

Summary
The cell wall-less prokaryote Mycoplasma pneumoniae causes tracheobronchitis and primary atypical pneumonia in humans. Colonization of the respiratory epithelium requires proper assembly of a complex, multifunctional, polar terminal organelle. Loss of a predicted J-domain protein also having domains unique to mycoplasma terminal organelle proteins (TopJ) resulted in a non-motile, adherence-deficient phenotype. J-domain proteins typically stimulate ATPase activity of Hsp70 chaperones to bind nascent peptides for proper folding, translocation or macro-molecular assembly, or to resolve stress-induced protein aggregates. By Western immunoblotting all defined terminal organelle proteins examined except protein P24 remained at wild-type levels in the topJ mutant; previous studies established that P24 is required for normal initiation of terminal organelle formation. Nevertheless, terminal organelle proteins P1, P30, HMW1 and P41 failed to localize to a cell pole, and when evaluated quantitatively, P30 and HMW1 foci were undetectable in >40% of cells. Complementation of the topJ mutant with the recombinant wild-type topJ allele largely restored terminal organelle development, gliding motility and cytadherence. We propose that this J-domain protein, which localizes to the base of the terminal organelle in wild-type M. pneumoniae, functions in the late stages of assembly, positioning, or both, of nascent terminal organelles.
Introduction
Mycoplasma pneumoniae is a cell wall-less obligate parasite of the human respiratory tract causing tracheobronchitis and atypical or ‘walking’ pneumonia, predominantly among children and young adults. Chronic infection has been linked to recurring asthma and permanent lung damage, with extrapulmonary complications reported as well (Waites and Talkington, 2004). Attachment to host mucosal epithelium (cytadherence) is pivotal for initiation of infection (Sobeslavsky et al., 1968), and therefore, considerable research has focused on the characterization of attachment-deficient mutants to elucidate the mechanism(s) of cytadherence with hopes of identifying potential therapeutic targets and vaccine candidates.
With a genome of only 816 kb, M. pneumoniae lacks the genes to produce a cell wall, synthesize nucleotides and amino acids de novo, or regulate transcription via two-component systems (Himmelreich et al., 1996; Dandekar et al., 2000; Seto et al., 2001). However, this minimal microbe is deceptively complex, assembling an elaborate cytoskeleton that includes a differentiated terminal organelle that functions in host cell attachment, cell division and gliding motility (Bredt, 1968; Biberfeld and Biberfeld, 1970; Collier et al., 1971; Krause and Balish, 2001). Despite significant progress in elucidating terminal organelle architecture (Henderson and Jensen, 2006), how a microbe with a minimal genome organizes and assembles this complex higher-ordered structure is poorly understood.
Mutant analysis has identified a number of terminal organelle proteins, many of which are associated directly or indirectly with cytadherence, including the high-molecular-weight proteins HMW1–HMW3, P1, B, C, P200, P30, P65, P28, P24 and P41 (Feldner et al., 1982; Baseman et al., 1987; Stevens and Krause, 1991; 1992; Layh-Schmitt and Herrmann, 1992; Jordan et al., 2001; 2007; Balish et al., 2003; Krause and Balish, 2004; Hasselbring and Krause, 2007a). Most are cytoskeletal proteins, several of which share common domains unique to mycoplasmas. Proteins HMW1, HMW3, P200 and P65 each contain an acidic and proline-rich (APR) domain, while HMW1 and P200 also have a conserved region enriched in aromatic and glycine residues termed the EAGR box (Balish et al., 2001). The role of these domains, identified solely in mycoplasma terminal organelle proteins, remains unclear.
Paraformaldehyde cross-linking of intact mycoplasma cells indicates that the Hsp70 class of chaperone, DnaK, may interact with the primary adhesin P1, raising questions about the role of chaperones in terminal organelle assembly (Layh-Schmitt et al., 2000). In Escherichia coli, the Hsp40 J-domain chaperone DnaJ folds nascent proteins and responds to cellular stress by presenting a non-native or denatured polypeptide to DnaK for restoration to its native conformation. The N-terminal J-domain stimulates DnaK to hydrolyse ATP, inducing a conformational change that securely binds DnaK to the presented polypeptide, thereby inhibiting premature protein folding. The DnaK/ADP/substrate complex releases the polypeptide in the presence of the nucleotide exchange factor GrpE. Continued binding and release of the polypeptide may occur until the mature protein reaches its native state (Hendrick and Hartl, 1995; Frydman, 2001). Homologues of the conserved DnaK/DnaJ/GrpE chaperone system exist in all domains of life, functioning in a variety of cellular processes including disassembly of protein complexes, translocation of proteins across membranes and stabilization of adjacent hydrophobic protein complexes (Jensen and Johnson, 1999; Lemmon, 2001). An attractive hypothesis based on this model is that a chaperone system of DnaK/a J-domain protein/GrpE participates in P1 trafficking to and/or translocation across the membrane (Layh-Schmitt et al., 2000); however, assembly and/or stabilization of other terminal organelle protein(s) also remains a possibility.
Mycoplasma pneumoniae ORF MPN119 encodes a novel protein having a predicted J-domain as well as the aforementioned EAGR and APR domains of certain terminal organelle proteins (Fig. 1). In the current study, disruption of MPN119 resulted in loss of the corresponding protein, termed TopJ for terminal organelle protein with a J-domain, and was accompanied by reduced steady-state levels of a GrpE homologue (MPN120) and terminal organelle protein P24 (MPN312). The topJ mutant was defective in cytadherence, gliding motility and cell division, yet all known terminal organelle-associated proteins except P24 remained at wild-type levels. Complementation of the mutant with the recombinant wild-type MPN119 allele by transposon delivery largely restored cytadherence, motility and P24 levels. However, GrpE levels were only restored by complementation with both recombinant MPN119 and MPN120 alleles, although no phenotypic difference was seen beyond complementation with MPN119 alone. TopJ localized to the proximal end of the terminal organelle in wild-type cells, similar to P24 and P41 (Hasselbring and Krause, 2007a), when analysed by immunofluorescence microscopy. Cytadhesin P1 exhibited reduced polar localization and more diffuse distribution in the topJ mutant compared with wild type. P41 and P30 localized as paired foci, although not typically at polar sites, while P30 and HMW1 foci were undetectable in > 40% of the cells examined. Based on these observations, we propose that the J-domain protein TopJ is required for the proper late-stage assembly of the M. pneumoniae terminal organelle, including correct polar positioning of this structure.
Fig. 1.
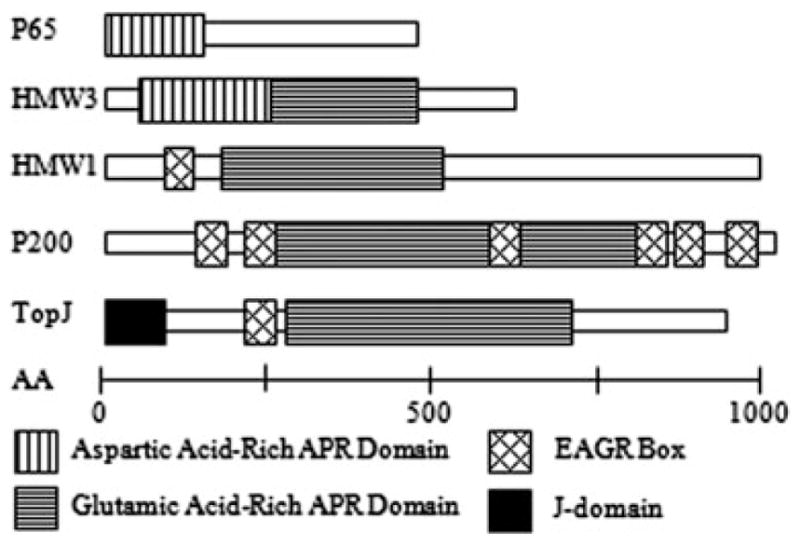
Schematic alignment of acidic- and proline-rich (APR) and enriched in aromatic and glycine residue (EAGR) domains of M. pneumoniae proteins (modified from Balish and Krause, 2002). Scale bar, number of amino acid residues.
Results
Tn4001 insertional inactivation of an ORF encoding a putative J-domain protein
As part of a previous study (Willby et al., 2004), a modified Tn4001 (Tn4001mod) transformant was identified that exhibited poor attachment to plastic, typically indicative of a cytadherence deficiency. The transposon insertion was mapped to nucleotide residue 50 of MPN119, encoding a putative Hsp40 class J-domain protein. A PredictProtein (Rost et al., 2004) sequence analysis of TopJ predicted four consecutive helices encoded within the N-terminal domain including a positively charged second helix. Specifically, a histidine–proline–aspartic acid tripeptide and a pair of basic amino acid residues immediately follow the positively charged second helix, which is essential for interaction with the negatively charged ATPase domain of an Hsp70 and defines a J-domain (Greene et al., 1998; Genevaux et al., 2002). Interestingly, this J-domain protein also has terminal organelle-associated EAGR and APR domains (Balish et al., 2001; Fig. 1). An 8 bp duplication in the MPN119 sequence adjacent to the inverted repeat of IS256 from the transposon accompanied insertion. A clonal population was achieved by filter-cloning and verified by PCR amplification, and Southern hybridization confirmed that no other copies of the transposon or IS256 were detectable in the genome (data not shown). The topJ gene in M. pneumoniae is the first ORF in a likely operon that includes grpE 22 bp downstream and an ORF encoding a hypothetical protein (Fig. 2); topJ is preceded by the gene for a putative ribonuclease in the opposite orientation.
Fig. 2.

Comparison of the topJ operon in M. pneumoniae with orthologues from the closely related M. genitalium (Peterson et al., 1993) identified by basic local alignment search tool (BLAST). The black arrow preceding MG200 indicates the promoter for the operon as identified by primer extension (Musatovova et al., 2006). The inverted triangle indicates the transposon insertion in MPN119 in the topJ mutant. The dotted line represents reading frames not included in the schematic. Open reading frames are not to scale. Primer pairs corresponding to a, b and a, c were used to amplify MPN119 or MPN119 + MPN120, respectively, for complementation studies. The underlined sequence corresponds to an engineered SmaI restriction site. Hyp, hypothetical protein.
a. 5′-AAAGCTATCACCCGGGCCAGATTCATCACT-3′
b. 5′-CACCATGTTAAACCCGGGGGTATAGT-3′
c. 5′-CCTTCAAGGCTTCCCGGGCTTTTTCTAAATCC-3′.
To determine if loss of TopJ was directly responsible for the observed phenotype, the topJ mutant was transformed with recombinant wild-type MPN119 cloned into a modified Tn4001 [Tn4001cat (Hahn et al., 1999)]. The topJ mutant was complemented with either recombinant wild-type MPN119 + MPN120 or MPN119 alone to determine if the reduction in GrpE contributed to the observed phenotype (Fig. 2). Transformants were isolated, and all exhibited wild-type attachment to plastic.
When cultured in a medium containing 3% gelatin, wild-type M. pneumoniae cells exhibit satellite growth as a function of gliding motility (Hasselbring et al., 2005). We examined the satellite growth phenotype of the topJ mutant with and without the recombinant wild-type MPN119 or MPN119 + MPN120 alleles to assess the effects of loss of TopJ on gliding motility (Fig. 3). The topJ mutant cells formed compact clumps with occasional extensions of cell chains from the colony, unlike the typical spread of wild-type cells, indicating a loss of motility. No gliding by individual cells was observed (Table 1). The accumulation or stacking of cells extending from micro-colonies suggested a cell division defect, as cells failed to separate normally (Hasselbring et al., 2006a). Delivery of recombinant MPN119 or MPN119 + MPN120 restored gliding frequency and velocity to 85–92% of wild-type cells (Table 1) as well as normal cell morphology, satellite growth and division (Fig. 3).
Fig. 3.

Satellite growth by wild-type M. pneumoniae (A), topJ mutant (B), and topJ transformants with MPN119 alone (C) or MPN119 + MPN120 (D). Mycoplasmas were incubated in SP4 medium with 3% gelatin for 72 h. Bar, 15 μm. Inset, enlarged view of the indicated region.
Table 1.
Comparison of cell gliding in wild-type M. pneumoniae, the topJ mutant and transformant strains.
| Cell gliding frequency (% WT) | Mean cell velocity μm s−1 ± SEM (% WT) | |
|---|---|---|
| Wild type | 12.0 (100) | 0.202 ± 0.003 (100) |
| topJ | 0 (0) | 0 (0) |
| topJ + MPN119 | 10.4 (87.3) | 0.185 ± 0.003 (91.6) |
| topJ + MPN119/MPN120 | 11.0 (92.0) | 0.171 ± 0.003 (84.7) |
SEM, standard error of the mean.
TopJ is required to stabilize P24
Immunoblot analysis confirmed the loss of TopJ with disruption of MPN119, while most currently known terminal organelle-associated proteins (P1, B, C, HMW1, HMW2, HMW3, P200, P30 and P65) remained at wild-type levels (Fig. 4 and data not shown). GrpE, the product of MPN120, immediately downstream of MPN119, exhibited a threefold reduction, assessed by immunoblot comparison of serially diluted wild-type protein (data not shown), and was restored to slightly above wild-type levels with recombinant wild-type MPN119 + MPN120, but not MPN119 alone (Fig. 4). We speculate that an internal, outward-reading promoter in IS256 (Byrne et al., 1989) enabled limited grpE transcription. Interestingly, the cell division-associated terminal organelle protein P24 (Hasselbring and Krause, 2007a), encoded by MPN312, exhibited a fivefold reduction with the loss of TopJ, while P41, predicted to be translationally coupled to P24 (Hasselbring and Krause, 2007b), remained at wild-type levels (Fig. 4). Wild-type M. pneumoniae cells pause gliding during terminal organelle duplication, then reinitiate gliding by the existing terminal organelle to displace the nascent terminal organelle to the opposite pole (Hasselbring et al., 2006b). The loss of P24 results in delayed displacement of the terminal organelle to the opposite pole and delayed reacquisition of motility (Hasselbring and Krause, 2007a). Sequence analysis of MPN312 and surrounding DNA in the topJ mutant revealed no DNA mutations (data not shown). Additionally, transformation of the topJ mutant with the recombinant wild-type MPN119 allele, but not MPN312 (Hasselbring and Krause, 2007b), restored P24 stability (Fig. 4).
Fig. 4.

Western immunoblot analysis of wild type, topJ mutant and transformant M. pneumoniae strains. Proteins were separated by 4–12% gradient SDS-PAGE and analysed by immunoblotting with the indicated antisera.
TopJ is essential for cytadherence
Both qualitative and quantitative analysis of haemadsorption (HA) were employed to evaluate the impact of loss of TopJ on cytadherence (Fig. 5). Qualitatively, the topJ mutant exhibited similar binding as the negative control, and HA was restored to wild type by transformation with either MPN119 or MPN119 + MPN120 (Fig. 5A). In quantitative analysis of HA the topJ mutant attached to erythrocytes at levels only 3% of wild type, but somewhat surprisingly, recombinant MPN119 or MPN119 + MPN120 consistently restored HA to only 66–75% of wild type (Fig. 5B). Cytadherence was also assessed by using the A549 human lung adenocarcinoma cell line in submerged culture. Loss of TopJ resulted in attachment levels only 14% of wild type, while recombinant MPN119 or MPN119 + MPN120 again largely restored attachment (80% of wild type; Fig. 5C).
Fig. 5.

Cytadherence capacity of wild type, topJ mutant and transformant M. pneumoniae strains.
A. Qualitative assessment of HA. WT, wild type; II-3, negative control. Bar, 30 μm.
B and C. Quantitative assessment of HA and attachment to A549 cells, respectively, normalized to wild-type binding. III-4, negative control. Error bars, standard deviation.
TopJ is required for proper terminal organelle placement
Colocalization of proteins in overlapping subcellular compartments, identified by phase contrast-fluorescence microscopy, can allow association with a common function. In wild-type M. pneumoniae, P1 localizes primarily to the distal end of the terminal organelle with a scattered distribution along the cell body (Baseman et al., 1982; Feldner et al., 1982; Seto et al., 2001). In contrast, P24 and P41 colocalize to the base of the terminal organelle (Kenri et al., 2004; Hasselbring and Krause, 2007a). Here we observed that, like P41 and P24, TopJ localizes to the base of the terminal organelle in wild-type cells (Fig. 6A). Although P1 formed foci with some diffusion in the absence of TopJ, these were rarely seen at cell poles (Fig. 6B). Similarly, P30 formed foci, typically paired with P41, but not localized to a cell pole (Fig. 6D). As seen in wild-type M. pneumoniae (Hasselbring et al., 2006b), P41 foci unpaired with P30 were also observed in the topJ mutant (Fig. 6D, panel 2). What appeared to be unseparated nucleoids were visible with DAPI staining in the topJ mutant, suggesting a defect in cytokinesis (Fig. 6B, panel 6). However, we have not been able to establish by way of a fluorescent membrane stain whether septation is apparent.
Fig. 6.

Immunolocalization of terminal organelle proteins in wild-type M. pneumoniae and the topJ mutant.
A. Localization of P1 and TopJ: panel 1, phase-contrast; panel 2, merged-phase and fluorescence images; panel 3, schematic representation; panel 4, TopJ false-coloured yellow; panel 5, P1 false-coloured red; panel 6 (wild type), merge of wild type panels 4 and 5; panel 6 (topJ), DAPI staining.
B. Localization of P30 and P41: panel 1, phase-contrast; panel 2, merged-phase and fluorescence images; panel 3, schematic representation; panel 4, P41 false-coloured yellow; panel 5, P30 false-coloured red; panel 6, merge of panels 4 and 5. Yellow arrow, P41 focus unpaired with P30.
Bar, 1 μm.
To further investigate terminal organelle protein organization in the topJ mutant we also examined quantitatively the colocalization of P30 and HMW1, both of which are primarily polar in wild-type M. pneumoniae (Stevens and Krause, 1991; Krause and Balish, 2001; Seto et al., 2001; Willby et al., 2004). Only cells visible by both phase-contrast microscopy and fluorescence microscopy following DAPI staining were included. Fluorescence images were captured at various focal depths that included upper and lower focal extremes for each field. For the topJ mutant, some foci were visible in one focal plane but absent in another, presumably due to cell stacking, and these images were subsequently merged to create a complete representation of fluorescence for quantification of foci localization. HMW1 and P30 fluorescent foci colocalizing at the end of a cell were considered polar and paired (Fig. 7A, panel 2, white arrow). Chains of cells extending from microcolonies were common in the topJ mutant, unlike in wild-type M. pneumoniae (Fig. 7), preventing accurate determination of the cell pole; therefore, only single cells, or cells terminating chains were evaluated for the presence and localization of foci (Fig. 7B, panel 2, white arrows). Cells within chains were not quantified. Two major observations emerged from this analysis (Table 2). First, 94% of wild-type cells but only 53% of topJ mutant cells examined exhibited paired HMW1 and P30. The remaining topJ mutant cells had no detectable HMW1 or P30 focus nor detectable diffuse fluorescence, despite the fact that immunoblot analysis indicated wild-type levels of each. Only rarely was an unpaired polar focus of HMW1 or P30 observed in the topJ mutant or in wild type. Second, of the cells examined, < 1% of paired HMW1 and P30 foci failed to localize to a cell pole in wild-type cells, compared with 22% in the topJ mutant cells.
Fig. 7.

Immunolocalization of P30 and HMW1 in wild-type M. pneumoniae and the topJ mutant. Panel 1, phase-contrast; panel 2, merged-phase and fluorescence images; panel 3, schematic representation; panel 4, HMW1 false-coloured yellow; panel 5, P30 false-coloured red; panel 6, merge of panels 4 and 5. Wild type: white arrow, fluorescent polar foci. topJ: white arrow, pole of terminal cell in chain with no fluorescent focus; white arrowhead, HMW1 and P30 pair within cell chain having no clearly polar focus in terminal cell; yellow arrowhead, unpaired HMW1 focus within cell chain; red arrowhead, unpaired P30 focus within cell chain. Bar, 2 μm.
Table 2.
Quantitative assessment of HMW1 and P30 localization in wild-type M. pneumoniae, the topJ mutant and transformant strains.
| WT (%) | topJ (%) | topJ + MPN119 (%) | topJ + MPN119/ MPN120 (%) | |
|---|---|---|---|---|
| Total cells or cell chainsa | 231 | 196 | 203 | 197 |
| Total paired P30/HMW1 | 218 (94.4) | 104 (53.1) | 185 (91.1) | 183 (92.8) |
| Polar foci | 217 (99.5) | 81 (77.9) | 184 (99.5) | 182 (99.5) |
| Non-polar foci | 1 (0.5) | 23 (22.1) | 1 (0.5) | 1 (0.5) |
| No foci | 4 (1.7) | 82 (41.8) | 2 (1.0) | 6 (3.0) |
| Total unpaired foci at cell pole | ||||
| HMW1 alone | 6 (2.6) | 5 (2.6) | 10 (4.9) | 4 (2.0) |
| P30 alone | 3 (1.3) | 5 (2.6) | 6 (3.0) | 4 (2.0) |
Only single cells (all strains) or cells terminating a chain extending from a colony (topJ) were quantified.
Discussion
Transmission electron microscopy of Triton X-100-fractionated M. pneumoniae cells reveals the complex architecture of the terminal organelle that contributes to its asymmetrical morphology and gliding capability. With cell membrane removed, a distinct electrondense core is visible, along with fibrous strands extending from its proximal end into the cell body (Meng and Pfister, 1980; Gobel et al., 1981). This electrondense core is comprised of a larger and smaller segmented rod, with additional distinct substructural elements at their proximal and distal ends (Henderson and Jensen, 2006). Mutant studies have identified a number of terminal organelle proteins required for cytadherence (Balish and Krause, 2002), while two-dimensional gel electrophoresis and mass spectrometry have established the complexity of the Triton-insoluble fraction (Regula et al., 2001). Biochemical analysis of various mutants defective in terminal organelle function predicts an assembly sequence during terminal organelle formation, where loss of specific components required early in assembly results in reduced stability of proteins incorporated subsequently (Balish and Krause, 2002). To date, results from analysis using fluorescent protein fusions and time-lapse imaging are consistent with predictions of assembly order (Hasselbring et al., 2006b; Hasselbring and Krause, 2007a). Nevertheless, despite significant progress in the characterization of terminal organelle architecture, elucidation of the regulation of assembly, including temporal and spatial parameters, remains elusive.
Here, loss of a predicted J-domain protein also having domains found only in terminal organelle proteins impacted terminal organelle function in attachment, gliding motility and cell division, all of which were largely restored by the recombinant wild-type allele. In the related Mycoplasma genitalium, which lacks a P24 homologue, targeted disruption of the topJ homologue, mg200, results in a 95% reduction in gliding frequency and speed. HA of mg200 mutant cells, assessed qualitatively, is marginally reduced. Transformation with the recombinant wild-type allele restores gliding, although a quantitative assessment of gliding parameters was not reported; furthermore, HA of transformant strains was not tested (Pich et al., 2006). In the current study the reduction in attachment to erythrocytes and A549 cells was approximately 10-fold. Multiple transformants were tested in complementation studies, all of which were consistently HA-positive qualitatively but only 75% of wild type in quantitative assays. Likewise, gliding frequency and velocity were restored to >85% in the topJ mutant complemented with the recombinant wild-type allele. Thus, it is not clear whether the phenotypic differences observed between the mg200 mutant of M. genitalium (Pich et al., 2006) and the topJ mutant of M. pneumoniae (this study) are a function of the lack of P24 in M. genitalium, merely a reflection of the lack of quantitative analysis of HA and gliding in the former study, or perhaps both.
At least three possibilities might account for the failure of recombinant MPN119 to fully rescue cytadherence and gliding motility. First, recombinant TopJ levels may differ from wild type, but not sufficiently so to be detectable by immunoblot analysis. Significantly, while the recombinant MPN119 allele included the putative promoter region identified previously for the corresponding gene in M. genitalium (Musatovova et al., 2006), other regulatory elements thought to affect chaperone levels in M. genitalium and M. pneumoniae (Weiner et al., 2003; Musatovova et al., 2006; Kannan et al., 2008) may nevertheless be missing. Second, as GrpE levels remained below wild type after complementation with MPN119 alone and appeared slightly above wild type with the additional copy of MPN120 (Figs 2 and 4), improper stoichiometry could impact DnaK function. Elevated GrpE levels have been shown to alter the functionality of the DnaK/DnaJ/GrpE system in E. coli, leading to cell division defects (Sugimoto et al., 2008). If DnaK indeed interacts directly with the major adhesin P1 (Layh-Schmitt et al., 2000), then improper stoichiometry with its nucleotide exchange factor GrpE may affect cytadherence. Third, and perhaps the simplest explanation, the double selection for both gentamicin and chloramphenicol resistance required for the complemented mutant may impact cytadherence and gliding motility. We have observed similar results in other complemented mutant strains (our manuscript in preparation).
Terminal organelle duplication precedes cell division in wild-type M. pneumoniae, often with several terminal organelles forming before cytokinesis is observed (Hasselbring et al., 2006b). Duplication of the terminal organelle correlates with an increase in genomic content, suggesting coupling of terminal organelle formation and chromosome replication (Seto et al., 2001). Cell division concludes as the parent cell resumes gliding, displacing the nascent terminal organelle to the opposite pole and leading to cytokinesis (Hasselbring et al., 2006b). The topJ mutant is defective in cytadherence, gliding and cell division, yet only the terminal organelle-associated protein P24 is diminished. Mutant cells lacking P24 form terminal organelles that bind to glass and host cells at wild-type levels, glide at wild-type speeds, and are properly positioned, but the frequencies of nascent terminal organelle formation and gliding initiation in the P24 mutant are <40% of wild type, suggesting stable terminal organelle assembly and largely normal function except in the co-ordination of cell division-associated events (Hasselbring and Krause, 2007a). Therefore, failure to properly assemble and localize functional terminal organelles with disruption of MPN119 appears to be specific to loss of TopJ. Whether the cell division defect in the mutant resulted from a failure to glide away from daughter cells to complete cytokinesis or from improper terminal organelle segregation is not yet known.
If TopJ functions to fold and stabilize cytadherence-associated proteins properly, then reduced levels of one or more of those proteins might be expected in its absence, due to proteolytic turnover. For example, the co-stabilizing proteins HMW1 and HMW2 each exhibit accelerated turnover in the absence of the other (Willby et al., 2004), P65 levels are reduced in cells lacking P30 (Jordan et al., 2001), and proteins B and C are unstable in the absence of P1 (Waldo et al., 2005). Here, however, only protein P24 was reduced in the topJ mutant, and preliminary studies of terminal organelle proteins fused with yellow fluorescent protein in topJ mutant cells revealed persistence of foci within cells for several hours, arguing against terminal organelle complex collapse due to instability (data not shown). The presence of a predicted J-domain in a protein does not necessarily dictate a role in non-native protein folding. M. pneumoniae encodes two other putative J-domain proteins in addition to TopJ (MPN002 and MPN021); of the three, the deduced MPN021 protein has the greatest similarity to E. coli DnaJ, which functions in non-native protein refolding in response to cellular stress. Perhaps significantly, both M. pneumoniae DnaJ (MPN021) and GroEL (MPN573), a 14-mer barrel complex that also responds to misfolded proteins in E. coli (Hartl et al., 1992), have been isolated in Triton X-100-insoluble extractions (Regula et al., 2001; Bose, 2007). Alternatively, or in addition, M. pneumoniae trigger factor may function similarly to its E. coli counterpart by substituting for DnaK and folding nascent polypeptides at the ribosome exit site, independent of a J-domain protein (Deuerling et al., 1999; 2003).
J-domain proteins also appear to participate in more complex functions in addition to protein folding, including macromolecular stability, assembly and regulation. For example, prevention of protein aggregation due to intracellular crowding is achieved with chaperone complexes binding to adjacent hydrophobic regions, thus stabilizing protein complexes (Hartl and Hayer-Hartl, 2002). A J-domain co-chaperone in Chlamydomonas reinhardtii assists with thylakoid membrane assembly in chloroplasts (Liu et al., 2007). Disassembly of clathrin-coated vesicles in yeast is achieved with the assistance of J-domain protein auxilin (Lemmon, 2001). Finally, the E. coli DnaJ homologue CbpA is a late-stationary-phase DNA-binding protein involved in cell growth and division and possibly affects transcription regulation (Bird et al., 2006). Thus, loss of TopJ in M. pneumoniae may affect terminal organelle assembly, placement and/or stability leading to cascading defects in cytadherence, motility and cell division. Regardless of the specific mechanism, the stability of what are considered early terminal organelle components (Krause and Balish, 2004) in the topJ mutant is consistent with a late-stage defect in the assembly process.
The key observation that polar foci of terminal organelle proteins P1, P30 and P41 were rare in the topJ mutant prompted a more quantitative assessment leading to a clearer understanding of TopJ association with the terminal organelle. Non-polar, paired P30/HMW1 foci were observed in 22% of topJ mutant cells compared with < 1% in wild-type cells. Furthermore, nearly half of the topJ mutant cells examined lacked P30 and HMW1 foci altogether, compared with < 2% of wild-type cells (Table 2), and yet the lack of detectable HMW1 and P30 foci in nearly half the cells was not accompanied by reduced steady-state levels of these proteins in Western immunoblots, leading us to believe that in topJ mutants these terminal organelle components form intermediate structures that are stabilized against the turnover that normally occurs in the absence of required binding partners (Popham et al., 1997). We speculate further that those intermediate complexes fail to form higher-ordered structures with wild-type efficiency, and as a result remain too dispersed, antibody inaccessible and/or improperly localized, without TopJ function. Initial studies with fluorescent protein fusions of P41 and P30 in topJ mutant cells revealed faint, diffuse fluorescence in cells lacking a fluorescent focus, indicating the presence, but epitope inaccessibility, limited sensitivity of detection, or both, of each protein by immunofluorescence in topJ mutant cells (data not shown). Furthermore, preliminary data by immunoelectron microscopy are consistent with a defect in terminal organelle localization in the topJ mutant, which differed in both terminal organelle presence and position compared with wild-type cells (data not shown). Finally, identification of specific domains of TopJ involved in assembly/positioning of the terminal organelle is likely to expand the defined roles of J-domain proteins.
Experimental procedures
Strains and culture conditions
The wild-type M. pneumoniae strain used in this study was M129-B18 (Lipman and Clyde, 1969). Spontaneous HA− strains used in attachment studies were M129 derivatives II-3 and III-4 (Krause et al., 1982). Mycoplasmas were cultured in tissue culture flasks with SP4 medium (Tully et al., 1977) at 37°C until mid-log phase. Gentamicin (18 μg ml−1) was included for transformants containing an ISM2062 derivative carrying the modified transposon, Tn4001mod (Knudtson and Minion, 1993), while chloramphenicol (24 μg ml−1) was included for transformants with Tn4001cat for delivery of recombinant alleles (Hahn et al., 1999).
Wild-type M129 was transformed with a Tn4001 derivative TN4001mod as part of a previous study (Willby et al., 2004). A clonal population was obtained after two rounds of filter-cloning of single colonies grown on PPLO agar (Tully, 1983; Hahn et al., 1996). Insertion mapping of the transposon required digestion of genomic DNA with HindIII, serial dilution of the digest and religation to circularize the DNA. The outward-directed primer 5′-GGGTCATGTAAAAGTCCTCCT GGGTA-3′ anneals to the IS256 inverted repeats of Tn4001 allowing amplification of the circularized DNA for cloning into the vector pCRII® (Invitrogen, Carlsbad, CA) utilizing the 3′ thymidine overhangs. Primers annealing to the pCRII® vector were used for sequencing of the cloned insert to localize the transposon insertion, which mapped to residue 17 of MPN119 and was accompanied by an 8 bp duplication. Southern blot hybridization confirmed the presence of a single copy of Tn4001mod; clonality was established by PCR.
Protein profiling by SDS-PAGE and Western immunoblotting
Total protein of M. pneumoniae cultures was quantified by the bicinchoninic acid assay (Pierce, Rockford, IL) prior to SDS-PAGE separation and Western immunoblotting (Hahn et al., 1996; Hasselbring et al., 2005). Rabbit polyclonal antibodies were used at the following dilutions: anti-HMW1, 1:10 000; anti-HMW2, 1:1000; anti-HMW3, 1:8000; anti-P200, 1:10 000; anti-TopJ, 1:2000; anti-GrpE, 1:500; anti-P1, 1:1000; anti-B, 1:10 000; anti-C, 1:1000; anti-P65, 1:3000; anti-P30, 1:100; anti-P41, 1:1000; anti-P24, 1:250; and anti-P28, 1:1000. Anti-rabbit alkaline phosphatase-conjugated secondary antibody (Promega, Madison, WI) was used at 1:7500. Polyclonal TopJ-specific antiserum was developed using two separate 20-residue peptides (60-NPKKRANYDKYGHDGVDNEG and 147-EDYPPQSDYPDDIPDVDARI) conjugated individually to Keyhole Limpet Haemocyanin by Antibodies (Davis, CA). Polyclonal anti-GrpE was developed similarly using the peptide 180-PSDQPANTVVKVSKSGYKLH.
Microcolony satellite growth and motility analysis
Mycoplasmas were cultured in four-well borosilicate glass chamber slides (Nunc Nalgene, Naperville, IL) containing SP4 medium + 3% gelatin, with gentamicin and chloramphenicol included as appropriate for transformants. Time-lapse images of colony growth and morphology were recorded at 12 h intervals (Hasselbring et al., 2005). Average gliding frequencies and velocities were quantified, as described previously (Hasselbring et al., 2005) except that wild-type and transformant cultures were incubated for 4–6 h and the topJ mutant was incubated overnight. Over 1200 cells per strain were evaluated for gliding frequency with speeds quantified for approximately 70–90 cells per strain.
Quantitative HA and A549 binding
Mycoplasma pneumoniae attachment to erythrocytes correlates with successful colonization of respiratory epithelium (Sobeslavsky et al., 1968) and serves as a convenient indicator of cytadherence capacity. Qualitative and quantitative HA analysis followed previously described procedures [Krause and Baseman (1982), and Krause and Baseman (1983) and Fisseha et al. (1999) respectively], except cells were cultured in SP4 medium. Radiolabelled mycoplasmas were also employed for quantitative assessment of adherence to the A549 human lung adenocarcinoma cell line as described elsewhere (Jordan et al., 2007) except that cells were fed apically in 24-well tissue culture plates (Corning, Corning, NY). M. pneumoniae M129-derived mutants II-3 and III-4 (Krause et al., 1982) were used as negative controls for HA and A549 attachment studies respectively.
Immunofluorescence microscopy
Mycoplasmas were grown as described above for motility assays except four-well borosilicate glass chamber slides (Nunc Nalgene, Naperville, IL) were coated in 0.01% poly-L-lysine. Samples were prepared as previously described (Jordan et al., 2001) with minor revisions: cells were fixed with 2.0% paraformaldehyde-0.2% glutaraldehyde in PBS, washed with PBS-0.5% Tween (TPBS), and incubated overnight in 5% skim milk + 0.5% Tween (blocking solution), then 1 h incubation at room temperature with primary antibody in blocking solution (rabbit polyclonal anti-TopJ, 1:500; rabbit polyclonal anti-HMW1, 1:100; rabbit polyclonal anti-P41, 1:100; mouse monoclonal anti-P1, 1:20; and mouse monoclonal anti-P30, 1:50). Images were captured as previously described (Hasselbring et al., 2005), digitized and superimposed with OpenLab Imaging Software (version 5.01, Lexington, MA).
Acknowledgments
We thank E.S. Sheppard for technical assistance. This work was supported by Public Health Service research Grant AI22362 from the National Institute of Allergy and Infectious Diseases to D.C.K.
References
- Balish MF, Krause DC. Cytadherence and the cytoskeleton. In: Herrmann SRaR., editor. Molecular Biology and Pathogenicity of the Mycoplasmas. New York: Kluwer Academic/Plenum Publishers; 2002. pp. 491–518. [Google Scholar]
- Balish MF, Hahn TW, Popham PL, Krause DC. Stability of Mycoplasma pneumoniae cytadherence-accessory protein HMW1 correlates with its association with the triton shell. J Bacteriol. 2001;183:3680–3688. doi: 10.1128/JB.183.12.3680-3688.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balish MF, Santurri RT, Ricci AM, Lee KK, Krause DC. Localization of Mycoplasma pneumoniae cytadherence-associated protein HMW2 by fusion with green fluorescent protein: implications for attachment organelle structure. Mol Microbiol. 2003;47:49–60. doi: 10.1046/j.1365-2958.2003.03282.x. [DOI] [PubMed] [Google Scholar]
- Baseman JB, Cole RM, Krause DC, Leith DK. Molecular basis for cytadsorption of Mycoplasma pneumoniae. J Bacteriol. 1982;151:1514–1522. doi: 10.1128/jb.151.3.1514-1522.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baseman JB, Morrison-Plummer J, Drouillard D, Puleo-Scheppke B, Tryon VV, Holt SC. Identification of a 32-kilodalton protein of Mycoplasma pneumoniae associated with hemadsorption. Isr J Med Sci. 1987;23:474–479. [PubMed] [Google Scholar]
- Biberfeld G, Biberfeld P. Ultrastructural features of Mycoplasma pneumoniae. J Bacteriol. 1970;102:855–861. doi: 10.1128/jb.102.3.855-861.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird JG, Sharma S, Roshwalb SC, Hoskins JR, Wickner S. Functional analysis of CbpA, a DnaJ homolog and nucleoid-associated DNA-binding protein. J Biol Chem. 2006;281:34349–34356. doi: 10.1074/jbc.M603365200. [DOI] [PubMed] [Google Scholar]
- Bose SR. Microbiology and Cell Biology Doctoral dissertation. University of Georgia; 2007. Structure/function analysis of HMW2 and proteomic analysis of the electrondense core in Mycoplasma pneumoniae. [Google Scholar]
- Bredt W. Motility and multiplication of Mycoplasma pneumoniae. A phase contrast study. Pathol Microbiol (Basel) 1968;32:321–326. doi: 10.1159/000162074. [DOI] [PubMed] [Google Scholar]
- Byrne ME, Rouch DA, Skurray RA. Nucleotide sequence analysis of IS256 from the Staphylococcus aureusgentamicin-tobramycin-kanamycin-resistance transposon Tn4001. Gene. 1989;81:361–367. doi: 10.1016/0378-1119(89)90197-2. [DOI] [PubMed] [Google Scholar]
- Collier AM, Clyde WA, Jr, Denny FW. Mycoplasma pneumoniae in hamster tracheal organ culture: immunofluorescent and electron microscopic studies. Proc Soc Exp Biol Med. 1971;136:569–573. doi: 10.3181/00379727-136-35313. [DOI] [PubMed] [Google Scholar]
- Dandekar T, Huynen M, Regula JT, Ueberle B, Zimmermann CU, Andrade MA, et al. Re-annotating the Mycoplasma pneumoniae genome sequence: adding value, function and reading frames. Nucleic Acids Res. 2000;28:3278–3288. doi: 10.1093/nar/28.17.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuerling E, Schulze-Specking A, Tomoyasu T, Mogk A, Bukau B. Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature. 1999;400:693–696. doi: 10.1038/23301. [DOI] [PubMed] [Google Scholar]
- Deuerling E, Patzelt H, Vorderwulbecke S, Rauch T, Kramer G, Schaffitzel E, et al. Trigger factor and DnaK possess overlapping substrate pools and binding specificities. Mol Microbiol. 2003;47:1317–1328. doi: 10.1046/j.1365-2958.2003.03370.x. [DOI] [PubMed] [Google Scholar]
- Feldner J, Gobel U, Bredt W. Mycoplasma pneumoniae adhesin localized to tip structure by monoclonal antibody. Nature. 1982;298:765–767. doi: 10.1038/298765a0. [DOI] [PubMed] [Google Scholar]
- Fisseha M, Gohlmann HW, Herrmann R, Krause DC. Identification and complementation of frame-shift mutations associated with loss of cytadherence in Mycoplasma pneumoniae. J Bacteriol. 1999;181:4404–4410. doi: 10.1128/jb.181.14.4404-4410.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- Genevaux P, Schwager F, Georgopoulos C, Kelley WL. Scanning mutagenesis identifies amino acid residues essential for the in vivo activity of the Escherichia coli DnaJ (Hsp40) J-domain. Genetics. 2002;162:1045–1053. doi: 10.1093/genetics/162.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobel U, Speth V, Bredt W. Filamentous structures in adherent Mycoplasma pneumoniae cells treated with nonionic detergents. J Cell Biol. 1981;91:537–543. doi: 10.1083/jcb.91.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene MK, Maskos K, Landry SJ. Role of the J-domain in the cooperation of Hsp40 with Hsp70. Proc Natl Acad Sci USA. 1998;95:6108–6113. doi: 10.1073/pnas.95.11.6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn TW, Krebes KA, Krause DC. Expression in Mycoplasma pneumoniae of the recombinant gene encoding the cytadherence-associated protein HMW1 and identification of HMW4 as a product. Mol Microbiol. 1996;19:1085–1093. doi: 10.1046/j.1365-2958.1996.455985.x. [DOI] [PubMed] [Google Scholar]
- Hahn TW, Mothershed EA, Waldo RH, 3rd, Krause DC. Construction and analysis of a modified Tn4001 conferring chloramphenicol resistance in Mycoplasma pneumoniae. Plasmid. 1999;41:120–124. doi: 10.1006/plas.1998.1387. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Martin J, Neupert W. Protein folding in the cell: the role of molecular chaperones Hsp70 and Hsp60. Annu Rev Biophys Biomol Struct. 1992;21:293–322. doi: 10.1146/annurev.bb.21.060192.001453. [DOI] [PubMed] [Google Scholar]
- Hasselbring BM, Krause DC. Proteins P24 and P41 function in the regulation of terminal-organelle development and gliding motility in Mycoplasma pneumoniae. J Bacteriol. 2007a;189:7442–7449. doi: 10.1128/JB.00867-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselbring BM, Krause DC. Cytoskeletal protein P41 is required to anchor the terminal organelle of the wall-less prokaryote Mycoplasma pneumoniae. Mol Microbiol. 2007b;63:44–53. doi: 10.1111/j.1365-2958.2006.05507.x. [DOI] [PubMed] [Google Scholar]
- Hasselbring BM, Jordan JL, Krause DC. Mutant analysis reveals a specific requirement for protein P30 in Mycoplasma pneumoniae gliding motility. J Bacteriol. 2005;187:6281–6289. doi: 10.1128/JB.187.18.6281-6289.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselbring BM, Page CA, Sheppard ES, Krause DC. Transposon mutagenesis identifies genes associated with Mycoplasma pneumoniae gliding motility. J Bacteriol. 2006a;188:6335–6345. doi: 10.1128/JB.00698-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselbring BM, Jordan JL, Krause RW, Krause DC. Terminal organelle development in the cell wall-less bacterium Mycoplasma pneumoniae. Proc Natl Acad Sci USA. 2006b;103:16478–16483. doi: 10.1073/pnas.0608051103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson GP, Jensen GJ. Three-dimensional structure of Mycoplasma pneumoniae’s attachment organelle and a model for its role in gliding motility. Mol Microbiol. 2006;60:376–385. doi: 10.1111/j.1365-2958.2006.05113.x. [DOI] [PubMed] [Google Scholar]
- Hendrick JP, Hartl FU. The role of molecular chaperones in protein folding. FASEB J. 1995;9:1559–1569. doi: 10.1096/fasebj.9.15.8529835. [DOI] [PubMed] [Google Scholar]
- Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24:4420–4449. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RE, Johnson AE. Protein translocation: is Hsp70 pulling my chain? Curr Biol. 1999;9:R779–R782. doi: 10.1016/S0960-9822(00)80012-3. [DOI] [PubMed] [Google Scholar]
- Jordan JL, Berry KM, Balish MF, Krause DC. Stability and subcellular localization of cytadherence-associated protein P65 in Mycoplasma pneumoniae. J Bacteriol. 2001;183:7387–7391. doi: 10.1128/JB.183.24.7387-7391.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan JL, Chang HY, Balish MF, Holt LS, Bose SR, Hasselbring BM, et al. Protein P200 is dispensable for Mycoplasma pneumoniae hemadsorption but not gliding motility or colonization of differentiated bronchial epithelium. Infect Immun. 2007;75:518–522. doi: 10.1128/IAI.01344-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan TR, Musatovova O, Gowda P, Baseman JB. Characterization of unique ClpB protein of Mycoplasma pneumoniae and its impact on growth. Infect Immun. 2008;76:5082–5092. doi: 10.1128/IAI.00698-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenri T, Seto S, Horino A, Sasaki Y, Sasaki T, Miyata M. Use of fluorescent-protein tagging to determine the subcellular localization of Mycoplasma pneumoniae proteins encoded by the cytadherence regulatory locus. J Bacteriol. 2004;186:6944–6955. doi: 10.1128/JB.186.20.6944-6955.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudtson KL, Minion FC. Construction of Tn4001lac derivatives to be used as promoter probe vectors in mycoplasmas. Gene. 1993;137:217–222. doi: 10.1016/0378-1119(93)90009-r. [DOI] [PubMed] [Google Scholar]
- Krause DC, Balish MF. Structure, function, and assembly of the terminal organelle of Mycoplasma pneumoniae. FEMS Microbiol Lett. 2001;198:1–7. doi: 10.1111/j.1574-6968.2001.tb10610.x. [DOI] [PubMed] [Google Scholar]
- Krause DC, Balish MF. Cellular engineering in a minimal microbe: structure and assembly of the terminal organelle of Mycoplasma pneumoniae. Mol Microbiol. 2004;51:917–924. doi: 10.1046/j.1365-2958.2003.03899.x. [DOI] [PubMed] [Google Scholar]
- Krause DC, Baseman JB. Mycoplasma pneumoniae proteins that selectively bind to host cells. Infect Immun. 1982;37:382–386. doi: 10.1128/iai.37.1.382-386.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause DC, Baseman JB. Inhibition of Mycoplasma pneumoniae hemadsorption and adherence to respiratory epithelium by antibodies to a membrane protein. Infect Immun. 1983;39:1180–1186. doi: 10.1128/iai.39.3.1180-1186.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause DC, Leith DK, Wilson RM, Baseman JB. Identification of Mycoplasma pneumoniae proteins associated with hemadsorption and virulence. Infect Immun. 1982;35:809–817. doi: 10.1128/iai.35.3.809-817.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layh-Schmitt G, Herrmann R. Localization and biochemical characterization of the ORF6 gene product of the Mycoplasma pneumoniae P1 operon. Infect Immun. 1992;60:2906–2913. doi: 10.1128/iai.60.7.2906-2913.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layh-Schmitt G, Podtelejnikov A, Mann M. Proteins complexed to the P1 adhesin of Mycoplasma pneumoniae. Microbiology. 2000;146:741–747. doi: 10.1099/00221287-146-3-741. [DOI] [PubMed] [Google Scholar]
- Lemmon SK. Clathrin uncoating: auxilin comes to life. Curr Biol. 2001;11:R49–R52. doi: 10.1016/s0960-9822(01)00010-0. [DOI] [PubMed] [Google Scholar]
- Lipman RP, Clyde WA., Jr The interrelationship of virulence, cytadsorption, and peroxide formation in Mycoplasma pneumoniae. Proc Soc Exp Biol Med. 1969;131:1163–1167. doi: 10.3181/00379727-131-34061. [DOI] [PubMed] [Google Scholar]
- Liu C, Willmund F, Golecki JR, Cacace S, Hess B, Markert C, Schroda M. The chloroplast HSP70B-CDJ2-CGE1 chaperones catalyse assembly and disassembly of VIPP1 oligomers in Chlamydomonas. Plant J. 2007;50:265–277. doi: 10.1111/j.1365-313X.2007.03047.x. [DOI] [PubMed] [Google Scholar]
- Meng KE, Pfister RM. Intracellular structures of Mycoplasma pneumoniae revealed after membrane removal. J Bacteriol. 1980;144:390–399. doi: 10.1128/jb.144.1.390-399.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musatovova O, Dhandayuthapani S, Baseman JB. Transcriptional heat shock response in the smallest known self-replicating cell, Mycoplasma genitalium. J Bacteriol. 2006;188:2845–2855. doi: 10.1128/JB.188.8.2845-2855.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SN, Hu PC, Bott KF, Hutchison CA., 3rd A survey of the Mycoplasma genitalium genome by using random sequencing. J Bacteriol. 1993;175:7918–7930. doi: 10.1128/jb.175.24.7918-7930.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pich OQ, Burgos R, Ferrer-Navarro M, Querol E, Pinol J. Mycoplasma genitalium mg200 and mg386 genes are involved in gliding motility but not in cytadherence. Mol Microbiol. 2006;60:1509–1519. doi: 10.1111/j.1365-2958.2006.05187.x. [DOI] [PubMed] [Google Scholar]
- Popham PL, Hahn TW, Krebes KA, Krause DC. Loss of HMW1 and HMW3 in noncytadhering mutants of Mycoplasma pneumoniae occurs post-translationally. Proc Natl Acad Sci USA. 1997;94:13979–13984. doi: 10.1073/pnas.94.25.13979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regula JT, Boguth G, Gorg A, Hegermann J, Mayer F, Frank R, Herrmann R. Defining the mycoplasma ‘cytoskeleton’: the protein composition of the Triton X-100 insoluble fraction of the bacterium Mycoplasma pneumoniae determined by 2-D gel electrophoresis and mass spectrometry. Microbiology. 2001;147:1045–1057. doi: 10.1099/00221287-147-4-1045. [DOI] [PubMed] [Google Scholar]
- Rost B, Yachdav G, Liu J. The PredictProtein server. Nucleic Acids Res. 2004;32:W321–W326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto S, Layh-Schmitt G, Kenri T, Miyata M. Visualization of the attachment organelle and cytadherence proteins of Mycoplasma pneumoniae by immunofluorescence microscopy. J Bacteriol. 2001;183:1621–1630. doi: 10.1128/JB.183.5.1621-1630.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobeslavsky O, Prescott B, Chanock RM. Adsorption of Mycoplasma pneumoniae to neuraminic acid receptors of various cells and possible role in virulence. J Bacteriol. 1968;96:695–705. doi: 10.1128/jb.96.3.695-705.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens MK, Krause DC. Localization of the Mycoplasma pneumoniae cytadherence-accessory proteins HMW1 and HMW4 in the cytoskeleton-like Triton shell. J Bacteriol. 1991;173:1041–1050. doi: 10.1128/jb.173.3.1041-1050.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens MK, Krause DC. Mycoplasma pneumoniae cytadherence phase-variable protein HMW3 is a component of the attachment organelle. J Bacteriol. 1992;174:4265–4274. doi: 10.1128/jb.174.13.4265-4274.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto S, Saruwatari K, Higashi C, Sonomoto K. The proper ratio of GrpE to DnaK is important for protein quality control by the DnaK–DnaJ–GrpE chaperone system and for cell division. Microbiology. 2008;154:1876–1885. doi: 10.1099/mic.0.2008/017376-0. [DOI] [PubMed] [Google Scholar]
- Tully JG. New laboratory techniques for isolation of Mycoplasma pneumoniae. Yale J Biol Med. 1983;56:511–515. [PMC free article] [PubMed] [Google Scholar]
- Tully JG, Whitcomb RF, Clark HF, Williamson DL. Pathogenic mycoplasmas: cultivation and vertebrate pathogenicity of a new spiroplasma. Science. 1977;195:892–894. doi: 10.1126/science.841314. [DOI] [PubMed] [Google Scholar]
- Waites KB, Talkington DF. Mycoplasma pneumoniae and its role as a human pathogen. Clin Microbiol Rev. 2004;17:697–728. doi: 10.1128/CMR.17.4.697-728.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo RH, 3rd, Jordan JL, Krause DC. Identification and complementation of a mutation associated with loss of Mycoplasma pneumoniae virulence-specific proteins B and C. J Bacteriol. 2005;187:747–751. doi: 10.1128/JB.187.2.747-751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner J, 3rd, Zimmerman CU, Gohlmann HW, Herrmann R. Transcription profiles of the bacterium Mycoplasma pneumoniae grown at different temperatures. Nucleic Acids Res. 2003;31:6306–6320. doi: 10.1093/nar/gkg841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willby MJ, Balish MF, Ross SM, Lee KK, Jordan JL, Krause DC. HMW1 is required for stability and localization of HMW2 to the attachment organelle of Mycoplasma pneumoniae. J Bacteriol. 2004;186:8221–8228. doi: 10.1128/JB.186.24.8221-8228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]