

This study links a noncoding DNA variant to long range regulation of IL-32 isoform expression, modulating susceptibility to HIV.
Abstract
We integrated data obtained from HIV-1 genome-wide association studies with T cell–derived epigenome data and found that the noncoding intergenic variant rs4349147, which is statistically associated with HIV-1 acquisition, is located in a CD4+ T cell–specific deoxyribonuclease I hypersensitive region, suggesting regulatory potential for this variant. Deletion of the rs4349147 element in Jurkat cells strongly reduced expression of interleukin-32 (IL-32), approximately 10-kb upstream, and chromosome conformation capture assays identified a chromatin loop between rs4349147 and the IL-32 promoter validating its function as a long-distance enhancer. We generated single rs4349147-A or rs4349147-G allele clones and demonstrated that IL-32 enhancer activity and interaction with the IL-32 promoter are strongly allele dependent; rs4349147 −/A cells display reduced IL-32 expression and altered chromatin conformation as compared to rs4349147 G/− cells. Moreover, RNA sequencing demonstrated that rs4349147 G/− cells express a lower relative ratio of IL-32α to non-α isoforms than rs4349147 −/A cells and display increased expression of lymphocyte activation factors rendering them more prone to infection with HIV-1. In agreement, in primary CD4+ T cells, both treatment with recombinant IL-32γ (rIL-32γ) but not rIL-32α, and exogenous lentiviral overexpression of IL-32γ or IL-32β but not IL-32α resulted in a proinflammatory T cell cytokine environment concomitant with increased susceptibility to HIV infection. Our data demonstrate that rs4349147-G promotes transcription of non–IL-32α isoforms, generating a proinflammatory environment more conducive to HIV infection. This study provides a mechanistic link between a HIV-associated noncoding DNA variant and the expression of different IL-32 isoforms that display discrete anti-HIV properties.
INTRODUCTION
Host genetic variation has long been recognized to play a major role in HIV-1 infection susceptibility and disease progression (1). Although it is widely accepted that genetic variants in loci encoding class I human leukocyte antigens (HLAs) and the chemokine receptor CCR5 have a major impact on HIV infection, the contribution of additional genetic variants remains elusive. In the past decade, genome-wide association studies (GWASs) have attempted to identify single-nucleotide polymorphisms (SNPs) that are correlated with various aspects of HIV disease acquisition or progression. In general, a major limitation of GWAS is that they are geared to identify genetic variants with relatively high minor allele frequencies and moderate to high effects, particularly when the sample size is small. This is especially the case for HIV-1 GWAS (1) where collection of large cohorts is challenging. As a consequence, common variants with weak effect size and rare variants fall below the stringent threshold for genome-wide significance (P < 5 × 10−8) and are missed. In addition, the functional significance of identified HIV-associated genetic variants is often unclear because most of the correlated SNPs locate to noncoding regions of the genome with unknown function (1). As a consequence, HIV-centered GWASs fail to deliver distinct candidate loci, which hampers further efforts to characterize the contribution of human genetic variation to HIV susceptibility and disease progression. Integration of additional approaches to prioritize candidate loci is therefore of utmost importance to push the field of HIV-1 host genetics forward.
Recently, it is becoming clear that inclusion of genome-wide functional data into the analysis can circumvent the caveats of GWAS, allowing the identification of candidate noncoding SNPs most likely to be biologically relevant. As we have shown for the pigmentation-associated variant rs12913832 (2), function can be inferred for noncoding SNPs by examining co-occurrence with chromatin marks generally associated with regulatory DNA elements using resources that include the Encyclopedia of DNA Elements (ENCODE) and the Human Epigenome Atlas, among others (3). Thus, epigenomic data are used to identify variants with potential function and strongly reduce the list of noncoding SNPs to analyze. This analysis can also identify the relevant cell types and candidate genes to be investigated. Promising candidate variants are subsequently scrutinized by detailed molecular analysis to elucidate their biological function. Crucially, it was recently demonstrated that such an approach can also prioritize so-called “subthreshold” variants that fail to reach the widely accepted threshold of P < 5 × 10−8 statistical significance and assign biological function to them (3). A total of 72% of the subthreshold loci that were prioritized in this way contained an allele-specific enhancer, and they were also more likely to replicate in subsequent GWASs than subthreshold loci that do not overlap active enhancers.
Here, we integrated data obtained from HIV-1 GWASs with T cell–derived epigenome data to identify noncoding HIV-1–associated SNPs with potential regulatory function. We performed a detailed molecular analysis of one of the identified variants, rs4349147, which is statistically associated at a subthreshold level with HIV-1 acquisition (4), and demonstrated that this variant is located in a lymphoid-specific enhancer for the interleukin-32 (IL-32) cytokine gene. Haplotypes of rs4349147 not only modulate the transcription level of IL-32 but also alter the ratio of isoforms expressed. This altered expression of IL-32 isoforms modulates the expression of proinflammatory factors that modify susceptibility to HIV-1 infection. Our study demonstrates that the integration of GWAS data with epigenome data can assign biological function to subthreshold HIV-1–associated variants, such as rs4349147, and that allele-dependent differential expression of the noncanonical IL-32 cytokine gene directs HIV-1 susceptibility.
RESULTS
Deoxyribonuclease I hypersensitive region encompassing rs4349147 is a long-distance enhancer of IL-32
As a starting point for this study, we collected published HIV-associated variants (P < 9 × 10−6) from the GWAS catalog (www.ebi.ac.uk/gwas/; accessed on May 2014), together with SNPs in strong linkage (r2 > 0.8). Because deoxyribonuclease I (DNase I) “hypersensitivity” is a feature of active cis-regulatory sequences in a given cell type, we compared the HIV-associated variants to DNase I hypersensitivity data obtained from CD4+ T helper naïve (TH0) and TH1 cells [ENCODE data set; University of California, Santa Cruz (UCSC) genome browser]. This analysis yielded a list of 21 HIV-associated SNPs that are located in potential regulatory regions (table S1). Most of these SNPs are located on chromosome 6 with the majority spread out more than a 1.3-Mb region encompassing the HLA loci, a thoroughly studied region and well known to be associated with HIV infection. Four SNPs are located in a histone cluster, which we reasoned to be of less interest. Two SNPs are located in a 500-kb gene desert on chromosome 1 and two in a 175-kb gene desert on chromosome 8. The remaining three variants are located in a gene-dense region on chromosome 16, and we decided to investigate this region in more detail. The three variants located on chromosome 16 (rs12447486, rs2015620, and rs4349147) are in perfect linkage (HapMap release 22; CEU), and rs4349147 has been reported to be associated at a subthreshold level (P = 7.91 × 10−6) with HIV-1 acquisition in a cohort of African HIV-1 serodiscordant heterosexual couples (4). rs4349147 is located in a region that is strongly DNase I–hypersensitive in the T cell–derived cell lines Jurkat and SupT1 but not in cells not belonging to the immune compartment (Fig. 1A), whereas the other two variants are located in regions with minimal DNase I hypersensitivity. In agreement, formaldehyde-assisted isolation of regulatory elements (FAIRE) assays demonstrated strong enrichment of the rs4349147 DNA region in the T cell lines but not in the unrelated G361 melanoma cells (fig. S1A), highlighting the presence of an active regulatory element and supporting a regulatory role for the rs4349147 region in T cells. To gain more insight into its function, we deleted the DNase I hypersensitive (DHS) region containing rs4349147 using an upstream- and downstream-targeted CRISPR (clustered regularly interspaced short palindromic repeats)–Cas9 (CRISPR-associated protein 9) gene editing approach (Fig. 1B, top). Polymerase chain reaction (PCR) confirmed homozygous deletion of the rs4349147 DHS (Fig. 1B, bottom), and a clonal Jurkat line containing a full knockout (KO) of the rs4349147 DHS was generated. Sequencing confirmed the deletion of the specific region (fig. S1B).
Fig. 1. DNA region encompassing rs4349147 is a strong enhancer element for the IL-32 gene in T cells.
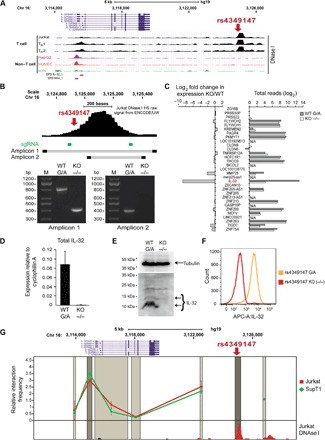
(A) UCSC genome browser tracks show that the HIV-associated variant rs4349147 is located in a T cell–specific DHS region. Each track represents the DNase I accessibility profile for a given cell line. Promoters as defined by the Eukaryotic Promoter Database (EPD; http://epd.vital-it.ch) are indicated as red bars. HUVEC, human umbilical cord endothelial cell. (B) Deletion of rs4349147 DHS. Location of small-guiding RNAs (sgRNAs) used in the CRISPR-Cas9 KO strategy is indicated. Panels show results from two different PCR assays generating amplicons 1 or 2, as indicated, that confirm homozygous deletion of rs4349147 DHS. (C) RNA expression levels of genes in a 500-kb window surrounding rs4349147 demonstrate a strong reduction in IL-32 expression. For each gene considered in the analysis, right panel shows total number of reads, whereas left panel shows fold change in expression in rs4349147 DHS KO cells compared to WT. N/A, not available. (D to F) Strong reduction of IL-32 expression is confirmed using real-time quantitative PCR (qPCR) (D), Western blotting using an antibody against IL-32 (E), and intracellular staining with biotin-conjugated antibody against IL-32, followed by fluorescence-activated cell sorting (FACS) analysis (F). (G) 3C analysis of the IL-32 locus demonstrates chromatin loop formation between rs4349147 and the IL-32 promoter region in Jurkat and SupT1 cells. The analyzed region of the human IL-32 locus is depicted on the top of each graph. X axis shows the approximate position on chromosome 16 (Chr 16) (UCSC genome browser GRCh37/hg19 assembly). Dark gray shading shows the position and size of the “fixed” Dpn II restriction fragment. Light gray shading indicates position and size of other Dpn II restriction fragments analyzed. The Dpn II restriction fragment containing the IL-32 promoter is indicated in a slightly darker gray color.
To determine which genes are regulated by this DHS region, we performed high-throughput sequencing of RNA isolated from wild-type (WT) and rs4349147 DHS KO Jurkat cells. We found that of all genes within a 500-kb region centered on rs4349147, specifically, the expression of IL-32 is severely reduced upon KO of the rs4349147 DHS (Fig. 1C and fig. S1C), whereas the expression of surrounding genes remains essentially unchanged (Fig. 1C and fig. S1D). We confirmed this observation by reverse transcription PCR (RT-PCR) (Fig. 1D). Western blotting (Fig. 1E) and intracellular flow cytometry (Fig. 1F) demonstrated that IL-32 expression at the protein level is, as expected, likewise reduced to undetectable levels in the rs4349147 DHS KO cells. In addition, targeting of a dead Cas9 (dCas9)–Kruppel-associated box (KRAB) fusion protein, which is a strong repressor of enhancer function (5), to the rs4349147 DHS in WT Jurkat cells also resulted in significant reduction of IL-32 RNA expression (fig. S1E), supporting our observations in the rs4349147 KO cells.
Distal regulatory elements, such as enhancers, are known to communicate with their target genes through chromatin loop formation (6). To determine whether chromatin at the rs4349147 DHS directly contacts the IL-32 promoter, we performed chromosome conformation capture (3C) analysis, which revealed the presence of a chromatin loop between rs4349147 DHS and the IL-32 promoter in WT Jurkat and SupT1 T cells (Fig. 1G). Together, these results demonstrate that the DNA element encompassing rs4349147 is a strong long-distance enhancer essential for the expression of IL-32 in CD4+ T cells.
IL-32 enhancer activity is dependent on the rs4349147 haplotype
Jurkat cells are heterozygous rs4349147-G/A that allowed us to generate heterozygous lines harboring a deletion of either the rs4349147-G or rs4349147-A allele enhancer and thus generate single rs4349147-G (G/−) or rs4349147-A (−/A) allele clones. rs4349147-G/A is in perfect linkage disequilibrium (LD) with rs4786376-G/C, which is also situated in the IL-32 enhancer region (Fig. 2A). rs4786376-G/C can be used to genotype clones by restriction fragment length polymorphism because the rs4786376-C allele generates a unique Nla III restriction site (Fig. 2, A to C). RNA and protein expression levels of IL-32 in the single rs4349147 G/− cells were found to be similar to WT levels, whereas rs4349147 −/A cells display significant reduction of IL-32 both at the RNA and protein level, as determined by RT-PCR, Western blotting, and intracellular flow cytometry (Fig. 2, D to F, and fig. S2A). Moreover, 3C analysis revealed that the long-range interaction of the enhancer region with the IL-32 promoter region is significantly altered in rs4349147 −/A cells compared to rs4349147 G/− Jurkat cells (Fig. 2G), suggesting a change in chromatin conformation. To get a better understanding of which variant, rs4349147-G/A or rs4786376-G/C, is responsible for the differential IL-32 expression, we cloned enhancer fragments with different genotypes in an IL-32 promoter–driven luciferase reporter construct and determined their activity. The rs4349147-G allele induces luciferase expression 3.5-fold when compared to the other haplotypes (Fig. 2H), suggesting that this variant is responsible for the main activity of this regulatory element. To further investigate the influence that the rs4349147 and rs4786376 variants have on the activity of the IL-32 regulatory element, we subjected the rs4349147 −/A clone to another round of CRISPR/Cas9 genome editing and obtained a clone designated −/A Δrs4349147 that harbored a 180–base pair (bp) deletion that removed the rs4349147-A variant but left the rs4786376-C variant in place (Fig. 2I and fig. S2B). Real-time PCR analysis of IL-32 expression in this clone revealed a significant 4.5-fold drop in expression as compared to the parental rs4349147 −/A clone (Fig. 2J) and confirmed that rs4349147 is the variant responsible for differential IL-32 expression. We conclude that the activity and physical interaction of the long-distance IL-32 enhancer element are strongly dependent on the allelic status of rs4349147.
Fig. 2. Activity of the rs4349147 enhancer element is dependent on the haplotype.

(A) Schematic representation of the region encompassing the rs4786376-G/C and rs4349147-G/A SNPs, indicating the size of the PCR products obtained using primers outside (amplicon 1) or inside (amplicon 2) the region targeted for KO. Amplicon 2 contains three Nla III restriction sites (asterisks). The central NIa III site (blue asterisk) includes the rs4786376-G/C SNP, which is in perfect linkage with rs4349147-G/A and can be used to genotype the heterozygous lines. (B) Simultaneous presence of two PCR products of different sizes indicates the occurrence of heterozygous rs4349147 KO lines. (C) Results of restriction reaction of amplicon 2 in WT (heterozygous rs4349147-G/A), rs4349147 −/A, G/−, or rs4349147 enhancer KO Jurkat lines. (D) Expression of IL-32 mRNA in the four lines was measured by real-time PCR. Cells homozygous for rs4349147-A express lower IL-32 RNA levels as compared to those homozygous for rs4349147-G. (E) IL-32 protein levels were determined by Western blotting using an antibody against IL-32. (F) Intracellular staining with biotin-conjugated antibody against IL-32, followed by FACS analysis, shows lower levels of IL-32 in rs4349147 −/A Jurkat cells and rs4349147 KO Jurkat cells as compared to WT and rs4349147 G/− Jurkat cells (Fig. 1F). (G) 3C analysis of the IL-32 locus demonstrates chromatin loop formation between rs4349147-G and the IL-32 promoter region in rs4349147 G/− Jurkat cells and a significant reduction in enhancer-promoter interaction in the rs4349147 −/A Jurkat cells. Ratio of interaction in rs4349147 −/A and G/− Jurkat cells is indicated. For further explanation of the figure, see Fig. 1G. (H) Activity of 476-bp enhancer fragments with different rs4349147 and rs4786376 genotypes in an IL-32 promoter–driven luciferase reporter construct. The rs4349147-G allele results in a 3.5-fold induction of luciferase expression when compared to the other haplotypes. The rs4349147 and rs4786376 alleles present in the reporter construct are indicated below the graph. (I) Schematic depicting the 158-bp deletion in the −/A Δrs4349147 clone. (J) The 158-bp deletion in the −/A Δrs4349147 clone reduces IL-32 expression 4.5-fold as compared to IL-32 expression in the parental rs4349147 −/A Jurkat clone.
rs4349147 G/− Jurkat cells are more conductive to HIV infection than rs4349147 −/A cells
When infected with a minimal HIV-derived virus, rs4349147 G/− Jurkat cells are more prone to infection than rs4349147 −/A Jurkat cells (Fig. 3A). The observed differences in infection remained similar over time and with different virus concentrations (fig. S3, A and B). These results support the observations reported in a GWAS, indicating that the rs4349147-G allele confers susceptibility to HIV infection (4). Remarkably, deletion of the rs4349147 enhancer region did not result in decreased susceptibility to HIV infection but displayed infection levels similar to the rs4349147 G/A and G/− Jurkat cells (Fig. 3A). This phenotype was not due to a clonal effect in the rs4349147 −/− cells because deletion of a 180-bp fragment that contains rs4349147-A variant from the rs4349147 −/A Jurkat cells (−/A Δrs4349147) reverted the low–rs4349147 −/A HIV susceptibility phenotype to a high-susceptibility phenotype, similar to the rs4349147 G/A, G/−, and −/− Jurkat cells (Fig. 3A).
Fig. 3. The rs4349147-G allele generates a proinflammatory activated T cell environment conducive to HIV infection.
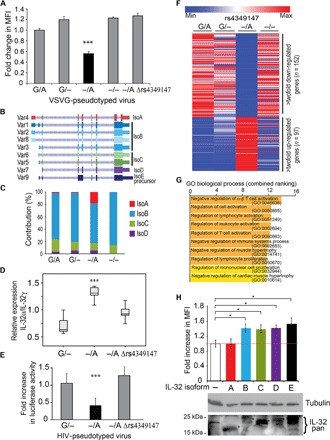
(A) Levels of infection, measured as fold increase in the mean fluorescence intensity (MFI), in rs4349147 G/A, −/A, G/−, −/−, and −/A Δrs4349147 Jurkat lines after challenge with minimal HIV vesicular stomatitis virus glycoprotein (VSVG)–pseudotyped virus. rs4349147 −/A Jurkat cells have reduced susceptibility to infection compared to Jurkat cells with other rs4349147 genotypes. (B) Nine RNA variants of IL-32 have been reported that encode for five different IL-32 protein isoforms [isoform A (IsoA), indicated in red; isoform B (IsoB), blue shades; isoform C (IsoC), green shades; isoform D (IsoD), purple; and a precursor isoform E (IsoE precursor), black]. (C) IL-32 isoform A expression is relatively elevated in rs4349147 −/A cells as compared to rs4349147 WT(G/A), G/−, or KO(−/−) cells. Bar charts show the contribution of each isoform to the total levels of IL-32 expression. (D) RT-PCR demonstrating a reduced ratio of IL-32α over IL-32γ transcripts upon deletion of the 180 bp rs4349147 containing region from the IL-32 regulatory element. (E) Levels of infection in rs4349147 −/A, G/−, and −/A Δrs4349147 Jurkat lines after challenge with HIV Env-pseudotyped virus. Luciferase activity obtained in rs4349147 G/− cells was set to 1. (F) RNA-seq heat map representation of differentially expressed genes in rs4349147 G/A, G/−, −/A, and −/− Jurkat cell lines. High-throughput RNA-seq reveals that 97 genes are up-regulated, whereas 152 genes are down-regulated at least twofold in rs4349147 −/A Jurkat cells when compared to rs4349147 G/− and WT (G/A) Jurkat cells. (G) GO-based functional classification of genes down-regulated in rs4349147 −/A Jurkat cells compared to rs4349147 G/− Jurkat cells. (H) Levels of infection, measured as fold increase in the MFI, following exogenous expression of IL-32 isoforms A to E in rs4349147 enhancer KO cells. Color coding is as for (B). Bottom: Exogenous expression of IL-32 as determined by Western blotting using the pan–IL-32 antibody.
Alternative rs4349147 alleles differentially modulate the relative expression of IL-32 isoforms
The observation that the rs4349147 KO cells −/− and −/A Δrs4349147 display similar susceptibility to infection as G/A and G/− cells, despite the fact that IL-32 expression is strongly reduced, compared to the less susceptible rs4349147 −/A cells strongly suggested that the absolute levels of IL-32 expression is not the key factor that determines susceptibility to HIV infection. Multiple protein variants of IL-32 with different but poorly characterized functions have been documented (Fig. 3B) (7, 8). The five major protein isoforms of IL-32 (isoforms A to E) are encoded by multiple RNA splice variants (variants 1 to 9). It is known that these IL-32 isoforms affect the immune system and susceptibility to HIV in different ways (8). In general, IL-32 isoforms are categorized as proinflammatory (represented mainly by IL-32β and IL-32γ) or nonproinflammatory (mainly represented by IL-32α). Controlling the expression of these distinct isoforms, two different promoters are assigned to the IL-32 gene by the EPD (http://epd.vital-it.ch). The upstream promoter is designated EPD NK4_1, whereas the downstream promoter is designated EPD IL32_1 (Fig. 1A). Promoter EPD NK4_1 generates long transcripts such as RNA variant 4 encoding the A isoform of IL-32 (IL-32α) and RNA variants 1, 2, and 8 encoding the B isoform of IL-32 (IL-32β), whereas the downstream promoter EPD IL32_1 generates shorter transcripts such as RNA variant 3 encoding the main IL-32β, RNA variants 6 and 5 encoding the C isoform of IL-32, RNA variant 7 encoding the D isoform, and RNA variant 9 encoding the isoform E precursor (IL-32γ) (Figs. 1A and 3B).
The fact that alternative IL-32 isoforms with different effects on the immune system are potentially expressed from two distinct promoters, together with the observation that reduction in absolute IL-32 expression levels is likely not the key determinant of reduced HIV infection susceptibility, we wondered whether the rs4349147 regulatory element could differentially regulate the IL-32 promoters. Therefore, we analyzed the relative expression levels of distinct IL-32 RNA variants in data obtained from high-throughput RNA sequencing (RNA-seq) in rs4349147 G/A, G/−, or −/A and −/− cells (Fig. 3C). This analysis revealed that, concurrent with a decrease in expression of IL-32, a switch occurs in the ratio of isoforms; rs4349147 −/A cells display a shift from “short” IL-32 variants originating from the EPD IL32_1 promoter to the “longer” RNA variants originating from the EPD NK4_1 promoter, resulting in a higher relative expression of the IL-32α–encoding RNA variant 4 (Fig. 3C). The higher relative expression of IL-32α is confirmed by RT-PCR using primers specific for the IL-32α and non–IL-32α isoforms (fig. S3D) and is matched by a relative increase in protein levels of IL-32α in rs4349147 −/A cells, as demonstrated by Western blotting using an IL-32α–specific antibody for detection (fig. S3, E and F). The relative ratio of IL-32α over non–IL-32α expression does not change after HIV infection of rs4349147 G/− and −/A cells (fig. S3G).
Relative IL-32 isoform expression in rs4349147 KO cells −/− showed a lower relative ratio of IL-32α expression, consistent with its similar propensity to HIV infection to G/A and G/− cells but not to −/A cells (Fig. 3, A to C). In agreement, in the −/A Δrs4349147 KO line, both the lower relative IL-32α–to–IL-32γ ratio and the higher susceptibility to HIV infection are similar to the rs4349147 G/− cells but not to the −/A cells (Fig. 3D and fig. S3, D and E). The observed difference in infection efficiency is independent from the envelope (Env) protein used to generate the pseudoviral particles because rs4349147 −/A cells infected with HIV-1 Env-pseudotyped virus still display a reduced infection rate as compared to rs4349147 G/− or −/A Δrs4349147 Jurkat cells (Fig. 3E). Therefore, the allelic contribution to susceptibility does not involve the interaction between gp120/gp41 and cellular receptor and co-receptors but rather is mediated by a more general mechanism.
rs4349147 G/− Jurkat cells generate a proinflammatory T cell–activating environment more conducive to HIV infection
To gain further insight into the mechanism by which rs4349147-G promotes HIV infection, we further analyzed the RNA-seq data obtained from unstimulated, uninfected rs4349147 G/A, G/−, −/A, and −/− cells (Fig. 3, F and G). We focused our analysis on genes differentially expressed specifically between rs4349147 −/A, G/−, and G/A Jurkat cells. We found that 97 genes are at least twofold up-regulated, whereas 152 genes are at least twofold down-regulated (Fig. 3F and table S2) in rs4349147 −/A Jurkat cells as compared to rs4349147 G/A and G/− Jurkat cells. Gene ontology (GO) biological process analysis of down-regulated genes revealed overrepresentation of factors involved in the regulation of cell activation (lymphocyte, leukocyte, and T cell), consistent with a relatively lower expression of proinflammatory IL-32 isoforms (Fig. 3G and table S3). RT-PCR confirmed the increased expression of T cell activation genes such as IL-4R (IL-4 receptor), TFFRSF14, STAT5A (signal transducer and activator of transcription 5A), LGALS1, and ANXA1 (fig. S4A) in the WT and rs4349147 G/− cells. Consistent with its reverted higher propensity for infection and increased relative ratio of the proinflammatory IL-32 isoforms, the proinflammatory gene expression profile is restored in the rs4349147 −/− KO cells (Fig. 3F and fig. S4A). An enrichment in GO biological process analysis was observed for the Wnt signaling pathway in up-regulated genes (fig. S4, B and C). Factors previously reported to interact with HIV-1 (www.ncbi.nlm.nih.gov/genome/viruses/retroviruses/hiv-1/interactions/) were also overrepresented (fig. S4D) in the list of down-regulated genes. Cytokine array profiling using supernatants of rs4349147 G/− or −/A Jurkat cultures demonstrates that, in line with the known immune modulatory role of proinflammatory IL-32 isoforms, rs4349147 G/− cells exhibit elevated secretion of several interleukins and other cytokines (fig. S4E and table S4). Together, these experiments demonstrate that Jurkat cells containing the rs4349147 G/− allele display an increased expression of proinflammatory genes and genes involved in T cell activation and proliferation when compared to rs4349147 −/A cells.
We determined the functional consequence of the observed shift in IL-32 isoform expression in context of HIV infection by overexpression of the distinct IL-32 isoforms in rs4349147 enhancer KO Jurkat cells, which express low amounts of endogenous IL-32 (Figs. 1, D to F, and 2, D to F), followed by infection with vesicular stomatitis virus (VSV)–pseudotyped HIV-derived virus (Fig. 3H). Expression of the distinct IL-32 isoforms was confirmed by Western blotting (Fig. 3H, bottom) and RT-PCR (fig. S4F). Exogenous expression of the IL-32 isoforms B to E resulted in an increase in HIV infectability (Fig. 3H). In contrast, when IL-32α (isoform A) was exogenously expressed, no increase in HIV infection was observed. A similar pattern of susceptibility to infection was observed when IL-32 isoforms were overexpressed in WT Jurkat cells expressing high levels of IL-32 (fig. S4G), in line with the notion that the relative ratio of IL-32 isoforms, but not their absolute expression levels, determine susceptibility to infection.
Relative ratio of IL-32 isoforms regulates the proinflammatory cytokine environment and influences susceptibility of primary CD4+ T cells to HIV infection
Our results thus far demonstrated that the rs4349147 SNP resides within an enhancer region, which, via long-distance interaction, regulates the expression of the IL-32 promoter and controls the expression of distinct isoforms of IL-32 at the RNA level. We also showed that the rs4349147 SNP–driven distinct expression profile of IL-32 RNA isoforms influences HIV infection susceptibility in the Jurkat model system. However, Jurkat cells have been reported to undergo posttranscriptional processing of several IL-32 isoforms at the protein level, resulting in generation of smaller IL-32 peptides (Fig. 1E and fig. S3F) (9), although the expression of IL-32α is not effected (fig. S3F). Therefore, to validate our results and to examine the functional consequence of the different IL-32 isoforms on HIV infection susceptibility in a more physiologically relevant system, we moved to the in vivo relevant primary CD4+ T cell targets of HIV.
It has previously been reported that stimulation of activated primary CD4+ T cells with purified IL-32γ, but not with IL-32α, results in an increased production of proinflammatory cytokines (10). We therefore asked whether supplementing primary CD4+ T cells with recombinant IL-32γ (rIL-32γ) or rIL-32α would have an effect on susceptibility to HIV infection. CD4+ T cells were isolated from eight healthy donors and stimulated with CD3-CD28–coated beads in the absence or presence of either rIL-32α or rIL-32γ. After stimulation, luciferase activity was evaluated as a measure of infection efficiency (Fig. 4A). Our experiments in Jurkat cells indicated that rs4349147 has only moderate effects on susceptibility to infection. To maximize the window of opportunity that would allow minor variations in infection susceptibility to be observed while minimizing the influence of factors associated with viral replication, cells were infected with single-round infectious viral particles. Furthermore, to recapitulate, at least in part, the complex mixture of viral variants present at the infection, we used viral particles pseudotyped with an X4 variant derived from the reference strain HXB2 and an R5 Env variant derived from a primary isolate. Similar to what was observed after exogenous expression of IL-32 isoforms in rs4349147 KO Jurkat cells (Fig. 3H), we found that luciferase activity was significantly increased in primary cells stimulated in the presence of rIL-32γ, whereas infection levels similar to untreated control cells were obtained after stimulation with rIL-32α (Fig. 4B). As expected, treatment of primary CD4+ T cells with rIL-32γ, but not with rIL-32α, resulted in significant increase in the transcription of the proinflammatory cytokine IL-8 (Fig. 4C). These data strongly support our observations in the Jurkat T cell line, where the increased relative expression of IL-32α over non-α isoforms reduced susceptibility to HIV infection. However, the cellular receptor for IL-32 is not yet identified. Therefore, we could not exclude the possibility that the rIL-32α is unable to enter into the cells. To confirm that the observed HIV-1 infection phenotype is linked to the exogenously provided distinct IL-32 isoforms and to ensure similar exposure to IL-32 isoforms, primary CD4+ T cells were lentivirally transduced to overexpress IL-32α, as well as the non-α isoforms β or γ, and subsequently infected with HIV according to the indicated schematic (Fig. 4D and fig. S5B). We examined the proinflammatory environment caused by exogenous overexpression of the specific IL-32 isoforms α and γ using both intracellular cytokine staining (fig. S5C) and cytokine arrays of the primary cell supernatants (fig. S5D and table S5). Cytokine arrays demonstrated significantly increased levels of IL-32 (approximately 10- and 6-fold overexpression over control for IL-32α and IL-32γ, respectively) in the culture supernatant, confirming the IL-32 lentiviral overexpression system (fig. S5D). Although the level of IL-32α was higher in the culture supernatants only, exogenous expression of IL-32γ resulted in the up-regulation of proinflammatory cytokines, including IL-8, IL-6, IL-10, TNFα (tumor necrosis factor–α), and IFN-γ (interferon-γ) (fig. S5D). Consistent with our data using rIL-32 isoforms, the proinflammatory cytokine environment generated by IL-32γ overexpression resulted in a significant increase in susceptibility to HIV infection (shown for five subjects). Similar to IL-32γ, overexpression of the non–IL-32α isoform β also resulted in increased susceptibility to HIV infection (fig. S5A).
Fig. 4. Relative ratio of IL-32 isoforms influences susceptibility of primary CD4+ T cells to HIV infection and alters the proinflammatory cytokine environment.
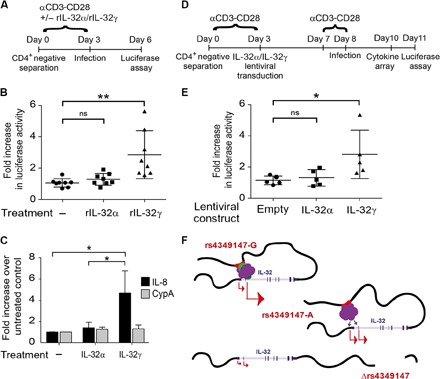
(A) CD4+ T cells isolated from eight healthy donors were stimulated with CD3-CD28–coated beads in the presence or absence of rIL-32α or rIL-32γ and infected with an HIV virus harboring a luciferase reporter, as shown in the schematic. Efficiency of infection was determined by measuring luciferase activity 72 hours after infection. (B) CD4+ T cells exogenously incubated with rIL-32γ are more susceptible to HIV infection than mock-treated cells and cells treated with rIL-32α. ns, not significant. (C) Real-time PCR analysis demonstrates that CD4+ T cells incubated with rIL-32γ, but not those treated with rIL-32α, show increased IL-8 expression. CypA, cyclophilin A. (D) CD4+ T cells isolated from five healthy donors (rs4349147-G/A) were stimulated with CD3-CD28–coated beads and infected with a control lentiviral vector or a lentiviral vector expressing IL-32α or IL-32γ. After 4 days, the cells were restimulated for 24 hours and infected with an HIV virus harboring a luciferase reporter, as shown in the schematic. Efficiency of infection was determined by measuring luciferase activity 72 hours after infection. (E) Exogenous lentiviral overexpression of IL-32γ results in an increase in HIV infection as compared to CD4+ T cells overexpressing IL-32α or control cells infected with an empty lentiviral construct. (F) Model depicting rs4349147 regulation of IL-32 expression. See Discussion for explanation.
Thus, in remarkable similarity to treatment of primary CD4+ T cells with rIL-32α and rIL-32γ, as well as to infections performed after exogenous expression of IL-32α and non–IL-32α isoforms in Jurkat cell lines, lentiviral overexpression of non–IL-32α isoforms γ and β, but not α, rendered the cells more susceptible to HIV infection (Fig. 4 and fig. S5). These results are consistent with the increased susceptibility to infection observed in rs4349147 G/− cells, which exhibit a lower expression of IL-32α and higher expression of proinflammatory IL-32 isoforms, and suggest that the rs4349147-A allele generates a subdued proinflammatory environment compared to rs4349147-G, which leads to reduced susceptibility to HIV infection.
DISCUSSION
Here, we demonstrate that the noncoding variant rs4349147, associated with HIV susceptibility, resides in a DNA region that functions as a long-distance enhancer element for the IL-32 gene, located approximately 10-kb downstream. Deletion of this DNA region results in abrogation of IL-32 gene expression in Jurkat cells, whereas 3C experiments demonstrated direct long-distance interaction between this enhancer region and the IL-32 promoter. Because Jurkat cells heterozygously harbor both rs4349147-A and rs4349147-G alleles, we were able to examine the effect of each allele on IL-32 expression and HIV infection efficiency by generating single-allele KO clones using the CRISPR-Cas9 technology. The presence of the rs4349147-A allele alone was associated with an increased relative expression of nonproinflammatory α IL-32 isoform, whereas the rs4349147-G allele cells displayed a switch in the relative expression of IL-32 isoforms to the proinflammatory non-α isoforms. We found that the functional consequence of this switch in IL-32 isoforms is enhanced lymphocyte activation, which directs increased susceptibility to HIV infection.
IL-32 isoforms and HIV infection
IL-32 has previously been reported to play a role in HIV infection and disease progression, although its precise role has remained controversial (7, 8). Comparison of IL-32 expression levels in HIV-infected patients and healthy individuals indicated a rise in IL-32 levels in peripheral blood mononuclear cells (PBMCs), gut, and lymph nodes in different stages of HIV infection (11–13). IL-32 was suggested to mediate immune suppression by promoting the expression of immunosuppressive molecules, leading to decreased immune activation and, as a consequence, increased HIV replication (13). However, several pieces of evidence also pointed to an inhibitory role for IL-32 in HIV replication; small interfering RNA (siRNA) depletion of IL-32 in Jurkat, human embryonic kidney (HEK) 293T, and latently infected U1 macrophage cell lines and infected PBMCs led to an increase in HIV replication (11, 14). IL-32 depletion also caused a reduction in levels of TH1 cells and proinflammatory cytokines including IL-6 and TNFα (14). Resolving these seemingly contradictory observations on the function of IL-32 in the context of HIV replication, IL-32 was proposed to play a dual immune regulatory role (13), inducing an antiviral immune response primarily via type I and type III IFNs (12, 15, 16) but also moderating immune activation during HIV infection (13). The siRNA depletion studies relied on depletion of all isoforms of IL-32 (11, 14), and many studies have used RT-PCR primers and antibodies that recognize common IL-32 sequences or epitopes and do not distinguish between different IL-32 isoforms and their potentially distinct functional roles in immune regulation.
Key to deciphering the role of IL-32 in immune function and regulation, IL-32 exists in multiple isoforms, which, it is becoming increasingly clear, are functionally distinct (17). IL-32α, which we found to be predominantly expressed in rs4349147-A allele Jurkat cells, has been in particular and in agreement with our findings, shown to be less potent than either IL-32β and IL-32γ isoforms in inducing a proinflammatory state in PBMCs (18, 19). A recent study has shown that plasma levels of the proinflammatory non–IL-32α isoforms are correlated with higher viral loads and significantly increased in HIV-infected slow progressors that experience virological failure (10). In addition, consistent with our data, IL-32γ was shown to be a much more potent inducer of IL-6 and IL-17F in CD4+ T cells than IL-32α (10). TH17 cells are known to be a preferential target susceptible to HIV infection (20), although it is long known that IL-6, alone or in combination with other cytokines, promotes HIV replication in macrophages and latently infected CD4+ T cells (21–23). Thus, as suggested by our cytokine array data comparing the expression of proinflammatory cytokines induced by exogenous expression of IL-32α and IL-32γ, one can envision that in vivo, the rs4349147-G allele by its induction of IL-32γ could augment the production of IL-6 and other proinflammatory cytokines, producing a sustained inflammatory environment ideal for HIV replication. rs4349147 is not readily picked up with high significance in HIV acquisition GWASs, which indicates that the effect of rs4349147 is small and it is not the main driver of HIV resistance. Likewise in our study, looking only at single rs4349147 alleles, we observe modest effects on IL-32 isoform ratio and infection susceptibility, although the fact that these effects may be enhanced in individuals that carry homozygous G/G or A/A alleles of rs4349147 cannot be excluded. We therefore envision that rs4349147 nuances the combinatorial effect that includes the contribution of additional host factors toward resistance to HIV acquisition.
Transcriptional regulation of IL-32 isoform expression
How the different alleles of rs4349147 (or the associated variant rs4786376) modulate the activity of the IL-32 enhancer remains to be understood. Bioinformatic transcription factor consensus site analysis of the region using PROMO (http://alggen.lsi.upc.es/) indicated that the rs4349147-G allele is part of a potential binding site for YY1 (Yin Yang 1), STAT, and ETS (E26 transformation–specific) family transcription factors, which are compromised when the rs4349147-A allele is present. Because similar analysis of rs4786376 did not yield a potential transcription factor binding site that is altered, rs4349147, and not the linked rs4786376, is likely to be the functionally relevant allele within the IL-32 enhancer. This is consistent with our data obtained in the luciferase reporter assays where only the rs4349147-G variant is increasing the expression from the reporter construct. Data from genome-wide binding studies performed in T cells indicate that STAT5, ETS, and other factors of the TAL1 (T cell acute lymphocytic leukemia 1) complex (HEB, E2A, LMO1, RUNX1, and GATA3) bind to the rs4349147 DHS (24–27). YY1 has been implicated in long-range chromatin interaction within the TH2 cytokine locus and V(D)J recombination in immature B cells (28, 29). Which of the above transcription factors are involved in the long-range regulation of IL-32 expression is the subject of an ongoing investigation.
The observed rs4349147 allele–specific switch in ratio of IL-32 isoform expression is in agreement with data from the International Human Epigenome Consortium that identified the IL-32 gene (among others) as having a significant genetic and epigenetic contribution to its expression levels and to its isoform expression in CD4+ T cells (30). Our data provide mechanistic insight into this phenomenon: In cells carrying only rs4349147-G, expression of “short” IL-32 variants presumably originating from the downstream EPD IL32_1 promoter is favored (most notably, variants that encode the proinflammatory IL-32 isoforms). Conversely, expression of “long” IL-32 variants originating from the upstream EPD NK4_1 promoter is favored in cells that contain the rs4349147-A allele [variants 2 and 8 encoding the B isoform of IL-32 (IL-32β) and variant 4 encoding the A isoform of IL-32 (IL-32α)]. This notion that these “long” and “short” IL-32 RNA transcripts are transcribed using different promoters is supported by the presence of a dual DNase I hypersensitivity peak at the IL-32 proximal upstream region in T cells (fig. S6). The alternative use of promoters has long been described to be a mechanism to generate diversity in transcriptional output, as well as complexity and nuance in the regulation of gene expression (31). We envision three models that can explain the observed IL-32 isoform switching. First, depending on its allelic nature, the enhancer (rs4349147-G) contacts either the EPD IL32_1 “short” isoform promoter or the EPD NK4_1 promoter of the long IL-32 RNA isoforms (rs4349147-A). Second, the rs4349147-G enhancer interacts with the EPD IL32_1 promoter of the short IL-32 RNA isoforms, and no interaction with either promoter is established when the rs4349147-A allele is present. Third, the rs4349147-A enhancer interacts with the EPD NK4_1 promoter of the long isoforms, and this interaction is lost with rs4349147-G. We deem the latter scenario unlikely because a strong reduction in IL-32 RNA expression is observed in rs4349147 −/A Jurkat cells, suggesting that the enhancer element is no longer fully functional. Unfortunately, 3C experiments lack the resolution to distinction between the two remaining models. However, 3C experiments indicate a significant reduction in the interaction between the enhancer and the general IL-32 promoter region, indicating a major change in the chromatin conformation in rs4349147 −/A Jurkat cells. In cell types that are not DNase I–hypersensitive at the rs4349147 region but still express low levels of IL-32, a major DHS peak is observed at the EPD NK4_1 promoter, which suggests that this is a “default” promoter for IL-32 expression (fig. S6). However, in cell lines lacking the rs4349147 regulatory element, IL-32 expression is strongly reduced compared to cells containing the rs4349147-A allele, which indicates that in these cells, the rs4349147 regulatory element still contains significant enhancer activity. The switch in IL-32 promoter usage in rs4349147 −/A cells suggests that this residual enhancer activity is now less focused, potentially by loss of binding of a tethering transcription factor, on the EPD IL32_1 short isoform promoter, which allows the EPD NK4_1 long isoform promoter to profit from the now more promiscuous regulatory element. Therefore, our results are consistent with the model depicted in Fig. 4F, where the rs4349147-G enhancer interacts with and specifically activates the EPD IL32_1 promoter while this interaction is altered in rs4349147 −/A Jurkat cells, resulting in relatively increased IL-32 expression from the EPD NK4_1 long isoform promoter and a relative increase in the expression of IL-32α. In the absence of the rs4349147 regulatory element, IL-32 expression from both promoters is strongly reduced (Fig. 4F).
Implications for HIV GWASs
We took a three-step approach to demonstrate that HIV-1–associated variants that fall below the accepted statistical threshold for genome-wide significance can still have biological relevance. We collected HIV-1–associated variants and compared them to epigenomic features to extract the variants with potential biological function. This integration of GWAS data with epigenomic data allowed us to discriminate true biological signals from noise. Promising candidate variants are subsequently scrutinized by detailed molecular analysis to elucidate their biological function. GWASs are geared to identify genetic variants with relatively high minor allele frequencies and moderate to high effects, especially when the sample size is small. As generally seen in HIV-1 GWASs (1), which are underpowered because of the difficulty in collection of large cohorts, rare variants and common variants with weak effect size are missed because they fall below the stringent threshold for genome-wide significance (P < 5 × 10−8). The study of Lingappa et al. (4), which initially pointed out the association between the rs4349147 and HIV-1 susceptibility in discordant couples, was likely affected by the low number of individuals included in the study. Furthermore, our data underline a modest effect of rs4349147 on HIV-1 susceptibility, which has likely further hampered the achievement of level of significance. The general approach to identify rarer variants with a low effect size is to enlarge the sample size (for example, by replication studies and/or meta-analysis), which is not always practical and still can only provide a statistical association and not actual biological relevance. Our current study, as well as the work of others (3), provides an additional approach to identify and characterize biologically important subthreshold variants. Using our approach, functional data can be extracted from previous GWASs, which is of utmost importance to push the field of HIV-1 host genetics forward.
In summary, we identify here, for the first time, the molecular mechanism by which a GWAS-identified HIV susceptibility allele functions. Our findings underscore the power of using a combined approach in which GWAS data and genome-wide functional data, together with functional molecular and phenotypic analysis, are leveraged to understand and unravel the underlying biological basis of disease. Our data provide the key mechanistic insight that allelic differences in a distal enhancer can direct a switch in promoter usage and, as a consequence, modulate differential isoform expression. This can subsequently lead to complex phenotypic changes, such as, as we show for IL-32, differential susceptibility to HIV infection but also potentially for other IL-32–related pathologies such as viral and bacterial infections, cancer, rheumatoid arthritis, and atherosclerosis (7). Our methodological approach can be expanded to identify additional loci associated with HIV susceptibility, pathogenesis, and latency, allowing the development of therapies and interventions tailored to specific patient populations.
MATERIALS AND METHODS
Cell culture and plasmids
Jurkat cells (a human T cell–derived line) were cultured in RPMI 1640 (Sigma-Aldrich) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin-streptomycin. rIL-32α and rIL-32γ were purchased from R&D Systems and resuspended according to the manufacturer’s instructions. For the IL-32 overexpression constructs, gBlocks gene fragments (Integrated DNA Technologies) corresponding to the complementary DNA (cDNA) sequence of the corresponding IL-32 isoforms were cloned into IRES-puro-GFP (a gift from B. Vogelstein; plasmid #16616, Addgene). Plasmids were sequence-verified. pHR-SFFV-KRAB-dCas9-P2A-mCherry was a gift from J. Weissman (plasmid #60954, Addgene).
Analysis of GWAS SNPs
HIV/AIDS implicated DNA variants were retrieved from the GWAS catalog (www.ebi.ac.uk/gwas/; June 2014). A total of 117 SNPs from 16 studies were recovered, 59 of which were classified as intergenic/noncoding. SNPs in strong LD (r2 > 0.8) were obtained from the CEU population panel of the 1000 Genomes Pilot 1 and HapMap3 (release 2) data sets using SNAP (SNP Annotation and Proxy Search) (http://archive.broadinstitute.org/mpg/snap/; distance limit, 500 kb). Human hg18 genome coordinates were lifted to hg19 using the lift-over function in the standard Galaxy tools of the Hypergenome browser (http://hyperbrowser.uio.no/3d). CD4+ TH0 and TH1 DNase I hypersensitivity peaks (http://genome.ucsc.edu/ENCODE/; wgEncodeAwgDnaseDukeTh0UniPk and wgEncodeAwg DnaseDukeTh1UniPk) containing HIV-associated SNPs were identified using the intersect function under Operate on Genomic intervals in the standard Galaxy tool, returning the overlapping intervals. Fifteen and 16 DHS regions were recovered for TH0 and TH1 cells, respectively. Subsequently, the SNPs overlapping these DHS regions were retrieved using the join function under Operate on Genomic intervals in the standard Galaxy tool, resulting in 17 SNPs for TH0 cells and 19 SNPs for TH1 cells, totaling 21 unique SNPs for both cell types (table S1).
Formaldehyde-assisted identification of regulatory elements
FAIRE was performed as described previously (32), except that selected genomic sites were analyzed by quantitative real-time PCR using GoTaq qPCR Master Mix (Promega) under the following cycling conditions: 95°C for 5 min, 45 cycles of 10 s at 95°C, and 30 s at 60°C, followed by a melting curve analysis. Enrichment was calculated relative to Axin2, and values were normalized to input measurements. PCR primers are available on request.
Amaxa nucleofection
Nucleofection of Jurkat cell clones was conducted as described previously (33). Cells were split to 3 × 105 cells/ml 24 hours before Amaxa nucleofection. Five to 8 million cells were centrifuged at 1000 rpm for 10 min at room temperature, resuspended in 100 μl of solution R, and nucleofected with 2 μg of expression plasmid using program O28. Nucleofected cells were resuspended in 500 μl of prewarmed, serum-free RPMI 1640 lacking antibiotics and allowed to recover at 37°C in a 5% CO2 incubator for 15 min. Prewarmed complete RPMI 1640 (4 ml) was then added to the cells.
Generation of rs4349147 KO cells using CRISPR-Cas9
The sgRNA cassette, the puro selection marker containing Cla I/Kpn I fragment from pLKO.1-puro U6 sgRNA BfuAI large stuffer, was cloned into pBluescript to yield pBS-puro U6 sgRNA BfuAI large stuffer. Oligos for guide RNAs targeting the rs4349147 containing DHS region were designed using ZiFiT (Zinc Finger Targeter) (http://zifit.partners.org/ZiFiT/CSquare9Nuclease.aspx) (34) and cloned into the BfuAI sites of pBS-puro U6 sgRNA BfuAI large stuffer. Sequences for the oligos used are available in table S6. sgRNA plasmids were conucleofeted with pSpCas9(BB)-2A-Puro (PX459) into Jurkat cells. Nucleofected cells were treated with puromycin (1 μg/ml) for 24 hours. After recovery, the polyclonal population was single cell–sorted using a BD FACSAria III cell sorter. Clones were screened for deletion of the rs4349147 DHS using PCR, and breakpoints were sequenced. Selected clones displayed similar growth characteristics.
pLKO.1-puro U6 sgRNA BfuAI large stuffer was a gift from S. Wolfe (plasmid #52628, Addgene). pSpCas9(BB)-2A-puro (PX459) was a gift from F. Zhang (plasmid #48139, Addgene) (35).
Primer sequences
Primer sequences used in this study are available in table S6.
RNA sequencing
RNA was isolated using TRI Reagent (Sigma-Aldrich). RNA-seq was performed according to the manufacturer’s instructions (Illumina) using the the TruSeq Stranded mRNA Library Prep kit. The resulting DNA libraries were sequenced according to the Illumina TruSeq v3 protocol on an Illumina HiSeq 2500 sequencer. Reads of 50 bp in length were generated. Reads were mapped against the UCSC genome browser hg19 reference genome with TopHat (version 2.0.10 or 2.0.13). Gene expression was quantified using both Cufflinks (version 2.1.1) and HTSeq-count (version 0.6.1). For the WT Jurkat and rs4349147 DHS KO samples, 13 to 18 million reads were generated, whereas approximately 20 million reads were generated for the Jurkat rs4349147 −/A and G/− lines. In all cases, more than 97% of these reads aligned to the hg19 genome. Differential expression analysis of the RNA-seq data was performed using edgeR package run under Galaxy (https://bioinf-galaxian.erasmusmc.nl/galaxy/). False discovery rate cutoff was set to 0.05.
Heat maps were generated using MORPHEUS (https://software.broadinstitute.org/morpheus/index.html). Enrichment of GO terms for the differentially expressed genes was assessed using Enrichr (http://amp.pharm.mssm.edu/Enrichr/) (36).
Western blotting
Whole-cell extracts were fractionated by SDS–polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane according to the manufacturer’s guidelines (Bio-Rad). Membranes were blocked in 5% nonfat milk in PBST [phosphate-buffered saline (PBS), 0.1% Tween 20] for 30 min and then washed three times in PBST. Membranes were incubated with antibodies against IL-32 (513501, BioLegend; or MAB30401, R&D Systems), IL-32α (AF3040, R&D Systems), and tubulin (1:1000; TS168, Sigma-Aldrich) overnight at 4°C. After washing the membranes three times for 10 min, they were incubated with horseradish peroxidase–conjugated anti-mouse (A9044, Sigma-Aldrich), anti-rat (A 9037, Sigma-Aldrich), or anti-goat (V8051, Promega) antibodies for at least 1 hour. Membranes were washed three times with TBST (tris-buffered saline, 0.1% Tween 20) and imaged using ImageQuant LAS 4000 Mini (GE Healthcare) with SuperSignal West Pico Chemiluminescent Substrate or SuperSignal Weste Femto Maximum Sensitivity Substrate (34080 and 34095, respectively, Thermo Fisher Scientific).
cDNA analysis
Total cellular RNA was isolated from the different cell lines with TriPure Isolation Reagent according to the manufacturer’s instructions (Roche Diagnostics). Subsequent DNase I digestion was performed with amplification grade DNase I (Invitrogen). The reverse-transcriptase reaction was performed using SuperScript II RNase H reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. Quantitative real-time PCR reactions for gene expression analysis were performed on a CFX96TM Real-Time System (Bio-Rad) with the GoTaq qPCR Master Mix (Promega) using the following parameters: denaturation at 95°C for 5 min, followed by 40 cycles at 95°C for 10 s, and 60°C for 30 s, followed by a melting curve analysis. The reference gene β2-macroglobulin or cyclophilin A was used to normalize the amplification signal between the samples of different cell lines, differences in treatment, and amount of input cDNA.
Intracellular stain
WT, rs4349147 −/A, rs4349147 G/−, and rs4349147 DHS KO Jurkat clones were harvested, counted, and directly stained for intracellular IL-32 expression. Briefly, 1 × 106 cells were first washed with Hanks’ balanced salt solution (HBSS)/3% FBS/0.015 M NaN3 and then fixed with IC Fixation Buffer (eBioscience). After washing with HBSS/3% FBS/0.015 M NaN3, cells were incubated with biotinylated anti-human IL-32 antibody (clone Ku32-52, BioLegend) for 60 min at 4°C, followed by two washes with permeabilization buffer (eBioscience) and incubation with allophycocyanin (APC)–conjugated streptavidin (Thermo Fisher Scientific) for 60 min at 4°C. Cells were also incubated with only APC-conjugated streptavidin as control. Cells were washed twice with permeabilization buffer and resuspended in 1% paraformaldehyde. A total of 100,000 cells were collected per sample on an LSRFortessa flow cytometry instrument (BD Biosciences) and analyzed using FlowJo software (Tree Star).
3C analysis
3C analysis was performed as described previously (37, 38) using Dpn II as the restriction enzyme. Quantitative real-time PCR (CFX96TM Real-Time System, Bio-Rad) was performed using GoTaq qPCR Master Mix (Promega) under the following cycling conditions: 50°C for 2 min, 95°C for 10 min, 45 cycles of 15 s at 95°C, and 1 min at 60°C, followed by a melting curve analysis. A random template was generated as described previously (39) using gBlocks gene fragments (Integrated DNA Technologies). PCR primers are available in table S6.
Luciferase reporter assays
Custom 500-bp gBlocks gene fragments (Integrated DNA Technologies) containing different haplotypes of the enhancer region were cloned into a modified pGL3-promoter vector [in which the SV40 promoter and the SV40 3′ untranslated region (3′UTR) were replaced by the IL-32 promoter and an HSP 3′UTR] (Promega). Jurkat cells are heterozygous for two additional common variants within the hypersensitive region: rs12598118 and rs12917910 (see fig. S1B). These variants are in high linkage to each other but are linked to rs4349147 at a much lower level, and there is no association with HIV acquisition reported for these SNPs. The genotypes for rs12598118 and rs12917910 were fixed to their ancestral C alleles while we varied the alleles for rs4349147 and rs4786376. Inserts in each construct were verified by sequencing (BaseClear). Constructs were nucleofected into Jurkat cells using the Cell Line Nucleofector Kit R (Lonza) and an Amaxa Nucleofector device (Lonza). Luciferase expression was measured after 24 and 48 hours in a GloMax 96 microplate luminometer (Promega) and normalized to Renilla luciferase expression. Data represent at least three independent experiments. Student’s two-tailed t test was used to determine statistical significance.
Virus production
HIV Env-pseudotyped particles were obtained by cotransfecting Env together with the HIV-1 backbone plasmid (pNL4.3.Luc.R-E-) into HEK 293T cells using FuGENE transfection reagent (Promega). To produce X4 tropic–pseudotyped viruses, pNL4.3.Luc.R-E- was cotransfected with HBXB2-Env expression construct, whereas cotransfection with pTRO11 Env expression vector resulted in production R5 tropic pseudoviruses. Twenty-four, 36, and 60 hours after transfection, the pseudovirus-containing supernatant was collected, filtered through a 0.45-μm filter, aliquoted, and stored at −80°C. HIV-1 molecular clone pNL4.3.Luc.R-E- and HIV-1 HXB2-Env expression vector were provided by the Centre for AIDS Reagents, National Institute for Biological Standards and Control. HIV-1 molecular clone pNL4.3.Luc.R-E-, HIV-1 HXB2-Env, and pTRO11 Env expression vector were donated by N. Landau; K. Page and D. Littman; and D. Montefiori, F. Gao, and M. Li, respectively.
HIV-derived virus particles were generated as described previously (40). Briefly, HEK293T cells were transfected with VSVG, the NL4.3 packaging vector R8.9, and the minimal HIV retroviral vector LTR-Tat-IRES-EGFP (pEV731). Virus was harvested every 12 hours, starting at 24 hours after transfection, filtered through a 0.45-μm filter, aliquoted, and stored at −80°C. Plasmids used to generate HIV-derived virus particles [VSV Env (VSVG), the NL4-3 packaging vector (R8.91), and the retroviral vector LTR-Tat-IRES-EGFP (pEV731)] were described previously (40).
HIV infection of Jurkat rs4349147 clones
Jurkat rs4349147 clones were infected with the minimal HIV VSVG-pseudotyped virus at low multiplicity of infection such that less than 10% of cells were infected. Samples were analyzed on a Fortessa flow cytometer with FACSDiva software (Becton Dickinson). The live population was defined by forward versus side scatter profiles. Cells were further gated by using forward scatter versus green fluorescent protein (GFP) to differentiate between GFP-positive and GFP-negative cells. GFP expression in the Jurkat rs4349147 clones was analyzed by FACS 24 and 48 hours after infection.
For HXB2 HIV Env–pseudotyped virus infection, 500,000 cells were infected by spinoculation (1200g, 90 min, 37°C), incubated in a humidified 37°C, 5% CO2 incubator for 2 hours, washed, and cultured for 48 hours in RPMI 1640/10% fetal calf serum (FCS). To evaluate infection levels, luciferase activity was measured using Luciferase Assay System (Promega). Relative light units were normalized to protein content determined by Bradford assay (Bio-Rad).
Primary CD4+ T cell isolation and infection
Primary CD4+ T cells were isolated from buffy coats from healthy donors by Ficoll gradient, followed by magnetic separation with Rosette Sep Human CD4+ T cell Enrichment Cocktail (STEMCELL Technologies) according to the manufacturer’s instructions. Cells were kept in culture in RPMI 1640/10% FCS for 1 week and subsequently stimulated for 3 days with anti–CD3-CD28–coated beads and IL-2 in the absence or presence of 500 ng/ml (10) of either rIL-32α or rIL-32γ (R&D Systems). After stimulation, cells were infected with a mixture of R5 and X4 Env-pseudotyped virus by spinoculation (1200g, 90 min, 37°C), incubated in a humidified 37°C, 5% CO2 incubator for 2 hours, washed, and then cultured in RPMI 1640/10% FCS supplemented with IL-2. Seventy-two hours after infection, cells were harvested and washed once with PBS, and luciferase activity was measured using Luciferase Assay System (Promega). Relative light units were normalized to protein content determined by Bradford assay (Bio-Rad).
Lentiviral overexpression of IL-32 isoforms
cDNAs encoding different isoforms of IL-32 were PCR-cloned into Mlu I/Spe I restriction sites of the lentiviral vector pLenti6.3/V5-DEST (Invitrogen). Virus particles were produced using the ViraPower Lentiviral Packaging Mix (Invitrogen) according to the supplier’s recommendations. Primary CD4+ T cells were isolated from buffy coats from healthy donors by Ficoll gradient, followed by magnetic separation with Rosette Sep Human CD4+ T cell Enrichment Cocktail (STEMCELL Technologies) according to the manufacturer’s instructions. Cells were subsequently stimulated for 3 days with anti–CD3-CD28–coated beads and IL-2. After stimulation, cells were infected by spinoculation with IL-32α–, IL-32β–, or IL-32γ–expressing lentivirus. After 4 days, cells were restimulated for 24 hours and infected with an HIV virus harboring a luciferase reporter, as described above. Seventy-two hours after infection, cells were harvested and washed once with PBS, and luciferase activity was measured using Luciferase Assay System (Promega). Relative light units were normalized to protein content determined by Bradford assay (Bio-Rad).
Intracellular cytokine detection
Purified CD4+ T cells were treated as described for the lentiviral IL-32 isoform overexpression. During the last 5 hours of the final bead activation step, a protein transport inhibitor (GolgiPlug, BD Biosciences) was added to prevent the release of cytokines. Cells were harvested and incubated with annexin V–phycoerythrin (PE) in FACS wash (HBSS, 3% FBS, 0.02% NaN3, and 2.5 nM CaCl2) for 20 min at 4°C in the dark. For intracellular staining for cytokines, cells were then fixed (IC Fixation Buffer, eBioscience), washed (permeabilization buffer, eBioscience), incubated with the antibodies (TNFα-APC, clone MAb11; IFN-γ–PE-Cy7, clone 4S.B3, eBioscience) for 45 min, washed two times with permeabilization buffer, and fixed with 1% paraformaldehyde. At least 100,000 events were collected per sample within 24 hours after staining on a LSRFortessa (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Proteome profiler Human XL Cytokine Array
Jurkat rs4349147 −/A and rs4349147 G/− clones or purified CD4+ cells were grown to equal density (8 × 105 cells/ml for Jurkat cells and 2 × 106 cells/ml for CD4+ cells). Cells were spun down, and 500 μl of supernatant was incubated with the Human XL Cytokine Array membrane (R&D systems). Membranes were processed according to the manufacturer’s instructions and imaged on a ImageQuant LAS 4000 (GE Healthcare). Signal was quantified using ImageQuant TL software (GE Healthcare).
Statistical analysis
All data are means ± SD of three or more independent biological replicates. For 3C and cDNA qPCR experiments, comparison of fold change data was performed by a Student’s two-tailed t test or by one-way analysis of variance (ANOVA).
For infection experiments, comparison of fold change data was performed by one-way ANOVA (Kruskal-Wallis test) using logarithmically transformed values. Statistical significance was reported as ns, P > 0.05, *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Supplementary Material
Acknowledgments
We would like to thank S. Kassem, G. Zonderland, and L. Gao for the excellent technical assistance. Funding: The research leading to these results has received funding from the European Research Council (ERC) under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC STG 337116 Trxn-PURGE, Dutch AIDS Fonds grants 2014021 and 2016014, and ErasmusMC mRACE research grant. Author contributions: R.-J.P. and E.d.C. designed and conducted the experiments, performed the analyses, and wrote the manuscript. Y.M.M. designed and conducted the experiments and performed the analysis. P.D.K. designed the experiments. M.D.R., T.W.K., and T.v.S. performed the experiments. W.v.I performed the analysis. T.M. designed the experiments and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/e1701729/DC1
fig. S1. Characterization of the rs4349147 DHS and the rs4349147 KO clone.
fig. S2. Characterization of the rs4349147 G/−, rs4349147 −/A, and −/A Δrs4349147 clones.
fig. S3. HIV infectability and differential expression of IL-32 isoforms between WT, rs4349147 G/−, and rs4349147 −/A clonal Jurkat cell lines.
fig. S4. Analysis of genes that are at least twofold differentially expressed in rs4349147 −/A compared to rs4349147 G/− and WT clonal Jurkat cell lines.
fig. S5. Endogenous lentiviral overexpression of IL-32 isoforms in CD4+ cells results in altered HIV susceptibility and proinflammatory cytokine expression.
fig. S6. UCSC genome browser graphic of DNase I hypersensitivity (upper track, black, blue, purple) and mRNA sequencing (lower track, green) data obtained from the Human Epigenome Atlas.
table S1. Overview of SNPs.
table S2. List of differentially regulated genes.
table S3. GO term analysis of −/A versus WT and G/− down-regulated genes.
table S4. Cytokine secretion in Jurkat cells as measured by Proteome Profiler Human XL Cytokine Array kit.
table S5. Cytokine secretion in CD4+ T cells after stimulation and infection with HIV as measured by Proteome Profiler Human XL Cytokine Array kit.
table S6. Primer sequences used.
REFERENCES AND NOTES
- 1.McLaren P. J., Carrington M., The impact of host genetic variation on infection with HIV-1. Nat. Immunol. 16, 577–583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Visser M., Kayser M., Palstra R.-J., HERC2 rs12913832 modulates human pigmentation by attenuating chromatin loop formation between a long-range enhancer and the OCA2 promoter. Genome Res. 22, 446–455 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X., Tucker N. R., Rizki G., Mills R., Krijger P. H. L., de Wit E., Subramanian V., Bartell E., Nguyen X.-X., Ye J., Leyton-Mange J., Dolmatova E. V., van der Harst P., de Laat W., Ellinor P. T., Newton-Cheh C., Milan D. J., Kellis M., Boyer L. A., Discovery and validation of sub-threshold genome-wide association study loci using epigenomic signatures. eLife 5, e10557 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lingappa J. R., Petrovski S., Kahle E., Fellay J., Shianna K., McElrath M. J., Thomas K. K., Baeten J. M., Celum C., Wald A., de Bruyn G., Mullins J. I., Nakku-Joloba E., Farquhar C., Essex M., Donnell D., Kiarie J., Haynes B., Goldstein D. the Partners in Prevention HSV/HIV Transmission Study Team , Genomewide association study for determinants of HIV-1 acquisition and viral set point in HIV-1 serodiscordant couples with quantified virus exposure. PLOS ONE 6, e28632 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thakore P. I., D’Ippolito A. M., Song L., Safi A., Shivakumar N. K., Kabadi A. M., Reddy T. E., Crawford G. E., Gersbach C. A., Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palstra R.-J., Close encounters of the 3C kind: Long-range chromatin interactions and transcriptional regulation. Brief. Funct. Genomics Proteomics 8, 297–309 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Joosten L. A. B., Heinhuis B., Netea M. G., Dinarello C. A., Novel insights into the biology of interleukin-32. Cell. Mol. Life Sci. 70, 3883–3892 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ribeiro-Dias F., Saar Gomes R., de Lima Silva L. L., Dos Santos J. C., Joosten L. A. B., Interleukin 32: A novel player in the control of infectious diseases. J. Leukoc. Biol. 101, 39–52 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Ko N.-Y., Chang S.-H., Lee J.-H., Kim N.-W., Kim Y.-M., Choi W.-S., Choi J.-D., Bae S.-Y., Hong J.-W., Jaekal J., Azam T., Her E., Kim S.-H., Unique expression of a small IL-32 protein in the Jurkat leukemic T cell line. Cytokine 42, 121–127 (2008). [DOI] [PubMed] [Google Scholar]
- 10.El-Far M., Kouassi P., Sylla M., Zhang Y., Fouda A., Fabre T., Goulet J.-P., van Grevenynghe J., Lee T., Singer J., Harris M., Baril J.-G., Trottier B., Ancuta P., Routy J.-P., Bernard N., Tremblay C. L. Investigators of the Canadian HIV+ Slow Progressor Cohort , Proinflammatory isoforms of IL-32 as novel and robust biomarkers for control failure in HIV-infected slow progressors. Sci. Rep. 6, 22902 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rasool S. T., Tang H., Wu J., Li W., Mukhtar M. M., Zhang J., Mu Y., Xing H. X., Wu J., Zhu Y., Increased level of IL-32 during human immunodeficiency virus infection suppresses HIV replication. Immunol. Lett. 117, 161–167 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Monteleone K., Di Maio P., Cacciotti G., Falasca F., Fraulo M., Falciano M., Mezzaroma I., D’Ettorre G., Turriziani O., Scagnolari C., Interleukin-32 isoforms: Expression, interaction with interferon-regulated genes and clinical significance in chronically HIV-1-infected patients. Med. Microbiol. Immunol. 203, 207–216 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Smith A. J., Toledo C. M., Wietgrefe S. W., Duan L., Schacker T. W., Reilly C. S., Haase A. T., The immunosuppressive role of IL-32 in lymphatic tissue during HIV-1 infection. J. Immunol. 186, 6576–6584 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nold M. F., Nold-Petry C. A., Pott G. B., Zepp J. A., Saavedra M. T., Kim S.-H., Dinarello C. A., Endogenous IL-32 controls cytokine and HIV-1 production. J. Immunol. 181, 557–65 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Zepp J. A., Nold-Petry C. A., Dinarello C. A., Nold M. F., Protection from RNA and DNA viruses by IL-32. J. Immunol. 186, 4110–4118 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Li Y., Xie J., Xu X., Liu L., Wan Y., Liu Y., Zhu C., Zhu Y., Inducible interleukin 32 (IL-32) exerts extensive antiviral function via selective stimulation of interferon λ1 (IFN-λ1). J. Biol. Chem. 288, 20927–20941 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khawar M. B., Abbasi M. H., Sheikh N., IL-32: A novel pluripotent inflammatory interleukin, towards gastric inflammation, gastric cancer, and chronic rhino sinusitis. Mediators Inflamm. 2016, 8413768 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim S.-H., Han S.-Y., Azam T., Yoon D.-Y., Dinarello C. A., Interleukin-32: A cytokine and inducer of TNFα. Immunity 22, 131–142 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Choi J.-D., Bae S.-Y., Hong J.-W., Azam T., Dinarello C. A., Her E., Choi W.-S., Kim B.-K., Lee C.-K., Yoon D.-Y., Kim S.-J., Kim S.-H., Identification of the most active interleukin-32 isoform. Immunology 126, 535–542 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Hed A., Khaitan A., Kozhaya L., Manel N., Daskalakis D., Borkowsky W., Valentine F., Littman D. R., Unutmaz D., Susceptibility of human Th17 cells to human immunodeficiency virus and their perturbation during infection. J. Infect. Dis. 201, 843–854 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poli G., Bressler P., Kinter A., Duh E., Timmer W. C., Rabson A., Justement J. S., Stanley S., Fauci A. S., Interleukin 6 induces human immunodeficiency virus expression in infected monocytic cells alone and in synergy with tumor necrosis factor alpha by transcriptional and post-transcriptional mechanisms. J. Exp. Med. 172, 151–158 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poli G., Kinter A. L., Fauci A. S., Interleukin 1 induces expression of the human immunodeficiency virus alone and in synergy with interleukin 6 in chronically infected U1 cells: Inhibition of inductive effects by the interleukin 1 receptor antagonist. Proc. Natl. Acad. Sci. U.S.A. 91, 108–112 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chun T.-W., Engel D., Mizell S. B., Ehler L. A., Fauci A. S., Induction of HIV-1 replication in latently infected CD4+ T cells using a combination of cytokines. J. Exp. Med. 188, 83–91 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanda T., Lawton L. N., Barrasa M. I., Fan Z. P., Kohlhammer H., Gutierrez A., Ma W., Tatarek J., Ahn Y., Kelliher M. A., Jamieson C. H. M., Staudt L. M., Young R. A., Look A. T., Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell 22, 209–221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollenhorst P. C., Chandler K. J., Poulsen R. L., Johnson W. E., Speck N. A., Graves B. J., DNA specificity determinants associate with distinct transcription factor functions. PLOS Genet. 5, e1000778 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei G. H., Badis G., Berger M. F., Kivioja T., Palin K., Enge M., Bonke M., Jolma A., Varjosalo M., Gehrke A. R., Yan J., Talukder S., Turunen M., Taipale M., Stunnenberg H. G., Ukkonen E., Hughes T. R., Bulyk M. L., Taipale J., Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 29, 2147–2160 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidl C., Hansmann L., Lassmann T., Balwierz P. J., Kawaji H., Itoh M., Kawai J., Nagao-Sato S., Suzuki H., Andreesen R., Hayashizaki Y., Forrest A. R. R., Carninci P., Hoffmann P., Edinger M., Rehli M. the FANTOM consortium , The enhancer and promoter landscape of human regulatory and conventional T-cell subpopulations. Blood 123, e68–e78 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Lee G. R., Role of YY1 in long-range chromosomal interactions regulating Th2 cytokine expression. Transcription 5, e27976 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebert A., Hill L., Busslinger M., Spatial regulation of V–(D)J recombination at antigen receptor loci. Adv. Immunol. 128, 93–121 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Chen L., Ge B., Casale F. P., Vasquez L., Kwan T., Garrido-Martín D., Watt S., Yan Y., Kundu K., Ecker S., Datta A., Richardson D., Burden F., Mead D., Mann A. L., Fernandez J. M., Rowlston S., Wilder S. P., Farrow S., Shao X., Lambourne J. J., Redensek A., Albers C. A., Amstislavskiy V., Ashford S., Berentsen K., Bomba L., Bourque G., Bujold D., Busche S., Caron M., Chen S.-H., Cheung W., Delaneau O., Dermitzakis E. T., Elding H., Colgiu I., Bagger F. O., Flicek P., Habibi E., Iotchkova V., Janssen-Megens E., Kim B., Lehrach H., Lowy E., Mandoli A., Matarese F., Maurano M. T., Morris J. A., Pancaldi V., Pourfarzad F., Rehnstrom K., Rendon A., Risch T., Sharifi N., Simon M.-M., Sultan M., Valencia A., Walter K., Wang S.-Y., Frontini M., Antonarakis S. E., Clarke L., Yaspo M.-L., Beck S., Guigo R., Rico D., Martens J. H. A., Ouwehand W. H., Kuijpers T. W., Paul D. S., Stunnenberg H. G., Stegle O., Downes K., Pastinen T., Soranzo N., Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167, 1398–1414.e24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davuluri R. V., Suzuki Y., Sugano S., Plass C., Huang T. H.-M., The functional consequences of alternative promoter use in mammalian genomes. Trends Genet. 24, 167–177 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Giresi P. G., Kim J., McDaniell R. M., Iyer V. R., Lieb J. D., FAIRE (formaldehyde-assisted isolation of regulatory elements) isolates active regulatory elements from human chromatin. Genome Res. 17, 877–885 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahmoudi T., Parra M., Vries R. G. J., Kauder S. E., Verrijzer C. P., Ott M., Verdin E., The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J. Biol. Chem. 281, 19960–19968 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Sander J. D., Maeder M. L., Reyon D., Voytas D. F., Joung J. K., Dobbs D., ZiFiT (Zinc Finger Targeter): An updated zinc finger engineering tool. Nucleic Acids Res. 38, W462–W468 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., McDermott M. G., Monteiro C. D., Gundersen G. W., Ma’ayan A., Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hagège H., Klous P., Braem C., Splinter E., Dekker J., Cathala G., de Laat W., Forné T., Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2, 1722–1733 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Palstra R.-J., Tolhuis B., Splinter E., Nijmeijer R., Grosveld F., de Laat W., The β-globin nuclear compartment in development and erythroid differentiation. Nat. Genet. 35, 190–194 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Visser M., Palstra R.-J., Kayser M., Allele-specific transcriptional regulation of IRF4 in melanocytes is mediated by chromatin looping of the intronic rs12203592 enhancer to the IRF4 promoter. Hum. Mol. Genet. 24, 2649–2661 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Jordan A., Defechereux P., Verdin E., The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J. 20, 1726–1738 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/e1701729/DC1
fig. S1. Characterization of the rs4349147 DHS and the rs4349147 KO clone.
fig. S2. Characterization of the rs4349147 G/−, rs4349147 −/A, and −/A Δrs4349147 clones.
fig. S3. HIV infectability and differential expression of IL-32 isoforms between WT, rs4349147 G/−, and rs4349147 −/A clonal Jurkat cell lines.
fig. S4. Analysis of genes that are at least twofold differentially expressed in rs4349147 −/A compared to rs4349147 G/− and WT clonal Jurkat cell lines.
fig. S5. Endogenous lentiviral overexpression of IL-32 isoforms in CD4+ cells results in altered HIV susceptibility and proinflammatory cytokine expression.
fig. S6. UCSC genome browser graphic of DNase I hypersensitivity (upper track, black, blue, purple) and mRNA sequencing (lower track, green) data obtained from the Human Epigenome Atlas.
table S1. Overview of SNPs.
table S2. List of differentially regulated genes.
table S3. GO term analysis of −/A versus WT and G/− down-regulated genes.
table S4. Cytokine secretion in Jurkat cells as measured by Proteome Profiler Human XL Cytokine Array kit.
table S5. Cytokine secretion in CD4+ T cells after stimulation and infection with HIV as measured by Proteome Profiler Human XL Cytokine Array kit.
table S6. Primer sequences used.