

Abstract
Errors during mRNA translation can lead to a reduction in the levels of functional proteins and an increase in deleterious molecules. Advances in next-generation sequencing have led to the discovery of rare genetic disorders, many caused by mutations in genes encoding the mRNA translation machinery, as well as a better understanding of translational dynamics through ribosome profiling. Here, we discuss multiple neurological disorders linked to errors in tRNA aminoacylation and ribosome decoding. We draw on studies from genetic models, including yeast and mice, to enhance our understanding of the translational defects observed in these diseases. Finally, we emphasize the importance of tRNA and their associated enzymes and the inextricable link between accuracy and efficiency in the maintenance of translational fidelity.
Keywords: mistranslation, elongation factor, ribosome stalling, tRNA modifications, neurodegeneration
Translational Infidelity and Disease
The fidelity of mRNA translation is essential for maintenance of the genetic code. Errors during translation elongation that result in incorporation of an incorrect amino acid, frameshifting (see Glossary), readthrough of stop codons, or premature termination can produce proteins that deviate from the encoded amino acid sequence. Depending on the nature of the error, these proteins can have reduced or aberrant function. In addition, abnormal folding of these aberrant proteins can lead to protein aggregation and cytotoxicity, further amplifying the cellular consequences of translational infidelity [1].
Growing evidence indicates that unicellular organisms can tolerate relatively high levels of translational errors, mitigating their effects through upregulation of the proteasome system or the heat shock response [2,3]. In contrast, mammals are exquisitely sensitive to even subtle disruptions of mRNA translation. Over the last few years, rapid advances in high-throughput genome- and exome-sequencing technologies have greatly amplified our ability to understand the genetic basis of disease and have led to the discovery of numerous causal mutations in components of the translational machinery. New technologies for global analysis of translation, such as ribosome profiling, along with insight gained from recent structural and biochemical studies have provided increased precision to dissect the molecular mechanisms underlying these diseases [4,5]. In this review, we will explore neurological diseases linked to impaired translational fidelity, highlighting the exceptional sensitivity of neurons to increased translational error rate. We will discuss diseases resulting from failures at major translational quality checkpoints, emphasizing recent studies that have implicated the complex regulation of tRNAs as a key player in the maintenance of faithful protein synthesis.
Mechanisms of Translational Fidelity
Protein synthesis is a complex, multistep process, and its fidelity derives largely from the combined accuracy of three major processes–the formation of aminoacyl tRNAs (aa-tRNAs), the selection of the appropriate aa-tRNA by the ribosome, and translocation of the ribosome along the mRNA. The production of an accurately charged aa-tRNA requires the aminoacyl tRNA synthetase (aaRS) to select both the correct tRNA from a large pool of diverse tRNA molecules and the appropriate amino acid from nineteen other proteinogenic amino acids and other structurally similar molecules (Figure 1). The active site of these enzymes screens the substrate pool with great accuracy, distinguishing cognate from non-cognate amino acids based on their size and structure. However, due to similarities between amino acids, non-cognate amino acids can occasionally be activated. The incorrect amino acid can be hydrolyzed by the synthetase prior to tRNA charging (pre-transfer editing) or after tRNA charging (post-transfer editing). In addition, mischarged tRNAs that escape these proofreading steps may be edited in trans after release from the synthetase, either by the synthetase itself or other stand-alone editing factors. After charging, aa-tRNAs and GTP are bound by the eukaryotic elongation factor (eEF)1A to form the ternary complex. The bacterial ortholog of eEF1A, EF-Tu, interacts with both the tRNA body and the amino acid, and thus may be able to identify misaminoacylated tRNAs [6]. When misaminoacylated tRNAs escape these editing mechanisms, they can result in the production of incorrect proteins [7,8]. Our understanding of how the ribosome faithfully decodes mRNA comes largely from structural studies of bacterial translation, and this process is highly conserved in eukaryotes. Briefly, aa-tRNAs are primarily distinguished by the ribosome based on their anticodon sequence (Figure 2). Initial selection begins with the binding of the aa-tRNA to the ribosome in complex with EF-Tu/eEF1A and GTP, followed by the rapid “sampling” of the interaction between the mRNA codon and the tRNA anticodon. Non-cognate and most near-cognate ternary complexes are rejected prior to GTP hydrolysis. Binding of the cognate tRNA and certain near-cognate tRNAs induces subtle conformational changes in the small ribosomal subunit, constricting the decoding center of the ribosome and triggering GTP hydrolysis. At this stage, near-cognate tRNAs are rejected because of the high free energy cost of forcing canonical Watson-Crick base pairing of the anticodon and mRNA codon. In contrast, the cognate tRNA is efficiently base-paired, leading to dissociation of EF-Tu•GDP and peptide bond formation. Additional proofreading of the codon-anticodon interaction may also occur in the P-site after peptide bond formation, leading to instability and termination of translation in the case of mistranslation. Multiple sampling of the codon-anticodon interaction maximizes the impact of free energy differences between cognate and near cognate matches, ensuring faithful translation [5,9,10]. The ribosome undergoes spontaneous and reversible rotation after peptide bond formation, and the associated tRNAs transition to a hybrid state, with their anticodons in the A and P sites and their acceptor stems in the P and E sites, respectively. Complete translocation of the ribosome on the mRNA requires the catalytic action of EF-G (eEF2 in eukaryotes). Binding of EF-G to the ribosome stabilizes the hybrid state of the tRNAs, and the insertion of the highly conserved domain IV of EF-G into the decoding center of the ribosome triggers translocation and return of the ribosome to the non-rotated state. This translocation requires the synchronized movement of both the mRNA and the bound tRNAs to ensure maintenance of the reading frame. Thus, accurate decoding involves a complex ballet between the ribosome, elongation factors and tRNA molecules, as well as the mRNA transcript.
Figure 1. tRNA Aminoacylation and Editing by Aminoacyl tRNA Synthetases.
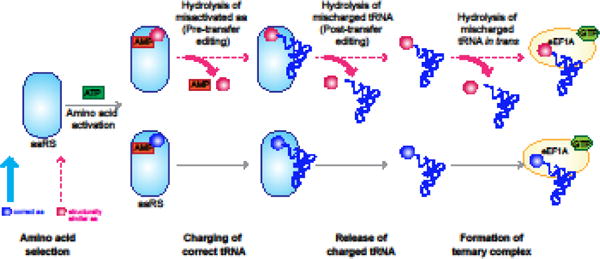
Aminoacyl tRNA synthetases (aaRS) activate an amino acid via ATP hydrolysis to form an aminoacyl adenylate. These enzymes then ligate the activated amino acid to the 3′ end of their cognate tRNA to generate an aminoacylated tRNA (aa-tRNA). Usually, aaRSs efficiently select the correct amino acid from the cellular pool, correctly discriminating between it and other related amino acids. However, if the non-cognate amino acid is activated, it can be hydrolyzed either directly or after ligation to the tRNA. Misaminoacylated tRNAs that escape these proofreading mechanisms may be edited after release from the synthetase (i.e., in trans) by the appropriate aaRS.
Figure 2. Ribosome Decoding and Translocation during Translation Elongation.
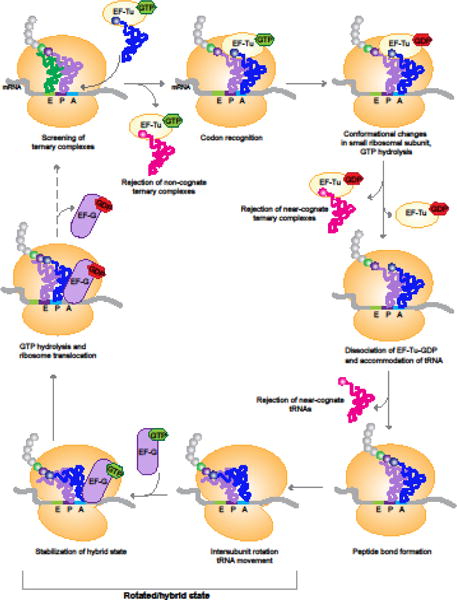
There are multiple opportunities for rejection of incorrect tRNAs during the selection of the cognate aminoacyl-tRNA for the codon in the ribosomal A site. Non-cognate and most near-cognate tRNAs are rejected during initial screening of EF-Tu•aminoacyl-tRNA•GTP ternary complexes. Near-cognate complexes that make it past initial screening can be rejected after GTP hydrolysis, or even after the release of EF-Tu•GDP. Peptide bond formation leads to a spontaneous rotation of the ribosomal subunits to form the rotated state. Binding of EF-G stabilizes this rotated state, and GTP hydrolysis catalyzes the translocation of the ribosome, and its return to the non-rotated state.
Mischarging by Aminoacyl tRNA Synthetases and Neurodegeneration
The aminoacylation of tRNAs by their cognate aaRS is the first quality control checkpoint in the maintenance of translation fidelity. Both dominant and recessive mutations in cytoplasmic tRNA synthetases have been identified in patients with neurological and neurodegenerative disorders [11]. Recently, compound heterozygosity for mutations in alanyl tRNA synthetase (AARS) were found in patients with microcephaly, hypomyelination, and epilepsy (Table 1) [12]. One of the mutant alleles virtually abolished the editing activity of AARS in vitro, impairing its ability to hydrolyze Ser-tRNAAla. However, this mutation also reduced aminoacylation activity in vitro, and patient-derived cell lines had significantly reduced levels of AARS, making the exact contribution of the editing defect to the disease progression unclear.
Table 1.
Mutations Associated with Impairment of Translational Fidelity and Neurological Disease
| Gene | Protein | Clinical phenotype | Mutations identified | Observed/predicted effect on fidelity | System used | Refs |
|---|---|---|---|---|---|---|
| AARS | Cytoplasmic alanyl tRNA synthetase | Progressive microcephaly, hypomyelination, and epilepsy | Compound het: missense/frameshift | Frameshift mutation increased mischarging of tRNAAla with serine | In vitro | [12] |
| EEF1A2 | Eukaryotic elongation factor 1 alpha 2 | Epilepsy, intellectual disability, Rett-syndrome -like phenotype, and autistic behavior | Multiple de novo missense mutations | One patient mutation increased suppression of frameshift and nonsense mutations | Reporter assays in yeast | [18–24] |
| EEF2 | Eukaryotic elongation factor 2 | Spinocerebellar ataxia 26 | Heterozygous missense | Patient mutation increased -1 frameshifting | Reporter assay in yeast | [29] |
| DPH1 | Diphthamide biosynthesis 1 | Intellectual disability, developmental delay, and brain malformations | Homozygous missense | Loss of diphthamide in eEF2 increases - 1 frameshifting | Reporter assay in embryonic fibroblasts from eEF2G71R/G71R mice, where eEF2 cannot be modified | [30,32, 33] |
| RPS23 | Ribosomal protein uS12 | Microcephaly, hearing loss, and dysmorphic features | De novo missense mutations | Patient mutation increased -1 frameshifting, missense suppression, and nonsense suppression | Reporter assay in patient cells and yeast | [35] |
| RPL10 | Ribosomal protein uL16 | X-linked intellectual disability, microcephaly, and autistic behavior | Multiple missense mutations | Unknown | N/A | [39–42] |
| KAE1 | Kinase associated endopeptidase 1 | Global developmental delay, microcephaly, and renal problems | Homozygous missense mutation | Loss of t6A modification in tRNA, altered cognate codon occupancy, and increased translation initiation at upstream non-AUG codons | Ribosome profiling on t6A- deficient yeast cells | [48, 49] |
| PUS3 | Pseudouridylate synthase 3 | Intellectual disability | Homozygous nonsense mutation | Loss of Pus3 decreased +1 frameshifting and reduced readthrough of stop codons by natural nonsense suppressor tRNAs | Reporter assay in pus3-deficient yeast cells | [50, 51] |
| ELP1-4 | Elongator acetyltransferase complex subunits 1-4 | Familial dysautonomia, intellectual disability, Rolandic epilepsy, amyotrophic lateral sclerosis | A variety of different mutations including missense and splice site mutations | Loss of Elongator-catalyzed tRNA U34 modification leads to slower decoding of cognate codons | Ribosome profiling in elongator complex mutants in yeast, and Elp3 knockout mouse forebrain | [52–55,58, 60] |
The role of mistranslation in neurodegeneration is more clearly defined in genetic models. In mice, a mutation in the editing domain of AARS that doubles the extremely low level of endogenous mischarging of tRNAAla with serine causes progressive Purkinje cell degeneration [13]. Intriguingly, while this particular mutation in AARS only affects the survival of Purkinje cells, mutations that resulted in more severe defects in AARS editing caused more widespread phenotypes, highlighting the extreme sensitivity of neurons to mistranslation [14]. Similarly, mutations in the phenylalanyl tRNA synthetase in Drosophila melanogaster that increased mischarging of tRNAPhe with tyrosine also caused neurodegeneration [8]. Strikingly, both of these mutations that reduce the fidelity of tRNA charging resulted in increased expression of molecular chaperones, formation of protein inclusions and aggregates, and increased endoplasmic stress, indicative of the production of incorrect proteins.
Elongation Factors and Neurological Disorders
The elongation factors play an essential role in ribosome decoding and the maintenance of the translational reading frame. Studies in Escherichia coli indicate that the affinity of EF-Tu binding to aa-tRNAs is tuned for optimal decoding. If the affinity of this interaction is too strong, peptide bond formation is impaired; alternatively, if the interaction is too weak, an incorrect aa-tRNA may get deposited in the ribosome A site [15,16]. Vertebrates have two genes encoding eEF1A that have non-overlapping expression in postnatal animals, with EEF1A2 expression restricted to adult neurons and muscle [17]. Exome sequencing has linked de novo missense mutations in EEF1A2 to diverse neurodevelopmental syndromes that are characterized by epilepsy, ataxia, intellectual disability, and autistic behavior [18–23]. One of these patient mutations was previously found to significantly increase both frameshift and nonsense suppression when introduced into yeast EEF1A, linking the pathogenesis of these disorders to translation infidelity [24]. Interestingly, while complete loss of Eef1a2 expression in mice leads to motor neuron degeneration and muscle wasting after weaning, heterozygous null mice are normal, in contrast to the devastating phenotype seen in patients [25,26]. This suggests that patient mutations do not simply result in haploinsufficiency of EEF1A2, but instead compromise its function, which would be consistent with an increase in translation errors. However, the effect of many of these patient mutations on translational fidelity remains to be characterized. In addition, eEF1A has been implicated in several other cellular pathways, including cytoskeleton organization and nuclear export [27], and thus the underlying pathogenic mechanism of these neurodevelopmental disorders may also involve disruption of these non-canonical functions.
The bacterial homolog of eEF2, EF-G, has been shown to accelerate the translocation rate of the ribosome by 104-106 fold, and mutations in conserved residues of domain IV of EF-G dramatically reduce the rate of translocation [28]. A mutation altering a conserved proline residue (P596H) within the homologous domain of eEF2 has been identified in patients with autosomal dominant spinocerebellar ataxia (SCA26), a neurodegenerative disorder characterized by Purkinje cell loss [29]. The replacement of proline by histidine is likely to increase steric hindrance, interfering with ribosome translocation. Indeed, mutation of an equivalent residue in yeast resulted in increased -1 frameshifting on a programmed ribosomal frameshifting (PRF) reporter sequence. PRF has historically been associated with viral translation. However, it is possible that mutation of eEF2 may disrupt PRF on a gene(s) necessary for Purkinje cell homeostasis. Intriguingly, this domain of eEF2 contains a unique posttranslationally modified histidine residue known as diphthamide that has not been found in any other protein. Diphthamide modification involves a multi-step biosynthetic pathway, involving at least 7 genes (DPH1-7 in yeast) that are highly conserved within eukaryotes. Mutation of this pathway demonstrated that this modification is essential for the maintenance of translational frame, and mice that are deficient in dipthamide biosynthesis or that have a point mutation in Eef2 that prevents this modification have gross delays and abnormalities in development [30,31]. Two recent studies have linked homozygous missense mutations in DPH1 to human neurodevelopmental abnormalities characterized by intellectual disability, central nervous system malformations, and craniofacial abnormalities [32,33], reminiscent of the phenotype of the mutant mice. Together, these studies highlight the essential role of eEF2 in ribosome translocation and neuronal function.
Error-prone Ribosomes and Neurodevelopmental Diseases
Mutations in ribosomal proteins, or in the components required for ribosome biosynthesis and maturation, cause ribosomopathies, a diverse group of disorders characterized by hypoproliferative phenotypes, such as anemia and bone marrow failure, or craniofacial developmental abnormalities. Although the specific mechanisms underlying the various ribosomopathies remain unclear, the field has generally converged around a loss of function mechanism, in which these mutations affect ribosome biogenesis, resulting in reduced protein synthesis [34].
However, mistranslation has been implicated in the pathogenesis of some of the neurodevelopmental disorders caused by mutations in ribosomal proteins. De novo missense mutations in RPS23, which encodes the small ribosomal protein uS12 have been linked to a syndrome characterized by microcephaly, hearing loss, and intellectual disability [35]. uS12 is present in the ribosome decoding center where it acts to stabilize the ribosome conformation induced by codon recognition [36]. Substitution of the R67K patient mutation into the yeast RPS23 gene dramatically reduced translational fidelity, increasing frameshifting and missense/nonsense suppression of reporter genes. Similarly, patient fibroblasts also showed a significant increase in stop codon readthrough [35]. Interestingly, the R67K mutation disrupts the posttranslational hydroxylation of a neighboring proline residue by OGFOD1 (2-oxoglutarate and iron dependent oxygenase domain containing 1), a modification that has previously been shown to modulate translational accuracy, in particular, the recognition of stop codons [37,38]. Mutant uS12 was severely underrepresented in polysomes from patient cells, suggesting that these defective ribosomes may be somewhat excluded from the polysomal pool. Together, these findings suggest that mutations in ribosomal proteins may result in decoding-deficient ribosomes and that even low levels of faulty ribosomes may have severe consequences.
Missense mutations in RPL10, which encodes the large ribosomal protein uL16, have been linked to autism, X-linked intellectual disability, and cerebellar hypoplasia [39–42]. While some of the characterized patient mutations were found to alter the polysome profile, suggesting altered translation may play a role in these diseases, the exact translational defect in these patients remains unclear. uL16 is located at the core of the large ribosomal subunit and a number of mutations in RPL10 have been shown to impair translational fidelity in yeast [43] Thus, it is also possible that mistranslation may play a role in the pathogenesis of some RPL10-linked neurodevelopmental phenotypes.
tRNA Modification, Decoding Errors, and Neurological disease
The interaction between three different RNA molecules: mRNA, rRNA, and tRNA, plays an essential role in the maintenance of accurate translation. These RNA molecules undergo posttranscriptional modifications that are thought to be crucial for their function. Although neurological disorders have been associated with defects in rRNA and mRNA modification, the precise contribution of impaired translational fidelity to the pathogenic mechanism is not clear (Box 1). In contrast, the relationship between modifications of tRNA and translational accuracy is clearer, and modified nucleotides within the anticodon stem loop (ASL), especially at the wobble position, have been shown to promote both correct and efficient codon-anticodon basepairing (Figure 3) [44]. Recently, mutations in enzymes that catalyze these diverse ASL modifications have been linked to multiple neurological disorders, highlighting the essential role of tRNA modification mediated translational fidelity in neurons [45,46]. However, yeast studies suggest that the effect of ASL modifications on decoding accuracy may be quite complex, with identical modifications having opposing effects on accuracy of different tRNAs [47,48] making it difficult to predict the exact translational defect in these disorders in vivo.
Box 1. mRNA Modifications, Translational Fidelity, and Neurological Disease.
The dynamic regulation of mRNA modifications has been implicated in multiple neurological disorders. The m6A (6-methyladenosine) modification is the most abundant internal posttranscriptional modification on eukaryotic mRNAs, with each transcript containing three to five m6A residues on average. Loss of function mutations in the m6A demethylase FTO (α-ketoglutarate-dependent dioxygenase) cause microcephaly and developmental delay [81,82]. In addition, genome wide association studies have associated FTO variants with ADHD (attention deficit hyperactive disorder), Alzheimer’s disease risk, and major depressive disorder [83]. m6A modifications have been implicated in practically every stage of a transcript’s lifecycle, including mRNA folding, maturation, stability, nuclear export, and cap-dependent initiation [84]. Intriguingly, single molecule imaging of translating E. coli ribosomes revealed that the presence of m6A modifications in the coding sequence of a transcript could disrupt the interaction between the codon and tRNA anticodon leading slower elongation [85]. Changes in translational fidelity may thus play a role in the pathogenesis of FTO-associated disorders.
Another modification that may play a role in the regulation of translational fidelity is the deamination of adenosine to inosine by ADARs (adenosine deaminases). Disruption of ADAR editing has been implicated in several neurological disorders including amyotrophic lateral sclerosis and schizophrenia [86]. When present in the coding sequence, this modification can alter the amino acid sequence of the encoded protein, as inosine is read as guanosine during translation elongation. Although only a very small number of transcripts are edited by ADAR within the coding sequence, this recoding can have a profound effect on neuronal function. For example, ADAR editing of subunits of both the AMPA and kainate glutamate receptors leads to the substitution of a glutamine for an arginine at the inner channel pore, and renders them impermeable to calcium ions. Defective ADAR editing of the AMPA subunit GluA2 has been linked to epilepsy and lethality in mice [87].
Figure 3. Modifications in the tRNA Anticodon Stem Loop Modulate Translational Fidelity.
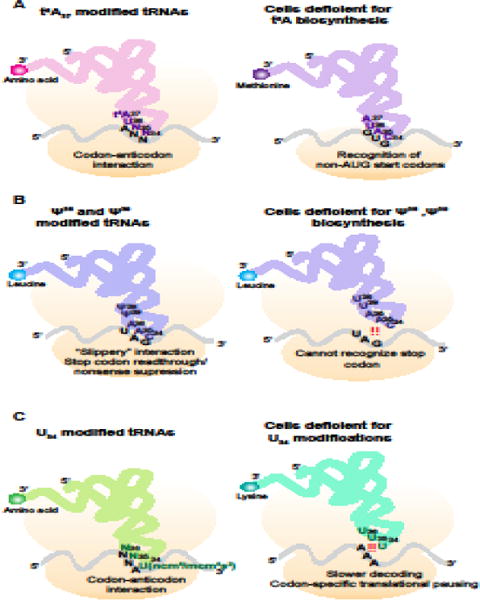
(A) tRNAs decoding ANN codons (where N is any nucleotide) have the t6A modification at nucleotide 37. Loss of this modification leads to recognition of non-AUG codons by the initiator methionyl tRNA.
(B) The modification of uridine to pseudouridine (Ψ) at nucleotides 38 and 39 broadens the decoding capacity of tRNAs. Modified tRNALeuCAA can decode stop (UAG) codons, leading to stop codon readthrough. Loss of this modification at these nucleotides narrows the decoding ability of the tRNA, preventing basepairing with UAG stop codons.
(C) Uridine at the wobble position (nucleotide 34) of most tRNAs is modified to ncm5U, mcm5U, or mcm5s2U. Loss of these modifications disrupts decoding of the cognate codon, and leads to codon specific ribosome pausing.
The advent of ribosome profiling, which allows for in vivo examination of translation at a codon specific level, has enhanced our understanding of the complex regulatory role of these modifications and has shed light on the molecular mechanism of these disorders. For example, homozygous mutations in the KAE1 (kinase-associated endopeptidase, OSGEP), which play an essential role in the biosynthesis of the t6A modification of adenosine (threonylcarbamoyl adenosine), have been linked to a neurodegenerative disorder accompanied by renal defects [49]. The t6A modification is found at position 37 in nearly all tRNAs decoding ANN codons (where N is any nucleotide), and the absence of this modification leads to severe growth defects in yeast that cannot be rescued by overexpression of ANN-decoding tRNAs lacking t6A modifications. Ribosome profiling of yeast strains with defective t6A biosynthesis demonstrated that loss of this modification altered ribosome occupancy of cognate codons, and in particular, impaired the recognition of start (AUG) codons, leading to increased translation initiation from upstream non-AUG codons. These data suggest that the t6A modification on tRNAMeti may normally prevent it from recognizing near-cognate AUGs [48].
ASL modifications have also been linked to cognitive and neurodevelopmental disorders. Homozygous loss of function mutations in PUS3, which encodes an enzyme that catalyzes the isomerization of uridine to pseudouridine, were identified in patients with intellectual disability [50]. The pseudouridine modification is one of the most commonly observed in tRNA and is found at multiple positions including the ASL. PUS3 catalyzes this modification at positions 38 and 39, and loss of PUS3 impairs growth in both bacteria and yeast. Surprisingly, loss of PUS3 reduced both +1 frameshifts and stop codon readthrough on reporter constructs, suggesting that this modification may normally reduce fidelity [51]. It remains unknown whether the cognitive impairments caused by PUS3 mutations result from defects in the recoding of a particular gene or from more global changes in translational speed and fidelity.
Similarly, mutations in many members of the multiprotein Elongator complex have been linked to neurological disorders, including intellectual disability, amyotrophic lateral sclerosis, Rolandic epilepsy, and familial dysautonomia [52–55]. Growing evidence indicates that the key role of the Elongator complex is the modification of uridine residues in the wobble position of tRNAs (nucleotide 34) to mcm5-uridine (5-methoxycarbonylmethyl) and ncm5-uridine (5-carbamoylmethyl) [56,57]. In certain tRNAs, mcm5-U34 is further modified by thiolation to form mcm5s2-U34. While loss of U34 modifications does not significantly alter the stability of these tRNAs, a recent study found that these modifications were essential for efficient decoding of cognate codons in both yeast and Caenorhabditis elegans [58]. The slower decoding observed in both yeast deficient for the mcm5-U34 modification (elp6 mutants) and yeast and C. elegans deficient for thiolation (nsc2 mutants) was linked to protein aggregation and activation of the unfolded protein response. Intriguingly, overexpression of the appropriate unmodified cognate tRNAs in elp6- or nsc2- deficient yeast strains or treatment of elp6/nsc2-deficient yeast with a drug that increases acceptance of near-cognate tRNAs was sufficient to rescue these defects, indicating that absence of U34 modifications likely impairs the kinetics of the interaction between the codon and anticodon during translation elongation. Slower decoding at cognate codons has also been suggested to result in other translation abnormalities, such as increased frameshifting [59]. Further supporting the key role of U34 modifications in translation fidelity and neuronal homeostasis, conditional deletion of the Elongator complex component Elp3 in mouse cortical progenitors impairs neurogenesis, resulting in microcephaly. Loss of Elp3 resulted in activation of the unfolded protein response in the developing mouse forebrain and ribosome profiling demonstrated increased ribosome occupancy at codons read by U34-containing tRNAs [60]. In addition to a general increase in aberrantly folded proteins, the change in decoding efficiency caused by loss of U34 modifications may alter the levels of specific proteins, such as those that are enriched in lysine residues encoded by AAA codons [61,62].
Dynamics of Ribosome Elongation and Translational Fidelity
It is generally established that there is a tradeoff between speed and accuracy during mRNA translation, and that the ribosome is optimized for high speed of translation. However, aberrant reductions in elongation rate, such as those caused by loss of U34 modifications, may impair translational fidelity. Indeed, reduction in the availability of particular charged tRNAs has been reported to increase mistranslation. For example, histidine deprivation results in increased misincorporation of glutamine at histidine codons in both bacterial and mammalian cells [63]. In addition, unusual translational events, such as PRF or translational bypassing are preceded by ribosomes pausing in their rotated state for an abnormally length of time, bolstering a link between abnormal translational pausing and translational infidelity [64,65].
Clearly, the availability of the cognate tRNA plays a key role in determining the efficiency with which a ribosome can decode a particular codon. A recent in vitro study found that frameshifting along the expanded CAG tract of a huntingtin reporter gene could be modulated by either the expression level of the cognate glutaminyl-tRNA, tRNAGlnCUG, or the cognate tRNA for the -1 frameshifted repeat, tRNAAlaUGC, demonstrating a clear link between tRNA levels and the maintenance of translational frame [66]. In mice, a loss of function mutation in a brain-specific arginine tRNA gene results in ribosome stalling on the cognate AGA codons [67]. These stalls are resolved by GTPBP2, a ribosome rescue protein, but in its absence, they persist, and cause widespread neurodegeneration and ataxia. Ribosome stalling has also been linked to human disease, as loss of function mutations in GTPBP2 have recently been identified in a family with ataxia, mental deficiency, and cerebellar and retinal degeneration [68].
The mechanism by which impaired ribosome elongation affects neuronal function and causes cell death remains unclear. Defects in translational elongation and fidelity activate the integrated stress response, leading to translational reprogramming, and chronic activation of this otherwise adaptive response may contribute to cellular dysfunction (Box 2). In addition, growing evidence indicates that changes in elongation speed, such as codon-specific pausing, can affect co-translational folding and thus protein function, even when the identity of the amino acid is unchanged [69,70]. Such misfolded proteins could aggregate and have deleterious effects. Another intriguing possibility is that ribosome stalling results in premature translation termination and the production of incomplete and possibly harmful peptides. Indeed, truncated proteins were found in bacteria in which ribosome stalling was induced by amino acid starvation [71]. The devastating consequences of production of these prematurely terminated proteins are highlighted by the lister mouse, which has profound neurodegeneration and motor dysfunction. Lister mice have a hypomorphic loss of function mutation in listerin (Ltn1) which encodes an E3 ubiquitin ligase responsible for the ubiquitination, and thus degradation, of aberrant nascent polypeptides resulting from stalled ribosomes [72]. Loss of Ltn1 in yeast causes the formation of protein aggregates that sequester molecular chaperones which likely results in global disruptions in proteostasis, further supporting a toxic role for aberrant nascent polypeptides [73].
Box 2. Translational Infidelity and the Integrated Stress Response.
mRNA translation is energetically demanding, and must be tightly regulated. Cells respond to a variety of stressors by inhibiting initiation, and one of the best-characterized mechanisms for this inhibition is the integrated stress response (ISR). The ISR is initiated by activation of stress-specific protein kinases (GCN2, PKR, PERK and HRI, in mammals) that phosphorylate the alpha subunit of eukaryotic initiation factor-2 (eIF2α), inhibiting ternary complex formation and thus repressing global translation (Figure I). Importantly, a subset of mRNAs escapes repression and is selectively translated. These transcripts, including that encoding the well-studied transcription factor ATF4, contain upstream open reading frames (uORFs) in their 5′ UTRs. Under normal conditions, translation of the main ORF is inhibited by these uORFs; however, upon phosphorylation of eIF2α, translation from the main ORF increases, resulting in the production of proteins that promote recovery from stress. Interestingly, recent studies indicate that translational infidelity may also activate this stress response, sometimes in non-canonical ways. Translation of GCN4, the yeast homolog of ATF4, is derepressed in cells deficient in the t6A tRNA modification, resulting in upregulation of GCN4 target genes. However, this translational activation of GCN4 occurs independently of GCN2, the sole yeast eIF2α kinase, and instead may be caused by leaky scanning bypass of the start codons of the regulatory uORFs by t6A-deficient tRNAMeti [48,88,89]. A similar result is observed in yeast deficient in either mcm5 or s2 modifications of tRNA wobble nucleotide, U34 [58,90]. In contrast, deletion of Elp3 in the mouse cortex, which catalyzes the U34 modifications, resulted in increased ribosome pausing on cognate codons, activation of PERK, the eIF2α kinase activated by ER stress, increased levels of phosphorylated eIF2α, and caused upregulation of ATF4 and its target genes. Interestingly, depletion of Atf4 in Elp3 conditional knockout mice rescued the impaired neurogenesis observed in these animals, suggesting that activation of this pathway contributes to the neurodevelopmental defects observed in these mice [60]. In contrast, activation of the integrated stress response was neuroprotective in a mouse model with ribosome stalling in neurons. Ribosome stalling in mice deficient in GTPBP2 and tRNAArgUCU was accompanied by GCN2-dependent activation of the ISR prior to neurodegeneration. GCN2 activation occurred independently of increases in uncharged tRNA, the canonical activator of GCN2 [91], and loss of GCN2 exacerbated cell death in these mice [92]. Together, these studies illustrate the complexity of the stress response in models of translational infidelity and disease.
Figure I. The Integrated Stress Response, and its Activation by Translational Infidelity.
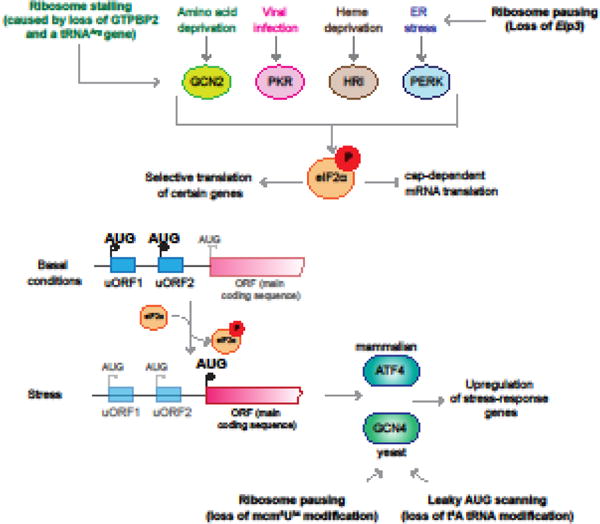
Activation of the eIF2α kinases by different stressors leads to repression of global translation and selective upregulation of translation from transcripts such as ATF4 (in mammals; GCN4 in yeast) that are regulated by uORFs. Translational infidelity caused by changes in either tRNA modifications or levels can activate this stress response in both canonical and non-canonical ways.
Concluding Remarks and Perspectives
The convergence of next-generation sequencing with methods for examining global translation on a codon-specific basis have implicated altered translational fidelity as a molecular mechanism underlying the pathogenesis of numerous neurological and neurodegenerative disorders. The discovery of these often rare genetic conditions highlights the extreme sensitivity of neurons to a chronic loss in translational fidelity. Intriguingly, studies of translational fidelity in the long-lived rodent species, the naked mole rat, suggest a link between the basal rate of translational errors and organismal lifespan, raising the question of the role of translational fidelity in age-related neurodegenerative disorders [74,75]. Many major questions on the influence of translational infidelity on disease remain unanswered, including the selective vulnerability of certain cell types to translational errors, and the mechanisms by which these errors lead to cell death (see Outstanding Questions).
Outstanding Questions.
Why are neurons particularly vulnerable to translational infidelity? Does their post-mitotic nature or polarized shape make clearance of misfolded proteins and resulting aggregates more difficult? Given that there is a tradeoff between speed and fidelity during translation, does the rapid protein synthesis required for synaptic plasticity place additional stress on the neuronal translation quality control system?
Many components of the translational machinery have been implicated in diverse cellular pathways beyond translation; for instance, certain ribosomal proteins have been implicated in immune signaling. Are these non-canonical functions involved in disease pathogenesis? Could the tissue/cell-type specificity of pathology result in part from these non-canonical functions?
What underlies the temporal heterogeneity of disorders linked to translational infidelity? For instance, loss of the post-translational modification of a particular histidine residue in eEF2 results in neurodevelopmental defects, but mutation of a neighboring residue causes neurodegeneration despite having an apparent similar effect on translational fidelity.
Does disease pathogenesis result from the disruption of the translation of a specific gene or subset of genes, or from more global processes, such as the activation of a cellular stress response?
How does the cell distinguish a physiological pause in translation that is linked to co-translational folding from an abnormal pause caused by deficiency in, or impaired modification of, charged tRNA? Does ribosome stalling result in unusual translational events such as frameshifting in vivo?
How do mutations in translational components, such as ribosomal proteins, affect the regulation of beneficial mistranslation? Does disruption of adaptive mistranslation play a role in the pathogenesis of these disorders?
Is the baseline rate of translational error the same or lower in neurons compared to other cell types? Does this baseline rate vary between neuronal types? Are translational quality control mechanisms that determine this rate regulated by neuronal activity?
While many neurological disorders are linked to increased translational errors, intellectual disability-linked mutations in PUS3 actually increase translational fidelity. This counterintuitive finding highlights the exquisite fine-tuning of translational fidelity in mammals, indicating that maximum accuracy in translation and the strict maintenance of the genetic code may sometimes be detrimental to cellular function. Indeed, the regulated misincorporation of methionine into proteins has been implicated in the oxidative stress response in mammalian cells. The beneficial and adaptive roles of regulated mistranslation have been explored in recent in-depth reviews, and will not be further discussed here [63,76].
Until relatively recently, the study of translational fidelity was hampered by the lack of techniques to quantitatively assess translational errors. Traditionally, analysis of fidelity has relied on reporter assays where misincorporation of an amino acid or a change in reading frame during translation leads to the expression or gain of function of a protein. These assays only monitor a fraction of the possible translational errors, and the development of unbiased approaches such as ribosome profiling has greatly enhanced our knowledge of the fundamental process of ribosome decoding. More recently, groups have developed techniques to examine global tRNA aminoacylation and misaminoacylation in vivo, approaches that will be beneficial for study of diseases caused by mutations in the tRNA synthetases [77,78]. Furthermore, changes in the proteome resulting from mistranslation have begun to be assessed using sophisticated mass spectrometry techniques [79,80]. We hope that in the coming years, these, and additional, methods will further inform our understanding of the link between translational infidelity and neurological disease.
Trends.
Faithful translation of mRNA into the corresponding protein requires accurate tRNA charging, and precise ribosome decoding and translocation.
The requirement for translational quality control varies between cell types and organisms, with neurons being particularly sensitive to translational infidelity.
Mutations in components of the translational machinery, including ribosomal proteins and elongation factors, have been linked to neurological and neurodegenerative disorders.
The regulation of tRNA expression and modification modulates translational fidelity, and defects in these processes impair neuronal function and survival.
The development of high-throughput and global techniques for the analysis of translation elongation in vivo has greatly facilitated our understanding of the molecular mechanism of many of these disorders.
Acknowledgments
We would like to thank Caitlin E. Monaghan and Markus Terrey for their suggestions on this manuscript. This work was supported by National Institutes of Health grants NS094637 and NS096600. S.L.A. is an investigator of the Howard Hughes Medical Institute.
Glossary
- Aminoacyl tRNA
a tRNA to which the appropriate amino acid has been ligated to the 3′-end by an aminoacyl tRNA synthetase. Also referred to as a charged tRNA. A tRNA in which an incorrect amino acid has been attached is misaminoacylated or mischarged
- Anticodon
The three consecutive nucleotides of the tRNA that basepair with the mRNA codon. Numbered (34-36) in the 5′ to 3′ direction, with nucleotide 36 basepairing with the first nucleotide of the codon
- Codon
A set of three consecutive nucleotides in an mRNA that specify a particular amino acid
- Cognate
related or connected; used to describe the appropriate tRNA synthetase for a given tRNA or amino acid and vice-versa. Also refers to tRNA with an anticodon perfectly complementary to the mRNA codon in the A site. Near-cognate tRNAs contain a single mismatch between their anticodon and the mRNA codon, usually at codon positions 1 or 2
- Decoding center
The functional part of the ribosome responsible for monitoring the codon-anticodon interaction. Located within the small ribosomal subunit, at its interface with the large subunit. Formed by evolutionarily conserved residues of the rRNAs and ribosomal proteins
- Frameshifting
Slippage of the ribosome along the mRNA transcript during translation by one or more nucleotides in either the 5′ or 3′ direction, resulting in translation of a different set of codons. Frameshifting induced by structural elements within the mRNA is known as programmed ribosomal frameshifting (PRF)
- Mistranslation
The insertion of an incorrect amino acid in a manner not defined by the genetic code
- Nonsense suppression
The decoding of a nonsense or stop codon as a sense codon (encoding an amino acid). Also referred to as stop codon readthrough
- Recoding
An alternative decoding event, usually restricted to a subset of mRNAs, which results in the production of a functional, but distinct, protein
- Ribosome profiling
Provides a “global snapshot” of the distribution of ribosomes along mRNA at a nucleotide resolution. This method is based on deep sequencing of a library produced from the mRNA fragments protected by ribosomes from nuclease treatment
- Ternary complex
Any complex consisting of two substrate molecules and a protein
- Translational bypassing
A recoding event where ribosomes pause translation at a particular codon and resume downstream, skipping translation of the intervening nucleotides, forming a contiguous protein
- Watson-Crick basepairing
Canonical basepairing: adenine basepairs with uracil using two hydrogen bonds, and guanine basepairs with cytosine using three hydrogen bonds
- Wobble position
Nucleotide 34 of the tRNA anticodon, which can form a non-Watson-Crick base pair with the third nucleotide of the mRNA codon during decoding. One of the most common wobble base pairs is guanine and uridine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Allan Drummond D, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet. 2009;10:715–724. doi: 10.1038/nrg2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruan B, et al. Quality control despite mistranslation caused by an ambiguous genetic code. Proc Natl Acad Sci. 2008;105:16502–16507. doi: 10.1073/pnas.0809179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalapis D, et al. Evolution of Robustness to Protein Mistranslation by Accelerated Protein Turnover. PLoS Biol. 2015;13:1–28. doi: 10.1371/journal.pbio.1002291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ingolia NT. Ribosome Footprint Profiling of Translation throughout the Genome. Cell. 2016;165:22–33. doi: 10.1016/j.cell.2016.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rozov A, et al. New Structural Insights into Translational Miscoding. Trends Biochem Sci. 2016;41:798–814. doi: 10.1016/j.tibs.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Ling J, et al. Aminoacyl-tRNA synthesis and translational quality control. Annu Rev Microbiol. 2009;63:61–78. doi: 10.1146/annurev.micro.091208.073210. [DOI] [PubMed] [Google Scholar]
- 7.Ling J, et al. Phenylalanyl-tRNA synthetase editing defects result in efficient mistranslation of phenylalanine codons as tyrosine. Rna. 2007;13:1881–1886. doi: 10.1261/rna.684107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu J, et al. Double-sieving-defective aminoacyl-tRNA synthetase causes protein mistranslation and affects cellular physiology and development. Nat Commun. 2014;5:5650. doi: 10.1038/ncomms6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prabhakar A, et al. Dynamic basis of fidelity and speed in translation: Coordinated multistep mechanisms of elongation and termination. Protein Sci. 2017;26:1352–1362. doi: 10.1002/pro.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaher HS, Green R. Quality control by the ribosome following peptide bond formation. Nature. 2009;457:161–6. doi: 10.1038/nature07582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer-Schuman R, Antonellis A. Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum Mol Genet. 2017;26:R114–R127. doi: 10.1093/hmg/ddx231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakayama T, et al. Deficient activity of alanyl-tRNA synthetase underlies an autosomal recessive syndrome of progressive microcephaly, hypomyelination, and epileptic encephalopathy. Hum Mutat. 2017;38:1348–1354. doi: 10.1002/humu.23250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JW, et al. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, et al. Deficiencies in tRNA synthetase editing activity cause cardioproteinopathy. Proc Natl Acad Sci. 2014;111:17570–17575. doi: 10.1073/pnas.1420196111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schrader JM, et al. Tuning the affinity of aminoacyl-tRNA to elongation factor Tu for optimal decoding. Proc Natl Acad Sci U S A. 2011;108:5215–20. doi: 10.1073/pnas.1102128108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ieong KW, et al. Two proofreading steps amplify the accuracy of genetic code translation. Proc Natl Acad Sci U S A. 2016;113:13744–13749. doi: 10.1073/pnas.1610917113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S, et al. Differential expression of S1 and elongation factor-1α during rat development. J Biol Chem. 1993;268:24453–24459. [PubMed] [Google Scholar]
- 18.Nakajima J, et al. De novo EEF1A2 mutations in patients with characteristic facial features, intellectual disability, autistic behaviors and epilepsy. Clin Genet. 2015;87:356–361. doi: 10.1111/cge.12394. [DOI] [PubMed] [Google Scholar]
- 19.Inui T, et al. Two cases of early-onset myoclonic seizures with continuous parietal delta activity caused by EEF1A2 mutations. Brain Dev. 2016;38:520–524. doi: 10.1016/j.braindev.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Veeramah KR, et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia. 2013;54:1270–81. doi: 10.1111/epi.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Ligt J, et al. Diagnostic Exome Sequencing in Persons with Severe Intellectual Disability. N Engl J Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 22.Lam WWK, et al. Novel de novo EEF1A2 missense mutations causing epilepsy and intellectual disability. Mol Genet genomic Med. 2016;4:465–74. doi: 10.1002/mgg3.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopes F, et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J Med Genet. 2016;53:190–9. doi: 10.1136/jmedgenet-2015-103568. [DOI] [PubMed] [Google Scholar]
- 24.Sandbaken MG, Culbertson MR. Mutations in elongation factor EF-1 alpha affect the frequency of frameshifting and amino acid misincorporation in Saccharomyces cerevisiae. Genetics. 1988;120:923–934. doi: 10.1093/genetics/120.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chambers DM, et al. The lethal mutation of the mouse wasted (wst) is a deletion that abolishes expression of a tissue-specific isoform of translation elongation factor 1, encoded by the Eef1a2 gene. Proc Natl Acad Sci. 1998;95:4463–4468. doi: 10.1073/pnas.95.8.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griffiths LA, et al. Haploinsufficiency for translation elongation factor eEF1A2 in aged mouse muscle and neurons is compatible with normal function. PLoS One. 2012;7 doi: 10.1371/journal.pone.0041917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mateyak MK, Kinzy TG. eEF1A: Thinking outside the ribosome. J Biol Chem. 2010;285:21209–21213. doi: 10.1074/jbc.R110.113795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu G, et al. EF-G catalyzes tRNA translocation by disrupting interactions between decoding center and codon–anticodon duplex. Nat Struct Mol Biol. 2014;21:817–824. doi: 10.1038/nsmb.2869. [DOI] [PubMed] [Google Scholar]
- 29.Hekman KE, et al. A conserved eEF2 coding variant in SCA26 leads to loss of translational fidelity and increased susceptibility to proteostatic insult. Hum Mol Genet. 2012;21:5472–5483. doi: 10.1093/hmg/dds392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu S, et al. Diphthamide modification on eukaryotic elongation factor 2 is needed to assure fidelity of mRNA translation and mouse development. Proc Natl Acad Sci U S A. 2012;109:13817–13822. doi: 10.1073/pnas.1206933109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaffrath R, et al. The diphthamide modification pathway from Saccharomyces cerevisiae - revisited. Mol Microbiol. 2014;94:1213–1226. doi: 10.1111/mmi.12845. [DOI] [PubMed] [Google Scholar]
- 32.Loucks CM, et al. Matching Two Independent Cohorts Validates DPH1 as a Gene Responsible for Autosomal Recessive Intellectual Disability with Short Stature, Craniofacial, and Ectodermal Anomalies. Hum Mutat. 2015;36:1015–1019. doi: 10.1002/humu.22843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alazami AM, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10:148–161. doi: 10.1016/j.celrep.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 34.Armistead J, Triggs-Raine B. Diverse diseases from a ubiquitous process: The ribosomopathy paradox. FEBS Lett. 2014;588:1491–1500. doi: 10.1016/j.febslet.2014.03.024. [DOI] [PubMed] [Google Scholar]
- 35.Paolini NA, et al. A Ribosomopathy Reveals Decoding Defective Ribosomes Driving Human Dysmorphism. Am J Hum Genet. 2017;100:506–522. doi: 10.1016/j.ajhg.2017.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shao S, et al. Decoding Mammalian Ribosome-mRNA States by Translational GTPase Complexes. Cell. 2016;167:1229–1240.e15. doi: 10.1016/j.cell.2016.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loenarz C, et al. Hydroxylation of the eukaryotic ribosomal decoding center affects translational accuracy. Proc Natl Acad Sci. 2014;111:4019–4024. doi: 10.1073/pnas.1311750111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singleton RS, et al. OGFOD1 catalyzes prolyl hydroxylation of RPS23 and is involved in translation control and stress granule formation. Proc Natl Acad Sci. 2014;111:4031–4036. doi: 10.1073/pnas.1314482111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brooks SS, et al. A novel ribosomopathy caused by dysfunction of RPL10 disrupts neurodevelopment and causes X-linked microcephaly in humans. Genetics. 2014;198:723–733. doi: 10.1534/genetics.114.168211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thevenon J, et al. RPL10 mutation segregating in a family with X-linked syndromic Intellectual Disability. Am J Med Genet Part A. 2015;167:1908–1912. doi: 10.1002/ajmg.a.37094. [DOI] [PubMed] [Google Scholar]
- 41.Klauck SM, et al. Mutations in the ribosomal protein gene RPL10 suggest a novel modulating disease mechanism for autism. Mol Psychiatry. 2006;11:1073–1084. doi: 10.1038/sj.mp.4001883. [DOI] [PubMed] [Google Scholar]
- 42.Zanni G, et al. A Novel Mutation in RPL10 (Ribosomal Protein L10) Causes X-Linked Intellectual Disability, Cerebellar Hypoplasia, and Spondylo-Epiphyseal Dysplasia. Hum Mutat. 2015;36:1155–1158. doi: 10.1002/humu.22860. [DOI] [PubMed] [Google Scholar]
- 43.Sulima SO, et al. Eukaryotic rpL10 drives ribosomal rotation. Nucleic Acids Res. 2014;42:2049–2063. doi: 10.1093/nar/gkt1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ranjan N, Rodnina MV. tRNA wobble modifications and protein homeostasis. Translation. 2016;4:e1143076. doi: 10.1080/21690731.2016.1143076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bednářová A, et al. Lost in Translation: Defects in Transfer RNA Modifications and Neurological Disorders. Front Mol Neurosci. 2017;10:1–8. doi: 10.3389/fnmol.2017.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Torres AG, et al. Role of tRNA modifications in human diseases. Trends Mol Med. 2014;20:306–14. doi: 10.1016/j.molmed.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 47.Manickam N, et al. Effects of tRNA modification on translational accuracy depend on intrinsic codon-anticodon strength. Nucleic Acids Res. 2015;44:1871–1881. doi: 10.1093/nar/gkv1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thiaville PC, et al. Global translational impacts of the loss of the tRNA modification t(6)A in yeast. Microb cell. 2016;3:29–45. doi: 10.15698/mic2016.01.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edvardson S, et al. tRNA N6-adenosine threonylcarbamoyltransferase defect due to KAE1/TCS3 (OSGEP) mutation manifest by neurodegeneration and renal tubulopathy. Eur J Hum Genet. 2017;25:545–551. doi: 10.1038/ejhg.2017.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shaheen R, et al. A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum Genet. 2016;135:707–13. doi: 10.1007/s00439-016-1665-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lecointe F, et al. Lack of Pseudouridine 38/39 in the anticodon arm of yeast cytoplasmic tRNA decreases in vivo recoding efficiency. J Biol Chem. 2002;277:30445–30453. doi: 10.1074/jbc.M203456200. [DOI] [PubMed] [Google Scholar]
- 52.Cohen JS, et al. ELP2 is a novel gene implicated in neurodevelopmental disabilities. Am J Med Genet Part A. 2015;167:1391–1395. doi: 10.1002/ajmg.a.36935. [DOI] [PubMed] [Google Scholar]
- 53.Simpson CL, et al. Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum Mol Genet. 2009;18:472–481. doi: 10.1093/hmg/ddn375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reinthaler EM, et al. Analysis of ELP4, SRPX2, and interacting genes in typical and atypical rolandic epilepsy. Epilepsia. 2014;55:e89–93. doi: 10.1111/epi.12712. [DOI] [PubMed] [Google Scholar]
- 55.Karlsborn T, et al. Familial dysautonomia (FD) patients have reduced levels of the modified wobble nucleoside mcm(5)s(2)U in tRNA. Biochem Biophys Res Commun. 2014;454:441–5. doi: 10.1016/j.bbrc.2014.10.116. [DOI] [PubMed] [Google Scholar]
- 56.Karlsborn T, et al. Elongator, a conserved complex required for wobble uridine modifications in eukaryotes. RNA Biol. 2014;11:1519–28. doi: 10.4161/15476286.2014.992276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kojic M, Wainwright B. The Many Faces of Elongator in Neurodevelopment and Disease. Front Mol Neurosci. 2016;9:1–10. doi: 10.3389/fnmol.2016.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nedialkova DD, Leidel SA. Optimization of Codon Translation Rates via tRNA Modifications Maintains Proteome Integrity. Cell. 2015;161:1606–1618. doi: 10.1016/j.cell.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klassen R, et al. Independent suppression of ribosomal +1 frameshifts by different tRNA anticodon loop modifications. RNA Biol. 2016;6286:1–8. doi: 10.1080/15476286.2016.1267098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laguesse S, et al. A Dynamic Unfolded Protein Response Contributes to the Control of Cortical Neurogenesis. Dev Cell. 2015;35:553–567. doi: 10.1016/j.devcel.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 61.Fernández-Vázquez J, et al. Modification of tRNA(Lys) UUU by elongator is essential for efficient translation of stress mRNAs. PLoS Genet. 2013;9:e1003647. doi: 10.1371/journal.pgen.1003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rezgui VAN, et al. tRNA tKUUU, tQUUG, and tEUUC wobble position modifications fine-tune protein translation by promoting ribosome A-site binding. Proc Natl Acad Sci U S A. 2013;110:12289–94. doi: 10.1073/pnas.1300781110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schwartz MH, Pan T. Function and origin of mistranslation in distinct cellular contexts. Crit Rev Biochem Mol Biol. 2017;52:205–219. doi: 10.1080/10409238.2016.1274284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen J, et al. Dynamic pathways of -1 translational frameshifting. Nature. 2014;512:328–32. doi: 10.1038/nature13428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen J, et al. Coupling of mRNA Structure Rearrangement to Ribosome Movement during Bypassing of Non-coding Regions. Cell. 2015;163:1267–1280. doi: 10.1016/j.cell.2015.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Girstmair H, et al. Depletion of cognate charged transfer RNA causes translational frameshifting within the expanded CAG stretch in huntingtin. Cell Rep. 2013;3:148–59. doi: 10.1016/j.celrep.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 67.Ishimura R, et al. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345:455–9. doi: 10.1126/science.1249749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jaberi E, et al. Identification of mutation in GTPBP2 in patients of a family with neurodegeneration accompanied by iron deposition in the brain. Neurobiol Aging. 2016;38:216.e11–216.e18. doi: 10.1016/j.neurobiolaging.2015.10.034. [DOI] [PubMed] [Google Scholar]
- 69.Kirchner S, et al. Alteration of protein function by a silent polymorphism linked to tRNA abundance. PLoS Biol. 2017;15:e2000779. doi: 10.1371/journal.pbio.2000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu CH, et al. Codon Usage Influences the Local Rate of Translation Elongation to Regulate Co-translational Protein Folding. Mol Cell. 2015;59:744–754. doi: 10.1016/j.molcel.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ferrin MA, Subramaniam AR. Kinetic modeling predicts a stimulatory role for ribosome collisions at elongation stall sites in bacteria. Elife. 2017;6:1–19. doi: 10.7554/eLife.23629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chu J, et al. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc Natl Acad Sci U S A. 2009;106:2097–103. doi: 10.1073/pnas.0812819106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choe YJ, et al. Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature. 2016;531:191–5. doi: 10.1038/nature16973. [DOI] [PubMed] [Google Scholar]
- 74.Azpurua J, et al. Naked mole-rat has increased translational fidelity compared with the mouse, as well as a unique 28S ribosomal RNA cleavage. Proc Natl Acad Sci. 2013;110:17350–17355. doi: 10.1073/pnas.1313473110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ke Z, et al. Translation fidelity coevolves with longevity. Aging Cell. 2017;16:988–993. doi: 10.1111/acel.12628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mohler K, Ibba M. Translational fidelity and mistranslation in the cellular response to stress. Nat Microbiol. 2017;2:17117. doi: 10.1038/nmicrobiol.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schwartz MH, Pan T. Determining the fidelity of tRNA aminoacylation via microarrays. Methods. 2017;113:27–33. doi: 10.1016/j.ymeth.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mohler K, et al. Isoacceptor specific characterization of tRNA aminoacylation and misacylation in vivo. Methods. 2017;113:127–131. doi: 10.1016/j.ymeth.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cvetesic N, et al. Proteome-wide measurement of non-canonical bacterial mistranslation by quantitative mass spectrometry of protein modifications. Sci Rep. 2016;6:28631. doi: 10.1038/srep28631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moghal A, et al. Mistranslation of the genetic code. FEBS Lett. 2014;588:4305–4310. doi: 10.1016/j.febslet.2014.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Daoud H, et al. Identification of a pathogenic FTO mutation by next-generation sequencing in a newborn with growth retardation and developmental delay. J Med Genet. 2016;53:200–7. doi: 10.1136/jmedgenet-2015-103399. [DOI] [PubMed] [Google Scholar]
- 82.Boissel S, et al. Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am J Hum Genet. 2009;85:106–11. doi: 10.1016/j.ajhg.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hess ME, Brüning JC. The fat mass and obesity-associated (FTO) gene: Obesity and beyond? Biochim Biophys Acta. 2014;1842:2039–47. doi: 10.1016/j.bbadis.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 84.Zhao BS, et al. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2016;18:31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Choi J, et al. N(6)-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat Struct Mol Biol. 2016;23:110–5. doi: 10.1038/nsmb.3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gallo A, et al. ADAR RNA editing in human disease; more to it than meets the I. Hum Genet. 2017;136:1265–1278. doi: 10.1007/s00439-017-1837-0. [DOI] [PubMed] [Google Scholar]
- 87.Behm M, Öhman M. RNA Editing: A Contributor to Neuronal Dynamics in the Mammalian Brain. Trends Genet. 2016;32:165–175. doi: 10.1016/j.tig.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 88.Lin CA, et al. The Sua5 Protein Is Essential for Normal Translational Regulation in Yeast. Mol Cell Biol. 2010;30:354–363. doi: 10.1128/MCB.00754-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Daugeron MC, et al. Gcn4 misregulation reveals a direct role for the evolutionary conserved EKC/KEOPS in the t6A modification of tRNAs. Nucleic Acids Res. 2011;39:6148–60. doi: 10.1093/nar/gkr178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zinshteyn B, Gilbert WV. Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS Genet. 2013;9:e1003675. doi: 10.1371/journal.pgen.1003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dong J, et al. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell. 2000;6:269–79. doi: 10.1016/s1097-2765(00)00028-9. [DOI] [PubMed] [Google Scholar]
- 92.Ishimura R, et al. Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. Elife. 2016;5 doi: 10.7554/eLife.14295. pii: e14295. [DOI] [PMC free article] [PubMed] [Google Scholar]