation is needed before establishing a biochemical target-based enzymatic high throughput screen. Usually the process begins with evaluation of target essentiality. A critical metabolic pathway is identified. Targets within that pathway are evaluated. Gene knockouts should result in loss of cell viability. In addition, it is desirable that the gene is unique to Mtb, with no close human homologues.
To have sufficient target protein for biochemical screening, the protein needs to be expressed in a bacterial host. Identification and synthesis of the enzyme substrate is required for the assay. This requires molecular knowledge of the role the encoded protein plays in the entire pathway, including its relationship with other proteins encoded by adjacent genes. A robust activity assay needs to be established that provides sufficient signal, usually optical, in a screening assay. After finding a hit in a screen, an X-ray crystal structure of the target protein and protein-hit complex are desired to guide rational optimization of the hit into a lead compound.
Designing a drug that targets a single protein in an exclusive, highly specific manner has been a goal of chemotherapeutic developers since Ehrlich first introduced the idea of a magic bullet.89 With the extensive advances in biophysical methods over the last few decades, one might assume that rational design of inhibitors using protein-compound cocrystal structures and structure–activity relationship studies in combination with biochemical target-based enzymatic screening would have yielded inhibitors with tight binding of and high specificity of their targets. Disappointingly, after more than a decade’s efforts, target based enzymatic screens have not yielded a successful anti-TB drug candidate.90,91 Translating biochemical enzyme inhibitors into whole-cell active antimicrobials turned out to be far more difficult than anticipated. The reasons for the failure are multiple.
5.1. Binding Affinity Is Insufficient for Drug Activity
The biochemical assays are driven by binding affinity for a single target. Failure of inhibitors to permeate the bacterial cell is the primary problem of compounds selected in enzymatic screens. Many libraries of compounds originally screened were designed for use against mammalian cells.91 Considering the unique mycobacterial cell wall that includes mycolic acid, the cell wall is extremely difficult to penetrate.92 Although care can be taken to monitor biophysical properties of the selected compounds,93,94 these properties are more difficult to engineer after a hit is identified.
5.2. Cellular Efflux Can Eliminate Excellent Inhibitors from the Cell
Enzymatic screens neglect cellular efflux issues. Even if compounds can pass through the cell membrane, it is likely that efflux pumps can pump them out of the cytosol, thereby reducing the cellular concentration of the compound to which the target is exposed. Efflux pump activity is a common source of intrinsic, i.e, endogenous, resistance of bacteria to new compounds.95−97 Hence, the balance between the rate of compound uptake (via cell permeation or bacterial transporter) and efflux out of the cell is critical for establishing sufficient accumulation of the compound in the cytosol to achieve whole-cell antibacterial activity.
5.3. Host or Bacterial Metabolism Can Rapidly Inactivate Excellent Inhibitors
Compounds selected through enzymatic or biochemical screens can be inactivated in vivo by Mtb or host metabolism.9 Biochemical assays do not select against compounds that are easily inactivated by either type of metabolism. Based on experience and drug development precedent, a medicinal chemist can often recognize and avoid host metabolic liabilities. Predicting microbial metabolism of new chemical entities is more challenging.
5.4. Unfavorable Binding Kinetics Can Reduce the Efficacy of Excellent Inhibitors
Typically utilized biochemistry assays measure the binding affinity (IC50 as a surrogate for Ki) at constant compound concentration. Thermodynamic equilibrium parameters do not fully reflect the target occupancy which is achieved in the human body. The human circulatory system represents a dynamic environment where drug concentration is time-dependent due to metabolism, distribution, and execretion.98−100 If the compound’s rate of dissociation from the target is comparatively slow, activity can be retained despite clearance of the drug from the circulatory system. Ignoring drug residence time on the target is postulated to be another primary shortcoming of reliance on biochemical screens.101
5.5. Pro-Drugs Are not Readily Identified in Biochemical Screens
Biochemical target-based enzymatic screens lack a system for the identification of pro-drugs. As outlined in section 3, many current TB drugs are pro-drugs which become activated once inside the mycobacterium. Specific activating enzymes not currently used by TB drugs, and thus less likely to encounter resistance, have not been systematically identified. Thus, testing in a whole-cell context is currently a more expeditious avenue to identify pro-drugs that may undergo transformation to an active drug once inside the mycobacterium.
6. Fragment Screens: An Intermediate Step to Whole-Cell Screens
Because of the limitations outlined in section 5, TB drug discovery efforts began to revisit empirical or target-based whole-cell screens. Fragment-based approaches have been applied to both enzymatic and whole-cell screens. For the enzymatic target-based approach, inhibitors of a selected protein target are developed with the help of computational simulation, X-ray crystallography, and biophysical approaches. A primary fragment can be built upon using fragment growing and fragment linking computational approaches. The resulting compound is evaluated for its target-binding affinity. Co-crystallization with the protein can provide further guidance for compound development. Although the inhibitor design process is logical and rationally based on structural biology information, the target protein is totally isolated from its physiologically relevant environment.102−105 Thus, the limitations of enzymatic screening still apply.
One of the strategies employed to overcome the shortcomings of in vitro enzymatic screens is screening fragments against whole bacterial cells. A fragment-based whole-cell screen usually starts with screening assays to find weak binding affinity small molecules. If the target of the weak binding molecule can be identified, further structural biology based modification may be undertaken. The modification of the small molecule should enable filling of the binding pocket and enhancement of the interaction between the small molecule and the residues in the binding pocket. In the case of two small molecules binding with the same target in different sites, these two fragments could be linked through a chemical linker to gain binding affinity.106,107 The advantage of small fragments is that they have better PK properties. They are small molecules (<300 Da), and they have better water solubility, which leads to better tissue distribution. In addition, they are moderately lipophilic, which allows good cell penetration. The combination can overcome the major downside of enzymatic screening, if care is taken to maintain these properties during modification of the initial fragment.
Fragments are likely to be taken inside the bacilli and acted upon by native enzymes inside the cell. In this fashion, the fragment is a pro-drug that can potentially hit multiple targets.106 The pro-drug mechanism of action is complex, but this strategy has been proven to be effective in the drug discovery of antimicrobials. Old anti-TB drugs, like INH and PZA, are dirty fragments: they are small molecules, they hit multiple targets, and they are metabolized inside bacilli to become biologically active.
Finally, fragments selected from screening can also be tested in animal models directly in hopes of finding the next PZA, and medicinal chemistry may be applied later to optimize the PK profile. In this case, one hopes for in vivo conversion of a fragment into a target-binding compound.
7. Target-Based Whole-Cell Screening
Fragment-based whole-cell screening was designed to solve compound permeability problems of enzymatic screens and potentially to select compounds acting on multiple targets. In contrast, target-based whole-cell screening is designed to identify compounds that can penetrate the cell envelope and specifically inhibit the target protein within the cellular environment. Target-based whole-cell screening utilizes conditional mutant strains in which the cellular concentration of a target gene is adjusted to bias compound selection for molecules that bind the desired target. Oftentimes, but not always, Mtb surrogates are used to eliminate biosafety concerns.
7.1. Mtb Surrogates for Whole-Cell Screening
For any type of screening for anti-TB drugs based on millions of compounds, it is risky and highly regulated to handle large volumes of Mtb cultures. Infectious cultures, robotics, and microplates must be handled at biosafety level 3. Considering the slow growth rate, and more importantly, how readily Mtb cultures are aerosolized, several microbes have been explored as surrogates for anti-TB drug screening.108,109
Mycobacterium smegmatis (M. smeg) and Mycobacterium bovis BCG (M. bovis BCG) are the most common surrogates for Mtb. Using M. smeg or M. bovis BCG as a surrogate has been widely explored in both academic and industrial laboratories. M. smeg and M. bovis BCG are genomically related to Mtb. In fact, M. bovis BCG has a genome that is >99.9% identical to Mtb and M. smeg shares 70% genomic identity with Mtb.110,111 Unlike Mtb, both microbes have low pathogenicity. M. smeg has a shorter generation time, so only 1–2 days are required for inhibitor screening. Likewise, when using ATP content as a readout for M. bovis BCG whole-cell screens, the incubation time can also be as short as 48 h.7 Due to the higher genomic similarity between Mtb and M. bovis BCG than between Mtb and M. smeg, M. bovis BCG has a higher sensitivity for anti-TB drug screening and is a better Mtb surrogate.
There are multiple studies comparing the use of M. bovis BCG and M. smeg as surrogates for screening for Mtb growth inhibitors. In one study, a parallel screen with the three microbes was conducted. Of the approximately 2000 compounds screened, ∼50% of the compounds active against Mtb were not active against M. smeg, while ∼20% were not active against M. bovis BCG.112 The percentage of undetected compounds is related to the assay cutoffs selected. Moderate variations in the percentage of growth inhibition at the compound concentration used could result in detection of activity in Mtb but not in a surrogate strain.
This difference is illustrated by GlaxoSmithKline’s (GSK) antimycobacterial screening campaign. GSK found that 55% of the hits that are active against M. bovis BCG are active in Mtb at a compound concentration of 10 μM. However, if the compound concentration is raised to 25 μM, the percentage of hits that translate from M. bovis BCG to Mtb increases to 86%.113
Conversely, Stanley et al. found that among the Mtb hits that they had obtained from a whole-cell screen, only 20% were active in M. smeg.114 The overlap with M. bovis BCG was slightly better, but it was still low.114 Therefore, the correlation of cross-species hits depends on the entities in the compound collection as well as the concentration of compound selected for screening.
One may conclude that M. smeg is never as good a surrogate as M. bovis BCG. However, we remind the reader that bedaquiline, the first FDA-approved anti-TB drug in 50 years, was identified through screening a compound library against M. smeg.(59)
The possibility of using M. smeg as a surrogate for MDR Mtb clinical isolates has also been explored. M. smeg has low susceptibility to RIF and INH, two frontline anti-TB drugs.115 Therefore, compounds which target InhA or RNA polymerase should be eliminated if M. smeg is screened in place of MDR Mtb. Among 50 compounds that are active against drug-sensitive Mtb with an MIC < 12.5 μg/mL, 27 were found to be active against MDR Mtb. Of these 50, 21 were active against M. smeg, and all 21 actives were from the MDR Mtb-active subset of 27 compounds.116 Thus, the specificity of M. smeg-based screening for MDR Mtb inhibitor screening is 100% and the sensitivity is 78%. This small test set suggests that M. smeg maybe a good choice as a surrogate of MDR Mtb for drug screening to eliminate the risks from using the difficult to treat infectious MDR Mtb. However, the assay was tested based on compounds that were already known to be active against drug-sensitive Mtb. If M. smeg was used in a primary assay for inhibitor screens, the specificity and sensitivity might be significantly lower.
Another less explored approach is to use a Mtb auxotroph that can be grown at biosafety level 2. One example, Mtb mc26206 which is an auxotroph in pantotheine biosynthesis (ΔpanCD, ΔleuCD) and is nonvirulent117 is described in further detail in section 8.2.3.
7.2. Approaches to Target-Based Whole-Cell Screening
Compounds may be selected against specific targets inside the cell using a variety of methods. Some methods lower the amount of protein target in the cell to sensitize the cell to any compound that targets the protein of interest, generally through inducible transcriptional control. Other methods rely on cellular enzyme activity reporter systems. Additionally, screens targeting specific promoters or regulons have been used. Because the method selected is very target dependent, we describe several case studies below to exemplify the types of approaches that can be taken.
7.2.1. Reduction of Target Level
Under-expression or overexpression mutant strains are expected to sensitize or desensitize Mtb’s response to the inhibitor that is acting on the specific protein expressed. Most commonly, a tetracycline-sensitive promoter is utilized to turn transcription on or off,118,119 depending on the system selected. Alternatively, an antisense interference system may be used.120
Abrahams et al. chose three genes as their targets—panC, lysA, and icl1—to explore the feasibility of target-based whole-cell screening on Mtb.119 These three targets were chosen for their known roles in CoA biosynthesis, lysine biosynthesis, and energy generation through lipid metabolism, respectively. These targets are considered essential for Mtb survival.82,121−124 Experiments with PanC were carried through to high-throughput screening (HTS) as proof of concept.
Conditional gene expression systems based on the tetracycline (Tet)-regulatable promoter were generated: Tet-ONs for overexpression and Tet-OFFs for under-expression. Known PanC inhibitors were tested on a panC Tet-OFF strain. Hypersensitivity to the known inhibitors was observed due to the silencing of target genes and correlated with reduced levels of target in the cell.
To make the mutant strain directly employable in high-throughput screening, they constructed conditional mutants that also express gfp to quantify bacterial growth by fluorescence readout. The addition of GFP did not affect the growth rate of Tet-OFF mutants, and the fluorescence correlated well with bacterial growth. The fluorescence-based panC Tet-OFFGFP was exploited for a HTS in a 384-well microplate format.
A set of compounds was tested against WT and panC Tet-OFFGFP. Compounds which showed greater potency against panC Tet-OFFGFP than WT were identified as primary hits. Primary hits were counter screened by testing against panC Tet-OFFGFP in the presence and absence of anhydrotetracycline. Only hits that displayed increased potency in the group with the presence of anhydrotetracycline were selected as PanC specific inhibitors. Although inhibition by these compounds could be rescued by addition of pantothenate, none of them showed activity in enzymatic assays with isolated PanC enzyme. Thus, the inhibitors appear to target the pantothenate pathway, without inhibiting the PanC enzyme.
However, use of inducible promoters should not be limited to the tetracycline system. The tetracycline-inducible system can be unstable in a high-throughput format. During culturing to accumulate sufficient biomass, strains can accumulate mutations that lead to the loss of promoter induciblility.125 In the case of LepB, it was necessary to test several native promoters to find a suitable system.125
LepB catalyzes the cleavage of the N-terminal signal peptide from preproteins. LepB is a key enzyme in the general protein secretion pathway of Mtb and its depletion affects Mtb cell survival and growth. Bonnett et al.125 generated eight strains in which expression of their target gene lepB is under the control of different native Mtb promoters to find a strain suitable for high-throughput screening. The most suitable strain for high-throughput screening was chosen by optimizing maximum depletion of lepB expression and minimum influence on growth rate as compared to WT Mtb expression and growth. The high-throughput screening was conducted using a dual read-out approach to avoid off-target hits on signal generation. A pCherry10 expression vector was introduced into different strains in order that bacteria growth could be monitored simultaneously by optical density and fluorescence.126
7.2.2. Counter Screening
Park et al.127 chose Mtb biotin synthesis as their target. BioA is a 5′-pyridoxal phosphate (PLP)-dependent aminotransferase that catalyzes the reductive amination of 7-keto-8-aminopelaragonic acid to 7,8-diaminopelargonic acid.128 BioD, a downstream enzyme in the pathway, catalyzes the carboxylation of 7,8-diaminopelargonic acid to dethiobiotin.129 Biotin biosynthesis has been validated genetically to be essential for Mtb’s replication and persistence in vivo.130 BioA was chosen as the target because its essentiality and vulnerability in biotin biogenesis had been established.130
From biochemical target-based screens, 298 noncytotoxic hits were identified through a fluorescence-based BioA enzymatic assay followed by a counter screen to exclude compounds that also inhibit BioD. Subsequently, biochemical hits were tested against WT Mtb with or without biotin supplementation to identify which compounds had activity that could be bypassed by exogenous biotin. These hits were assumed to be targeting biotin synthesis or biotin dependent proteins.
Hits that passed the biotin supplement secondary screen were further counterscreened in BioA under-expression and overexpression strains. This secondary screen was used to prioritize hits based on on-target whole-cell activity. Those hits that had improved MICs in the hypersensitive under-expression strain and decreased MICs in the hyposensitive overexpression strain were judged to be acting on the BioA target. Several promising hits were cocrystallized with BioA and these structure-based tools provided a basis for efficient and rational optimization in lead compound development to improve target engagement, a process that is more difficult with empirical whole-cell screens.
This work demonstrated a thorough biochemical target-based screening approach followed by pathway whole-cell counter screens for on target hit confirmation and structural characterization of several hit-target complexes to provide a starting point for hit optimization. These development efforts have resulted in a nanomolar inhibitor, compound 36, that remains on target (Figure 4).131
Figure 4.

Hit compounds (A) identified in target-based whole-cell screens and (B) identified in whole-cell screen under growth culture conditions and discussed in the text. Also see Table 2.
7.2.3. Off-Target Hits Can Be Essential
Intracellular pH homeostasis is important for Mtb survival in the phagoendosome of the macrophage. In addition, hypoxia is thought to lead to acidification of Mtb through increased reliance of Mtb on glycolysis and subsequent accumulation of glycolytic products.153 MarP is a transmembrane serine protease that is required for Mtb to maintain a neutral pH intracellularly in the presence of external acidity.162 Darby et al. utilized a target-based screen for disruptors of intrabacterial pH homeostasis that relied on a pH-sensitive fluorescent reporter protein.159 However, none of the compounds identified targeted the essential gene encoding MarP.
Therefore, Zhao et al.153 performed a comprehensive lead generation and characterization campaign against MarP protein. They demonstrated that lead BO43 (Figure 4), a benzoxazinone class of compounds, acted on the desired target, MarP. However, they observed that BO43 retained efficacy in a marP-deficient Mtb strain. Using an alkyne version of B043 and affinity purification through conjugation to an azido-biotin handle they successfully identified a second target protein, HtrA1. BO43 binds to both MarP and HtrA1. HtrA1 is high temperature requirement A1 homologue Mb1255, and its function is not fully understood. However, htrA1 is an essential gene in Mtb that had not been previously implicated in pH-homeostasis through genetic essentiality screens. The presumption is that the gene was not represented in the gene mutant libraries used because it is essential for Mtb growth.
The work on MarP highlights the importance of using complementary approaches for target identification, even when a presumptive target has already been identified for use in a compound screen. Despite using a target-based enzymatic screen, after hit or lead selection, much work must be done to determine whether the lead maintains target fidelity and to establish the mechanism of action of the lead in the cell. BO43 exemplifies that inhibitor action on several targets may be required for efficient antimycobacterial cell killing.
7.2.4. Target Mechanism-Based Screens
A target-based whole-cell screen can also utilize a specific enzymatic function of the target protein to develop a cellular assay. Moreira et al. developed a target mechanism-based whole-cell screen approach to identify inhibitors of caseinolytic protease (ClpP1P2) inside the cell.163 ClpP1P2 is a serine protease found in a wide range of bacteria, and it is mainly responsible for the degradation of partially synthesized and misfolded proteins including regulatory transcription factor WhiB3.164
An engineered M. smeg strain expressing a caseinolytic-protease-specific (SsrA) degradation signal peptide fused to GFP was constructed. The ssrA tagged gfp gene was under the control of a tetracycline-inducible promoter. Under native conditions, ClpP1P2 degraded SsrA-GFP’s fluorescence to a background level. When ClpP1P2 was inhibited, there was an accumulation of SsrA-GFP in the cell which resulted in an increase of fluorescence signal. Thus, inhibition of the catalytic function of ClpP1P2 could be detected by fluorescence in a high-throughput screening format.
As one of many controls to determine if hits were target selective, a second fluorescent reporter strain was constructed using SsrA-fused to mCherry protein under the control of a constitutive p38 promoter. This strain was used to counterscreen and to find false positive hits which might result from interference with the tetracycline-dependent reporter system or green fluorescence. In a high-throughput screen of a set of ∼500 000 compounds, 86 of 89 early hits that showed dose-dependent inhibition in the GFP screen was lost in the mCherry based counterscreen, leaving only three hits. The high attrition rate between these two screening approaches indicates the care that must be taken to ensure reporter systems are not the target of compounds selected. Regardless, this work demonstrates that target mechanism-based whole-cell screens are a viable approach for antimycobacterial drug discovery.
In a second example, Rybniker et al.165 targeted structural and regulatory EXS-1 components using a whole Mtb cell screen in combination with a mammalian cell survival readout. EXS-1 is a major virulence protein type VII secretion system, that is comprised of regulatory and structural proteins as well as ATPases. ESX-1 plays an important role in Mtb cell invasion and replication. The ESX-1 locus is missing in M. bovis BCG, a strain which is sufficiently attenuated to use as a vaccine.166 Correspondingly, ESX-1 is not essential for bacterial growth under liquid culture conditions, and thus it will not be targeted by conventional whole-cell screen formats.
Rybniker et al. developed a whole-cell-based high throughput screen that tests the toxicity of secreted virulence factors against mammalian cells to identify compounds that target ESX-1 function. They compared the cell viability of MRC-5 lung fibroblasts infected with WT Mtb that had been treated with 10 880 different compounds and counterscreened to eliminate compounds with antimycobacterial activities. They found that 90% of the compounds that protected the lung fibroblast cells from death upon Mtb infection do not inhibit Mtb growth in culture. Of the 91 hits, 55 maintained activity when rescreened in the lung fibroblast assay, and they did not affect mycobacterial growth directly.165 This assay has the additional advantage of directly selecting against hits that show mammalian cytotoxicity and directly selecting for hits that are active intracellularly in the primary screen.
7.2.5. Native Promoter Response
Another example of a target-based approach for antimycobacterial development is whole-cell screening for compounds targeting Mtb cell wall synthesis. iniBAC is a promoter that is specifically induced by inhibitors of cell wall synthesis.167 By fusing a luciferase with the promoter, the induction level of iniBAC can be monitored, and as a result, compounds whose mechanism of action is related to cell wall synthesis can be selected. This approach has led to the discovery of SQ109 and thiophenes as Mtb cell wall synthesis inhibitors (Figure 4).132,137,168
In the respiratory pathway of Mtb, the cydAB operon encodes the two subunits of cytochrome bd oxidase and is upregulated upon Mtb encountering respiratory stress and low oxygen tension.169,170 An engineered M. bovis BCG strain was created by fusing an mWasabi reporter to the putative promoter (PcydAB) for the Mtb cydAB operon. With this reporter strain, inhibitors potentially targeting respiration and the electron transport chain are detected by monitoring the fluorescence signal from the reporter.138 DG70 was identified and selected through a screen of 168 compounds with known anti-Mtb activity due to its prominent induction of the PcydAB promoter (Figure 4). Further work on target identification revealed that DG70 inhibits MenG, a demethylmenaquinone methyltransferase, in the menaquinone biosynthesis pathway. DG70 showed extraordinary bactericidal activity against nonreplicating persisters in a nutrient starvation model, which highlighted the importance of menaquinone biosynthesis and respiratory chain for the survival of nonreplicating Mtb persisters.138
7.3. Limitations of Target-Based Whole-Cell Screening
One limitation that is seen in target-based whole-cell screening utilizing under-expression strains is that primary hits are not very potent against wild-type Mtb. The higher expression level of the target gene in WT can abrogate activity. Thus, using under- and overexpression of a target’s gene may not yield the desired result. A major concern is that target vulnerability was not assessed for many of the targets screened before target selection. Target vulnerability and bactericidal potential need to be fully evaluated before proceeding to target-based whole-cell screens to improve success.
In some cases, the compound that appears to act on a target intracellularly may not bind to the same target in vitro. Furthermore, a lot of work remains to improve compound potency before delivering a drug candidate. Optimization of hits without structure-based guidance is more challenging.
The phenotype that results from compound treatment depends on the mechanism of action. Although based on the premise of biasing toward a single target, target-based whole-cell screening does not guarantee selection of compounds that bind to only one protein. Whether this is a limitation or advantage is compound specific. However, the target-based assay is a useful means of determining how a specific compound works later in drug development.171
With a dearth of hits being transformed into drug leads that have in vivo efficacy, screening efforts have turned to whole-cell screening systems that better mimic the in vivo environment with a reduction of focus on prior target identification.
8. Replicating Infection Conditions in Whole-Cell Screening
When mycobacteria are engulfed by a macrophage through phagocytosis and delivered to the phagoendosome, Mtb arrests phagosome maturation. Mtb can reside intracellularly in host macrophages for decades and escape killing by the host immune system. As the granuloma is formed by recruitment of additional immune cells to wall off the infected macrophage, many macrophages in the granuloma accumulate lipid bodies and become foamy. Phagosomal mycobacteria reside in juxtaposition to these lipid bodies (Figure 5).172 Mtb accumulates lipids and cholesterol, particularly during dormancy in the granuloma,172−174 and uses these carbon sources in vivo for energy generation.175 Mtb has dedicated metabolic pathways expressly for catabolizing these lipids.176,177
Figure 5.
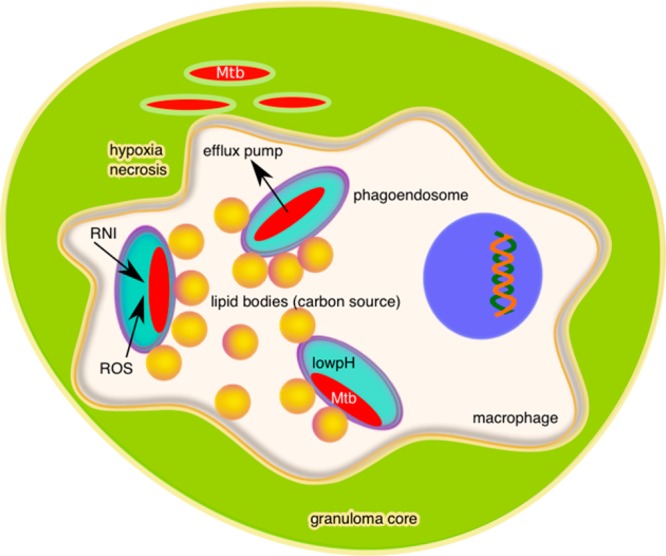
Diagram of an Mtb-infected macrophage within the necrotic core of a granuloma illustrating the intracellular and extracellular microenvironments which Mtb encounters.
In the infected macrophage and granuloma, Mtb encounters different types of stresses from host immune system defense against infection and mechanisms to eradicate infection (Figure 5). These stresses include growth limiting nutrient conditions in the macrophage, low pH in the phagoendosome, a hypoxic environment due to restriction of circulation in the tissue, accumulation of acidic metabolites due to reliance on glycolysis in a hypoxic environment, and the production of reactive oxygen species (ROS) and reactive nitrogen intermediates (RNI) as macrophage defense against bacteria. To survive, a subpopulation of Mtb reversibly adopts a nonreplicating persistent state.
These nonreplicating subpopulations are metabolically resistant to most current TB antibiotics178 and are thought to be one reason for the long treatment times required to sterilize Mtb infection in the patient. Nonreplicating persistent Mtb can be defined as two classes.179 Class I Mtb persisters are defined as cells that are reversibly tolerant to one or more antibiotics but not necessarily to their combination.179 Class II persisters are cells formed under externally applied nonreplicating conditions that exhibit drug tolerance to almost all antibiotics.179 Screens that target Mtb in this nonreplicating state provide an important opportunity for finding better chemotherapeutics that will shorten treatment times by sterilization of persistent mycobacterial subpopulations. An extensive review has recently been published describing TB drug development efforts against nonreplicating persister subpopulations.179 Here we focus on types of screens that are utilized to mimic in vivo conditions, including the nonreplicating state. These examples provide context and insight into where future screening efforts might focus. Strategies that have been taken focus on exposing mycobacteria to a single stress or multiple stresses in culture conditions to replicate the environment in the granuloma and more elaborate in vitro infection models that can be adapted to chemical screens.
8.1. Single-Condition Whole-Cell Screening
The culture medium that provides optimal growth of Mtb in the laboratory does not replicate the conditions encountered by Mtb in vivo. Different methods to mimic the environment Mtb encounters in the granuloma with liquid culture have been undertaken. These conditions include utilizing the lipid carbon sources primarily available to Mtb in the granuloma and exposing Mtb to stresses delivered by the immune system and macrophage, such as low pH and reactive oxygen or nitrogen species. Models for the nonreplicating population of persistent Mtb include cultures depleted of oxygen,180−183 starved of nutrients, or using a lipid carbon source,184−188 acidic culture conditions,153,159,162,189 biofilms,145,190,191 and a streptomycin dependent model.192,193
8.1.1. Fatty Acid Carbon Sources
Mtb can metabolize different carbon sources simultaneously.194,195 In the lipid-rich intracellular environment, Mtb metabolism adapts to the available nutrients. Fatty acid and cholesterol metabolism genes are upregulated and many are required for intracellular survival and persistence.196−200 Mtb utilizes host-derived lipids such as fatty acid and cholesterol as carbon sources.14,177,200,201 Therefore, numerous studies have utilized fatty acids such as palmitate,202,203 butyrate,160 acetate,114,150 and cholesterol150 as the major or primary carbon source in in vitro screening assays to imitate the host environment.
Early et al. conducted a high-throughput screen using butyrate medium to identify growth inhibitors of Mtb.160 They tested the growth rate of Mtb in media with different short chain fatty acids used as the primary carbon source, e.g., acetic acid, butyric acid, isovaleric acid, and palmitic acid. Of these fatty acids, butyrate gives the most robust growth. After a screen of ∼87 000 compounds in butyrate medium, the primary hits were counter screened in glucose medium to identify compounds with butyrate-dependent activity. Among the 166 confirmed hits, a set of compounds was identified to be members of the oxadiazole family (Figure 6). The oxadiazole class of compounds was already known for its broad antimicrobial activities.
Figure 6.

Hit compounds identified in whole-cell screens under stress culture conditions and discussed in the text. Also see Table 2.
Other longer chain and sterol carbon sources have been underutilized due to the difficulties in using these lipids in culture solution in high-throughput screening formats. However, see section 8.3 for approaches which circumvent these limitations.
8.1.2. Hypoxia
The classic model of hypoxia was developed by Wayne and Hayes.181 This model relies on a low dissolved oxygen saturation (0.1–0.06%) to mimic the hypoxic center of the granuloma in which persistent Mtb is thought to reside. The low oxygen culture solution is generated by stirring mycobacteria in airtight culture and waiting until oxygen is consumed. This approach is not viable in a high-throughput screen format given the variability in waiting time required to generate hypoxic conditions and the reliance on measuring colony forming units (a 3-week analysis using solid media to culture remaining bacteria) to determine MICs.
The Wayne oxygen depletion method was adapted to a fermentor system with continuous oxygen monitoring to systematize when to harvest mycobacterial cells for transfer to high-throughput screening plates.182,183 The system was further enhanced by use of a luciferase reporter strain which facilitates determination of cell number remaining after compound treatment. Luciferase directly reports on cellular ATP level and thus is not reliable for determining cell numbers in nonreplicating populations. However, short (1–2 day) incubations of cells under ambient oxygen conditions allows outgrowth of remaining live mycobacteria and quantitative enumeration with little interference from the remaining compound. However, some classes of compounds interfere with the luciferase system, e.g., fluoroquinolones. Therefore, assay hits should be confirmed by direct measurement of colony forming units before proceeding to optimize lead structures. This assay known as the low-oxygen recovery assay, or LORA, has become a staple of screening for Mtb drugs and has been adopted as part of hit to lead advancement criteria in the field.69
Grant et al. found that only a 20% drop in the dissolved oxygen saturation could result in a stable population of persister Mtb.180 Antibiotic efficacy is reduced against this population. They also found that, upon treatment with bactericidal antibiotics, the level of dissolved oxygen in the culture directly affects the survival rate of the persister subpopulation through the concentration of hydroxyl radicals generated by bactericidal antibiotics. This observation provided a new approach to drug targeting through potentiating the hydroxyl radical formation with bactericidal antibiotics or by inhibiting the Mtb hydroxyl radical defense system to sterilize persistent infection.
Besides being used as a surrogate of replicating Mtb, M. bovis BCG has also been developed as a surrogate of dormant tubercle bacilli for high-throughput screening (HTS) of inhibitors against nonreplicating Mtb. These assays are all variations on the original observation that hypoxic conditions result in a nonreplicating state and that M. bovis BCG behaves similarly.158,204,205 The chief difficulty for all these assays is how to enumerate remaining bacilli at the end of the assay. The levels of cellular ATP are too low to detect directly.
Nitrate reductase is induced during oxygen depletion. One approach to enumerate remaining dormant M. bovis BCG is through monitoring the enzymatic activity of nitrate reductase.204,205 This method improved the efficiency and capability of oxygen depletion dormancy models by replacing CFU counting with enzyme activity monitoring. However, this method could also lead to screening of nitrate reductase inhibitors instead of dormant tubercle bacilli growth inhibitors. As a result, redox dye based assays have been used to assess the drug susceptibility of mycobacteria in aerobic and anaerobic conditions, such as the Alamar Blue assay, the Menadione-Tetrazolium assay, and the Resazurin assay.182,206−210
Another approach is to add nitrate as an electron acceptor to increase the intracellular level of ATP in the dormant state to facilitate detection of ATP with a luciferase reporter system.158 The latter approach required introducing the Mtb narGHJI locus into M. bovis BCG to circumvent a promoter mutation in M. bovis BCG and to increase nitrate reductase activity to improve detection. Despite increased levels of nitrate reductase, this nonreplicating M. bovis BCG strain still has a low cellular concentration of ATP. Low levels of cellular ATP sensitize bacilli to disruption of ATP synthesis, and compounds that cause further ATP depletion can be bactericidal.
Novartis performed a screen using the M. bovis BCG hypoxia ATP assay.158 They further adapted the assay to a 1536-well format and screened 600 000 compounds. A total of 866 hits were confirmed to reduce intracellular ATP levels in a dose-dependent manner in M. bovis BCG. However, when retested in a Mtb hypoxic ATP IC50 assay, only 283 (∼35%) hits were active. The relatively low rate of reconfirmation in Mtb was attributed to genomic differences between these two microbes. Even though the two genomes are 99% identical, there are more than 2400 single nucleotide polymorphisms (SNPs) between M. bovis BCG and Mtb that may result in differential expression of potential drug targets and alter drug susceptibilities. After additional screening and cytocixity testing, three new chemical clusters, benzimidazole (BZ), thiophene (TH), and imidazopyridines (IP), were selected for further study and development (Figure 6).
As evidenced by the discovery of bedaquiline, targeting ATP synthesis and maintenance under hypoxic nongrowing condition is a promising strategy for anti-TB drug discovery. These hypoxic screens provide entry into identification and future development of bactericidal compounds that target persistent nonreplicating subpopulations of Mtb.
8.1.3. Low pH
The acidic microenvironment Mtb encounters in phagosomes is about pH 4.5–5.5. Mtb maintains a neutral intracellular pH through homeostasis mechanisms.211,212 Mildly acidic media is used to mimic the pH in phagosomes of immunologically activated macrophages or inflammatory sites of lesions.
Darby et al. developed a whole-cell-based assay to identify compounds that interfere with Mtb’s acid–base homeostasis.159 This screen shed light on which pathways Mtb utilizes to maintain intracellular pH and survive in the acidic conditions of the host. When a marP- (gene encoding pH homeostatic protein MarP) Mtb strain is exposed to low pH 4.5 buffer, the intracellular pH is lowered to 6.5 and a 1.5 log decrease in cell count is observed. Using these cutoffs, a Mtb strain expressing a pH-sensitive protein fluorophore (pHGFP) was used to detect shifts in intracellular pH from neutral to below pH 6.5 upon compound treatment. The assay does not require a long incubation time of compounds with bacilli, as the disruption of intracellular pH can be as quick as 4 h.
Several important controls or conditions were applied in the assay to ensure screening remained on target. Phosphate citrate buffer, pH 4.5 with tyloxapol detergent was used as the medium instead of conventional 7H11 medium which includes Tween and albumin. This was done to avoid the generation of toxic fatty acids from hydrolysis of Tween detergent and albumin at low pH. Secondary screens were performed to identify and eliminate compounds that perturbed intracellular pH by disrupting the bacterial membrane or acting as protonophores. In a test of current anti-TB drugs in this low pH (4.5) assay, only PZA reduced the intracellular pH of Mtb to below 6.5. This result supports a mechanism of action for PZA that includes disrupting Mtb pH homeostasis, and explains the requirement for low pH screening conditions to observe PZA bactericidal activity in vitro.34
The PhoPR regulon plays an essential role in sensing external pH and adapting Mtb to the acidic environment in macrophages.197,213,214 The PhoPR regulon is essential for Mtb growth in in vivo models of infection. However, it is not required for growth in vitro; a phoPR mutant grows well in rich medium at acidic pH.215 The Mtb aprABC locus is induced by acidic pH both in vitro and in macrophages.213 Using the aprA promoter, a pH-inducible fluorescent reporter strain (CDC1551(aprA’::GFP)) was generated. The induction of gfp is fully dependent on PhoPR.213,215 Johnson et al. conducted a high-throughput screen from ∼220 000 compounds to select compounds that inhibit pH-inducible fluorescence, but not growth, which were anticipated to be inhibitors of PhoPR pathway.215 The screening assay was conducted at pH 5.7 in 7H9 rich medium. Ethoxzolamide (ETZ) is a carbonic anhydrase (CA) inhibitor and is approved by the FDA for the treatment of glaucoma. Further transcriptional profiling work verified that ETZ is an inhibitor of the core PhoPR regulon.
8.1.4. Biofilm Model
Mtb forms biofilms as a form of persistence, and these biofilms contain highly drug-tolerant cells. Therefore, Mtb biofilm development may present a promising drug target to eradicate persistence.191 Wang et al. developed a high-throughput screening assay based on in vitro biofilm formation to identify compounds against drug resistant and nonreplicating persistent Mtb.145
M. smeg was utilized as a surrogate for Mtb biofilm formation. M. smeg in biofilm forming medium was incubated with ∼70 000 compounds for 3 days, and optical density was used as an end point readout. The primary hits were tested in a secondary screen for their ability to inhibit in vitro biofilm growth in Mtb. TCA1 (Figure 6) was selected for its potent inhibitory activity against Mtb under both biofilm and planktonic culture conditions. TCA1 exhibits excellent bactericidal activity against both drug-susceptible and drug-resistant Mtb strains. It has also been shown that TCA1 kills nonreplicating persistent Mtb and is efficacious in both acute and chronic murine models of TB infection.145
TCA1 inhibits cell wall synthesis and its target was identified as DprE1 by genome sequencing of spontaneous TCA1 resistant mutants. However, TCA1 was still active against a DprE1 overexpression strain under nutrient starvation conditions and could still potentiate INH and RIF activities in this overexpression strain. These activities indicated TCA1 could potentially act on an additional target. Therefore, an affinity pull-down assay was performed with Mtb cell lysates. MoeW, a protein in the molybdenum cofactor biosynthetic pathway, was identified. A molybdenum cofactor is essential for nitrate respiration of Mtb. Upon treatment with TCA1, Mtb completely abolishes the biosynthesis of molybdenum cofactor, which confirms that MoeW is a second cellular target of TCA1.
8.1.5. Carbon Starvation Model
Mtb enters a nonreplicating state and becomes antibiotic tolerant upon nutrient starvation.186,187 Nutrient starvation can be achieved by culturing bacteria in phosphate-buffered saline (PBS)186 or in minimal salt medium (7H9) that has not been supplemented with a carbon source.184 However, Mtb clumps in PBS culture which prevents its use in high-throughput screening. Minimal salt medium supplemented with 0.05% tyloxapol detergent is suitable for high-throughput screening of Mtb in culture.184 From a high-throughput screen of ∼300 000 compounds in this carbon starvation model, 116 compounds were selected and found to only have activity against nonreplicating Mtb.
Importantly, in a comparison of hits between LORA and the minimal salt carbon starvation model, only 9 of 52 hit compounds were active in both assays.184 Four compounds that are highly active in both these assays were selected and tested in two additional nonreplicating Mtb models, a stochastic hypoxic persister model based on 20% dissolved oxygen180 and a streptomycin-dependent model.192,193 Only one compound showed significant activity at a higher concentration in the stochastic hypoxic persister model, and only two compounds exhibited significant activity in streptomycin-dependent model at a higher concentration.
It is worth noting that there is only a minimal overlap of compounds identified in these three models. The reason could be attributed to the diversity of Mtb persister metabolic states under different nonreplicating conditions. It has been shown by transcriptome studies that there is a very little overlap of differentially regulated genes and transcriptomes between different persister models.179,216,217
8.1.6. Serial Campaigns
Recently, researchers at GlaxoSmithKline (GSK) conducted several large phenotypic screening campaigns based on more than 2 million compounds in hopes of identifying novel antimycobacterial chemical series for further antimycobacterial development.113 Initial efforts focused on performing large phenotypic inhibition of growth assays in surrogate strain M. bovis BCG to enable a screening campaign with more than 2 million compounds. The capacity and efficiency of the screening was as high as 16 runs, ∼125 000 compounds per run, and an average throughput of 90 plates per day.113 The initial screen was followed by hit confirmation in Mtb. Then the primary hits were further tested against the HepG2 liver cell line for cytotoxicity, against Mtb growing in alternative carbon sources and under nonreplicating conditions. After these counter screens, a set of 177 potent noncytotoxic Mtb hits was identified. Among these 177 hits, 7 chemical structural clusters were identified and prioritized based on Mtb MIC and the number of analogues in the hit list. These seven clusters were further evaluated by MIC determination for additional M. tuberculosis strains, MIC determination for intracellular Mtb, activity profiling against eight different bacteria for early assessment of the potential for wide-spectrum activity, a full panel of ADME assessment, and liver cytochrome P450 (CYP) inhibition profiling. Based on these results, one family of compounds, the tetrahydropyrazolopyrimidines (THPP), was advanced into a full lead optimization program (Figure 4).
In GSK’s second antitubercular screening campaign, another 250 000 compounds were screened.218 However, this time, compounds were screened against M. bovis BCG and Mtb in parallel. Compounds showing activity in the primary ATP based assay were further tested in a resazurin-dye reduction assay (MABA) to determine the MIC for Mtb. At this stage, using the criterion of 90% inhibition with 10 μM compound, only ∼20% of the primary hits were detected. The large portion of abandoned primary hits highlights the disconnect between two readouts, ATP concentration determination (Glo assay) and resazurin, and emphasizes that secondary screening must be undertaken to select inhibitors of the bacterium not the reporter system.
Using extensive biological and biophysical evaluation assays, and a series of orthogonal computational approaches based on structural similarity to proprietary GSK historical assay data, 50 of the most attractive compounds were narrowed down to ten compounds that do not share mechanism of action with existing TB drugs. Of these ten compounds, only three were active in the original M. bovis BCG assay. Thus, the majority of novel chemical entities with novel targets would have been eliminated if a surrogate screen was the sole criterion used in the first phase of screening. This study suggests that, despite previous successes, focus on Mtb screening models is paramount to finding new drugs with new mechanisms of action.
It is unclear and difficult to establish which models best represent the real metabolic state of nonreplicating Mtb in humans. The complexity and heterogeneity of both the host environment and Mtb metabolic state call into question using a single condition in culture to represent the in vivo state. Researchers have turned to more sophisticated in vitro assay conditions for answers to increase the likelihood that hits that selected from those models may also work in the host.179
8.2. Multistress Conditions Used in Whole-Cell Screening
Recognizing that the in vivo environment in which Mtb resides is much more complex than a single stressing condition and with the successes in establishing high throughput screens under stressing conditions, compound screening strategies rapidly turned to stressing Mtb with multiple conditions simultaneously.
As outlined in section 8.1, there are different nonreplicating persister Mtb culture models, and different models tend to have different susceptibilities to the panopoly of TB drugs.184 The key focus remains which model will lead to the identification of compounds that kills persistent Mtb in the host.179
8.2.1. Hypoxia and NO-Inducible GFP Reporter Strain
DosRST is a two-component gene regulator that plays an important role in Mtb survival during nonreplicating persistence.219 Through targeting the DosRST regulator or DosRST regulon, the persistence of Mtb may be perturbed. dosR is a nonessential gene for Mtb in vitro growth.220 Therefore, by using a fluorescent reporter strain which is engineered to exhibit DosR-dependent GFP fluorescence that is induced by hypoxia and NO,221,222 inhibitors of the DosRST regulon under nonreplicating conditions can be identified.
A high throughput screen was conducted in mild hypoxic culture conditions with ∼540 000 compounds. Compounds that inhibited DosR-dependent fluorescence but not Mtb growth were selected as putative inhibitors of DosRST regulon inhibitors. Interestingly, artemisinin, a drug for malaria treatment, was discovered to inhibit the persistence of Mtb.222
To confirm the hits are on target, comprehensive RNA-seq-based transcriptional profiling compared the transcription of a ΔdosR Mtb strain with wild type transcription upon compound treatment. This work is an excellent example of how to use RNA-seq-based transcriptional profiling to elucidate or confirm mechanism of action, a method which has become a mainstay of mechanism investigation since its introduction to Mtb research in 2004.169
8.2.2. Carbon Starvation, Hypoxia, and low pH
In a combined stress model developed by Deb et al.,223 cultures were grown in low-nutrient media with 5% O2, 10% CO2, and acidic pH (pH 5.0) for 18 days, after which Mtb was found to enter a lipid-loaded, drug-tolerant nonreplicating dormant state.223 Transcriptional profiling of Mtb under the combined-stress model showed that genes in the glyoxylate cycle such as isocitrate lyase and citrate synthase were significantly up-regulated and genes in ATP synthesis such as cytochrome c complex and ATP synthase subunit were repressed. Triacylglycerol synthase encoding genes were also upregulated. Storage lipid accumulation is one of the major features of in vivo dormancy. The comparison of transcription profiles of Mtb in combined-stress model and single stress models, such as nutrient starvation, acidic pH, and hypoxia, revealed that nutrient starvation accounted for the major changes in gene transcription. However, the gene expression overlap among the three single stress models was very limited.223 This combined-stress model was further adapted to a high- throughput model for drug screening by coupling with an outgrowth phase assay for 5 days and the viable cells were quantified by Alamar blue assay.223 Using this multistress dormancy model, 4400 marine natural products were screened against dormant Mtb.161 A few marine natural products, such as puupehenone (Figure 6) metabolites, were identified with selective antimycobacterial activity against dormant Mtb.161
8.2.3. Carbon Starvation, Hypoxia, low pH, and ROS/RNI
Gold et al.224,225 developed a multistress model for high throughput screening against nonreplicating Mtb. The multistress model includes four conditions that Mtb is likely to encounter in the host simultaneously, including mild acidity (pH 5), hypoxia (1% O2, 5% CO2), a flux of nitric oxide and other RNI (0.5 mM), and fatty acid (0.05% butyrate) as a carbon source. As discussed in section 8.1.2, the problem with developing screening assays against nonreplicating Mtb is its low ATP level. Therefore, it is often accompanied by a prolonged outgrowth phase to improve the readout signal, which can be either a luciferase or intracellular ATP level as an indicator of biomass. In the nonreplicating phase of the assay, Mtb cultures under the multistress conditions were incubated with the compounds for 3–6 days, after which aliquots were taken and diluted into replicating conditions. After 7–10 days of outgrowth, the optical density of each well was read to determine the MIC of compounds against nonreplicating Mtb. To eliminate the compounds that are inactive against nonreplicating Mtb but are potent enough against replicating Mtb after dilution, they ran two assays in parallel: the nonreplicating Mtb assay and an assay under replicating growth conditions.
By adopting this model, GSK conducted a high-throughput screen against nonreplicating Mtb based on a library of 270 000 compounds. A total of 166 hits were confirmed among which 19 nonreplicating specific hits were characterized by evaluation of potency, specificity, cytotoxicity, and stability.147 Using the same screening assay, Gold et al. identified oxyphenbutazone (OPB), a nonsteroidal anti-inflammatory drug, as a conditionally bactericidal antibiotic to nonreplicating Mtb under mildly acidic conditions, and OPB activity was augmented by the addition of nitrite and fatty acid (butyrate).224 Further study revealed that, under acidic conditions (pH 5.5–4.5), OPB undergoes oxidation to become 4-OH-OPB that kills both replicating and nonreplicating Mtb, including drug-resistant strains.224 This work was conducted using the Mtb mc26206 strain at biosafety level 2.
Mtb mc26206 is an auxotroph (ΔpanCD and ΔleuCD) that is nonvirulent.117 In addition, knockout of panCD and leuCD does not affect susceptibility of the strain to major TB drugs.226 Thus, this is an auxotrophic Mtb strain that is an excellent replacement for surrogate strains to enable robotic screening to be performed at biosafety level 2 rather than level 3.
Six compounds (PBTZ-169,142 THPP,134 Spiro,134 indolcarboxamide,135 imidazopyridine,151 and TCA1145) identified in whole-cell screens were tested in animals. Three of them (PBTZ-169,142 indolcarboxamide,135 and TCA1145), including one identified in a stress screen (TCA1), are effective against both acute and chronic models of infection. PBTZ-169, which has low activity against nonreplicating Mtb in vitro, has been advanced to clinical trials. It is not clear whether the remainder of the identified compounds lacked efficacy and were not reported or if poor pharmacokinetics precluded testing them. It remains to be determined whether sterilization times in humans, and thus treatment times, will be shortened with the use of the identified compounds. With the limited testing that has been performed, it is unclear how effective a strategy screening compounds in stress models is for identifying compounds that will target nonreplicating and persistent populations in the host.
8.3. Intracellular Infection Screens
Chemical screening against the replication of Mtb within macrophages served as a secondary assay for intracellular evaluation of primary hits due to its limitations such as slow bacillary growth and requirements for enumerating remaining bacteria by solid media culture which takes 2–3 weeks (CFU counting). With development of high-content screening methods, the intracellular assay can be used to screen large numbers of compounds in a relatively short period of time. Chemical screening against Mtb-infected macrophages allows the direct identification of inhibitors in a complex, more physiologically relevant environment. This type of screening assay also facilitates the discovery of compounds that are acting directly on the function of the host cell to interfere and abolish the manipulation of host cell-signaling by Mtb and thereby boost the host response to eradicate the infection. At the same time, the macrophage serves as a filter to select compounds that can penetrate both the macrophage and the Mtb cell envelope. Moreover, macrophages can induce efflux pumps in Mtb. Screening compounds against infected macrophages helps to select compounds that are not removed from the cell by efflux.227 Additionally, screening directly in infected macrophages identifies compounds that are cytotoxic.
Because each macrophage is infected with 1–2 bacilli, detection of changes in Mtb number is a challenge. Therefore, microscopic imaging has been employed to perform high content screens.
8.3.1. Infected Raw 264.7 Macrophage high Content Screen
Christophe et al. developed a high-content screen utilizing a Mtb strain expressing fluorescent protein (Mtb-GFP) to screen compounds against infected macrophages.149 A human macrophage cell line (Raw 264.7 macrophages) was infected with Mtb-GFP at a multiplicity of infection (MOI) of 1:1 at 37 °C for 2 h. Remaining extracellular bacilli were removed by washing and treatment with amikacin. The growth of bacilli was monitored by fluorescence. The infected cells were incubated with compounds for 5 days after which the images were acquired by confocal fluorescent microscopy, and levels of fluorescent bacilli remaining were obtained from image analysis.
In a chemical screen based on ∼60 000 compounds, 135 noncytotoxic hits (hit rate: 0.2%) with high potency, MIC < 5 μM, were selected. Among them, dinitrobenzamide derivatives were identified as highly potent compounds against Mtb, including extensively drug-resistant Mtb strains. Dinitrobenzamide was also used as a tool compound to identify its target. This class of compounds inhibits arabinan biosynthesis by targeting DprE1, a decaprenylphosphoryl-β-d-ribose 2-epimerase. Together with the previous discovery that the mechanism of action of BTZ is also through the inhibition of DprE1,228 the intracellular vulnerability and drugability of DprE1 was confirmed as a promising drug target for drug-resistant TB treatment.
The same high-content screening method based on a set of 121 156 compounds led to the identification of a series of imidazopyridine amides (IPA) as promising anti-TB agents that target the respiratory cytochrome bc1:aa3 complex,152 specifically the B subunit (QcrB). A lead optimization program identified Q203 (Figure 7), a potent clinical candidate for TB treatment. Despite its nanomolar in vitro activity and excellent PK profile, Q203 only inhibits bacilli growth, that is, it is bacteriostatic, and does not kill persistent bacteria.
Figure 7.

Hit compound identified in intracellular whole-cell screens against Mtb-infected macrophages and discussed in the text. Also see Table 2.
Recent studies have shown that the alternative Mtb cytochrome bd oxidase compensates for cytochrome bc1:aa3 inhibition to maintain ATP synthesis at a level that keeps bacilli alive. Knockout of the cytochrome bd oxidase encoding gene cydAB is required to convert Q203 to a bactericidal drug.229 Therefore, these two respiratory oxidases for oxidative phosphorylation provide a pair of potential targets for combination drug therapy.
8.3.2. Infected J774 Macrophage high Content Screen
A similar high-content screening was conducted with a group of known bioactive compounds against Mtb infected macrophages. The aim was to identify host-targeted compounds that can block host responses utilized by Mtb for pathogenicity and persistence.230 Stanley et al. compared different cell lines for the best reproducibility and robustness as macrophage models. Among THP-1, Raw 264.7, and J774 cell lines, they found that J477 cells provided the most consistent growth of intracellular Mtb. Instead of 5 days postinfection in Raw 264.7 cells, they chose 3 days as the optimal time point for measuring bacterial growth in J774 cells, as the Mtb infection levels were homogeneous and no apparent cell death occurred.230
Their screen utilized ∼2000 compounds known to be bioactive in key mammalian signaling pathways. They included kinase, GPCR, ion channel, membrane transport, and inflammation inhibitors. They identified 133 hits that can be categorized into five groups based on their annotated biofunctions. These hits most likely function by interacting with host pathways to block Mtb intracellular replication and they serve as tool compounds for the identification of essential host pathways or targets that are modulated by Mtb as part of its intracellular virulence.
8.3.3. High-Throughput Screen of Infected Macrophages
Fully automated fluorescence microscopes in combination with fluorophore or luciferase-expressing bacilli can be used to monitor intracellular infection, bacilli localization, and viability. However, high-content imaging is relatively slow in terms of sample processing due to tedious sample image acquisition and data analysis. In addition, the microscopic imaging devices are expensive and space consuming.10 Thus, finding methods to adapt optical screening methods to Mtb-infected macrophage platforms is essential. Intensity-based high-throughput screening assays utilize a reporting system based on either fluorescence or luminescence to quantify the bacilli viability. It is low cost and therefore accessible to most laboratories. The limitation of the latter assay is that intracellular growth and macrophage viability are not monitored directly.
VanderVen et al. identified compounds that inhibit Mtb replication during infection of macrophages using a high-throughput screen that utilizes Mtb expressing fluorophore mCherry and infected J774 macrophages.150 Mtb utilizes lipids in vivo as nutrients for survival, virulence, and pathogenesis. Therefore, this group performed secondary screens against Mtb in culture to identify compounds that induce lipid-based toxicity. An Alamar Blue-based assay in 7H9 OADC medium identified 141 compounds that retained good potency (IC50 < 5 μM) against Mtb replication in 7H9 OADC and 132 conditionally active compounds that only inhibit Mtb growth in macrophages. Of these 132 conditionally active compounds, addition of cholesterol into the 7H12 culture medium converted 74 compounds into inhibitors. The remaining 58 compounds could not be converted into culture medium inhibitors with the addition of cholesterol or fatty acid.
The isocitrate lyase (icl) shunt is required for utilization of cholesterol metabolites in the TCA cycle. VanderVen et al. found that three of the 74 conditionally active compounds rescued growth of a Mtb icl1 knockout strain in cholesterol-containing media. This rescue phenotype indicated that the compounds directly inhibited production of toxic metabolites from cholesterol catabolism. One of the three compounds was additionally able to rescue Mtb icl1 knockout strain growth in propionate.
Further metabolic profiling and biochemical enzyme inhibition assays identified HsaAB and PrpC, from the cholesterol ring degradation pathway and methylcitrate cycle, respectively, as the targets of those three compounds. For those conditionally active hits that could not rescue bacterial growth in cholesterol containing media, VanderVen et al. observed that intracellular cyclic AMP production was increased. cAMP production down regulates cholesterol utilization. This study provided insights into how Mtb utilizes cholesterol and how Mtb regulates cholesterol catabolism during macrophage infection.
Sorrentino et al. developed an intracellular high-throughput screen against Mtb-infected human macrophages.136 In this assay, they utilized two Mtb strains, a luminescent strain and a fluorescent (GFP) strain, for primary identification and secondary confirmation, respectively. The Mtb infected THP-1 cells were produced in a large quantity by incubating the differentiated THP-1 cells with a Mtb single cell suspension in a roller bottle at 37 °C for 4 h followed by removal of extracellular bacilli and incubation with testing compounds for 5 days for primary screening and 4 days for secondary profiling. In a trial screen of 155 compounds that are active as in vitro growth inhibitors, 56 compounds were found to be more active against Mtb-infected macrophages than in vitro Mtb culture while 74 compounds had higher activity in vitro. Thus, different criteria and filters are selected by intracellular and in vitro assays.
Although the size and number of infected macrophage screens performed to date are limited, the early results are quite promising. Two compounds, Q203152 and TBA-7371,148 are in phase 1 clinical trials. Mechanism of action elucidation methods utilized are analogous to those used for whole-cell screening and, therefore, tractable. A shortcoming is that these types of screens focus on intracellular Mtb which is not the sole microenvironment encountered by Mtb in granulomas (Figure 1). The major technical limitations are the preparation of sufficient quantities of infected macrophages and the availability of BSL3 facilities in which large quantities of infected macrophages can be prepared and screened.
8.4. Granuloma Models
The macrophage-based intracellular assays mimic the host environment Mtb encounters at the single cell level. However, these assays do not represent the heterogeneity of the host microenvironment which Mtb encounters. Due to a granuloma’s closer similarity to an animal infection model with human-like lesions, the granuloma model is the next step in bridging the gap between in vitro extracellular assays and in vivo animal models of infection for drug screens. Moreover, the granuloma is a good predictor of a drug’s in vivo efficacy.226 However, it is important to be mindful that in vivo Mtb does not only reside inside the cellular granuloma. For example, extracellular Mtb is found in the caseum of the necrotic granuloma, and infection may be found outside the lung.
8.4.1. Human Cell Granuloma Model
Silva-Miranda et al. developed a high-content screening assay to evaluate compound activities against a human granuloma model.231 In vitro human granulomas were formed from human peripheral blood mononuclear cells infected with Mtb-GFP at 37 °C for 1 h. After washing with buffer, the cell suspension was incubated at 37 °C for 3 days to allow the formation of granulomas. The test compounds were incubated with granulomas for 5 days, and then the cell aggregates were fixed and stained for image acquisition. The granulomas were selected based on the size and form of the cells, and only intragranuloma bacilli were counted. Data analysis focused on counting the number of granulomas and the number of bacilli, and the number of bacilli per granuloma was used for the calculation of percentage of inhibition of each compound.
The MIC50s of INH, MOX, LZD, and PZA were tested in this artificial granuloma model and compared with their MIC50s in macrophage and extracellular assays. For INH, the MIC50s measured in all three models are similar, whereas for MOX and LZD there is an ∼15-fold increase in the MIC50 in the granuloma model. The decrease of drug potency in the granuloma model is explained by the different permeabilities of single cells and cell aggregates. The different metabolic state of Mtb in the granuloma environment is also a key factor.
8.4.2. Human Cell Line Granuloma Model
Instead of using human peripheral blood mononuclear cells which were donated by healthy volunteers,231 Schaaf at al. used THP-1 cells infected with Mtb-GFP mc26206 (ΔpanCD and ΔleuCD)117 at a high multiplicity of infection to form granulomas. It took 10–14 days for aggregates to form, which is longer than human peripheral blood mononuclear cells, during which time the cell medium had to be changed every 2–3 days. Its utilization allows the production of a large quantity of granulomas in a cost-effective way for high-throughput screening.
The morphologic and phenotypic characterization of THP-1 cell granulomas showed key features of a physiologically relevant Mtb lesion, such as, a tightly packed cell core with necrotic tissue and the presence of intracellular lipid bodies which may indicate a metabolic shift occurred in Mtb (Figures 1 and 5).226 RIF drug-resistance was also observed in this infection model.
Mtb in the granuloma model exhibited a more than 10-fold increase in tolerance to RIF compared to extracellular Mtb. This increased drug tolerance may be attributed to tissue penetration issues, increased efflux pump activity in the macrophage, and decreased drug uptake by Mtb in a metabolically latent stage.185,227,232 This work confirmed the finding of Silva-Miranda231 that the multiple layer cell structure of a granuloma presents a barrier to drug penetration. The fact that RIF has an ∼10-fold MIC increase in the granuloma model, compared to a 4-fold increase in the nutrient starvation latent model, suggests that the loss of potency is mainly due to the increased tissue penetration barriers presented by the structure of granuloma. For a TB drug to reach the intended molecular target, it must be carried and transported through blood to pulmonary lesions, diffuse into the caseum, and penetrate the cell wall to reach the target protein.233 Therefore, this granuloma model serves as a good predictor for the evaluation of drug in vivo efficacy, particularly tissue penetrability at the lesion site.226
8.4.3. Zebrafish Granuloma Model
The M. marinum infected zebrafish model has drawn a lot of interest as a useful tool for the study of the complexity and pathogenicity of Mtb infection. M. marinum infected zebrafish closely recapitulate the pathology of human Mtb infection, including macrophage aggregates and the formation of granulomas.234,235 In addition, the immune system of zebrafish is complex enough to mimic the human immune system.236 Therefore, the infected zebra fish can be used as a tool for identification of host-derived pathways that are important for progression of Mtb infection in humans. Due to the relatively easy husbandry of zebrafish larvae, which are optically transparent, and the ease of in situ imaging by automated plate fluorimetry, fluorescently labeled M. marinum infected zebrafish larval model has been developed into an in vivo high-throughput screening platform for antitubercular drug identification.237 This in vivo high-throughput screen allows the direct selection of compounds with in vivo efficacy that are not acutely toxic. The screen can also facilitate the selection of host-targeting compounds that mediate the host and pathogen interaction during the process of infection and progression.
This high-throughput screening assay evaluates two aspects of drug efficacy: quantification of bacterial burden and host survival. The 3-day postfertilization larvae can be maintained in 96-well plate format with no mortality for 10 days without water changes. Larvae are infected with fluorescently labeled M. marinum through caudal vein injection. Larvae at 1 day postinfection are kept in a 96-well plate in the presence of drugs and the viability and bacterial burden are monitored daily through automated plate fluorimetry. Before detection, larvae are reversibly anesthetized for immobilization. Viability is quantified by cardiac assessment and fluorescence intensity in the green channel of automated plate fluorimetry. Bacterial burden is measured by fluorescence intensity in a second fluorescence channel, the far-red range. The granuloma formation and heterogeneity after 3 days post infection does not affect the fluorescence measurement. For drug efficacy assessment of one compound, only a few zebrafish larvae are needed. Therefore, this in vivo assay can process extensive numbers of samples in a short time with limited amount of compound.
The original protocol for infection of zebrafish with M. marinum requires vein injection. Dalton et al. have developed a protocol to infect larvae by bathing them in luciferase-expressing M. marinum solution, followed by selection of infected larvae by fluorescent cell sorting.238 Despite this innovation, the model remains limited by the use of a surrogate bacterium in a surrogate organism.
9. Current Status of TB Drug Discovery
Based on two decades of screening by a variety of methods, lead compounds have begun to enter clinical trials. At present, approximately seven novel compounds are in early development (Figure 8 and Table 2). These lead compounds have been identified through a variety of different screens and methods as outlined below.
Figure 8.

TB compounds identified in the screens outlined in Table 2 that are currently in clinical trials.
Table 2. Summary of Hit Compounds, Their Targets, and the Type of Screen in which They Were Identifieda.
| target | repurposed compound screen | Target-based WCSb | WCS growth | single stress WCS | multistress WCS | infected macrophage |
|---|---|---|---|---|---|---|
| BioA | compound 36131 | |||||
| MmpL3 | SQ109(132,133) | THPP,113,134 spiro,113,134 AU1235,133 indolcarboxamide135 | benzoimidazolamine (C215)114 | THPP134,136 | ||
| Pks13 | thiophene-1137 | |||||
| MenG | DG70138 | |||||
| EchA6 | THPP113,139 | THPP136,139 | ||||
| DprE1 | OPC-167832(140,141) | PBTZ-169(144) | TCA1,145Benzylnitrotriazole (377790)114 | compound 14 (pyridobenzimidazolone)146,147 | TBA-7371(148) dinitrobenzamide (DNB1)149 | |
| MoeW | TCA1145 | |||||
| HsaA/HsaB | V-13–011503, V-13–012725150 | |||||
| PrpC | V-13-009920150 | |||||
| QcrB | imidazopyridine-1113,151 | Q203(152) | ||||
| MarP | BO43153 | |||||
| LeuRS | GSK-070(154−157) | |||||
| target unknown | benzimidazole,158 thiophene-2,158 imidazopyridine-2,158 4-methoxybenzoic acids,159 4-hydroxyfuranone,159 agrimophol,159 Oxadiazoles160 | compound 12 (Indazole),147puupehenone161 |
Compound names shown in bold are in clinical trials as per https://www.newtbdrugs.org/pipeline/clinical.
WCS: whole-cell screening.
9.1. SQ109
A combinatorial library based on 1,2-ethylenediamines like EMB was screened in two in vitro high-throughput screens. The first screening assay is a broth dilution to determine MIC against Mtb. The second assay is a iniBAC promoter based, cell wall inhibition bioluminescence high throughput screen.239,240 SQ109 was identified in this screen.239,240 However, SQ109’s mechanism of action, potency and activity are different than that of EMB. SQ109 acts by targeting MmpL3, a transmembrane transport protein that transports trehalose monomycolate during cell wall synthesis.132,133 SQ109 is bactericidal. It is active against extracellular and intracellular bacilli and works in both acute and chronic murine models of Mtb infection. SQ109 improved the drug efficacy of the current four first line TB drugs and shows good synergism with bedaquiline.239 SQ109 is currently in a phase II clinical trial.
9.2. Q203
Q203 is an imidazopyridine amide compound and was identified through an infected macrophage whole-cell screen. Q203 inhibits the cytochrome bc1 complex and disrupts the electron transport chain for ATP synthesis.152 Q203 shows potent inhibition of replicating bacilli and has an excellent PK profile. The phase I clinical trial of Q203 was successfully completed recently by Infectex.
9.3. TBA-7371
TBA-7371 is a member of an 1,4-azaindole series that was identified through a scaffold morphing strategy followed by a lead optimization program from an imidazopyridine compound.148,241 The parental imidazopyridine compound was identified from the infected RAW 264.7 macrophage-based phenotypic screen which also generated Q203.242 TBA-7371 noncovalently inhibits DprE1, a decaprenylphosphoryl-β-d-ribose2′-epimerase, in the arabinan biosynthesis pathway.148 TBA-7371 is bactericidal and is active in both acute and chronic Mtb infection murine models.148 TBA-7371 is in a phase I clinical trial.
9.4. PBTZ-169
PBTZ-169 and BTZ-043 belong to the class of benzothiazinones that were identified from an in vitro broth dilution assay screening for antibacterial and antifungal activities.228,243 Benzothiazinones target DprE1 by forming a covalent adduct irreversibly with DprE1 and block the production of d-arabinose for cell wall synthesis.144 Both PBTZ-169 and BTZ-043 display very potent bactericidal activity against replicating bacilli and MDR strains, although they have low activity against nonreplicating subpopulations and show comparable efficacies to INH and RIF in the chronic Mtb infection murine model. PBTZ-169 is in a phase I clinical trial.
9.5. OPC-167832
OPC-167832 is a 3,4-dihydrocarostyril derivative developed by Otsuka. OPC-167832 also targets DprE1 and shows bactericidal activity against replicating and intracellular Mtb bacilli. Otsuka has a long history of drug development with this compound class for various clinical indications. OPC-167832 displays especially good synergism in combination with delamanid. OPC-167832 is currently in a phase I clinical trial.140,141
9.6. GSK-070
GSK-070 is an oxaborole derivative that targets leucyl-tRNA synthetase (LeuRS). Oxaboroles block leucyl-tRNA synthesis and consequentially protein synthesis by forming an adduct with tRNA and trapping the enzyme-tRNA complex in the editing site.154 The benzoxaboroles were originally discovered as antifungal agents and were repurposed after testing against Mtb.155,156 The activity of GSK-070 has been demonstrated in both acute and chronic Mtb infection murine models.157 GSK-070 is currently in a phase I clinical trial.
9.7. Analysis of Current Status
The degree of target engagement that can be achieved in vivo, and the vulnerability of these four targets in human disease remain to be established. Until full human efficacy trials are under way, we will not know how valid these targets are for treating TB.
In addition to the six clinical leads discovered through original screens against Mtb, there are compounds identified as antifungal or antiprotozoal that are effective against Mtb and which are in clinical trials. Furthermore, there are second generation bedaquiline, clofazimine, and linezolid compounds in various stages of clinical trial.
Intriguingly, despite the use of a wide variety of compound screens (Table 2), the same targets have been identified through a disparate set of conditional in vitro compound screens. For example, consider the case of a new, apparently druggable, target DprE1, decaprenylphosphoryl-β-d-ribose 2′-epimerase, an enzyme in the arabinan synthesis pathway, which is required for cell wall biosynthesis. DprE1 has been identified through multiple different screens and is inhibited by several compound classes.114,143,146,228,244−246 The compounds act both through covalent mechanism-based inhibition as well as noncovalent competitive inhibition. It appears that DprE1 is easily targeted because it resides in the periplasmic space of the mycobacterium.247 The reader is referred to an extensive review of DprE1 drug development for further details.246
The reasons for the convergence on a small number of new targets are unclear. These targets may be central to Mtb survival in vivo and consequently susceptible to small molecule inhibition. Alternatively, the frequency of their selection may arise from similarities in compound libraries screened. Many of the compound libraries screened have been repurposed from pharmaceutical efforts in other arenas, primarily mammalian targets in human disease.91
10. Future Prospects
Many additional hit compounds beyond the six in clinical trials described above have been identified through creative and inventive screens to replicate the microenvironment in which Mtb resides. Extensive biochemistry remains to be performed to inform the mechanism of action, to enable lead development, and to ensure in vivo efficacy. The extensive failure rate in early screening programs certainly provides impetus to do more sophisticated screens sooner in the hit and lead selection process. However, no screen can replace the requirements of extensive follow-on experiments to determine specificity and the mechanism of action.
The next question then is which screen to use first. Closer examination of the hits identified through different types of phenotypic screens (Table 2) reveals that the infected macrophage screens yielded hits against nearly all of the targets currently under investigation. Thus, the use of an infected macrophage screen appears most general and has the advantage of reflecting physiologic conditions to the greatest extent currently possible.
The greater technical complexity of using infected macrophage screens means that simpler whole-cell screens will continue to be utilized. Therefore, it is important to consider which screening conditions and selection criteria are likely to yield the most useful hits. Regardless of screening conditions, whole-cell Mtb screens identified hits primarily against extracellular targets that reside in or on the cell membrane, although many targets have yet to be identified (Table 2). The preponderance of cell-surface targets suggests preferential selection for targets that are most accessible to compounds in this format. These selections may be a consequence of filtering, library compositions, or innate target vulnerability.
Generally, in a whole-cell screen, the highest potency hits are selected for investigation. Due to the static, equilibrium conditions of MIC determinations, these conditions may bias toward selection of extracellular targets. We propose that less stringent MIC cutoffs in initial whole-cell screens may lead to identification of different hits that can be developed into lead compounds with improved potency against vulnerable intracellular targets, much like a fragment-based approach (see section 6). Then medicinal chemistry efforts are essential to develop an initial hit into a lead compound for in vivo use. Compound pharmacokinetics must be such that penetration into the caseum core of a necrotic granuloma occurs. Recent studies have highlighted how compounds with low or no efficacy in vitro, like ethambutol and pyrazinamide, have excellent biodistribution properties that contributes to their in vivo efficacy.248,249
One approach to improve weaker hits may be to design pro-drugs that take advantage of Mtb transporters. For example, trehalose is a disaccharide important for Mtb virulence. LpqY-SugABC transports trehalose back into the mycobacterial cell after hydrolysis of trehalose monomycolate (TMM) which is exported by the MmpL3 transporter. The LpqY-SugABC transporter accepts structural analogs of trehalose250 suggesting that it may be used for drug delivery purposes. Existing synthetic methods and applications,251 as well as validation the pathway is required to maintain viability,252 suggest that trehalose conjugates could be a fruitful direction to pursue. Mtb expresses many additional transporters, including Mce1 and Mce4 to transport fatty acids and cholesterol, respectively,200,253 that could be considered for similar approaches.
Many of the most recently identified leads (Table 2) and most recently approved TB drugs (Figure 3) contain two or more aromatic moieties (Figures 4, 6, and 7) and are very hydrophobic (cLogPs > 5).8 In contrast, most existing TB drugs are very polar (cLogPs < 2).8 High hydrophobicities can be problematic due to drug–drug interactions, to human metabolic liabilities, and to formulation issues. Another approach to improve the diversity of hits and targets is to refocus on the use of natural products. Improved analytical methods in combination with modern bioinformatics and genetic engineering approaches have improved access to greater structural novelty and activity than obtained in earlier decades.254,255 Natural product antibiotics have the advantage of being selected through evolution to permeate bacterial cell walls and to be specific for their targets.255 For example, Rohde and co-workers have recently demonstrated the utility of screening marine natural product extracts against Mtb in a multistress culture condition.161 Although the target of their puupehenone metabolite hits has not been identified, the preponderance of evidence suggests that the target is NADH oxidase activity, a new target for Mtb drugs.
Ultimately, in developing and selecting a lead compound, additional screening assays will be performed to understand its mechanism of action, and it is desirable that a compound should work on the selected target under multiple physiologically relevant conditions. What is important is to identify new hit scaffolds for drug development and new targets that can provide different mechanisms of action from existing TB drugs.
The best prospect to identify future TB drugs is to continue the development of screens to their logical end point of replicating the Mtb microenvironment fully. Current granuloma models rely on a single cell type to mimic the aggregated complex that is formed. Tissue engineering methods may yield more heterogeneous, yet still organized, multicellular structures that better recapitulate the structure of human granulomas.256 These types of models will provide physiological relevance, unbiased selection of essential and vulnerable targets at a near organismal level, and simultaneously avoid compounds with unfavorable biophysical properties. Many of the simpler screens established and described above will still be essential for identifying targets and mechanism of action once hits have been identified.
However, identification of the specific target selected in such screens will be even more challenging than for current screening efforts. Whole genome sequencing of mutants resistant to hits from empirical screens followed by introducing a mutation into the target gene independently for confirmation is the most common approach to target identification and validation.257 With current screens, the slow replication rate of Mtb prolongs selection of mutants, and the resulting mutants often have reduced viability in liquid media. Alternative strategies used with success in other systems include comparative phenotypic profiling and gene under-expression to increase drug sensitivity.258 The former approach was used successfully by VanderVen and co-workers to identify the targets of hits obtained in an infected macrophage screen.150 These approaches can be challenging to implement, if the inhibition efficacy is condition dependent, and the challenges will be compounded with the use of more elaborate screens. Moreover, hits may have multiple targets, which is advantageous for slowing the development of resistance in the clinic, but increases the difficulty of understanding mechanism of action. In addition, hits that target the host may be generated, further complicating analysis. Thus, continued development of methods to identify targets are required.
Once targets are identified, methods to assess level of target engagement in vivo are needed.259 Lack of efficacy once reaching human clinical trials is the most frequent cause of compound attrition. Small differences in the in vivo drug concentration required for efficacy and the in vivo drug concentration which is toxic (therapeutic window) can make it difficult to achieve required levels of target engagement. Ultimately, the relation of the target to human disease in the human patient must be established to validate drugging the target. Human imaging technologies are advancing rapidly, with the greatest advances occurring in neuroscience and cancer. These same imaging modalities can be used to establish the degree of target occupancy in conjunction with biochemical experiments.259 Recently, methods have been developed that advance our understanding of drug kinetics in the granuloma.248,260−262 All of these types of experiments must be moved earlier into the TB drug development pipeline to ensure that druggable, vulnerable targets are being engaged appropriately for sterilization of Mtb in a diverse array of granuloma states.
Acknowledgments
We appreciate the support of the Stellenbosch Institute for Advanced Study (STIAS), Stony Brook University, and National Institutes of Health (NIH) Grants R01AI134054 and U01HL127522.
Glossary
Abbreviations
- ATP
adenosine-triphosphate
- BDQ
bedaquiline
- BioA
5′-pyridoxal phosphate (PLP)-dependent aminotransferase in biotin synthesis
- BioD
7,8-diaminopelargonic acid carboxylase
- BTZ
1,3-benzothiazin-4-ones
- CFU
colony forming units
- ClpP1P2
caseinolytic protease
- CYP2C8
cytochrome P450 C8
- CYP2C9
cytochrome P450 C9
- CYP3A4
cytochrome P450 A4
- DosRST
two component gene regulator of dormancy
- DprE1
decaprenylphosphoryl-β-d-ribose 2′-epimerase
- EBA
early bactericidal activity
- EchA6
enoyl-CoA hydratase 6
- EMA
European Medicines Agency
- EMB, E
ethambutol
- EthA
thionamide monooxygenase
- EthR
ethionamide repressor
- EXS-1
major virulence protein type VII secretion system
- ETZ
ethoxzolamide
- FDA
United States Food and Drug Administration
- GFP
green fluorescent protein
- GPCR
G-protein coupled receptor
- GSK
GlaxoSmithKline
- HsaA
steroid-degrading flavin monooxygenase
- HsaB
steroid-degrading flavin monooxygenase
- HTS
high-throuput screen
- HtrA1
high temperature required A1 homologue
- INH, H
isoniazid
- InhA
enoyl-ACP reductase
- IPA
imidazopyridine
- LORA
low oxygen recovery assay
- LepB
signal peptidase I
- LeuRS
leucyl-tRNA synthase
- LZD
linezolid
- MarP
transmembrane serine protease involved in pH homeostasis
- M. bovis BCG
Mycobacterium bovis bacille Calmette-Guerin
- M. marinum
Mycobacterium marinum
- M. smeg
Mycobacterium smegmatis
- MBC
minimal bactericidal concentration
- MDR
multidrug resistant
- MDR-TB
multidrug resistant tuberculosis
- MIC
minimal inhibitory concentration
- MIC50
minimal 50% inhibition concentration
- MmpL3
transmembrane transport protein that transports trehalose monomycolate
- MoeW
molybdenum cofactor biosynthesis gene product W
- MOI
multiplicity of infection
- MOX
moxifloxin
- Mtb
Mycobacterium tuberculosis
- NAD+
nicotinamide adenine dinucleotide
- NADH
nicotinamide adenine dinucleotide, hydrogen
- NMDA
N-methyl-d-aspartate
- OPB
oxyphenbutazone
- PBS
phosphate-buffered saline
- PD
pharmacodynamics
- PK
pharmacokinetic
- PrpC
2-methylcitrate synthase
- PZA, Z
pyrazinamide
- QcrB
Cytochrome b subunit of the cytochrome bc1 complex
- RIF, R
rifampicin, rifampin
- RNI
reactive nitrogen intermediates
- ROS
reactive oxygen species
- SsrA
caseinolytic-protease-specific degradation signal peptide
- TB
tuberculosis
- Tet
tetracycline
- TET-OFF
tetracycline-regulated under expression
- TET-ON
tetracycline-regulated overexpression
- TMM
trehalose monomycolate
- WCS
whole-cell screen
- WhiB3
iron–sulfur cluster redox sensor and transcription factor
- XDR
extensively drug resistant
- XDR-TB
extensively drug resistant tuberculosis
Biographies
Tianao Yuan received his B.S. degree in Chemistry from Nanjing University in 2013. Currently he is a graduate student in Chemical Biology under the guidance of N. S. Sampson at Stony Brook University. His research focuses on understanding and targeting cholesterol metabolism by Mycobacterium tuberculosis to discover and develop new TB drugs.
Nicole S. Sampson received a B.S. degree in Chemistry from Harvey Mudd College, obtained a Ph.D. in Chemistry with P. A. Bartlett at the University of California, Berkeley, and did postdoctoral research with J. R. Knowles at Harvard University. She has been on the faculty of Stony Brook University since 1993 and is currently a Professor of Chemistry and Co-Director of the Chemical Biology Training Program (NIH-T32). Her research interests include the design of chemical probes of mammalian fertilization, developing new methods for precision polymer synthesis, and mapping metabolic pathways that enable survival of Mycobacterium tuberculosis in the host. The latter forms the basis for the TB drug discovery and diagnosis efforts in her laboratory.
The authors declare the following competing financial interest(s): T. Y. and N. S. S. are named inventors on patents and patent applications related to this article.
References
- WHO “Global Tuberculosis Report 2016”, World Health Organization, 2017 ; http://www.who.int/tb/publications/global_report/en/ accessed: 29 September 2017.
- CDC “Tuberculosis Fact Sheets”, Centers for Disease Control and Prevention, 2014 ; https://www.cdc.gov/tb/publications/factsheets/general/ltbiandactivetb.htm accessed: 22 December 2017.
- WHO “Collaborative Framework for Care and Control of Tuberculosis and Diabetes”, World Health Organization , 2017; http://www.who.int/tb/publications/tb-diabetes-framework/en/ accessed: 5 January 2018. [PubMed]
- Pai M.; Behr M. A.; Dowdy D.; Dheda K.; Divangahi M.; Boehme C. C.; Ginsberg A.; Swaminathan S.; Spigelman M.; Getahun H.; et al. Tuberculosis. Nat. Rev. Dis. Primers 2016, 2, 16076. 10.1038/nrdp.2016.76. [DOI] [PubMed] [Google Scholar]
- WHO “Multidrug-Resistant Tuberculosis (MDR-TB) 2016 Update”, World Health Organization, 2016 ; http://www.who.int/tb/challenges/mdr/mdr_tb_factsheet.pdf accessed: 29 September 2017.
- Cole S. T.; Brosch R.; Parkhil J.; Garnier T.; Churcher C.; Harris D.; Gordon S. V.; Eiglmeier K.; Gas S.; Barry C. E. 3rd; et al. Deciphering the Biology of Mycobacterium tuberculosis from the Complete Genome Sequence. Nature 1998, 393, 537–544. 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Pethe K.; Sequeira P. C.; Agarwalla S.; Rhee K.; Kuhen K.; Phong W. Y.; Patel V.; Beer D.; Walker J. R.; Duraiswamy J. A Chemical Genetic Screen in Mycobacterium tuberculosis Identifies Carbon-Source-Dependent Growth Inhibitors Devoid of in vivo Efficacy. Nat. Commun. 2010, 1, 1–8. 10.1038/ncomms1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoagland D. T.; Liu J.; Lee R. B.; Lee R. E. New Agents for the Treatment of Drug-Resistant Mycobacterium tuberculosis. Adv. Drug Delivery Rev. 2016, 102, 55–72. 10.1016/j.addr.2016.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechartier B.; Rybniker J.; Zumla A.; Cole S. T. Tuberculosis Drug Discovery in the Post-Post-Genomic Era. EMBO Mol. Med. 2014, e201201772. 10.1002/emmm.201201772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.; Av-Gay Y. New Era of Tb Drug Discovery and Its Impact on Disease Management. Curr. Treat. Options Infect. Dis. 2016, 8, 299–310. 10.1007/s40506-016-0098-0. [DOI] [Google Scholar]
- Zumla A.; Nahid P.; Cole S. T. Advances in the Development of New Tuberculosis Drugs and Treatment Regimens. Nat. Rev. Drug Discovery 2013, 12, 388–404. 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- Franzblau S. G.; DeGroote M. A.; Cho S. H.; Andries K.; Nuermberger E.; Orme I. M.; Mdluli K.; Angulo-Barturen I.; Dick T.; Dartois V.; et al. Comprehensive Analysis of Methods Used for the Evaluation of Compounds against Mycobacterium tuberculosis. Tuberculosis (Oxford, U. K.) 2012, 92, 453–488. 10.1016/j.tube.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Russell D. G. Who Puts the Tubercle in Tuberculosis?. Nat. Rev. Microbiol. 2007, 5, 39–47. 10.1038/nrmicro1538. [DOI] [PubMed] [Google Scholar]
- VanderVen B. C.; Huang L.; Rohde K. H.; Russell D. G. The Minimal Unit of Infection: Mycobacterium tuberculosis in the Macrophage. Microbiol. Spectr. 2016, 4, 0025–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell D. G.; Barry C. E.; Flynn J. L. Tuberculosis: What We Don’t Know Can, and Does, Hurt Us. Science 2010, 328, 852–856. 10.1126/science.1184784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apt A.; Kramnik I. Man and Mouse TB: Contradictions and Solutions. Tuberculosis (Oxford, U. K.) 2009, 89, 195–198. 10.1016/j.tube.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon V. E.; Valbuena G.; Goez Y.; Judy B. M.; Huante M. B.; Sutjita P.; Johnston R. K.; Estes D. M.; Hunter R. L.; Actor J. K.; et al. A Humanized Mouse Model of Tuberculosis. PLoS One 2013, 8, e63331. 10.1371/journal.pone.0063331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A. M. Mouse Model of Tuberculosis. Cold Spring Harbor Perspect. Med. 2015, 5, a018556. 10.1101/cshperspect.a018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C. J.; Cadena A. M.; Leung V. W.; Lin P. L.; Maiello P.; Hicks N.; Chase M. R.; Flynn J. L.; Fortune S. M. Digitally Barcoding Mycobacterium tuberculosis Reveals in vivo Infection Dynamics in the Macaque Model of Tuberculosis. mBio 2017, 8, e00312–00317. 10.1128/mBio.00312-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan L.; Tang J.; Sun M.; Qin C.. Animal Models for Tuberculosis in Translational and Precision Medicine. Front. Microbiol. 2017, 8, 10.3389/fmicb.2017.00717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrichs T.; Kaufmann S. H. New Insights into the Function of Granulomas in Human Tuberculosis. J. Pathol. 2006, 208, 261–269. 10.1002/path.1906. [DOI] [PubMed] [Google Scholar]
- Russell D. G.; Cardona P.-J.; Kim M.-J.; Allain S.; Altare F. Foamy Macrophages and the Progression of the Human Tuberculosis Granuloma. Nat. Immunol. 2009, 10, 943–948. 10.1038/ni.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb V. C.; Chuturgoon A. A.; Moodley D. Mycobacterium tuberculosis: Manipulator of Protective Immunity. Int. J. Mol. Sci. 2016, 17, 131. 10.3390/ijms17030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via L. E.; Lin P. L.; Ray S. M.; Carrillo J.; Allen S. S.; Eum S. Y.; Taylor K.; Klein E.; Manjunatha U.; Gonzales J. Tuberculous Granulomas Are Hypoxic in Guinea Pigs, Rabbits, and Nonhuman Primates. Infect. Immun. 2008, 76, 2333–2340. 10.1128/IAI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. A.; Baumgardner J. E.; Good V. P.; Otto C. M. Physiological and Hypoxic O2 Tensions Rapidly Regulate NO Production by Stimulated Macrophages. Am. J. Physiol. Cell. Physiol. 2008, 294, C1079–1087. 10.1152/ajpcell.00469.2007. [DOI] [PubMed] [Google Scholar]
- Cadena A. M.; Fortune S. M.; Flynn J. L. Heterogeneity in Tuberculosis. Nat. Rev. Immunol. 2017, 17, 691–702. 10.1038/nri.2017.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier C.; Falkow S.; Ramakrishnan L. Host Evasion and Exploitation Schemes of Mycobacterium tuberculosis. Cell 2014, 159, 1497–1509. 10.1016/j.cell.2014.11.024. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Lienhardt C.; McIlleron H.; Nunn A. J.; Wang X. Global Tuberculosis Drug Development Pipeline: The Need and the Reality. Lancet 2010, 375, 2100–2109. 10.1016/S0140-6736(10)60359-9. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Heym B.; Allen B.; Young D.; Cole S. The Catalase-Peroxidase Gene and Isoniazid Resistance of Mycobacterium tuberculosis. Nature 1992, 358, 591–593. 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- Johnsson K.; King D. S.; Schultz P. G. Studies on the Mechanism of Action of Isoniazid and Ethionamide in the Chemotherapy of Tuberculosis. J. Am. Chem. Soc. 1995, 117, 5009–5010. 10.1021/ja00122a038. [DOI] [Google Scholar]
- Mitchison D. A. Role of Individual Drugs in the Chemotherapy of Tuberculosis. Int. J. Tuberc. Lung Dis. 2000, 4, 796–806. [PubMed] [Google Scholar]
- Hartmann G.; Honikel K. O.; Knusel F.; Nuesch J. The Specific Inhibition of the DNA-Directed RNA Synthesis by Rifamycin. Biochim. Biophys. Acta, Nucleic Acids Protein Synth. 1967, 145, 843–844. 10.1016/0005-2787(67)90147-5. [DOI] [PubMed] [Google Scholar]
- Diacon A. H.; Donald P. R. The Early Bactericidal Activity of Antituberculosis Drugs. Expert Rev. Anti-Infect. Ther. 2014, 12, 223–237. 10.1586/14787210.2014.870884. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Wade M. M.; Scorpio A.; Zhang H.; Sun Z. Mode of Action of Pyrazinamide: Disruption of Mycobacterium tuberculosis Membrane Transport and Energetics by Pyrazinoic Acid. J. Antimicrob. Chemother. 2003, 52, 790–795. 10.1093/jac/dkg446. [DOI] [PubMed] [Google Scholar]
- Shi W.; Zhang X.; Jiang X.; Yuan H.; Lee J. S.; Barry C. E.; Wang H.; Zhang W.; Zhang Y. Pyrazinamide Inhibits Trans-Translation in Mycobacterium tuberculosis. Science 2011, 333, 1630–1632. 10.1126/science.1208813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal P.; Nartey W.; Ragunathan P.; Sarathy J.; Kaya F.; Yee M.; Setzer C.; Manimekalai M. S. S.; Dartois V.; Gruber G.; et al. Pyrazinoic Acid Inhibits Mycobacterial Coenzyme A Biosynthesis by Binding to Aspartate Decarboxylase PanD. ACS Infect. Dis. 2017, 3, 807–819. 10.1021/acsinfecdis.7b00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimhony O.; Cox J. S.; Welch J. T.; Vilcheze C.; Jacobs W. R. Jr Pyrazinamide Inhibits the Eukaryotic-Like Fatty Acid Synthetase I (FasI) of Mycobacterium tuberculosis. Nat. Med. 2000, 6, 1043–1047. 10.1038/79558. [DOI] [PubMed] [Google Scholar]
- Kim H.; Shibayama K.; Rimbara E.; Mori S. Biochemical Characterization of Quinolinic Acid Phosphoribosyltransferase from Mycobacterium tuberculosis H37Rv and Inhibition of Its Activity by Pyrazinamide. PLoS One 2014, 9, e100062. 10.1371/journal.pone.0100062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon N. A.; Peterson N. D.; Rosen B. C.; Baughn A. D. Pantothenate and Pantetheine Antagonize the Antitubercular Activity of Pyrazinamide. Antimicrob. Agents Chemother. 2014, 58, 7258–7263. 10.1128/AAC.04028-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon N. A.; Peterson N. D.; Feaga H. A.; Keiler K. C.; Baughn A. D. Anti-Tubercular Activity of Pyrazinamide Is Independent of Trans-Translation and RpsA. Sci. Rep. 2017, 7, 6135. 10.1038/s41598-017-06415-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njire M.; Wang N.; Wang B.; Tan Y.; Cai X.; Liu Y.; Mugweru J.; Guo J.; Hameed H. M. A.; Tan S.; et al. Pyrazinoic Acid Inhibits a Bifunctional Enzyme in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2017, 61, e00070–17. 10.1128/AAC.00070-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorpio A.; Zhang Y. Mutations in PncA, a Gene Encoding Pyrazinamidase/Nicotinamidase, Cause Resistance to the Antituberculous Drug Pyrazinamide in Tubercle Bacillus. Nat. Med. 1996, 2, 662–667. 10.1038/nm0696-662. [DOI] [PubMed] [Google Scholar]
- Via L. E.; Savic R.; Weiner D. M.; Zimmerman M. D.; Prideaux B.; Irwin S. M.; Lyon E.; O’Brien P.; Gopal P.; Eum S.; et al. Host-Mediated Bioactivation of Pyrazinamide: Implications for Efficacy, Resistance, and Therapeutic Alternatives. ACS Infect. Dis. 2015, 1, 203–214. 10.1021/id500028m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K.; Kilburn J. O. Inhibition of Synthesis of Arabinogalactan by Ethambutol in Mycobacterium smegmatis. Antimicrob. Agents Chemother. 1989, 33, 1493–1499. 10.1128/AAC.33.9.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobino A.; Piccaro G.; Giannoni F.; Mustazzolu A.; Fattorini L. Activity of Drugs against Dormant Mycobacterium tuberculosis. Int. J. Mycobacteriol. 2016, 5, S94–S95. 10.1016/j.ijmyco.2016.09.061. [DOI] [PubMed] [Google Scholar]
- DeBarber A. E.; Mdluli K.; Bosman M.; Bekker L.-G.; Barry C. E. Ethionamide Activation and Sensitivity in Multidrug-Resistant Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 9677–9682. 10.1073/pnas.97.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willand N.; Dirié B.; Carette X.; Bifani P.; Singhal A.; Desroses M.; Leroux F.; Willery E.; Mathys V.; Déprez-Poulain R. Synthetic EthR Inhibitors Boost Antituberculous Activity of Ethionamide. Nat. Med. 2009, 15, 537–544. 10.1038/nm.1950. [DOI] [PubMed] [Google Scholar]
- Blondiaux N.; Moune M.; Desroses M.; Frita R.; Flipo M.; Mathys V.; Soetaert K.; Kiass M.; Delorme V.; Djaout K. Reversion of Antibiotic Resistance in Mycobacterium tuberculosis by Spiroisoxazoline Smart-420. Science 2017, 355, 1206–1211. 10.1126/science.aag1006. [DOI] [PubMed] [Google Scholar]
- Feng Z.; Barletta R. G. Roles of Mycobacterium smegmatis D-Alanine: D-Alanine Ligase and D-Alanine Racemase in the Mechanisms of Action of and Resistance to the Peptidoglycan Inhibitor D-Cycloserine. Antimicrob. Agents Chemother. 2003, 47, 283–291. 10.1128/AAC.47.1.283-291.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins C. A.; Cohen K. A.; Munsamy V.; Abeel T.; Maharaj K.; Walker B. J.; Shea T. P.; Almeida D. V.; Manson A. L.; Salazar A. Genomic and Functional Analyses of Mycobacterium tuberculosis Strains Implicate ald in D-Cycloserine Resistance. Nat. Genet. 2016, 48, 544–551. 10.1038/ng.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shandil R. K.; Jayaram R.; Kaur P.; Gaonkar S.; Suresh B.; Mahesh B.; Jayashree R.; Nandi V.; Bharath S.; Balasubramanian V. Moxifloxacin, Ofloxacin, Sparfloxacin, and Ciprofloxacin against Mycobacterium tuberculosis: Evaluation of in vitro and Pharmacodynamic Indices That Best Predict in vivo Efficacy. Antimicrob. Agents Chemother. 2007, 51, 576–582. 10.1128/AAC.00414-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald P. R.; Sirgel F. A.; Venter A.; Smit E.; Parkin D. P.; Van de Wal B. W.; Dore C. J.; Mitchison D. A. The Early Bactericidal Activity of Streptomycin. Int. J. Tuberc. Lung Dis. 2002, 6, 693–698. [PubMed] [Google Scholar]
- Heifets L.; Lindholm-Levy P. Comparison of Bactericidal Activities of Streptomycin, Amikacin, Kanamycin, and Capreomycin against Mycobacterium avium and M. tuberculosis. Antimicrob. Agents Chemother. 1989, 33, 1298–1301. 10.1128/AAC.33.8.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf K.; Hayley V.; Speer A.; Wolschendorf F.; Niederweis M.; Kutsch O.; Sun J. A Macrophage Infection Model to Predict Drug Efficacy against Mycobacterium tuberculosis. Assay Drug Dev. Technol. 2016, 14, 345–354. 10.1089/adt.2016.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechartier B.; Cole S. T. Mode of Action of Clofazimine and Combination Therapy with Benzothiazinones against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 4457–4463. 10.1128/AAC.00395-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano T.; Kassovska-Bratinova S.; Teh J. S.; Winkler J.; Sullivan K.; Isaacs A.; Schechter N. M.; Rubin H. Reduction of Clofazimine by Mycobacterial Type 2 NADH: Quinone Oxidoreductase a Pathway for the Generation of Bactericidal Levels of Reactive Oxygen Species. J. Biol. Chem. 2011, 286, 10276–10287. 10.1074/jbc.M110.200501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Lee J.; Carroll M. W.; Choi H.; Min S.; Song T.; Via L. E.; Goldfeder L. C.; Kang E.; Jin B. Linezolid for Treatment of Chronic Extensively Drug-Resistant Tuberculosis. N. Engl. J. Med. 2012, 367, 1508–1518. 10.1056/NEJMoa1201964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S.; Biukovic G.; Gruber G.; Dick T. Bedaquiline Targets the Epsilon Subunit of Mycobacterial F-ATP Synthase. Antimicrob. Agents Chemother. 2016, 60, 6977–6979. 10.1128/AAC.01291-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andries K.; Verhasselt P.; Guillemont J.; Göhlmann H. W.; Neefs J.-M.; Winkler H.; Van Gestel J.; Timmerman P.; Zhu M.; Lee E. A Diarylquinoline Drug Active on the ATP Synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- Feng X.; Zhu W.; Schurig-Briccio L. A.; Lindert S.; Shoen C.; Hitchings R.; Li J.; Wang Y.; Baig N.; Zhou T.; et al. Antiinfectives Targeting Enzymes and the Proton Motive Force. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E7073–7082. 10.1073/pnas.1521988112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul A.; Vranckx L.; Dendouga N.; Balemans W.; Van den Wyngaert I.; Vergauwen K.; Gohlmann H. W.; Willebrords R.; Poncelet A.; Guillemont J.; et al. Diarylquinolines Are Bactericidal for Dormant Mycobacteria as a Result of Disturbed ATP Homeostasis. J. Biol. Chem. 2008, 283, 25273–25280. 10.1074/jbc.M803899200. [DOI] [PubMed] [Google Scholar]
- Stover C. K.; Warrener P.; VanDevanter D. R.; Sherman D. R. A Small-Molecule Nitroimidazopyran Drug Candidate for the Treatment of Tuberculosis. Nature 2000, 405, 962–966. 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- Singh R.; Manjunatha U.; Boshoff H. I.; Ha Y. H.; Niyomrattanakit P.; Ledwidge R.; Dowd C. S.; Lee I. Y.; Kim P.; Zhang L. Pa-824 Kills Nonreplicating Mycobacterium tuberculosis by Intracellular NO Release. Science 2008, 322, 1392–1395. 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha U. H.; Boshoff H.; Dowd C. S.; Zhang L.; Albert T. J.; Norton J. E.; Daniels L.; Dick T.; Pang S. S.; Barry C. E. Identification of a Nitroimidazo-Oxazine-Specific Protein Involved in PA-824 Resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 431–436. 10.1073/pnas.0508392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha U.; Boshoff H. I.; Barry C. E. The Mechanism of Action of PA-824: Novel Insights from Transcriptional Profiling. Commun. Integr. Biol. 2009, 2, 215–218. 10.4161/cib.2.3.7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M.; Hashizume H.; Tomishige T.; Kawasaki M.; Tsubouchi H.; Sasaki H.; Shimokawa Y.; Komatsu M. OPC-67683, a Nitro-Dihydro-Imidazooxazole Derivative with Promising Action against Tuberculosis in vitro and in Mice. PLoS Med. 2006, 3, e466. 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diacon A.; Dawson R.; Hanekom M.; Narunsky K.; Venter A.; Hittel N.; Geiter L.; Wells C.; Paccaly A.; Donald P. Early Bactericidal Activity of Delamanid (OPC-67683) in Smear-Positive Pulmonary Tuberculosis Patients. Int. J. Tuberc. Lung Dis. 2011, 15, 949–954. 10.5588/ijtld.10.0616. [DOI] [PubMed] [Google Scholar]
- Gler M. T.; Skripconoka V.; Sanchez-Garavito E.; Xiao H.; Cabrera-Rivero J. L.; Vargas-Vasquez D. E.; Gao M.; Awad M.; Park S.-K.; Shim T. S. Delamanid for Multidrug-Resistant Pulmonary Tuberculosis. N. Engl. J. Med. 2012, 366, 2151–2160. 10.1056/NEJMoa1112433. [DOI] [PubMed] [Google Scholar]
- Katsuno K.; Burrows J. N.; Duncan K.; van Huijsduijnen R. H.; Kaneko T.; Kita K.; Mowbray C. E.; Schmatz D.; Warner P.; Slingsby B. T. Hit and Lead Criteria in Drug Discovery for Infectious Diseases of the Developing World. Nat. Rev. Drug Discovery 2015, 14, 751–758. 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- Budha N. R.; Lee R. E.; Meibohm B. Biopharmaceutics, Pharmacokinetics and Pharmacodynamics of Antituberculosis Drugs. Curr. Med. Chem. 2008, 15, 809–825. 10.2174/092986708783955509. [DOI] [PubMed] [Google Scholar]
- Vaddady P. K.; Lee R. E.; Meibohm B. In Vitro Pharmacokinetic/Pharmacodynamic Models in Anti-Infective Drug Development: Focus on TB. Future Med. Chem. 2010, 2, 1355–1369. 10.4155/fmc.10.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G. R.; Nuermberger E. Pharmacokinetics and Pharmacodynamics in the Development of Anti-Tuberculosis Drugs. Tuberculosis 2008, 88, S65–S74. 10.1016/S1472-9792(08)70037-4. [DOI] [PubMed] [Google Scholar]
- Combalbert J.; Fabre I.; Fabre G.; Dalet I.; Derancourt J.; Cano J. P.; Maurel P. Metabolism of Cyclosporin A. IV. Purification and Identification of the Rifampicin-Inducible Human Liver Cytochrome P-450 (Cyclosporin A Oxidase) as a Product of P450IIIA Gene Subfamily. Drug Metab. Dispos. 1989, 17, 197–207. [PubMed] [Google Scholar]
- La Porte C.; Colbers E.; Bertz R.; Voncken D.; Wikstrom K.; Boeree M.; Koopmans P.; Hekster Y.; Burger D. Pharmacokinetics of Adjusted-Dose Lopinavir-Ritonavir Combined with Rifampin in Healthy Volunteers. Antimicrob. Agents Chemother. 2004, 48, 1553–1560. 10.1128/AAC.48.5.1553-1560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronacher K.; van Crevel R.; Critchley J. A.; Bremer A. A.; Schlesinger L. S.; Kapur A.; Basaraba R.; Kornfeld H.; Restrepo B. I. Defining a Research Agenda to Address the Converging Epidemics of Tuberculosis and Diabetes: Part 2: Underlying Biologic Mechanisms. Chest 2017, 152, 174–180. 10.1016/j.chest.2017.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley K. E.; Chaisson R. E. Tuberculosis and Diabetes Mellitus: Convergence of Two Epidemics. Lancet Infect. Dis. 2009, 9, 737–746. 10.1016/S1473-3099(09)70282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi M.; Backman J. T.; Neuvonen M.; Neuvonen P. J.; Kivistö K. T. Effects of Rifampin on the Pharmacokinetics and Pharmacodynamics of Glyburide and Glipizide. Clin. Pharmacol. Ther. 2001, 69, 400–406. 10.1067/mcp.2001.115822. [DOI] [PubMed] [Google Scholar]
- Niemi M.; Backman J. T.; Neuvonen P. J. Effects of Trimethoprim and Rifampin on the Pharmacokinetics of the Cytochrome P450 2C8 Substrate Rosiglitazone. Clin. Pharmacol. Ther. 2004, 76, 239–249. 10.1016/j.clpt.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Niemi M.; Backman J. T.; Neuvonen M.; Neuvonen P. J. Effect of Rifampicin on the Pharmacokinetics and Pharmacodynamics of Nateglinide in Healthy Subjects. Br. J. Clin. Pharmacol. 2003, 56, 427–432. 10.1046/j.1365-2125.2003.01884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatorp V.; Hansen K. T.; Thomsen M. S. Influence of Drugs Interacting with Cyp3A4 on the Pharmacokinetics, Pharmacodynamics, and Safety of the Prandial Glucose Regulator Repaglinide. J. Clin. Pharmacol. 2003, 43, 649–660. 10.1177/0091270003253704. [DOI] [PubMed] [Google Scholar]
- Sassetti C. M.; Boyd D. H.; Rubin E. J. Comprehensive Identification of Conditionally Essential Genes in Mycobacteria. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 12712–12717. 10.1073/pnas.231275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti C. M.; Boyd D. H.; Rubin E. J. Genes Required for Mycobacterial Growth Defined by High Density Mutagenesis. Mol. Microbiol. 2003, 48, 77–84. 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Wei J.-R.; Rubin E. J. The Many Roads to Essential Genes. Tuberculosis 2008, 88, S19–S24. 10.1016/S1472-9792(08)70033-7. [DOI] [PubMed] [Google Scholar]
- Hingley-Wilson S. M.; Sambandamurthy V. K.; Jacobs W. R. Survival Perspectives from the World’s Most Successful Pathogen, Mycobacterium tuberculosis. Nat. Immunol. 2003, 4, 949–955. 10.1038/ni981. [DOI] [PubMed] [Google Scholar]
- Evans J. C.; Mizrahi V. The Application of Tetracyclineregulated Gene Expression Systems in the Validation of Novel Drug Targets in Mycobacterium tuberculosis. Front. Microbiol. 2015, 6, 812. 10.3389/fmicb.2015.00812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J.-R.; Krishnamoorthy V.; Murphy K.; Kim J.-H.; Schnappinger D.; Alber T.; Sassetti C. M.; Rhee K. Y.; Rubin E. J. Depletion of Antibiotic Targets Has Widely Varying Effects on Growth. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 4176–4181. 10.1073/pnas.1018301108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H.; O’Brien K. M.; Sharma R.; Boshoff H. I.; Rehren G.; Chakraborty S.; Wallach J. B.; Monteleone M.; Wilson D. J.; Aldrich C. C.; et al. A Genetic Strategy to Identify Targets for the Development of Drugs That Prevent Bacterial Persistence. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 19095–19100. 10.1073/pnas.1315860110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkup G. K.; You Z.; Ross P. L.; Allen E. K.; Daryaee F.; Hale M. R.; O’Donnell J.; Ehmann D. E.; Schuck V. J.; Buurman T. Translating Slow-Binding Inhibition Kinetics into Cellular and in vivo Effects. Nat. Chem. Biol. 2015, 11, 416–423. 10.1038/nchembio.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strebhardt K.; Ullrich A. Paul Ehrlich’s Magic Bullet Concept: 100 Years of Progress. Nat. Rev. Cancer 2008, 8, 473–480. 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- Sharma U. Current Possibilities and Unresolved Issues of Drug Target Validation in Mycobacterium tuberculosis. Expert Opin. Drug Discovery 2011, 6, 1171–1186. 10.1517/17460441.2011.626763. [DOI] [PubMed] [Google Scholar]
- Singh S. B. Confronting the Challenges of Discovery of Novel Antibacterial Agents. Bioorg. Med. Chem. Lett. 2014, 24, 3683–3689. 10.1016/j.bmcl.2014.06.053. [DOI] [PubMed] [Google Scholar]
- Brennan P. J.; Nikaido H. The Envelope of Mycobacteria. Annu. Rev. Biochem. 1995, 64, 29–63. 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- O’Shea R.; Moser H. E. Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 2008, 51, 2871–2878. 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Drug-Like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- Schmalstieg A. M.; Srivastava S.; Belkaya S.; Deshpande D.; Meek C.; Leff R.; van Oers N. S.; Gumbo T. The Antibiotic Resistance Arrow of Time: Efflux Pump Induction Is a General First Step in the Evolution of Mycobacterial Drug Resistance. Antimicrob. Agents Chemother. 2012, 56, 4806–4815. 10.1128/AAC.05546-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balganesh M.; Dinesh N.; Sharma S.; Kuruppath S.; Nair A. V.; Sharma U. Efflux Pumps of Mycobacterium tuberculosis Play a Significant Role in Antituberculosis Activity of Potential Drug Candidates. Antimicrob. Agents Chemother. 2012, 56, 2643–2651. 10.1128/AAC.06003-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi E. D.; Aínsa J. A.; Riccardi G. Role of Mycobacterial Efflux Transporters in Drug Resistance: An Unresolved Question. FEMS Microbiol. Rev. 2006, 30, 36–52. 10.1111/j.1574-6976.2005.00002.x. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Pompliano D. L.; Meek T. D. Drug−Target Residence Time and Its Implications for Lead Optimization. Nat. Rev. Drug Discovery 2006, 5, 730–739. 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Lu H.; Tonge P. J. Drug–Target Residence Time: Critical Information for Lead Optimization. Curr. Opin. Chem. Biol. 2010, 14, 467–474. 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A. The Dynamics of Drug−Target Interactions: Drug−Target Residence Time and Its Impact on Efficacy and Safety. Expert Opin. Drug Discovery 2010, 5, 305–310. 10.1517/17460441003677725. [DOI] [PubMed] [Google Scholar]
- Tonge P. J. Drug–Target Kinetics in Drug Discovery. ACS Chem. Neurosci. 2018, 9, 29–39. 10.1021/acschemneuro.7b00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung A. W.; Silvestre H. L.; Wen S.; Ciulli A.; Blundell T. L.; Abell C. Application of Fragment Growing and Fragment Linking to the Discovery of Inhibitors of Mycobacterium tuberculosis Pantothenate Synthetase. Angew. Chem., Int. Ed. 2009, 48, 8452–8456. 10.1002/anie.200903821. [DOI] [PubMed] [Google Scholar]
- Kavanagh M. E.; Chenge J.; Zoufir A.; McLean K. J.; Coyne A. G.; Bender A.; Munro A. W.; Abell C. Fragment Profiling Approach to Inhibitors of the Orphan M. tuberculosis P450 Cyp144A1. Biochemistry 2017, 56, 1559–1572. 10.1021/acs.biochem.6b00954. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Coyne A. G.; Hudson S. A.; Abell C. Fragment-Based Approaches in Drug Discovery and Chemical Biology. Biochemistry 2012, 51, 4990–5003. 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- Sledz P.; Silvestre H. L.; Hung A. W.; Ciulli A.; Blundell T. L.; Abell C. Optimization of the Interligand Overhauser Effect for Fragment Linking: Application to Inhibitor Discovery against Mycobacterium tuberculosis Pantothenate Synthetase. J. Am. Chem. Soc. 2010, 132, 4544–4545. 10.1021/ja100595u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal P.; Dick T. Reactive Dirty Fragments: Implications for Tuberculosis Drug Discovery. Curr. Opin. Microbiol. 2014, 21, 7–12. 10.1016/j.mib.2014.06.015. [DOI] [PubMed] [Google Scholar]
- Moreira W.; Lim J. J.; Yeo S. Y.; Ramanujulu P. M.; Dymock B. W.; Dick T. Fragment-Based Whole Cell Screen Delivers Hits against M. tuberculosis and Non-Tuberculous Mycobacteria. Front. Microbiol. 2016, 7, 1392. 10.3389/fmicb.2016.01392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung G.; Aktar Z.; Jackson S.; Duncan K. High-Throughput Screen for Detecting Antimycobacterial Agents. Antimicrob. Agents Chemother. 1995, 39, 2235–2238. 10.1128/AAC.39.10.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arain T. M.; Resconi A. E.; Hickey M. J.; Stover C. K. Bioluminescence Screening in vitro (Bio-Siv) Assays for High-Volume Antimycobacterial Drug Discovery. Antimicrob. Agents Chem. 1996, 40, 1536–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier T.; Eiglmeier K.; Camus J.-C.; Medina N.; Mansoor H.; Pryor M.; Duthoy S.; Grondin S.; Lacroix C.; Monsempe C. The Complete Genome Sequence of Mycobacterium bovis. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7877–7882. 10.1073/pnas.1130426100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan A.; Padiadpu J.; Baloni P.; Chandra N. Complete Genome Sequences of a Mycobacterium smegmatis Laboratory Strain (MC2 155) and Isoniazid-Resistant (4XR1/R2) Mutant Strains. Genome Announc. 2015, 3, e01520–01514. 10.1128/genomeA.01520-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altaf M.; Miller C. H.; Bellows D. S.; O’Toole R. Evaluation of the Mycobacterium smegmatis and BCG Models for the Discovery of Mycobacterium tuberculosis Inhibitors. Tuberculosis 2010, 90, 333–337. 10.1016/j.tube.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Ballell L.; Bates R. H.; Young R. J.; Alvarez-Gomez D.; Alvarez-Ruiz E.; Barroso V.; Blanco D.; Crespo B.; Escribano J.; González R. Fueling Open-Source Drug Discovery: 177 Small-Molecule Leads against Tuberculosis. ChemMedChem 2013, 8, 313–321. 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S. A.; Grant S. S.; Kawate T.; Iwase N.; Shimizu M.; Wivagg C.; Silvis M.; Kazyanskaya E.; Aquadro J.; Golas A.; et al. Identification of Novel Inhibitors of M. tuberculosis Growth Using Whole Cell Based High-Throughput Screening. ACS Chem. Biol. 2012, 7, 1377–1384. 10.1021/cb300151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace R. J. Jr; Nash D. R.; Tsukamura M.; Blacklock Z. M.; Silcox V. A. Human Disease Due to Mycobacterium smegmatis. J. Infect. Dis. 1988, 158, 52–59. 10.1093/infdis/158.1.52. [DOI] [PubMed] [Google Scholar]
- Chaturvedi V.; Dwivedi N.; Tripathi R. P.; Sinha S. Evaluation of Mycobacterium smegmatis as a Possible Surrogate Screen for Selecting Molecules Active against Multi-Drug Resistant Mycobacterium tuberculosis. J. Gen. Appl. Microbiol. 2007, 53, 333–337. 10.2323/jgam.53.333. [DOI] [PubMed] [Google Scholar]
- Sampson S. L.; Dascher C. C.; Sambandamurthy V. K.; Russell R. G.; Jacobs W. R.; Bloom B. R.; Hondalus M. K. Protection Elicited by a Double Leucine and Pantothenate Auxotroph of Mycobacterium tuberculosis in Guinea Pigs. Infect. Immun. 2004, 72, 3031–3037. 10.1128/IAI.72.5.3031-3037.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Zhang M.; Zhu L.; Liao R. P.; Mutai C.; Hafsat S.; Sherman D. R.; Wang M.-W. High-Throughput Screening and Sensitized Bacteria Identify an M. tuberculosis Dihydrofolate Reductase Inhibitor with Whole Cell Activity. PLoS One 2012, 7, e39961. 10.1371/journal.pone.0039961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams G. L.; Kumar A.; Savvi S.; Hung A. W.; Wen S.; Abell C.; Barry C. E.; Sherman D. R.; Boshoff H. I.; Mizrahi V. Pathway-Selective Sensitization of Mycobacterium tuberculosis for Target-Based Whole-Cell Screening. Chem. Biol. 2012, 19, 844–854. 10.1016/j.chembiol.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisa S.; Blokpoel M. C.; Robertson B. D.; Tyndall J. D.; Lun S.; Bishai W. R.; O’Toole R. Targeting the Chromosome Partitioning Protein ParA in Tuberculosis Drug Discovery. J. Antimicrob. Chemother. 2010, 65, 2347–2358. 10.1093/jac/dkq311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka M. S. Jr.; Jacobs W. R. Jr Comparison of the Construction of Unmarked Deletion Mutations in Mycobacterium smegmatis, Mycobacterium bovis Bacillus Calmette-Guerin, and Mycobacterium tuberculosis H37Rv by Allelic Exchange. J. Bacteriol. 1999, 181, 4780–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka M. S. Jr.; Chen B.; Kelley C. L.; Collins F. M.; Jacobs W. R. Jr Vaccine Efficacy of a Lysine Auxotroph of Mycobacterium tuberculosis. Infect. Immun. 2003, 71, 4190–4192. 10.1128/IAI.71.7.4190-4192.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Elias E. J.; McKinney J. D. Mycobacterium tuberculosis Isocitrate Lyases 1 and 2 Are Jointly Required for in vivo Growth and Virulence. Nat. Med. 2005, 11, 638–644. 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney J. D.; Honer zu Bentrup K.; Munoz-Elias E. J.; Miczak A.; Chen B.; Chan W. T.; Swenson D.; Sacchettini J. C.; Jacobs W. R. Jr.; Russell D. G. Persistence of Mycobacterium tuberculosis in Macrophages and Mice Requires the Glyoxylate Shunt Enzyme Isocitrate Lyase. Nature 2000, 406, 735–738. 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- Bonnett S. A.; Ollinger J.; Chandrasekera S.; Florio S.; O’Malley T.; Files M.; Jee J.-A.; Ahn J.; Casey A.; Ovechkina Y. A Target-Based Whole Cell Screen Approach to Identify Potential Inhibitors of Mycobacterium tuberculosis Signal Peptidase. ACS Infect. Dis. 2016, 2, 893–902. 10.1021/acsinfecdis.6b00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollinger J.; Bailey M. A.; Moraski G. C.; Casey A.; Florio S.; Alling T.; Miller M. J.; Parish T. A Dual Read-out Assay to Evaluate the Potency of Compounds Active against Mycobacterium tuberculosis. PLoS One 2013, 8, e60531. 10.1371/journal.pone.0060531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. W.; Casalena D. E.; Wilson D. J.; Dai R.; Nag P. P.; Liu F.; Boyce J. P.; Bittker J. A.; Schreiber S. L.; Finzel B. C.; et al. Target-Based Identification of Whole-Cell Active Inhibitors of Biotin Biosynthesis in Mycobacterium tuberculosis. Chem. Biol. 2015, 22, 76–86. 10.1016/j.chembiol.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann S.; Ploux O. 7, 8-Diaminoperlargonic Acid Aminotransferase from Mycobacterium tuberculosis, a Potential Therapeutic Target. FEBS J. 2006, 273, 4778–4789. 10.1111/j.1742-4658.2006.05479.x. [DOI] [PubMed] [Google Scholar]
- Gibson K. J.; Lorimer G. H.; Rendina A. R.; Taylor W. S.; Cohen G.; Gatenby A. A.; Payne W. G.; Roe D. C.; Lockett B. A. Dethiobiotin Synthetase: The Carbonylation of 7, 8-Diaminononanoic Acid Proceeds Regiospecifically via the N7-Carbamate. Biochemistry 1995, 34, 10976–10984. 10.1021/bi00035a003. [DOI] [PubMed] [Google Scholar]
- Park S. W.; Klotzsche M.; Wilson D. J.; Boshoff H. I.; Eoh H.; Manjunatha U.; Blumenthal A.; Rhee K.; Barry C. E. III; Aldrich C. C. Evaluating the Sensitivity of Mycobacterium tuberculosis to Biotin Deprivation Using Regulated Gene Expression. PLoS Pathog. 2011, 7, e1002264. 10.1371/journal.ppat.1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Dawadi S.; Maize K. M.; Dai R.; Park S. W.; Schnappinger D.; Finzel B. C.; Aldrich C. C. Structure-Based Optimization of Pyridoxal 5′-Phosphate-Dependent Transaminase Enzyme (BioA) Inhibitors That Target Biotin Biosynthesis in Mycobacterium tuberculosis. J. Med. Chem. 2017, 60, 5507–5520. 10.1021/acs.jmedchem.7b00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahlan K.; Wilson R.; Kastrinsky D. B.; Arora K.; Nair V.; Fischer E.; Barnes S. W.; Walker J. R.; Alland D.; Barry C. E. SQ109 Targets MmpL3, a Membrane Transporter of Trehalose Monomycolate Involved in Mycolic Acid Donation to the Cell Wall Core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzegorzewicz A. E.; Pham H.; Gundi V. A.; Scherman M. S.; North E. J.; Hess T.; Jones V.; Gruppo V.; Born S. E.; Korduláková J. Inhibition of Mycolic Acid Transport across the Mycobacterium tuberculosis Plasma Membrane. Nat. Chem. Biol. 2012, 8, 334–341. 10.1038/nchembio.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remuiñán M. J.; Pérez-Herrán E.; Rullás J.; Alemparte C.; Martínez-Hoyos M.; Dow D. J.; Afari J.; Mehta N.; Esquivias J.; Jiménez E. Tetrahydropyrazolo [1, 5-a] Pyrimidine-3-Carboxamide and N-Benzyl-6’, 7’-Dihydrospiro [Piperidine-4, 4’-Thieno [3, 2-c] Pyran] Analogues with Bactericidal Efficacy against Mycobacterium tuberculosis Targeting MmpL3. PLoS One 2013, 8, e60933. 10.1371/journal.pone.0060933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S. P.; Lakshminarayana S. B.; Kondreddi R. R.; Herve M.; Camacho L. R.; Bifani P.; Kalapala S. K.; Jiricek J.; Ma N. L.; Tan B. H. Indolcarboxamide Is a Preclinical Candidate for Treating Multidrug-Resistant Tuberculosis. Sci. Transl. Med. 2013, 5, 214ra168. 10.1126/scitranslmed.3007355. [DOI] [PubMed] [Google Scholar]
- Sorrentino F.; del Rio R. G.; Zheng X.; Matilla J. P.; Gomez P. T.; Hoyos M. M.; Herran M. E. P.; Losana A. M.; Av-Gay Y. Development of an Intracellular Screen for New Compounds Able to Inhibit Mycobacterium tuberculosis Growth in Human Macrophages. Antimicrob. Agents Chemother. 2016, 60, 640–645. 10.1128/AAC.01920-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R.; Kumar P.; Parashar V.; Vilchèze C.; Veyron-Churlet R.; Freundlich J. S.; Barnes S. W.; Walker J. R.; Szymonifka M. J.; Marchiano E. Antituberculosis Thiophenes Define a Requirement for Pks13 in Mycolic Acid Biosynthesis. Nat. Chem. Biol. 2013, 9, 499–506. 10.1038/nchembio.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukheja P.; Kumar P.; Mittal N.; Li S. G.; Singleton E.; Russo R.; Perryman A. L.; Shrestha R.; Awasthi D.; Husain S.; et al. A Novel Small-Molecule Inhibitor of the Mycobacterium tuberculosis Demethylmenaquinone Methyltransferase MenG Is Bactericidal to Both Growing and Nutritionally Deprived Persister Cells. mBio 2017, 8, e02022-16. 10.1128/mBio.02022-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J. A.; Abrahams K. A.; Alemparte C.; Ghidelli-Disse S.; Rullas J.; Angulo-Barturen I.; Singh A.; Gurcha S. S.; Nataraj V.; Bethel S.; et al. THPP Target Assignment Reveals EchA6 as an Essential Fatty Acid Shuttle in Mycobacteria. Nature Microbiol. 2016, 1, 15006. 10.1038/nmicrobiol.2015.6. [DOI] [PubMed] [Google Scholar]
- StopTBPartnership “Working Group on New TB Drugs”, 2017 ; http://www.newtbdrugs.org/ accessed: 26 September 2017.
- Workshop “Critical Path to TB Drug Regimens”, 2017 ; http://www.cptrinitiative.org/wp-content/uploads/2017/05/Jeffrey_Hafkin_CPTR2017_JH.pdf accessed: 1 October 2017.
- Makarov V.; Lechartier B.; Zhang M.; Neres J.; van der Sar A. M.; Raadsen S. A.; Hartkoorn R. C.; Ryabova O. B.; Vocat A.; Decosterd L. A. Towards a New Combination Therapy for Tuberculosis with Next Generation Benzothiazinones. EMBO Mol. Med. 2014, 6, e201303575. 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefzer C.; Škovierová H.; Buroni S.; Bobovská A.; Nenci S.; Molteni E.; Pojer F.; Pasca M. R.; Makarov V.; Cole S. T. Benzothiazinones Are Suicide Inhibitors of Mycobacterial Decaprenylphosphoryl-β -D-Ribofuranose 2’-Oxidase DprE1. J. Am. Chem. Soc. 2012, 134, 912–915. 10.1021/ja211042r. [DOI] [PubMed] [Google Scholar]
- Trefzer C.; Rengifo-Gonzalez M.; Hinner M. J.; Schneider P.; Makarov V.; Cole S. T.; Johnsson K. Benzothiazinones: Prodrugs That Covalently Modify the Decaprenylphosphoryl-β-D-Ribose 2’-Epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc. 2010, 132, 13663–13665. 10.1021/ja106357w. [DOI] [PubMed] [Google Scholar]
- Wang F.; Sambandan D.; Halder R.; Wang J.; Batt S. M.; Weinrick B.; Ahmad I.; Yang P.; Zhang Y.; Kim J. Identification of a Small Molecule with Activity against Drug-Resistant and Persistent Tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E2510–E2517. 10.1073/pnas.1309171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier T.; Kapilashrami K.; Argyrou A.; Ioerger T. R.; Little D.; Murphy K. C.; Nandakumar M.; Park S.; Gold B.; Mi J. N-Methylation of a Bactericidal Compound as a Resistance Mechanism in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 4523–4530. 10.1073/pnas.1606590113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier T.; Martinez-Hoyos M.; Marin-Amieva M.; Colmenarejo G.; Porras-De Francisco E.; Alvarez-Pedraglio A. I.; Fraile-Gabaldon M. T.; Torres-Gomez P. A.; Lopez-Quezada L.; Gold B.; et al. Identification of Novel Anti-Mycobacterial Compounds by Screening a Pharmaceutical Small-Molecule Library against Nonreplicating Mycobacterium tuberculosis. ACS Infect. Dis. 2015, 1, 580–585. 10.1021/acsinfecdis.5b00025. [DOI] [PubMed] [Google Scholar]
- Shirude P. S.; Shandil R.; Sadler C.; Naik M.; Hosagrahara V.; Hameed S.; Shinde V.; Bathula C.; Humnabadkar V.; Kumar N. Azaindoles: Noncovalent DprE1 Inhibitors from Scaffold Morphing Efforts, Kill Mycobacterium tuberculosis and Are Efficacious in vivo. J. Med. Chem. 2013, 56, 9701–9708. 10.1021/jm401382v. [DOI] [PubMed] [Google Scholar]
- Christophe T.; Jackson M.; Jeon H. K.; Fenistein D.; Contreras-Dominguez M.; Kim J.; Genovesio A.; Carralot J.-P.; Ewann F.; Kim E. H. High Content Screening Identifies Decaprenyl-Phosphoribose 2’ Epimerase as a Target for Intracellular Antimycobacterial Inhibitors. PLoS Pathog. 2009, 5, e1000645. 10.1371/journal.ppat.1000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderVen B. C.; Fahey R. J.; Lee W.; Liu Y.; Abramovitch R. B.; Memmott C.; Crowe A. M.; Eltis L. D.; Perola E.; Deininger D. D.; et al. Novel Inhibitors of Cholesterol Degradation in Mycobacterium tuberculosis Reveal How the Bacterium’s Metabolism Is Constrained by the Intracellular Environment. PLoS Pathog. 2015, 11, e1004679. 10.1371/journal.ppat.1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams K. A.; Cox J. A.; Spivey V. L.; Loman N. J.; Pallen M. J.; Constantinidou C.; Fernandez R.; Alemparte C.; Remuinan M. J.; Barros D.; et al. Identification of Novel Imidazo[1,2-a]Pyridine Inhibitors Targeting M. tuberculosis Qcrb. PLoS One 2012, 7, e52951. 10.1371/journal.pone.0052951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pethe K.; Bifani P.; Jang J.; Kang S.; Park S.; Ahn S.; Jiricek J.; Jung J.; Jeon H. K.; Cechetto J. Discovery of Q203, a Potent Clinical Candidate for the Treatment of Tuberculosis. Nat. Med. 2013, 19, 1157–1160. 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- Zhao N.; Darby C. M.; Small J.; Bachovchin D. A.; Jiang X.; Burns-Huang K. E.; Botella H.; Ehrt S.; Boger D. L.; Anderson E. D. Target-Based Screen against a Periplasmic Serine Protease That Regulates Intrabacterial pH Homeostasis in Mycobacterium tuberculosis. ACS Chem. Biol. 2015, 10, 364–371. 10.1021/cb500746z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock F. L.; Mao W.; Yaremchuk A.; Tukalo M.; Crépin T.; Zhou H.; Zhang Y.-K.; Hernandez V.; Akama T.; Baker S. J. An Antifungal Agent Inhibits an Aminoacyl-tRNA Synthetase by Trapping tRNA in the Editing Site. Science 2007, 316, 1759–1761. 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- Baker S. J.; Zhang Y. K.; Akama T.; Lau A.; Zhou H.; Hernandez V.; Mao W.; Alley M. R.; Sanders V.; Plattner J. J. Discovery of a New Boron-Containing Antifungal Agent, 5-Fluoro-1,3-Dihydro-1-Hydroxy-2,1- Benzoxaborole (AN2690), for the Potential Treatment of Onychomycosis. J. Med. Chem. 2006, 49, 4447–4450. 10.1021/jm0603724. [DOI] [PubMed] [Google Scholar]
- Palencia A.; Li X.; Bu W.; Choi W.; Ding C. Z.; Easom E. E.; Feng L.; Hernandez V.; Houston P.; Liu L.; et al. Discovery of Novel Oral Protein Synthesis Inhibitors of Mycobacterium tuberculosis That Target Leucyl-tRNA Synthetase. Antimicrob. Agents Chemother. 2016, 60, 6271–6280. 10.1128/AAC.01339-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Hernandez V.; Rock F. L.; Choi W.; Mak Y. S. L.; Mohan M.; Mao W.; Zhou Y.; Easom E. E.; Plattner J. J.; et al. Discovery of a Potent and Specific M. tuberculosis Leucyl-tRNA Synthetase Inhibitor: (S)-3-(Aminomethyl)-4-Chloro-7-(2-Hydroxyethoxy)Benzo[c][1,2]Oxaborol-1(3H)-ol (GSK656). J. Med. Chem. 2017, 60, 8011–8026. 10.1021/acs.jmedchem.7b00631. [DOI] [PubMed] [Google Scholar]
- Mak P. A.; Rao S. P.; Ping Tan M.; Lin X.; Chyba J.; Tay J.; Ng S. H.; Tan B. H.; Cherian J.; Duraiswamy J.; et al. A High-Throughput Screen to Identify Inhibitors of ATP Homeostasis in Non-Replicating Mycobacterium tuberculosis. ACS Chem. Biol. 2012, 7, 1190–1197. 10.1021/cb2004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby C. M.; Ingolfsson H. I.; Jiang X.; Shen C.; Sun M.; Zhao N.; Burns K.; Liu G.; Ehrt S.; Warren J. D.; et al. Whole Cell Screen for Inhibitors of pH Homeostasis in Mycobacterium tuberculosis. PLoS One 2013, 8, e68942. 10.1371/journal.pone.0068942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early J. V.; Casey A.; Martinez-Grau M. A.; Gonzalez Valcarcel I. C.; Vieth M.; Ollinger J.; Bailey M. A.; Alling T.; Files M.; Ovechkina Y.; et al. Oxadiazoles Have Butyrate-Specific Conditional Activity against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2016, 60, 3608–3616. 10.1128/AAC.02896-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues Felix C.; Gupta R.; Geden S.; Roberts J.; Winder P.; Pomponi S. A.; Diaz M. C.; Reed J. K.; Wright A. E.; Rohde K. H. Selective Killing of Dormant Mycobacterium tuberculosis by Marine Natural Products. Antimicrob. Agents Chemother. 2017, 61, e00743–17. 10.1128/AAC.00743-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandal O. H.; Pierini L. M.; Schnappinger D.; Nathan C. F.; Ehrt S. A Membrane Protein Preserves Intrabacterial pH in Intraphagosomal Mycobacterium tuberculosis. Nat. Med. 2008, 14, 849–854. 10.1038/nm.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira W.; Ngan G. J.; Low J. L.; Poulsen A.; Chia B. C.; Ang M. J.; Yap A.; Fulwood J.; Lakshmanan U.; Lim J. Target Mechanism-Based Whole-Cell Screening Identifies Bortezomib as an Inhibitor of Caseinolytic Protease in Mycobacteria. mBio 2015, 6, e00253–00215. 10.1128/mBio.00253-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju R. M.; Jedrychowski M. P.; Wei J. R.; Pinkham J. T.; Park A. S.; O’Brien K.; Rehren G.; Schnappinger D.; Gygi S. P.; Rubin E. J. Post-Translational Regulation Via Clp Protease Is Critical for Survival of Mycobacterium tuberculosis. PLoS Pathog. 2014, 10, e1003994. 10.1371/journal.ppat.1003994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybniker J.; Chen J. M.; Sala C.; Hartkoorn R. C.; Vocat A.; Benjak A.; Boy-Rottger S.; Zhang M.; Szekely R.; Greff Z.; et al. Anticytolytic Screen Identifies Inhibitors of Mycobacterial Virulence Protein Secretion. Cell Host Microbe 2014, 16, 538–548. 10.1016/j.chom.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Pym A. S.; Brodin P.; Brosch R.; Huerre M.; Cole S. T. Loss of RD1 Contributed to the Attenuation of the Live Tuberculosis Vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol. Microbiol. 2002, 46, 709–717. 10.1046/j.1365-2958.2002.03237.x. [DOI] [PubMed] [Google Scholar]
- Alland D.; Steyn A. J.; Weisbrod T.; Aldrich K.; Jacobs W. R. Characterization of the Mycobacterium tuberculosis iniBAC Promoter, a Promoter That Responds to Cell Wall Biosynthesis Inhibition. J. Bacteriol. 2000, 182, 1802–1811. 10.1128/JB.182.7.1802-1811.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R. E.; Protopopova M.; Crooks E.; Slayden R. A.; Terrot M.; Barry C. E. Combinatorial Lead Optimization of [1, 2]-Diamines Based on Ethambutol as Potential Antituberculosis Preclinical Candidates. J. Comb. Chem. 2003, 5, 172–187. 10.1021/cc020071p. [DOI] [PubMed] [Google Scholar]
- Boshoff H. I.; Myers T. G.; Copp B. R.; McNeil M. R.; Wilson M. A.; Barry C. E. 3rd The Transcriptional Responses of Mycobacterium tuberculosis to Inhibitors of Metabolism: Novel Insights into Drug Mechanisms of Action. J. Biol. Chem. 2004, 279, 40174–40184. 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
- Kana B. D.; Weinstein E. A.; Avarbock D.; Dawes S. S.; Rubin H.; Mizrahi V. Characterization of the cydAB-Encoded Cytochrome bd Oxidase from Mycobacterium smegmatis. J. Bacteriol. 2001, 183, 7076–7086. 10.1128/JB.183.24.7076-7086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A. C.; Kishony R. Opposing Effects of Target Overexpression Reveal Drug Mechanisms. Nat. Commun. 2014, 5, 4296. 10.1038/ncomms5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron P.; Vaubourgeix J.; Poquet Y.; Levillain F.; Botanch C.; Bardou F.; Daffe M.; Emile J. F.; Marchou B.; Cardona P. J.; et al. Foamy Macrophages from Tuberculous Patients’ Granulomas Constitute a Nutrient-Rich Reservoir for M. tuberculosis Persistence. PLoS Pathog. 2008, 4, e1000204. 10.1371/journal.ppat.1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel J.; Deb C.; Dubey V. S.; Sirakova T. D.; Abomoelak B.; Morbidoni H. R.; Kolattukudy P. E. Induction of a Novel Class of Diacylglycerol Acyltransferases and Triacylglycerol Accumulation in Mycobacterium tuberculosis as It Goes into a Dormancy-Like State in Culture. J. Bacteriol. 2004, 186, 5017–5030. 10.1128/JB.186.15.5017-5030.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb C.; Daniel J.; Sirakova T. D.; Abomoelak B.; Dubey V. S.; Kolattukudy P. E. A Novel Lipase Belonging to the Hormone-Sensitive Lipase Family Induced under Starvation to Utilize Stored Triacylglycerol in Mycobacterium tuberculosis. J. Biol. Chem. 2006, 281, 3866–3875. 10.1074/jbc.M505556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal W.; Bloch H. Biochemical Differentiation of Mycobacterium tuberculosis Grown in vivo and in vitro. J. Bacteriol. 1956, 72, 132–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Elias E. J.; McKinney J. D. Carbon Metabolism of Intracellular Bacteria. Cell. Microbiol. 2006, 8, 10–22. 10.1111/j.1462-5822.2005.00648.x. [DOI] [PubMed] [Google Scholar]
- Wipperman M. F.; Sampson N. S.; Thomas S. T. Pathogen Roid Rage: Cholesterol Utilization by Mycobacterium tuberculosis. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 269. 10.3109/10409238.2014.895700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry C. E.; Boshoff H. I.; Dartois V.; Dick T.; Ehrt S.; Flynn J.; Schnappinger D.; Wilkinson R. J.; Young D. The Spectrum of Latent Tuberculosis: Rethinking the Biology and Intervention Strategies. Nat. Rev. Microbiol. 2009, 7, 845–855. 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B.; Nathan C. Targeting Phenotypically Tolerant Mycobacterium tuberculosis. Microbiol. Spectr. 2017, 5, 0031–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant S. S.; Kaufmann B. B.; Chand N. S.; Haseley N.; Hung D. T. Eradication of Bacterial Persisters with Antibiotic-Generated Hydroxyl Radicals. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 12147–12152. 10.1073/pnas.1203735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne L. G.; Hayes L. G. An in vitro Model for Sequential Study of Shiftdown of Mycobacterium tuberculosis through Two Stages of Nonreplicating Persistence. Infect. Immun. 1996, 64, 2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.; Lee H. S.; Franzblau S. Microplate Alamar Blue Assay (MABA) and Low Oxygen Recovery Assay (LORA) for Mycobacterium tuberculosis. Methods Mol. Biol. 2015, 1285, 281–292. 10.1007/978-1-4939-2450-9_17. [DOI] [PubMed] [Google Scholar]
- Cho S. H.; Warit S.; Wan B.; Hwang C. H.; Pauli G. F.; Franzblau S. G. Low-Oxygen-Recovery Assay for High-Throughput Screening of Compounds against Nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1380–1385. 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant S. S.; Kawate T.; Nag P. P.; Silvis M. R.; Gordon K.; Stanley S. A.; Kazyanskaya E.; Nietupski R.; Golas A.; Fitzgerald M.; et al. Identification of Novel Inhibitors of Nonreplicating Mycobacterium tuberculosis Using a Carbon Starvation Model. ACS Chem. Biol. 2013, 8, 2224–2234. 10.1021/cb4004817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarathy J.; Dartois V.; Dick T.; Gengenbacher M. Reduced Drug Uptake in Phenotypically Resistant Nutrient-Starved Nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 1648–1653. 10.1128/AAC.02202-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts J. C.; Lukey P. T.; Robb L. C.; McAdam R. A.; Duncan K. Evaluation of a Nutrient Starvation Model of Mycobacterium tuberculosis Persistence by Gene and Protein Expression Profiling. Mol. Microbiol. 2002, 43, 717–731. 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- Xie Z.; Siddiqi N.; Rubin E. J. Differential Antibiotic Susceptibilities of Starved Mycobacterium tuberculosis Isolates. Antimicrob. Agents Chemother. 2005, 49, 4778–4780. 10.1128/AAC.49.11.4778-4780.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl J. L.; Kraus C. N.; Boshoff H. I.; Doan B.; Foley K.; Avarbock D.; Kaplan G.; Mizrahi V.; Rubin H.; Barry C. E. 3rd The Role of RelMtb-Mediated Adaptation to Stationary Phase in Long-Term Persistence of Mycobacterium tuberculosis in Mice. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 10026–10031. 10.1073/pnas.1631248100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandal O. H.; Nathan C. F.; Ehrt S. Acid Resistance in Mycobacterium tuberculosis. J. Bacteriol. 2009, 191, 4714–4721. 10.1128/JB.00305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackart D. F.; Hascall-Dove L.; Caceres S. M.; Kirk N. M.; Podell B. K.; Melander C.; Orme I. M.; Leid J. G.; Nick J. A.; Basaraba R. J. Expression of Antimicrobial Drug Tolerance by Attached Communities of Mycobacterium tuberculosis. Pathog. Dis. 2014, 70, 359–369. 10.1111/2049-632X.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojha A. K.; Baughn A. D.; Sambandan D.; Hsu T.; Trivelli X.; Guerardel Y.; Alahari A.; Kremer L.; Jacobs W. R. Jr.; Hatfull G. F. Growth of Mycobacterium tuberculosis Biofilms Containing Free Mycolic Acids and Harbouring Drug-Tolerant Bacteria. Mol. Microbiol. 2008, 69, 164–174. 10.1111/j.1365-2958.2008.06274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Sala C.; Hartkoorn R. C.; Dhar N.; Mendoza-Losana A.; Cole S. T. Streptomycin-Starved Mycobacterium tuberculosis 18b, a Drug Discovery Tool for Latent Tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 5782–5789. 10.1128/AAC.01125-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C.; Dhar N.; Hartkoorn R. C.; Zhang M.; Ha Y. H.; Schneider P.; Cole S. T. Simple Model for Testing Drugs against Nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2010, 54, 4150–4158. 10.1128/AAC.00821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho L. P.; Fischer S. M.; Marrero J.; Nathan C.; Ehrt S.; Rhee K. Y. Metabolomics of Mycobacterium tuberculosis Reveals Compartmentalized Co-Catabolism of Carbon Substrates. Chem. Biol. 2010, 17, 1122–1131. 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Rhee K. Y.; de Carvalho L. P.; Bryk R.; Ehrt S.; Marrero J.; Park S. W.; Schnappinger D.; Venugopal A.; Nathan C. Central Carbon Metabolism in Mycobacterium tuberculosis: An Unexpected Frontier. Trends Microbiol. 2011, 19, 307–314. 10.1016/j.tim.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D.; Ehrt S.; Voskuil M. I.; Liu Y.; Mangan J. A.; Monahan I. M.; Dolganov G.; Efron B.; Butcher P. D.; Nathan C.; et al. Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J. Exp. Med. 2003, 198, 693–704. 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde K. H.; Abramovitch R. B.; Russell D. G. Mycobacterium tuberculosis Invasion of Macrophages: Linking Bacterial Gene Expression to Environmental Cues. Cell Host Microbe 2007, 2, 352–364. 10.1016/j.chom.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Nesbitt N. M.; Yang X.; Fontán P.; Kolesnikova I.; Smith I.; Sampson N. S.; Dubnau E. A Thiolase of Mycobacterium tuberculosis Is Required for Virulence and Production of Androstenedione and Androstadienedione from Cholesterol. Infect. Immun. 2010, 78, 275–282. 10.1128/IAI.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Geize R.; Yam K.; Heuser T.; Wilbrink M. H.; Hara H.; Anderton M. C.; Sim E.; Dijkhuizen L.; Davies J. E.; Mohn W. W.; et al. A Gene Cluster Encoding Cholesterol Catabolism in a Soil Actinomycete Provides Insight into Mycobacterium tuberculosis Survival in Macrophages. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 1947–1952. 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A. K.; Sassetti C. M. Mycobacterial Persistence Requires the Utilization of Host Cholesterol. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 4376–4380. 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Nesbitt N. M.; Dubnau E.; Smith I.; Sampson N. S. Cholesterol Metabolism Increases the Metabolic Pool of Propionate in Mycobacterium tuberculosis. Biochemistry 2009, 48, 3819–3821. 10.1021/bi9005418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddry J. A.; Ananthan S.; Goldman R. C.; Hobrath J. V.; Kwong C. D.; Maddox C.; Rasmussen L.; Reynolds R. C.; Secrist J. A. 3rd; Sosa M. I.; et al. Antituberculosis Activity of the Molecular Libraries Screening Center Network Library. Tuberculosis (Oxford, U. K.) 2009, 89, 354–363. 10.1016/j.tube.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthan S.; Faaleolea E. R.; Goldman R. C.; Hobrath J. V.; Kwong C. D.; Laughon B. E.; Maddry J. A.; Mehta A.; Rasmussen L.; Reynolds R. C.; et al. High-Throughput Screening for Inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis (Oxford, U. K.) 2009, 89, 334–353. 10.1016/j.tube.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A.; Sarkar D. Identification of a Respiratory-Type Nitrate Reductase and Its Role for Survival of Mycobacterium smegmatis in Wayne Model. Microb. Pathog. 2006, 41, 90–95. 10.1016/j.micpath.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Khan A.; Sarkar D. A Simple Whole Cell Based High Throughput Screening Protocol Using Mycobacterium bovis BCG for Inhibitors against Dormant and Active Tubercle Bacilli. J. Microbiol. Methods 2008, 73, 62–68. 10.1016/j.mimet.2008.01.015. [DOI] [PubMed] [Google Scholar]
- Collins L.; Franzblau S. G. Microplate Alamar Blue Assay Versus Bactec 460 System for High-Throughput Screening of Compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chem. 1997, 41, 1004–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoro E.; Lemus D.; Echemendia M.; Martin A.; Portaels F.; Palomino J. C. Comparative Evaluation of the Nitrate Reduction Assay, the MTT Test, and the Resazurin Microtitre Assay for Drug Susceptibility Testing of Clinical Isolates of Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2005, 55, 500–505. 10.1093/jac/dki023. [DOI] [PubMed] [Google Scholar]
- Palomino J. C.; Martin A.; Camacho M.; Guerra H.; Swings J.; Portaels F. Resazurin Microtiter Assay Plate: Simple and Inexpensive Method for Detection of Drug Resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002, 46, 2720–2722. 10.1128/AAC.46.8.2720-2722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja N. K.; Tyagi J. S. Resazurin Reduction Assays for Screening of Anti-Tubercular Compounds against Dormant and Actively Growing Mycobacterium tuberculosis, Mycobacterium bovis BCG and Mycobacterium smegmatis. J. Antimicrob. Chemother. 2007, 60, 288–293. 10.1093/jac/dkm207. [DOI] [PubMed] [Google Scholar]
- Singh U.; Akhtar S.; Mishra A.; Sarkar D. A Novel Screening Method Based on Menadione Mediated Rapid Reduction of Tetrazolium Salt for Testing of Anti-Mycobacterial Agents. J. Microbiol. Methods 2011, 84, 202–207. 10.1016/j.mimet.2010.11.013. [DOI] [PubMed] [Google Scholar]
- Mehta M.; Rajmani R. S.; Singh A. Mycobacterium tuberculosis WhiB3 Responds to Vacuolar pH-Induced Changes in Mycothiol Redox Potential to Modulate Phagosomal Maturation and Virulence. J. Biol. Chem. 2016, 291, 2888–2903. 10.1074/jbc.M115.684597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde K. H.; Veiga D. F.; Caldwell S.; Balazsi G.; Russell D. G. Linking the Transcriptional Profiles and the Physiological States of Mycobacterium tuberculosis During an Extended Intracellular Infection. PLoS Pathog. 2012, 8, e1002769. 10.1371/journal.ppat.1002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramovitch R. B.; Rohde K. H.; Hsu F. F.; Russell D. G. AprABC: A Mycobacterium tuberculosis Complex-Specific Locus That Modulates pH-Driven Adaptation to the Macrophage Phagosome. Mol. Microbiol. 2011, 80, 678–694. 10.1111/j.1365-2958.2011.07601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters S. B.; Dubnau E.; Kolesnikova I.; Laval F.; Daffe M.; Smith I. The Mycobacterium tuberculosis PhoPR Two-Component System Regulates Genes Essential for Virulence and Complex Lipid Biosynthesis. Mol. Microbiol. 2006, 60, 312–330. 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- Johnson B. K.; Colvin C. J.; Needle D. B.; Mba Medie F.; Champion P. A.; Abramovitch R. B. The Carbonic Anhydrase Inhibitor Ethoxzolamide Inhibits the Mycobacterium tuberculosis PhoPR Regulon and Esx-1 Secretion and Attenuates Virulence. Antimicrob. Agents Chemother. 2015, 59, 4436–4445. 10.1128/AAC.00719-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren I.; Minami S.; Rubin E.; Lewis K. Characterization and Transcriptome Analysis of Mycobacterium tuberculosis Persisters. mBio 2011, 2, e00100–00111. 10.1128/mBio.00100-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjak A.; Uplekar S.; Zhang M.; Piton J.; Cole S. T.; Sala C. Genomic and Transcriptomic Analysis of the Streptomycin-Dependent Mycobacterium tuberculosis Strain 18b. BMC Genomics 2016, 17, 190. 10.1186/s12864-016-2528-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo-Lopez M. J.; Lelièvre J.; Alvarez-Gomez D.; Castro-Pichel J.; Martínez-Jiménez F.; Papadatos G.; Kumar V.; Colmenarejo G.; Mugumbate G.; Hurle M. Release of 50 New, Drug-Like Compounds and Their Computational Target Predictions for Open Source Anti-Tubercular Drug Discovery. PLoS One 2015, 10, e0142293. 10.1371/journal.pone.0142293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon C.; Dick T. Mycobacterium bovis BCG Response Regulator Essential for Hypoxic Dormancy. J. Bacteriol. 2002, 184, 6760–6767. 10.1128/JB.184.24.6760-6767.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leistikow R. L.; Morton R. A.; Bartek I. L.; Frimpong I.; Wagner K.; Voskuil M. I. The Mycobacterium tuberculosis Dosr Regulon Assists in Metabolic Homeostasis and Enables Rapid Recovery from Nonrespiring Dormancy. J. Bacteriol. 2010, 192, 1662–1670. 10.1128/JB.00926-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S.; Sukumar N.; Abramovitch R. B.; Parish T.; Russell D. G. Mycobacterium tuberculosis Responds to Chloride and pH as Synergistic Cues to the Immune Status of Its Host Cell.. PLoS Pathog. 2013, 9, e1003282. 10.1371/journal.ppat.1003282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H.; Colvin C. J.; Johnson B. K.; Kirchhoff P. D.; Wilson M.; Jorgensen-Muga K.; Larsen S. D.; Abramovitch R. B. Inhibitors of Mycobacterium tuberculosis DosRST Signaling and Persistence. Nat. Chem. Biol. 2017, 13, 218–225. 10.1038/nchembio.2259. [DOI] [PubMed] [Google Scholar]
- Deb C.; Lee C. M.; Dubey V. S.; Daniel J.; Abomoelak B.; Sirakova T. D.; Pawar S.; Rogers L.; Kolattukudy P. E. A Novel in vitro Multiple-Stress Dormancy Model for Mycobacterium tuberculosis Generates a Lipid-Loaded, Drug-Tolerant, Dormant Pathogen. PLoS One 2009, 4, e6077. 10.1371/journal.pone.0006077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B.; Pingle M.; Brickner S. J.; Shah N.; Roberts J.; Rundell M.; Bracken W. C.; Warrier T.; Somersan S.; Venugopal A. Nonsteroidal Anti-Inflammatory Drug Sensitizes Mycobacterium tuberculosis to Endogenous and Exogenous Antimicrobials. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 16004–16011. 10.1073/pnas.1214188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B.; Warrier T.; Nathan C. A Multi-Stress Model for High Throughput Screening against Non-Replicating Mycobacterium tuberculosis. Methods Mol. Biol. 2015, 1285, 293–315. 10.1007/978-1-4939-2450-9_18. [DOI] [PubMed] [Google Scholar]
- Schaaf K.; Hayley V.; Speer A.; Wolschendorf F.; Niederweis M.; Kutsch O.; Sun J. A Macrophage Infection Model to Predict Drug Efficacy against Mycobacterium tuberculosis. Assay Drug Dev. Technol. 2016, 14, 345–354. 10.1089/adt.2016.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams K. N.; Takaki K.; Connolly L. E.; Wiedenhoft H.; Winglee K.; Humbert O.; Edelstein P. H.; Cosma C. L.; Ramakrishnan L. Drug Tolerance in Replicating Mycobacteria Mediated by a Macrophage-Induced Efflux Mechanism. Cell 2011, 145, 39–53. 10.1016/j.cell.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V.; Manina G.; Mikusova K.; Mollmann U.; Ryabova O.; Saint-Joanis B.; Dhar N.; Pasca M. R.; Buroni S.; Lucarelli A. P.; et al. Benzothiazinones Kill Mycobacterium tuberculosis by Blocking Arabinan Synthesis. Science 2009, 324, 801–804. 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia N. P.; Hasenoehrl E. J.; Ab Rahman N. B.; Koh V. H.; Ang M. L.; Sajorda D. R.; Hards K.; Grüber G.; Alonso S.; Cook G. M. Exploiting the Synthetic Lethality between Terminal Respiratory Oxidases to Kill Mycobacterium tuberculosis and Clear Host Infection. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 7426–7431. 10.1073/pnas.1706139114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S. A.; Barczak A. K.; Silvis M. R.; Luo S. S.; Sogi K.; Vokes M.; Bray M. A.; Carpenter A. E.; Moore C. B.; Siddiqi N.; et al. Identification of Host-Targeted Small Molecules That Restrict Intracellular Mycobacterium tuberculosis Growth. PLoS Pathog. 2014, 10, e1003946. 10.1371/journal.ppat.1003946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Miranda M.; Ekaza E.; Breiman A.; Asehnoune K.; Barros-Aguirre D.; Pethe K.; Ewann F.; Brodin P.; Ballell-Pages L.; Altare F. High-Content Screening Technology Combined with a Human Granuloma Model as a New Approach to Evaluate the Activities of Drugs against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 693–697. 10.1128/AAC.03705-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Tan S.; Huang L.; Abramovitch R. B.; Rohde K. H.; Zimmerman M. D.; Chen C.; Dartois V.; VanderVen B. C.; Russell D. G. Immune Activation of the Host Cell Induces Drug Tolerance in Mycobacterium tuberculosis Both in vitro and in vivo. J. Exp. Med. 2016, 213, 809–825. 10.1084/jem.20151248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dartois V. The Path of Anti-Tuberculosis Drugs: From Blood to Lesions to Mycobacterial Cells. Nat. Rev. Microbiol. 2014, 12, 159–167. 10.1038/nrmicro3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllymäki H.; Bäuerlein C. A.; Rämet M. The Zebrafish Breathes New Life into the Study of Tuberculosis. Front. Immunol. 2016, 7, 196. 10.3389/fimmu.2016.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin D. M.; Ramakrishnan L. Comparative Pathogenesis of Mycobacterium marinum and Mycobacterium tuberculosis. Cell. Microbiol. 2008, 10, 1027–1039. 10.1111/j.1462-5822.2008.01133.x. [DOI] [PubMed] [Google Scholar]
- Trede N. S.; Langenau D. M.; Traver D.; Look A. T.; Zon L. I. The Use of Zebrafish to Understand Immunity. Immunity 2004, 20, 367–379. 10.1016/S1074-7613(04)00084-6. [DOI] [PubMed] [Google Scholar]
- Takaki K.; Cosma C. L.; Troll M. A.; Ramakrishnan L. An in vivo Platform for Rapid High-Throughput Antitubercular Drug Discovery. Cell Rep. 2012, 2, 175–184. 10.1016/j.celrep.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton J.; Uy B.; Okuda K.; Hall C.; Denny W.; Crosier P.; Swift S.; Wiles S. Screening of Anti-Mycobacterial Compounds in a Naturally Infected Zebrafish Larvae Model. J. Antimicrob. Chemother. 2017, 72, 421–427. 10.1093/jac/dkw421. [DOI] [PubMed] [Google Scholar]
- Sacksteder K. A.; Protopopova M.; Barry C. E.; Andries K.; Nacy C. A. Discovery and Development of SQ109: A New Antitubercular Drug with a Novel Mechanism of Action. Future Microbiol. 2012, 7, 823–837. 10.2217/fmb.12.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protopopova M.; Hanrahan C.; Nikonenko B.; Samala R.; Chen P.; Gearhart J.; Einck L.; Nacy C. A. Identification of a New Antitubercular Drug Candidate, SQ109, from a Combinatorial Library of 1, 2-Ethylenediamines. J. Antimicrob. Chemother. 2005, 56, 968–974. 10.1093/jac/dki319. [DOI] [PubMed] [Google Scholar]
- Shirude P. S.; Shandil R. K.; Manjunatha M.; Sadler C.; Panda M.; Panduga V.; Reddy J.; Saralaya R.; Nanduri R.; Ambady A. Lead Optimization of 1, 4-Azaindoles as Antimycobacterial Agents. J. Med. Chem. 2014, 57, 5728–5737. 10.1021/jm500571f. [DOI] [PubMed] [Google Scholar]
- Brodin P.; Cechetto J.; Christophe T.; Contreras D. M.; Ewann F. A.; Fenistein D. P. C.; Genovesio A.; Heo J.; Jeon H.; Kang S.; et al. Anti - Infective Pyrido (1,2-a) Pyrimidines. WO/2011/085990 A8, 2010.
- Makarov V.; Riabova O. B.; Yuschenko A.; Urlyapova N.; Daudova A.; Zipfel P. F.; Möllmann U. Synthesis and Antileprosy Activity of Some Dialkyldithiocarbamates. J. Antimicrob. Chemother. 2006, 57, 1134–1138. 10.1093/jac/dkl095. [DOI] [PubMed] [Google Scholar]
- Neres J.; Hartkoorn R. C.; Chiarelli L. R.; Gadupudi R.; Pasca M. R.; Mori G.; Venturelli A.; Savina S.; Makarov V.; Kolly G. S. 2-Carboxyquinoxalines Kill Mycobacterium tuberculosis through Noncovalent Inhibition of DprE1. ACS Chem. Biol. 2015, 10, 705–714. 10.1021/cb5007163. [DOI] [PubMed] [Google Scholar]
- Batt S. M.; Cacho Izquierdo M.; Castro Pichel J.; Stubbs C. J.; Vela-Glez Del Peral L.; Pérez-Herrán E.; Dhar N.; Mouzon B.; Rees M.; Hutchinson J. P. Whole Cell Target Engagement Identifies Novel Inhibitors of Mycobacterium tuberculosis Decaprenylphosphoryl-β-D-Ribose Oxidase. ACS Infect. Dis. 2015, 1, 615–626. 10.1021/acsinfecdis.5b00065. [DOI] [PubMed] [Google Scholar]
- Piton J.; Foo C. S.Y.; Cole S. T. Structural Studies of Mycobacterium tuberculosis DprE1 Interacting with Its Inhibitors. Drug Discovery Today 2017, 22, 526–533. 10.1016/j.drudis.2016.09.014. [DOI] [PubMed] [Google Scholar]
- Brecik M.; Centárová I.; Mukherjee R.; Kolly G. S.; Huszár S.; Bobovská A.; Kilacsková E.; Mokošová V.; Svetlíková Z.; Šarkan M.; et al. DprE1 Is a Vulnerable Tuberculosis Drug Target Due to Its Cell Wall Localization. ACS Chem. Biol. 2015, 10, 1631–1636. 10.1021/acschembio.5b00237. [DOI] [PubMed] [Google Scholar]
- Zimmerman M.; Lestner J.; Prideaux B.; O’Brien P.; Dias-Freedman I.; Chen C.; Dietzold J.; Daudelin I.; Kaya F.; Blanc L.; et al. Ethambutol Partitioning in Tuberculous Pulmonary Lesions Explains Its Clinical Efficacy. Antimicrob. Agents Chemother. 2017, 61, e00924–17. 10.1128/AAC.00924-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prideaux B.; Via L. E.; Zimmerman M. D.; Eum S.; Sarathy J.; O’Brien P.; Chen C.; Kaya F.; Weiner D. M.; Chen P. Y.; et al. The Association between Sterilizing Activity and Drug Distribution into Tuberculosis Lesions. Nat. Med. 2015, 21, 1223–1227. 10.1038/nm.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolber J. M.; Urbanek B. L.; Meints L. M.; Piligian B. F.; Lopez-Casillas I. C.; Zochowski K. M.; Woodruff P. J.; Swarts B. M. The Trehalose-Specific Transporter LpqY-SugABC Is Required for Antimicrobial and Anti-Biofilm Activity of Trehalose Analogues in Mycobacterium smegmatis. Carbohydr. Res. 2017, 450, 60–66. 10.1016/j.carres.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill M. K.; Piligian B. F.; Olson C. D.; Woodruff P. J.; Swarts B. M. Tailoring Trehalose for Biomedical and Biotechnological Applications. Pure Appl. Chem. 2017, 89, 1223–1249. 10.1515/pac-2016-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanna S.; Sucheck S. J. Targeting the Trehalose Utilization Pathways of Mycobacterium tuberculosis. MedChemComm 2016, 7, 69–85. 10.1039/C5MD00376H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarova E. V.; Montague C. R.; La T.; Wilburn K. M.; Sukumar N.; Lee W.; Caldwell S.; Russell D. G.; VanderVen B. C.. Rv3723/LucA Coordinates Fatty Acid and Cholesterol Uptake in Mycobacterium tuberculosis. eLife 2017, 6, 10.7554/eLife.26969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genilloud O. Actinomycetes: Still a Source of Novel Antibiotics. Nat. Prod. Rep. 2017, 34, 1203–1232. 10.1039/C7NP00026J. [DOI] [PubMed] [Google Scholar]
- Wright G. D. Opportunities for Natural Products in 21st Century Antibiotic Discovery. Nat. Prod. Rep. 2017, 34, 694–701. 10.1039/C7NP00019G. [DOI] [PubMed] [Google Scholar]
- Hosoya M.; Czysz K. Translational Prospects and Challenges in Human Induced Pluripotent Stem Cell Research in Drug Discovery. Cells 2016, 5, 46. 10.3390/cells5040046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioerger T. R.; O’Malley T.; Liao R.; Guinn K. M.; Hickey M. J.; Mohaideen N.; Murphy K. C.; Boshoff H. I.; Mizrahi V.; Rubin E. J. Identification of New Drug Targets and Resistance Mechanisms in Mycobacterium tuberculosis. PLoS One 2013, 8, e75245. 10.1371/journal.pone.0075245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farha M. A.; Brown E. D. Strategies for Target Identification of Antimicrobial Natural Products. Nat. Prod. Rep. 2016, 33, 668–680. 10.1039/C5NP00127G. [DOI] [PubMed] [Google Scholar]
- Durham T. B.; Blanco M. J. Target Engagement in Lead Generation. Bioorg. Med. Chem. Lett. 2015, 25, 998–1008. 10.1016/j.bmcl.2014.12.076. [DOI] [PubMed] [Google Scholar]
- Sarathy J. P.; Liang H. H.; Weiner D.; Gonzales J.; Via L. E.; Dartois V.. An in vitro Caseum Binding Assay That Predicts Drug Penetration in Tuberculosis Lesions. J. Visualized Exp. 2017, 10.3791/55559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pienaar E.; Sarathy J.; Prideaux B.; Dietzold J.; Dartois V.; Kirschner D. E.; Linderman J. J. Comparing Efficacies of Moxifloxacin, Levofloxacin and Gatifloxacin in Tuberculosis Granulomas Using a Multi-Scale Systems Pharmacology Approach. PLoS Comput. Biol. 2017, 13, e1005650. 10.1371/journal.pcbi.1005650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarathy J. P.; Zuccotto F.; Hsinpin H.; Sandberg L.; Via L. E.; Marriner G. A.; Masquelin T.; Wyatt P.; Ray P.; Dartois V. Prediction of Drug Penetration in Tuberculosis Lesions. ACS Infect. Dis. 2016, 2, 552–563. 10.1021/acsinfecdis.6b00051. [DOI] [PMC free article] [PubMed] [Google Scholar]