

Abstract
Rationale
Mutations in the LMNA gene, encoding nuclear inner membrane protein Lamin A/C, cause distinct phenotypes, collectively referred to as laminopathies. Heart failure, conduction defects, and arrhythmias are the common causes of death in laminopathies.
Objective
To identify and therapeutically target the responsible mechanism(s) for cardiac phenotype in laminopathies.
Methods and Results
Whole heart RNA sequencing was performed prior to the onset of cardiac dysfunction in the Lmna−/− and matched control mice. Differentially expressed transcripts and their upstream regulators were identified, validated, and targeted by AAV9-shRNA constructs. A total of 576 transcripts were upregulated and 233 were downregulated in the Lmna−/− mouse hearts (q<0.05). FOXO transcription factors (TFs) were the most activated, while E2Fs were the most suppressed transcriptional regulators. Transcript levels of FOXO targets were also upregulated in the isolated Lmna−/− cardiac myocytes and in the myocardium of human heart failure patients. Nuclear localization of FOXO1 and 3 was increased, whereas phosphorylated (inactive) FOXO1 and 3 levels were reduced in the Lmna−/− hearts. Gene Set Enrichment Analysis and Gene Ontology showed activation of apoptosis and inflammation and suppression of cell cycle, adipogenesis, and oxidative phosphorylation in the Lmna−/− hearts. AAV9-shRNA-mediated suppression of FOXO TFs rescued selected molecular signatures, improved apoptosis, and prolonged survival by ~2-fold.
Conclusions
FOXO TFs are activated and contribute to the pathogenesis of cardiac phenotype in laminopathies. Suppression of the FOXO TFs in cardiac myocytes partially rescues the phenotype and prolongs survival. The findings identify FOXO TFs as potential therapeutic targets for cardiac phenotype in laminopathies.
Keywords: Lamin A/C, transcriptome, transcription factors, FOXO transcription factors, gene therapy
Subject Terms: Animal Models of Human Disease, Basic Science Research, Gene Therapy, Myocardial Biology, Pathophysiology
INTRODUCTION
Heart failure is a major cause of mortality and morbidity, estimated to affect about 6.5 million Americans 20 years and older.1 The prevalence of heart failure, despite the remarkable progress in the management of patients with heart failure during the last three decades, continues to increase, in part because of the aging population. Heart failure is estimated to affect about 8 million Americans ≥ 18 years of age by the year 2030.2
Familial dilated cardiomyopathy (DCM) is a prototypic form of heart failure with reduced systolic function.3 The genetic basis of familial DCM is partially known. TTN and LMNA, encoding the giant sarcomere protein titin and nuclear inner membrane protein lamin A/C, respectively, are the two most common causal genes, accounting for approximately 1/3rd of the primary DCM.4–6 A notable feature of DCM caused by the LMNA mutations is a severe and progressive cardiac dysfunction, typically occurring in conjunction with conduction defects.4, 5
LMNA mutations, in addition to DCM and conduction defects, cause a dozen distinct phenotypes, including Hutchinson-Gilford Progeria Syndrome (HGPS), Emery-Dreifuss muscular dystrophy, familial partial lipodystrophy, arrhythmogenic right ventricular cardiomyopathy, restrictive dermopathy, and axonal Charcot-Marie-Tooth disease (type 2B1), which are collectively referred to as laminopathies.7–9 The diversity of the phenotype is in accord with the ubiquitous expression of the LMNA in almost all differentiated cells, including cardiac myocytes.10 DCM is the major cause of morbidity and mortality in laminopathies, particularly in the subset that predominantly involves the striated muscles.4, 5, 11, 12
The molecular mechanisms leading to DCM in laminopathies are largely unknown, but expected to be diverse, in keeping with the multiple functions of LMNA in cardiac myocytes.13, 14 Alterations in signaling pathways, including MAPK, AKT, NOTCH, and WNT pathways, have been implicated along with changes in mechanotransduction and the linker of nucleoskeleton and cytoskeleton complex (LINC).7, 13, 15, 16 Dysregulated gene expression, proposed by Hutchinson over a decade ago,14 has emerged as the prevailing mechanism to explain phenotypic plasticity of the LMNA mutations. The hypothesis is based on the known interactions of LMNA, directly or indirectly through other nuclear lamina proteins, with chromatin in over 1,000 genomic regions.17–19 The LMNA-associated domains (LADs), with an average length of about 500 kilobase pairs, are predominantly located in the heterochromatin and generally gene poor regions, but they also involve gene rich and the regulatory loci.18, 20–22 Consequently, LMNA is expected to influence expression of a diverse array of genes involved in various biological processes, which are likely to be pertinent to the pathogenesis of the phenotypes in laminopathies.17, 18 Thus, the purpose of the study was to identify the differentially expressed genes and their upstream (transcriptional) regulators, and to determine potential beneficial effects of targeting of the main dysregulated pathway in LMNA deficiency. The objectives are to gain new insights into the molecular pathogenesis of cardiac phenotypes in laminopathies and identify new therapeutic targets.
Methods
The data, methods used in the analysis, and materials used to conduct the research will be made available to any researcher for purposes of reproducing the results or replicating the procedure. The transcriptomic data will be deposited in NCBI GEO Profile, which will maintain the data indefinitely
A detailed Methods Section has been provided as Online Supplementary Material. The studies conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and were approved by the Animal Care and Use Committee and the Biological Safety Committee of the University of Texas Health Science Center-Houston.
Lmna−/− and wild type (WT) littermates
The phenotype in the Lmna−/− mice has been published.23, 24 Oligonucleotide primers used in PCR reactions are listed in Online Table I.
Gross morphology and survival
Body weight (BW) was recorded weekly and heart weight/body weight ratio was calculated in age- and sex-matched mice. Survival was analyzed by constructing Kaplan-Meier survival plots.
Echocardiography
Cardiac size and function were assessed in age- and sex-matched mice by 2D, M mode, and Doppler echocardiography using a Vevo 1100 ultrasound imaging system equipped with a 22-55 MHz MicroScan transducer (MS550D) (FUJIFILM VisualSonics Inc., Toronto, ON, Canada), as published.25–29
Isolation of adult cardiac myocytes
Adult cardiac myocytes were isolated as published26, 29 In brief, the excised heart was perfused with a digestion buffer containing collagenase II and the perfusion was continued until complete softening of the myocardium. The cells were dissociated from the digested hearts by gentle pipetting and were placed in a buffer containing 10% calf serum and 12.5 μM CaCl2 to stop the reaction. Cells were then filtered through a 100 μm nylon mesh, myocytes were left to sediment by gravity in the presence of 2 mM ATP and pelleted by centrifugation at 20g. Myocytes were re-introduced to calcium in step-wise concentrations of 100 μm, 400 μm and 900 μm, added to the stop buffer at 2 mM ATP. The isolated myocytes were resuspended in a plating media containing antibiotics, calf serum and ATP. The isolated myocytes were placed on laminin-coated cover glasses in a 2% CO2 incubator at 37 °C or were immediately frozen.
Isolation of neonatal mouse ventricular myocytes and transduction with adenoviruses
Cardiac cells were isolated from 1- to 2-day old WT mice by enzymatic dispersion with collagenase and pacreatin. Neonatal mouse cardiac myocytes were separated from the rest of the cells by differential attachment, and grown on chamber slides. On day 2, cells were infected with recombinant adenoviruses expressing a constitutively active form of FOXO (Ad-FOXOAAA) or a vector virus alone (Vector Biolabs, #1025 and #1060) for 48 hours with a multiplicity of infection of 20.30, 31. Neonatal mouse cardiac myocytes were treated for superoxide mitochondrial production detection by MitoSOX or were fixed for detection of apoptosis.
Histology
Fibrosis was assessed upon staining of thin myocardial sections with Masson trichrome and Sirius Red, as described.26, 29, 32 Collagen volume fraction (CVF) was calculated as published32
Detection of apoptosis
Apoptosis was detected by nick-end labelling of DNA with the TUNEL assay using In Situ Cell Death Detection Kit (Roche catalogue # 11684795910 and 12156792910), per manufacturer instructions and quantification of the transcript levels of genes involved in apoptosis, as published.26, 29, 33
Quantitative real-time PCR (qPCR)
Total RNA was extracted from myocardial tissue or from isolated cardiac myocytes using the Qiagen miRNeasy Mini Kit (catalogue # 217006).25, 28 Taqman probes and SYBR Green primers are detailed in Online Table I.
Immunoblotting of total and cellular protein fractions
Expression levels of the proteins of interest in mouse heart samples and isolated cardiac myocytes were detected and quantified by immunoblotting, as described.25, 26, 28 Nuclear and cytosolic proteins were extracted using a commercial kit (ProteoExtract®, Calbiochem, catalogue # 539790) and probes with the specific antibodies against proteins of interest.34 Immunofluoresence (IF): IF were performed on thin myocardial sections as published.25, 26, 29
RNA-Sequencing (RNA-Seq)
RNA-sequencing was performed in the whole heart ribosome-depleted RNA extracts from the 2-week old WT and Lmna−/− mice on an Illumina platform, as published with minor modifications.25, 28 In brief, ribosome-depleted cardiac RNA samples were analyzed for integrity on an Agilent Bioanalyser RNA chip. Samples with high integrity index were used to prepare sequencing library using the Illumina TruSeq stranded total RNA library preparation kit. The samples were sequenced on the Illumina HiSeq 4000 instrument using the paired-end sequencing reagents to generate 100 base pair runs.
Raw RNA sequencing reads were mapped to the Mouse reference genome build 10 (UCSC mm10/GRCm38) by Tophat2.35 Gene expression was assessed using Cufflinks2 and the GENCODE gene model.35, 36 Only transcripts that exceeded 1 fragments per kilobase of transcript per million mapped fragments (FPKM) in at least one sample were included in the analyses. Differentially expressed transcripts (DETs) were identified using the edgeR analysis package in R statistical program with the significance level set at q<0.05 and the fold change at > 1.25. Quality control and data visualization were assessed by principal components analysis and hierarchical clustering in R. GENE-E software (http://www.broadinstitute.org/cancer/software/GENE-E) was used to generate the heatmaps from the raw FPKM values and the Graph Pad Prism was used to generate the volcano plots. Enriched upstream regulators and transcription factor binding sequence motifs were inferred using Gene Set Enrichment (GSEA, version 2.2.3, http://software.broadinstitute.org/gsea/), Transcription factor binding motifs TRANSFAC,37, Molecular Signature Database (MSigDB),38–40 and the Ingenuity Pathway Analysis® (IPA, Qiagen). Gene Ontology (GO) analyses were conducted using ConsensusPathDB (release MM9-http://cpdb.molgen.mpg.de/MCPDB), (41, 42 43 and GO terms were gathered according to their similarities using REVIGO (http://revigo.irb.hr/).44 Circos plot was generated in R using the GO-Chord option.
AAV9-mediated In vivo suppression of FOXO TFs in the heart
A previously validated short hairpin RNA (shRNA) that targets FOXO1 and FOXO345, 46 was cloned into AAV9 vectors downstream to a U6 promoter (VectoBiolab). Control AAV9 vectors contained either a GFP or a scrambled shRNA expression cassette. Sequence of the shRNA against FOXO1 and 3 and scrambled shRNA are presented in Online Table I.
To knock down expression of FOXO1 and FOXO3 in the heart, recombinant AAV9 constructs at a titer of 0.5×1011 vector genomes per gram of body weight (vg/g) were injected subcutaneously into neonatal mice, sequentially at P2, P4, and P6 postnatal days, delivering a total of 1.5 × 1011 vg/g to each mouse. Control AAV9 vectors were injected at the same dosage and time points. The early time points were chosen to allow sufficient time for gene expression prior to the onset of cardiac phenotypes in the Lmna−/− mice.23, 24 Serial injections before complete immune competency are expected to enhance transduction efficiency by targeting the newly formed cardiac myocytes.47 The viral titer was chosen to induce an efficient transduction of cardiac myocytes without toxicity.48–50
Cytokines measurement
Cytokine and chemokine levels in the heart of 2 weeks old WT and Lmna−/− mice were measured using the Milliplex MAP Mouse Cytokine/Chemokine 32-plex assay (Millipore) according to manufacturer’s protocol (MCYTMAG-70K-PX32).
Mitochondrial Electron Chain Transport (ETC) Activity
Enzymatic activity of each of the complexes of mitochondrial ETC was assessed in the heart of 2-week old WT and Lmna−/− mice by adding the specific substrates to cardiac homogenates, as published.51 All activities were calculated as nmoles/min/mg protein, normalized to citrate synthase activity, and expressed as a percentage of WT activity.
Mitochondrial ETC complexes assembly immunoblotting
ETC complexes assembly was assessed by immunoblotting of specific subunit of each of the five ETC complexes using the Total OXPHOS Rodent WB Antibody Cocktail (Abcam, #ab110413) per manufacturer’s instruction.
Mitochondrial superoxide anion production detection by MitoSOX
mNVM were incubated with Red MitoSOX according to manufacturer’s instructions.
Statistical analyses
Data that followed a Gaussian distribution pattern were presented as mean ± SEM and were compared by t-test and one-way ANOVA. Otherwise, data were presented as the median values and compared by Kruskall-Wallis test, as were the categorical data. Survival rates were analyzed by constructing Kaplan-Meier survival plots and comparing the survival rate by Log-rank (Mantel-Cox) test.
Results
Cardiac phenotype in 2-week old Lmna−/− mice
To validate deletion of the Lmna gene, cardiac protein extracts from the WT and Lmna−/− mice were probed for the expression of the LMNA protein using an anti-LMNA antibody. As expected expression of the LMNA protein was not detected in the heart of the Lmna−/− mice (Online Figure IA). The Lmna−/− mice survived normally within the first 2 weeks, but exhibited growth retardation, as noted by the smaller body weight (Lmna−/− mice: 5.44±0.2 g, WT mice: 6.97±0.3 g, p<0.0001, Online Figure IB, and IC). Given that body weight is a major determinant of cardiac size, including echocardiographic indices of left ventricular dimension, these indices were corrected for the body weight. Accordingly, heart weight/body weight ratio was not significantly different between Lmna−/− and WT mice (Online Figure ID). To detect early transcriptomic changes that precede cardiac dysfunction, and therefore, to reduce the confounding changes, cardiac function was analyzed in 2-week old Lmna−/− and WT mice by echocardiography. Consistent with the previous data, there were no differences in the echocardiographic indices of cardiac size and function between 2-week old Lmna−/− and WT mice with the exception of the mean left ventricular end diastolic diameter corrected for body weight, which was slightly enlarged in the Lmna−/− mice (by 0.05 mm/g, p=0.032), while left ventricular fractional shortening was normal. (Online Table II).23, 24, 52 Histological examinations showed no discernible cardiac fibrosis in the Lmna−/− mice at 2 weeks of age (Online Figure IE and F). In accord with the histological findings, levels of Col1a1, Col1a2 and Tgfb1 transcripts, markers of fibrosis, as assessed by qPCR, were unchanged in the hearts of 2-week old Lmna−/− mice (Online Figure IG).
Differentially expressed cardiac transcripts in 2-week old Lmna−/− mice
Given the absence of discernible cardiac dysfunction in the Lmna−/− mice, RNA-Seq was performed in the ribosome-depleted cardiac transcripts isolated from the hearts of 2-week old WT and Lmna−/− mice (whole heart ribosome-depleted RNA). Of the 809 transcripts that were dysregulated in the hearts of 2-week old Lmna−/−, as compared to the WT mice, 233 were down-regulated and 576 upregulated. Volcano plots and heat maps of the DETs are shown in Figure 1, panels A and B.
Figure 1. Cardiac RNA-Seq data in 2-week old WT and Lmna−/− mice.
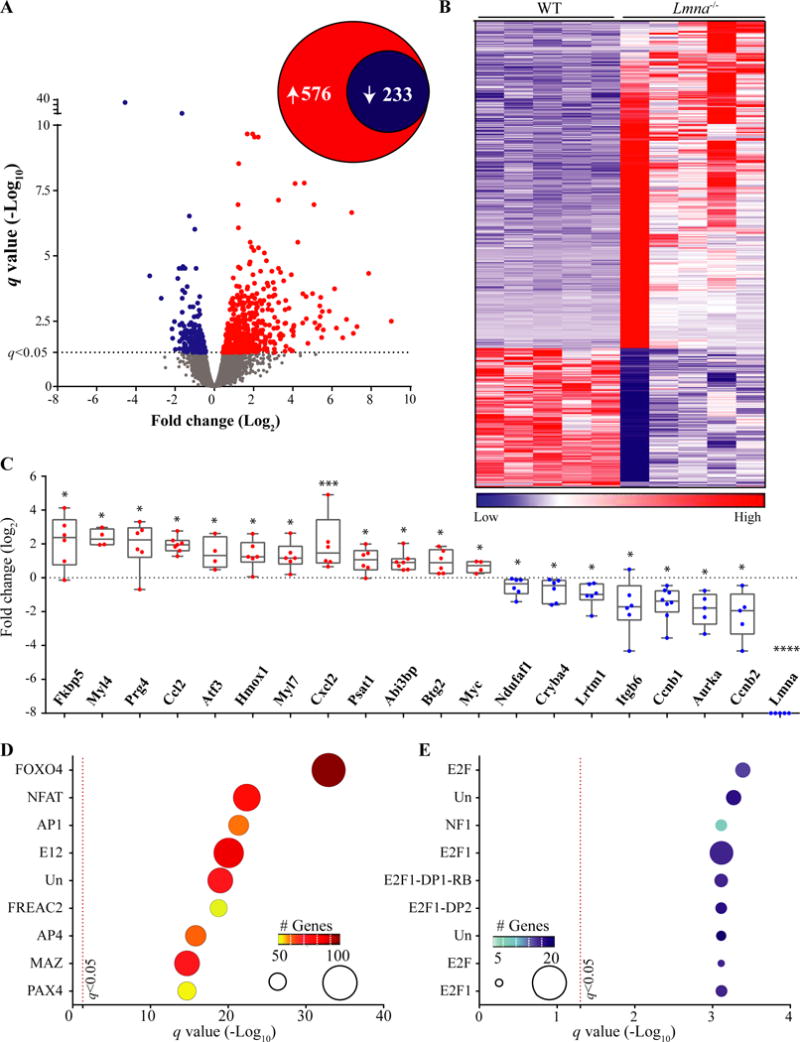
A. Volcano plot of all the transcripts detected by RNA-Seq. The significance level for the differentially expressed transcripts (DETs) is marked by a dashed line at the q value of 0.05. A total of 809 transcripts were differentially expressed between Lmna−/− and WT, comprised of 576 upregulated and 233 down-regulated transcripts (insert circles). B. Heat-map of the DETs. Transcripts were ranked, according to their differential expression across genotypes, from the most up-regulated genes to the most down-regulated in Lmna−/− hearts. C. Quantitative PCR validation of RNA-Seq data for twenty DETs, including Lmna, the latter as a control. The transcripts were randomly selected for validation among those showing about a 2-fold or higher change in RNA-Seq data and were differentially expressed at a q value of 0.05. Gene expression in Lmna−/− after normalization is presented here as Log2 of fold change relative to the expression levels in WT mice *: p<0.05, **: p<0.01, ***: p<0.001, ****: p<0.0001 D and E: Enrichment of conserved cis-regulatory motifs in DETs. DETs were analyzed using GSEA and the MSigDB for enrichment of transcription factor (TF) binding motifs by hypergeometric distribution. Top ten over-represented TFs among the up- (D) and down-regulated (E) TFs are presented. The number of the genes in the overlap is displayed by size of the ball on the graph along with the color coding indicated in the insert. Un: Unknown TF for the identified binding motif. Detailed analysis is provided in Online Table III.
To corroborate the DETs identified by RNA-Seq, transcript levels of randomly selected 20 dysregulated genes, showing at least a 2-fold or higher change (q<0.05), were analyzed by qPCR in the whole hearts in an independent set of WT and Lmna−/− mouse hearts. As shown in Figure 1C, the findings corroborated the RNA-Seq findings.
Dysregulated upstream transcriptional regulators and enriched TF binding sites in the DETs in the Lmna−/− hearts
To identify the dysregulated upstream transcriptional regulators in the Lmna−/− hearts, DETs were analyzed against the MSigDB to identify over-representation of the binding sites of the TFs in the promoter regions of the differentially expressed genes. The consensus binding motif for FOXO TFs (TTGTTT) was over-represented in the up-regulated genes, while the motif for E2 factors (E2Fs) was the most over-represented in genes whose transcript levels were reduced (Figure 1, panels D and E). In addition, motifs for the NFAT, AP1, and E12 TFs showed significant enrichment in the Lmna−/− hearts. Nevertheless, FOXO TFs had by far the most dysregulated targets (Figure 1D). Detailed results are provided in Online Table III.
IPA of the DETs also identified FOXO3 among the top transcriptional regulators in the Lmna−/− hearts (z score: 3.76, p=3.3E−07, Online Table IV). A total of 145 FOXO target genes were dysregulated in the heart of Lmna−/− mice, of which 128 were upregulated, and only 19 were downregulated (Figure 2A). The magnitude of the differential expression of FOXO TF targets is illustrated in the heat map (Figure 2B). Quantitative PCR analysis also showed increased transcript levels of Foxo3 in the Lmna−/− mouse hearts (Figure 2C). The FOXO family of TFs share TTGTTT as their common binding site motif. Therefore, the analysis of the differentially expressed genes does not enable distinction among the specific dysregulated FOXO TFs. Therefore, network analysis by IPA showed dysregulated transcript levels of several FOXO TF targets, which were shared among the three common FOXO TFs expressed in the heart (Figure 2D).53 To further corroborate activation of the FOXO TFs in the hearts in the Lmna−/− mice, transcript levels of a selected number of FOXO TF targets were analyzed by qPCR. The findings were notable for increased levels in the Lmna−/− as opposed to the WT hearts (Figure 2E).
Figure 2. Activation of FOXO transcription factors in Lmna−/− mice at 2 weeks of age.
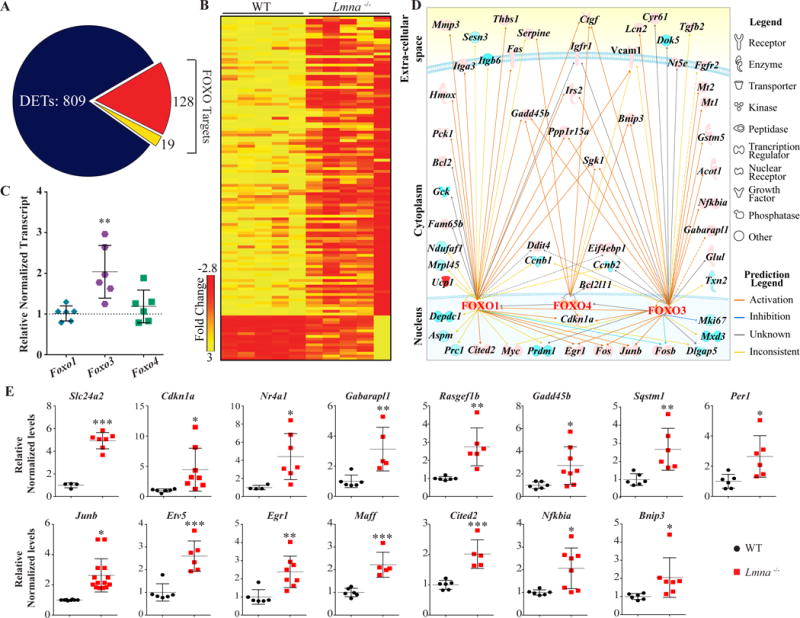
A. Pie-chart showing the fraction of DETs that are targets of FOXO TFs. A total of 128 targets out of 809 DETs were upregulated and 19 were down-regulated, as highlighted in red and yellow colors, respectively. B. Heat map of the DETs that are targets of the FOXO TFs shown in the WT and Lmna−/− mice. Heat-map colors (red, up-regulation; yellow, down-regulation) indicate transcript fold change obtained from the RNA-Seq data. The scale bar represents magnitude of the change. C. Transcript levels of Foxo1, Foxo3, and Foxo4 in the WT and Lmna−/− mouse hearts, as quantified by qPCR. Among the three isoforms, Foxo3 transcript levels were increased by 2.03±0.49–fold, as compared to WT mice (N=6, p=0.003). D. Upstream regulatory pathways altered in the heart in 2-week old Lmna−/− mice. Ingenuity Pathway Analysis (IPA) of DETs identified FOXO3 transcription factor among the most activated pathway (z-score: 3.75), as reflected in the increased transcript levels of more than 100 target genes. Interactive networks depicting increased (orange) or suppressed (blue) targets of FOXO1, 3 and 4 TFs. Solid and dashed lines represent direct and indirect interactions, respectively. The grey lines indicate predicted interactions without indication of the nature of the relationship, and the yellow interactions with opposite effects from the literature. E. Validation of the RNA-Seq data of FOXO TF targets by qPCR. Relative transcript levels (normalized to Gapdh) of over a dozen FOXO TF target genes, identified by GSEA and IPA analyses, in the Lmna−/− mouse hearts are depicted (N-6-8 mice per group). The findings are concordant with increased transcript levels in the RNA-Seq data.
Increased nuclear localization of FOXO1 and 3 in the Lmna−/− hearts
To further corroborate evidence for the activation of FOXO TFs, levels of FOXO1, 3, and 4 transcripts and proteins were determined by qPCR, immunoblotting, and immunofluorescence, respectively. Transcript levels of Foxo3, the most abundant FOXO TF in the heart, but not Foxo1 and Foxo4, were increased significantly in the heart, likely reflecting auto-regulation (Figure 2C). Immunoblotting of myocardial nuclear protein subfraction showed increased levels of FOXO1 and FOXO3 by 1.4±0.1 -fold (N=5, p=0.035) and 1.8±0.1 -fold (N=6, p=0.014 vs. WT), respectively, in the Lmna−/− as compared to WT mouse hearts, whereas FOXO4 levels were unchanged (Figure 3A-B). Levels of FOXO3 in the cytoplasmic protein fraction were unchanged, while those of FOXO1 were reduced by 46.0 ± 3.9% (N=5, p=0.002 vs. WT, Figure 3C-D). Levels of FOXO1 in the total protein extracts were increased 1.6 ± 0.2 -fold (N=8-9, p=0.0007), while FOXO3 and FOXO4 levels were unchanged (Figure 3E-F). Finally, because FOXO TFs are inactivated upon phosphorylation at threonine residues 24 and 32 (Thr24/32), pFOXO levels were determined in myocardial protein extracts using an antibody that recognizes phospho-FOXO1, 3 and 4 proteins. As shown, pFOXO1 and 3 levels were reduced by 63.4 ± 2.6% (N=4, p=0.028 vs. WT)(Figure 3, panels G-H). Consequently, the relative levels of active to inactive FOXO1 and 3 were increased in Lmna−/− as compared to control WT mouse hearts (Figure 3I). Finally, FOXO3 nuclear localization was analyzed by immunofluorescence co-staining of thin myocardial sections for FOXO3 and ACTN2, the latter as a marker for cardiac myocytes. The number of myocytes showing localization of FOXO3 in the nucleus was increased in the Lmna−/− mouse hearts as compared to WT (1.8± 0.2-fold in Lmna−/− mouse heart vs, WT hearts, N=3, p=0.006, Figure 3, panels J and K). Collectively, the findings indicate activation of FOXO1 and FOXO3 TFs in the hearts of the Lmna−/− mice.
Figure 3. Increased nuclear localization of FOXO transcription factors in cardiac myocytes in the Lmna−/− mice at 2 weeks of age.
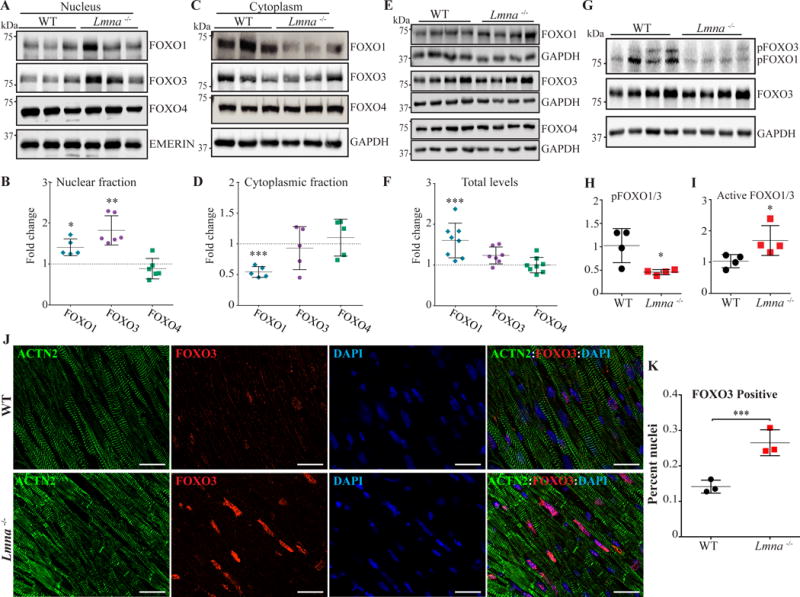
A-B. Sub-cellular localization of FOXO TFs in the heart. FOXO1, 3 and 4 proteins were detected by immunoblotting on nuclear and cytoplasmic protein fractions, as well as in the total myocardial proteins extracts from the Lmna−/− and WT mouse hearts (Panels E-F). Corresponding quantitative data show increased levels of FOXO3 in the nuclear protein sub-fraction by 1.8±0.14-fold (Panel B, N=6, p=0.014 vs WT). FOXO1 levels also showed a modest increase in the nuclear protein extracts in the Lmna−/− mouse heart (1.4±0.1 -fold, p=0.035), whereas FOXO4 levels were unchanged (Panel B). Level of FOXO1 in the cytoplasmic protein fraction was reduced by 46.0±3.9% (N=5, p=0.002 vs. WT), whereas levels of FOXO3 and FOXO4 were unchanged (panel C). A modest increase in levels of FOXO1 protein in the total myocardial protein extract (1.6±0.2 -fold vs. WT, N=8-9, p=0.0007) was also noted. There were no significant differences in levels of FOXO3 and FOXO4 in the total myocardial protein extracts between the groups (Panel E and F).
G-H. Immunoblot of phospho-Thr24,32-FOXO1 and 3 (pFOXO1/3), total FOXO1 and 3, and GAPDH, the latter as a loading control. Levels of pFOXO1 and 3 were reduced by 63.4±2.6% in the Lmna−/− mouse hearts (N=4, p=0.028 vs. WT, Panel G and H). Conversely, the estimated active to inactive FOXO1 and 3 levels (total FOXO1/3- pFOXO1/3), were increased by 2.76±0.62 fold in Lmna−/− mouse hearts (p=0.038 vs. WT, panel I). J-K. Representative immunofluorescence staining of FOXO3 and α-actinin (ACTN2), a myocyte marker, on thin myocardial section showing increased nuclear localization of FOXO3 in cardiac myocytes in the Lmna−/− mouse hearts compared to the WT (N=3, scale bar is 20 μm, panel J). Corresponding quantitative data, based on analysis of a minimum of 21820 nuclei per group, is shown in panel K (0.26 ± 0.02 % vs. 0.14 ± 0.01% myocytes in the Lmna−/− and WT mouse hearts, respectively, p=0.006). *: p<0.05, **: p<0.01, ***: p<0.001, ****: p<0.0001
To extend the findings on activation of FOXO TFs in the Lmna−/− mouse hearts to human hearts with DCM, publically available RNA-seq data from failing and non-failing human hearts (GSE46224) without defined mutations were analyzed by GSEA for the transcription factor binding motif enrichment. As shown in Online Figure II, the TTGTTT motif was significantly enriched in the human heart samples from patients with DCM. This finding corroborates activation of the FOXO TFs in the Lmna−/− mouse hearts.
Dysregulated biological processes
To identify the biological processes associated with the dysregulated transcriptional regulators, cardiac transcripts were analyzed by GSEA using the MSigDB. Genes involved in inflammation, hypoxia, and apoptosis were upregulated and those involved in oxidative phosphorylation, cell cycle progression, and adipogenesis were downregulated (Figure 4, panels A-B). Given that genes involved in the inflammatory responses were prominently depicted in the hearts of the Lmna−/− mice, GSEA plot and transcript levels of genes involved in the NFκB pathway were analyzed along with the protein levels of selected cytokines, the later by ELISA. GSEA plots showed marked enrichment of the NFκB pathway (Figure 4C). Likewise, mRNA levels of several NFκB target genes were increased significantly (Figure 4D). Moreover, myocardial levels of several cytokines, including CXCL1, CCL2, IL6, CXCL10, LIF, and IL9 were increased in the Lmna−/− mouse hearts as compared to WT mouse hearts (Figure 4E).
Figure 4. Dysregulated biological pathways in Lmna−/− mice at 2 weeks of age.
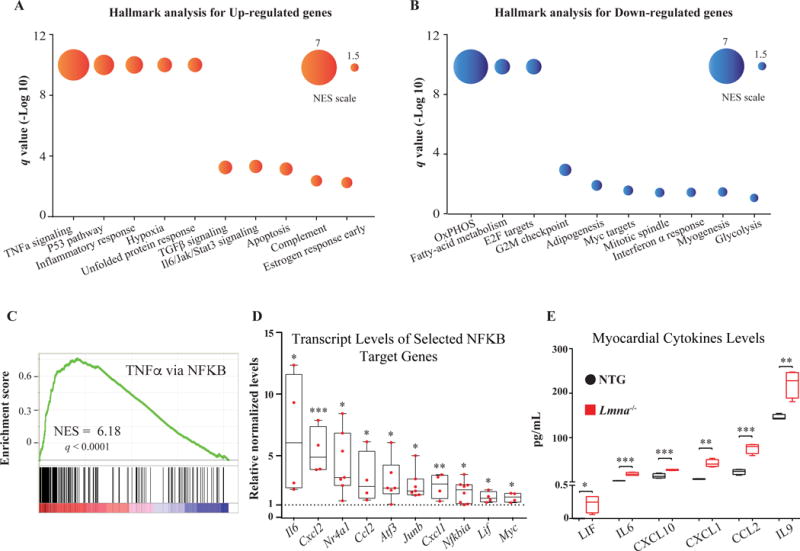
A and B. The list of dysregulated biological processes (upregulated in red and downregulated in blue colors) in the Lmna−/− mouse hearts (q value < 0.05), identified by the Hallmark gene set, are shown along with circle size indicators for the normalized enrichment score (NES). C. GSEA enrichment plot of “TNFα signaling via NFκB”, the most enriched Hallmark gene set in the Lmna−/− mouse heart, showing significant enrichment of the transcript levels contributing to activation of this pathway. Distribution of the genes involved in the pathway is plotted against their individual enrichment score amongst the total ranked list of all transcripts analyzed by GSEA. D. Validation of enrichment of the “TNFα signaling via NFκB” in Lmna−/− mouse hearts by qPCR for about a dozen genes involved in the pathway, showing increased transcript levels (N=4 to 8 per group). E. Levels of cytokines in the Lmna−/− mouse hearts. Levels of CXCL1, CCL2, IL6, CXCL10, LIF and IL9, measured by Luminex assays in the myocardial protein extracts, showing increased levels in the Lmna−/− mouse hearts as compared to WT (N=4 per group).
*: p<0.05, **: p<0.01, ***: p<0.001, ****: p<0.0001
To further characterize the potential impact of FOXO activation in the heart, biological pathways that are regulated by FOXO target genes were curated using Gene Ontology (GO). GO analysis of FOXO target genes identified programmed cell death as the most activated biological pathway in the Lmna−/− mouse hearts (Figure 5A). Consistent with this finding, GSEA of whole transcriptome showed enrichment of genes involved in apoptosis (Figure 5B). Transcript levels of a subset of FOXO TFs target genes involved in apoptosis were also quantified by qPCR and found to be significantly increased in the Lmna−/− mouse hearts (Figure 4C). In contrast, transcript levels of anti-apoptotic gene Bcl2 were unchanged. TUNEL assays performed on thin myocardial sections showed a 3.8 ± 0.5 -fold increase in the number of TUNEL positive nuclei in 2-week old Lmna−/− mouse hearts (N=4 mice per group, p=0.002, Figure 5D and E). Finally, adenoviral-mediated expression of a constitutive active form of FOXO3 (Ad-FOXO3AAA) in neonatal mouse ventricular myocytes was associated with an increased number of TUNEL positive nuclei (3.1± 0.1 –fold vs. control vector, p<0.0001, Figure 5, panels F and G). Altogether, the findings indicate a deleterious role for overactivation of FOXO TFs in the hearts of the Lmna−/− mice.
Figure 5. Activated biological pathways in the Lmna−/− mouse hearts.
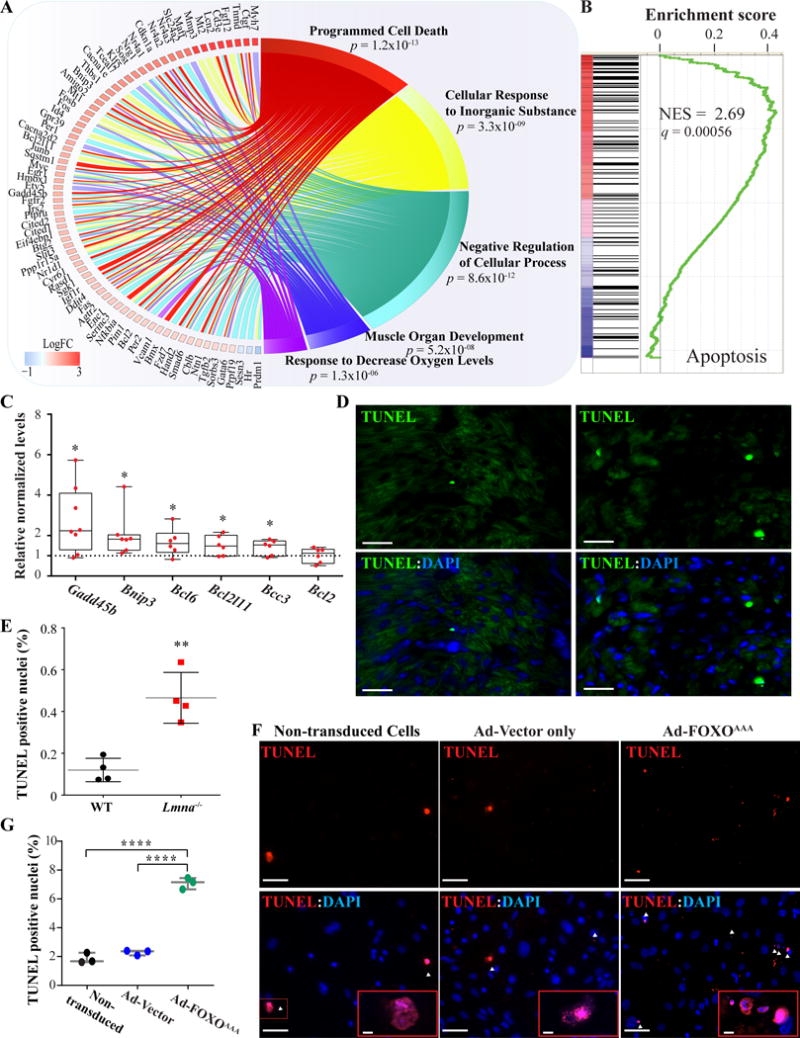
A. Circos plot depicting the top 5 enriched biological pathways associated with the differentially expressed FOXO TF target genes, as identified by Gene Ontology (GO) analysis and then compiled by relative similarity using Revigo. Expression level of genes involved in the pathway is indicated in log2 of fold change. As shown, programmed cell death (apoptosis) was the most dysregulated biological process. B. GSEA plot showing enrichment of genes involved in apoptosis. C. Quantitative RT-PCR showing increased transcript levels of selected genes involved in the apoptosis process. D-E. Apoptosis staining by TUNEL labeling of thin myocardial sections and the corresponding quantitative data showing increased number of TUNEL (green) positive DAPI-stained nuclei (blue) in the Lmna−/− mouse hearts by 3.8 ±0.5-fold (N=4, p=0.002 vs. WT). Scale bar is 10 μm F- G. TUNEL assay of mouse neonatal ventricular myocytes (mNVM) after infection with adenoviruses expressing a constitutively active form of FOXO3 (Ad-FOXO3AAA) and corresponding quantitative data showing an increased number of TUNEL (red) positive DAPI-stained nuclei (blue) compared to control adenovirus (N=3, 3.1 ± 0.1-fold, p<0.0001) or cell not infected (N=3, 3.8 ± 0.1-fold, p<0.0001). No significant changes in the number of TUNEL positive nuclei between the mNVM infected with the control adenovirus and the cells not infected (2.3 ± 0.1% vs 1.8 ± 0.2%, p=0.3). Scale bar is 50 μm and 5 μm in the insert.
Oxidative phosphorylation was the most suppressed biological function in the Lmna−/− mouse hearts (Figure 4B). Its suppression was demonstrated by GSEA, quantification of the transcript levels of selected mitochondrial genes and determination of mitochondrial electron transport chain activity (Online Figure III). Complex I enzymatic activity was decreased by 37.0 ± 7.8% (N=4, p=0.021 vs. WT) and the transcript levels of the corresponding genes involved in complex I were also reduced (Online Figure III). Likewise, suppression of the cell cycle regulatory pathways, namely E2F targets and G2M checkpoint, were confirmed in the Lmna−/− mouse hearts by GSEA along with quantification of transcript levels of selected genes in the pathways (Online Figure III).
Suppression of FOXO TFs in the Lmna−/− hearts
To establish the pathogenic role of activation of FOXO TFs in cardiac phenotypes in the Lmna−/− mice, recombinant AAV9 constructs were generated to target FOXO1 and 3 upon expression of an established sequence-specific shRNA (AAV9-FoxoshRNA).45, 46 A prelude to AAV9-mediated gene targeting, which predominantly transduces cardiac myocytes54, 55, is evidence of activation of FOXO TFs in cardiac myocytes. Accordingly, cardiac myocytes were isolated from the WT and Lmna−/− mouse hearts and analyzed for the transcript levels of 15 randomly selected FOXO TF targets by qPCR. The results (Figure 6A) showed increased transcript levels of FOXO TF target genes in cardiac myocytes isolated from the Lmna−/− mouse hearts, indicating activation of the FOXO TFs in the Lmna−/− cardiac myocytes.
Figure 6. Knock-down of FOXO TFs in Lmna−/− mouse heart.
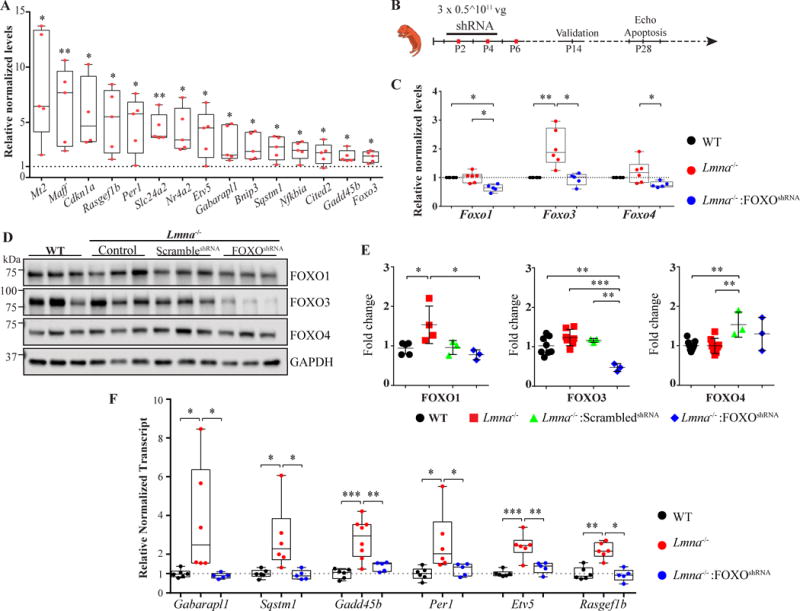
A. Validation of FOXO TFs activation in cardiac myocytes isolated from the Lmna−/− mouse hearts. Transcript levels of 15 FOXO TFs target genes were quantified by RT-qPCR on mRNA extracted from isolated cardiac myocytes from the WT and Lmna−/− mice. As shown, transcript levels were significantly increased in isolated cardiac myocytes form the Lmna−/− mice (N= 4 to 5 independent myocyte isolations per group). B. Schematic presentation of the gene therapy protocol to knock down FOXO TFs upon injection of the recombinant AAV9 constructs. Recombinant AAV9-FoxoshRNA viral vectors were injected sequentially at post-natal day 2, 4, and 6 (P2, P4 and P6) subcutaneously at 0.5 × 1011 viral genomes (vg) per gram. Knock down of FOXO TF was confirmed in 2-week old mice and phenotypic characterization was performed at 4 weeks of age.
C. Validation of FOXO TFs knock-down at the mRNA level in the Lmna−/− mouse hearts. Transcript levels of Foxo1, 3 and 4 were quantified by RT-qPCR on whole heart mRNA extracts from the WT and Lmna−/− and Lmna−/− mice injected with the recombinant AAV9 vectors. Foxo1 mRNA levels were decreased by 36.25 ± 5.57% in the AAV9-FoxoshRNA, as compared to control (not-injected) Lmna−/− mice (p=0.018) and to 36.3 ± 12% compared to the WT mice (N=5-6 per group, p=0.022). Foxo3 mRNA level was decreased to 53.7 ± 10% in the AAV9-FoxoshRNA treated mice compared to non-treated Lmna−/− (p=0.026). Foxo4 levels are reduced by 37.4 ± 18% in the AAV9 treated mice compared to Lmna−/− (un-injected) mice (N=5 to 6 mice per group, p=0.024) and were unchanged compared to WT mice. D- E. Knock down of FOXO proteins in Lmna−/− mouse hearts. Panel D shows immunoblots of FOXO1, 3 and 4 proteins in protein extracted from whole heart in the experimental groups. Quantitative data for FOXO1, 3 and 4 protein levels are depicted in panel E. GAPDH was used as loading control. F. Normalization of FOXO TFs target transcript levels upon administration of the recombinant AAV9-FoxoshRNA construct to the Lmna−/− mice. Transcript levels of selected FOXO TF target genes were quantified by RT-qPCR in cardiac mRNA extracts from the WT and Lmna−/− (untreated) and Lmna−/− mice treated with AAV9-FoxoshRNA. As shown, injection of AAV9-FoxoshRNA normalized increased transcript levels of the FOXO TF target genes.
*: p<0.05, **: p<0.01, ***: p<0.001, ****: p<0.0001
Recombinant AAV9-FoxoshRNA, AAV9-Gfp, or AAV9-scrambledshRNA were injected at P2, P4, and P6 days at a total 1.5 × 1011 vg/g particles subcutaneously, followed by testing for knock down of FOXO1 and FOXO3 TF at 2 weeks and phenotypic characterization at 4 weeks of age (Figure 6B). Treatment with the control AAV9 constructs (AAV9-Gfp and AAV9-scrambledshRNA) had no discernible effect on the survival rate (Online Figure IV). Therefore, in the subsequent studies AAV9-scrambledshRNA construct was used as a control group. Administration of AAV9-FoxoshRNA reduced Foxo3 transcript levels by 53.7±10% (p=0.026 compared to non-injected Lmna−/− mouse heart), which were increased in the Lmna−/− mouse hearts, effectively normalizing the levels (comparable to WT). Likewise, transcript levels of Foxo1 and Foxo4 were reduced significantly in the AAV9-FoxoshRNA group, as compared to non-injected Lmna−/− mice (37.8 ± 12%, p=0.017, and 37.4 ± 18%, p=0.024, respectively, Figure 6C). At the protein level, the most remarkable finding was reduced levels of FOXO3 upon delivery of AAV9-FoxoshRNA construct, which was reduced by 61.6 ± 4.5% compared to non-injected Lmna−/− mice (N=3, p=0.024, Figure 6, panels D and E). Likewise, levels of FOXO1 protein were also reduced (49.5 ± 13.9%, p=0.006 vs non-injected Lmna−/− mice) and were comparable to those in the WT mice, while levels of FOXO4 were unchanged (Figure 6, panels D and E). To further support effective knock down of FOXO TF activities upon administration of the AAV9-FoxoshRNA construct, transcript levels of several well-established FOXO TFs targets were quantified by qPCR. The results are remarkable for normalization of the transcript levels of FOXO TF targets in AAV9-FoxoshRNA group (Figure 6F).
Partial phenotypic rescue upon knock down of FOXO TFs
To determine whether knock down of FOXO TFs rescues the phenotype in the Lmna−/− mice, the effects of administration of the AAV9-FoxoshRNA construct on survival, cardiac function, and apoptosis were analyzed. Knock down of FOXO TFs in the heart was associated with about two-fold prolongation of survival in the Lmna−/− mice as compared to untreated Lmna−/− mice or Lmna−/− mice injected with the control AAV9-Gfp or AAV9-scrambledshRNA vector constructs (Figure 7A and Online Figure IV). Accordingly, the median survival time was increased from 29 days in the untreated Lmna−/− mice to 52 days in the AAV9-FoxoshRNA treated mice.
Figure 7. Phenotypic consequences of knock down of FOXO TFs in the Lmna−/− mouse hearts.
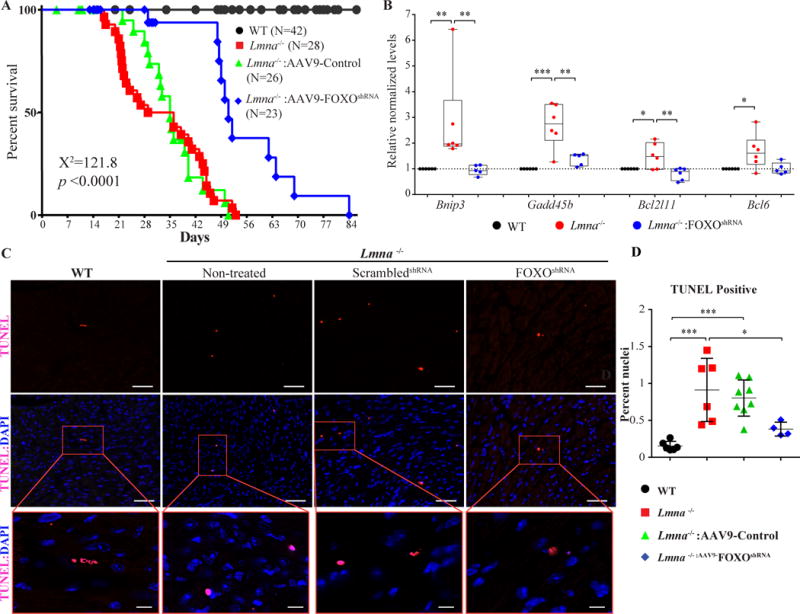
A. Kaplan-Meier survival plots of WT, Lmna−/− mice (not injected), Lmna−/− mice injected with AAV9-scrambledshRNA, or AAV9-Control (combined AAV9-scrambledshRNA and AAV9-Gfp) and Lmna−/− mice injected with the AAV9-FoxoshRNA constructs. The median survival time of Lmna−/− mice was increased from 29 days to 51 days upon administration of the AAV9-FoxoshRNA construct. The survival rates in the Lmna−/− mice injected with AAV9-Gfp or AAV9-scrambledshRNA constructs were similar to that in the (non-injected) Lmna−/− mice and are shown individually in Online FigureV. B. Transcript levels of selected markers of apoptosis, which are known to be FOXO TF targets, in the experimental groups. Transcript levels of BCL2 Interacting Protein 3 (Bnip3), Growth Arrest And DNA Damage Inducible Beta (Gadd45b), BCL2 Like 11 (Bcl2l11), and B-Cell CLL/Lymphoma 6 (Bcl6), quantified by RT-qPCR from mRNA extracted from 2-week old WT, Lmna−/− mice (not injected) and Lmna−/− mice injected with the AAV9-FoxoshRNA construct, were normalized in the treated group. C. Apoptosis assessments by TUNEL assay in the heart at 4 weeks of age. Representative TUNEL stained thin myocardial cross-section from 4-week old WT, Lmna−/− mice (not injected) and Lmna−/− mice injected either with AAV9-scrambledshRNA or the AAV9-FoxoshRNA constructs are shown. Upper panels show TUNEL staining in red and the middle panels show overlay of TUNEL (red) and DAPI-stained nuclei (blue) myocardial sections. The lower panels show magnification of selected areas displaying apoptotic cells. Bar is 50 μm in upper and middle panels, and 20 μm in the lower panels. Quantitative data of the TUNEL positive stained nuclei in the experimental groups are shown in panel D. The number of apoptotic cells was increased by 5.9 ± 1.13-fold in Lmna−/− mice (p=0.0008 vs WT); whereas the number in the AAV9-FoxoshRNA treated group was reduced by 58.2 ± 5.1 % (p=0.0247) compared to the non-treated Lmna−/− mice and tend to be decreased (p=0.071) compared to treated Lmna−/− mice. The number of apoptotic cells in the AAV9-FoxoshRNA treated Lmna−/− mice was not significant different from that in the WT mice (0.38 ± 0.05% vs. 0.15 ± 0.02%, respectively, p=0.55).
*: p<0.05, **: p<0.01, ***: p<0.001, ****: p<0.0001
Given marked activation of the apoptotic pathways in the Lmna−/− mouse hearts, effects of knock down of FOXO TFs on myocardial apoptosis were analyzed. Treatment with the AAV9-FoxoshRNA was associated with normalization of transcript levels of several genes involved in apoptosis, which are known to be FOXO TF targets (Figure 7B). Consistent with this finding, knock down of FOXO TFs in the heart was associated with a significant reduction in the number of TUNEL positive cells in the myocardium (58.2±18.6% p=0.025 vs non-injected Lmna−/− mice, Figure 7, panels C and D).
Administration of AAV9-FoxoshRNA did not have a significant effect on cardiac function (Online Table VI). However, because of the presence of significant differences in the body weight between the WT and Lmna−/− mice and body weight-dependence of the left ventricular dimensions, the effects on cardiac functions could not be reliably assessed. To determine whether knock down of FOXO TFs had an effect on mitochondrial functions, selected parameters of mitochondrial functions were determined in the experimental groups (Online Figure V). Integrity of the mitochondrial ETC complexes is reflected by the stable levels of known constituents of each complex, such as NDUFB8 for complex I and ATP5A for complex V. Therefore, levels of these labile protein constituents of mitochondrial ETC were quantified by immunblotting. As shown, only levels of NDUFB8 were reduced in the Lmna−/− mice (Online Figure V). Administration of AAV9-FOXOshRNA did not rescue reduced levels of NDUFB8 and had no effect on other labile protein constituents of ETC, indicating a mechanism independent of FOXO TFs (Online Figure V). Likewise, MitoSOX assay showed no significant effect on superoxide production in the neonatal mouse cardiac myocyte transduced with recombinant adenoviruses expressing constitutively active FOXO TFs (Online Figure V). Moreover, no significant differences in the percent CVF, indicative of interstitial fibrosis, were noted among the experimental groups (Online Figure VI). Finally, myocardial architecture, as assessed by staining for ACTN2 and JPH2 were not affected in the Lmna−/− mice treated with adenoviral vector alone and following knock down of FOXO TFs in the heart (Online Figure VII).
Discussion
We identified about 800 differentially expressed transcripts in the hearts of Lmna−/− mice, which occurred in the absence of and prior to the onset of cardiac dysfunction. Dysregulated gene expression is in accord with the known role of LMNA in influencing chromatin structure through the LADs and regulation of gene expression.17, 18, 20, 21 Most of the dysregulated transcripts were upregulated, which is consistent with the data suggesting that most genes at the LADs are transcriptionally silent or suppressed.17, 19, 20 Because the changes occurred in the absence of and preceded discernible cardiac dysfunction, they are likely pathogenic and partly responsible for the cardiac phenotype, as opposed to being secondary to cardiac dysfunction. Bioinformatics analyses complemented with biological validations identified FOXO TFs as among the most activated transcriptional regulators in LMNA-deficient hearts. Knock down of FOXO1 and FOXO3 using recombinant AAV9 expressing an established anti-FOXO1 and 3 shRNA indicated a pathogenic role for the FOXO TFs in induction of apoptosis and increased mortality in the Lmna−/− mice. Overall, these findings implicate FOXO TFs as an important transcriptional regulator in the pathogenesis of the cardiac phenotypes in laminopathies.
RNA-Seq was performed on ribosome-depleted whole heart transcriptomes. Given the ubiquitous expression of LMNA in multiple cardiac cell types, DETs are expected to originate from multiple cell types. Likewise, cardiac phenotypes in laminopathies are expected to arise from pathogenic involvement of multiple cell types. In the present study, upregulation of FOXO TF targets were verified in the heart as well as in cardiac myocytes isolated from the Lmna−/− mouse hearts. In addition, partial rescue of cardiac phenotype upon knockdown of FOXO TFs using recombinant AAV9, which predominantly target cardiac myocytes in the heart54, 55, signifies the biological effects of activation of FOXO TFs in cardiac myocytes in lamin-deficiency. Moreover, doubling of survival and marked reduction in apoptosis upon targeting of FOXO TFs in cardiac myocytes, emphasize the biological significance of this regulatory network in cardiac myocytes in laminopathies. The findings also point to the complexity of pathways involved in the pathogenesis of cardiac phenotypes in laminopathies. Hence, targeting of multiple pathways are expected to be required for therapeutic and preventive gains in this complex disorder.
Several measures were built into the study design in order to identify the primary transcriptomic changes and reduce the chance of fortuitous findings and the confounding factors. Transcriptomic changes were defined prior to and in the absence of discernible cardiac dysfunction, which enabled identification of dysregulated pathogenic pathways in the heart in laminopathies. Likewise, reversal of these molecular events along with partial phenotypic rescue support the pathogenic role of FOXO TFs in induction of cardiac phenotype in laminopathies. To reduce the possibility of fortuitous effects, two control viral vectors, one expressing a scrambled shRNA and the other expressing the GFP protein but not an shRNA were included as controls. To increase the transduction efficiency and hence garner robust data, recombinant AAV9 vectors were delivered in three sequential injections, with the last injection at P6. This approach is expected to transduce the newly formed cardiac myocytes, which occurs in the early post-natal days.47 In addition, sequential injections during the early post-natal days was designed to avoid potential neutralizing effects of the antibodies against AAV9 capsids. Overall, the design of the study enabled garnering less confounded data.
The study has a number of shortcomings. The mechanism(s) responsible for doubling of the survival upon knock down of FOXO TFs is unclear. Whereas the number of apoptotic cells in the myocardium was significantly reduced, cardiac function, as assessed by echocardiography, was not improved. The presence of significant differences in body weights, major determinants of echocardiographic indices of cardiac size and function, among the experimental groups, hindered robust conclusions on the effects on cardiac function. In addition to heart failure, cardiac arrhythmias are also important causes of death in patients with laminopathies.4, 5 Telemetric cardiac rhythm monitoring and electrophysiological studies were not performed in these mice, partly because of the small size of the Lmna−/− mice, to determine contributions of cardiac arrhythmias to premature death and improved survival in the experimental groups. It is also noteworthy that AAV9 constructs, despite their well-established tropism for cardiac myocytes,54, 55 also transduce skeletal muscles, including the diaphragm, and to a lesser extent the liver.56, 57 Consequently, the survival benefit observed in the Lmna−/− mice upon administration of AAV9-FoxoshRNA might be due to the effects of suppression of FOXO TF in other organs, such as the striated respiratory muscles, prevention of muscle atrophy, and the resultant improvement in respiratory efficiency, as shown in other models.58, 59
The observed beneficial effects of suppression of FOXO TFs on survival and apoptosis are in agreement with the previous data showing deleterious effects of activation of FOXO3 in the heart but in apparent discord with its cardioprotective role, reported in the knock out studies.60, 61 The key distinction, as opposed to the knock out studies, is that FOXO TFs were only partially suppressed and not totally abolished in our studies. While excessive suppression, such as in genetic knock out studies, as well as excessive over-expression of FOXO TFs in the heart are expected to be deleterious, partial suppression of the already activated FOXO TFs is likely to beneficial.62
The mechanistic basis of DETs and activation of FOXO TFs in the heart is unclear. Whether changes in the transcriptomics are the direct consequences of dysregulated LADs resulting from the deficiency of LMNA or are the consequences of impaired assembly of the other chromatin components to the LADs remain to be determined. Likewise, specific LADs in cardiac myocytes that are dysregulated in the absence of LMNA have yet-to-be identified. Therefore, direct data to show enrichment of LMNA and likewise, enrichment of the FOXO TFs on the promoter regions of their targets could not be demonstrated, due to an inadequate quality of the available anti LMNA and anti FOXO antibodies for chromatin immunoprecipitation studies. Nevertheless, activation of FOXO1 and FOXO3 was confirmed at multiple levels, including their nuclear localization and reduced FOXO3 phosphorylation, as well as increased transcript levels of its target genes. Moreover, specific targeting of FOXO TFs by a shRNA, whose effectiveness has been shown in independent studies45, 46, in cardiac myocytes using AAV9 constructs partially rescued the molecular (gene expression), biological (apoptosis), and clinical (mortality) phenotypes. Collectively, these findings lend further support to the pathogenic activation of FOXO TFs in induction of cardiac phenotypes in laminopathies.
Analysis of RNA-Seq data shows activation of FOXO TFs in human heart failure with undefined mutations. Although preliminary and requiring testing for replication in additional samples and validation by complementary techniques, activation of FOXO TFs might be a general feature of heart failure and not specific to laminopathies. Consequently, one might speculate that FOXO TFs might be therapeutic targets in garden variety form of heart failure.
In conclusion, the findings implicate LMNA in the transcriptional regulation of several hundred genes and over a dozen upstream transcriptional regulators, including FOXO TFs in the heart. Dysregulation of these networks of TFs collectively, and FOXO1 and FOXO3 particularly, are in part responsible for induction of cardiac phenotype and early mortality in laminopathies. The findings identify FOXO TFs as potential targets to prevent or attenuate cardiac phenotype in laminopathies.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Mutations in LMNA gene cause over a dozen distinct phenotypes, including dilated cardiomyopathy (DCM).
LMNA is among the common causal genes for hereditary DCM.
The molecular pathogenesis of DCM caused by the LMNA mutations is largely unknown and seems to involve a number of signaling pathways, including MAPK, NOTCH and Canonical Wnt.
Genetic deletion of the Lmna gene in mice leads to several cardiac dysfunction and premature death by 4-8 weeks of age.
What New Information Does This Article Contribute?
We sequenced ribosome-depleted whole heart transcripts (RNA-Seq) in 2-week old Lmna−/− mice, prior to the onset of cardiac dysfunction, and their wild type littermates.
We identified about 1,000 differentially expressed transcripts (DETs), the majority of which were upregulated in the Lmna−/− hearts.
Network analysis of DETs led to identification of over two-dozen deranged upstream regulators of gene expression.
FOXO transcription factors (TFs) were among the most activated upstream transcriptional regulators, which was validated by multiple methods.
Knock down of FOXO1 and FOXO3 in the heart using recombinant adeno-associated viruses (AAV9), expressing a sequence-specific shRNA, attenuated apoptosis and nearly doubled the survival rate in the Lmna−/− mice.
The findings of the present study point to multiplicity of the pathogenic pathways dysregulated in the heart in LMNA deficiency, including activation of FOXO 1 and 3 TFs. The findings also identify FOXO 1 and 3 as potential therapeutic targets in DCM caused by the LMNA mutations. Furthermore, the findings highlight the shortcomings of targeting of a single pathogenic pathway in fully rescuing the phenotype. Therefore, targeting of multiple pathogenic pathways might be required for superior therapeutic achievements in heart failure caused by the LMNA mutations.
Acknowledgments
SOURCES OF FUNDING
This work was supported in part by grants from NIH, National Heart, Lung and Blood Institute (NHLBI, R01 HL088498 and 1R01HL132401), Leducq Foundation (14 CVD 03), George and Mary Josephine Hamman Foundation, American Heart Association Beginning Grant in Aid (15BGIA25080008 to RL). XHTW was supported by grants from NIH (R01-HL089598, R01-HL091947, R01-HL117641, and R41-HL129570) and American Heart Association (13EIA14560061).
Nonstandard Abbreviations and Acronyms
- ACTN2
Actinin alpha 2
- AAV9
Adeno-associated virus serotype 9
- DCM
Dilated cardiomyopathy
- DETs
Differentially expressed transcripts
- E2Fs
E2 factors
- FOXO
Forkhead box O
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GO
Gene ontology
- GSEA
Gene set enrichment analysis
- IPA
Ingenuity pathway analysis
- LADs
LMNA-associated domains
- LMNA
Lamin A/C
- MSigDB
Molecular signature database
- RNA-Seq
RNA-sequencing
- shRNA
Short hairpin RNA
- TF
Transcription factor
- TUB1A1
Tubulin α 1a
- TTN
Titin
- WT
Wild Type
Footnotes
DISCLOSURE
None.
This manuscript was sent to Roger J. Hajjar, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
In December 2017, the average time from submission to first decision for all original research papers submitted to Circulation Research was 13.60 days.
References
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Statistics C, Stroke Statistics S Heart disease and stroke statistics-2017 update: A report from the american heart association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Pina IL, Trogdon JG, American Heart Association Advocacy Coordinating C, Council on Arteriosclerosis T, Vascular B, Council on Cardiovascular R, Intervention, Council on Clinical C, Council on E, Prevention, Stroke C Forecasting the impact of heart failure in the united states: A policy statement from the american heart association. Circulation. Heart failure. 2013;6:606–619. doi: 10.1161/HHF.0b013e318291329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McNally EM, Mestroni Luisa. Dilated cardiomyopathy: Genetic determinants and mechanisms. Circulation research. 2017 doi: 10.1161/CIRCRESAHA.116.309396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin a/c gene as causes of dilated cardiomyopathy and conduction-system disease. The New England journal of medicine. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 5.Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, Boucek MM, Lascor J, Moss AC, Li WL, Stetler GL, Muntoni F, Bristow MR, Mestroni L, Familial Dilated Cardiomyopathy Registry Research G Natural history of dilated cardiomyopathy due to lamin a/c gene mutations. Journal of the American College of Cardiology. 2003;41:771–780. doi: 10.1016/s0735-1097(02)02954-6. [DOI] [PubMed] [Google Scholar]
- 6.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. The New England journal of medicine. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schreiber KH, Kennedy BK. When lamins go bad: Nuclear structure and disease. Cell. 2013;152:1365–1375. doi: 10.1016/j.cell.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quarta G, Syrris P, Ashworth M, Jenkins S, Zuborne Alapi K, Morgan J, Muir A, Pantazis A, McKenna WJ, Elliott PM. Mutations in the lamin a/c gene mimic arrhythmogenic right ventricular cardiomyopathy. European heart journal. 2012;33:1128–1136. doi: 10.1093/eurheartj/ehr451. [DOI] [PubMed] [Google Scholar]
- 9.Liang JJ, Grogan M, Ackerman MJ. Lmna-mediated arrhythmogenic right ventricular cardiomyopathy and charcot-marie-tooth type 2b1: A patient-discovered unifying diagnosis. Journal of cardiovascular electrophysiology. 2016;27:868–871. doi: 10.1111/jce.12984. [DOI] [PubMed] [Google Scholar]
- 10.Rober RA, Weber K, Osborn M. Differential timing of nuclear lamin a/c expression in the various organs of the mouse embryo and the young animal: A developmental study. Development. 1989;105:365–378. doi: 10.1242/dev.105.2.365. [DOI] [PubMed] [Google Scholar]
- 11.Anselme F, Moubarak G, Savoure A, Godin B, Borz B, Drouin-Garraud V, Gay A. Implantable cardioverter-defibrillators in lamin a/c mutation carriers with cardiac conduction disorders. Heart rhythm : the official journal of the Heart Rhythm Society. 2013;10:1492–1498. doi: 10.1016/j.hrthm.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 12.van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbuchel H, de Visser M, Crijns HJ, Pinto YM. Meta-analysis of clinical characteristics of 299 carriers of lmna gene mutations: Do lamin a/c mutations portend a high risk of sudden death? Journal of molecular medicine. 2005;83:79–83. doi: 10.1007/s00109-004-0589-1. [DOI] [PubMed] [Google Scholar]
- 13.Carmosino M, Torretta S, Procino G, Gerbino A, Forleo C, Favale S, Svelto M. Role of nuclear lamin a/c in cardiomyocyte functions. Biol Cell. 2014;106:346–358. doi: 10.1111/boc.201400033. [DOI] [PubMed] [Google Scholar]
- 14.Hutchison CJ. Lamins: Building blocks or regulators of gene expression? Nat Rev Mol Cell Biol. 2002;3:848–858. doi: 10.1038/nrm950. [DOI] [PubMed] [Google Scholar]
- 15.Stroud MJ, Banerjee I, Veevers J, Chen J. Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circulation research. 2014;114:538–548. doi: 10.1161/CIRCRESAHA.114.301236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brayson D, Shanahan CM. Current insights into lmna cardiomyopathies: Existing models and missing lincs. Nucleus. 2017;8:17–33. doi: 10.1080/19491034.2016.1260798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paulsen J, Sekelja M, Oldenburg AR, Barateau A, Briand N, Delbarre E, Shah A, Sorensen AL, Vigouroux C, Buendia B, Collas P. Chrom3d: Three-dimensional genome modeling from hi-c and nuclear lamin-genome contacts. Genome Biol. 2017;18:21. doi: 10.1186/s13059-016-1146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lund EG, Duband-Goulet I, Oldenburg A, Buendia B, Collas P. Distinct features of lamin a-interacting chromatin domains mapped by chip-sequencing from sonicated or micrococcal nuclease-digested chromatin. Nucleus. 2015;6:30–39. doi: 10.4161/19491034.2014.990855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- 20.van Steensel B, Belmont AS. Lamina-associated domains: Links with chromosome architecture, heterochromatin, and gene repression. Cell. 2017;169:780–791. doi: 10.1016/j.cell.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gesson K, Rescheneder P, Skoruppa MP, von Haeseler A, Dechat T, Foisner R. A-type lamins bind both hetero- and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res. 2016;26:462–473. doi: 10.1101/gr.196220.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perovanovic J, Dell’Orso S, Gnochi VF, Jaiswal JK, Sartorelli V, Vigouroux C, Mamchaoui K, Mouly V, Bonne G, Hoffman EP. Laminopathies disrupt epigenomic developmental programs and cell fate. Science translational medicine. 2016;8:335ra358. doi: 10.1126/scitranslmed.aad4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of a-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. The Journal of cell biology. 1999;147:913–920. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, Rainer S, Stewart CL, Martin D, Feneley MP, Fatkin D. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin a/c-deficient mice. The Journal of clinical investigation. 2004;113:357–369. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gurha P, Chen X, Lombardi R, Willerson JT, Marian AJ. Knockdown of plakophilin 2 downregulates mir-184 through cpg hypermethylation and suppression of the e2f1 pathway and leads to enhanced adipogenesis in vitro. Circulation research. 2016;119:731–750. doi: 10.1161/CIRCRESAHA.116.308422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lombardi R, Chen SN, Ruggiero A, Gurha P, Czernuszewicz GZ, Willerson JT, Marian AJ. Cardiac fibro-adipocyte progenitors express desmosome proteins and preferentially differentiate to adipocytes upon deletion of the desmoplakin gene. Circulation research. 2016;119:41–54. doi: 10.1161/CIRCRESAHA.115.308136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruggiero A, Chen SN, Lombardi R, Rodriguez G, Marian AJ. Pathogenesis of hypertrophic cardiomyopathy caused by myozenin 2 mutations is independent of calcineurin activity. Cardiovascular research. 2013;97:44–54. doi: 10.1093/cvr/cvs294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen SN, Gurha P, Lombardi R, Ruggiero A, Willerson JT, Marian AJ. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circulation research. 2014;114:454–468. doi: 10.1161/CIRCRESAHA.114.302810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karmouch J, Zhou QQ, Miyake CY, Lombardi R, Kretzschmar K, Bannier-Helaouet M, Clevers H, Wehrens XHT, Willerson JT, Marian AJ. Distinct cellular basis for early cardiac arrhythmias, the cardinal manifestation of arrhythmogenic cardiomyopathy, and the skin phenotype of cardiocutaneous syndromes. Circulation research. 2017;121:1346–1359. doi: 10.1161/CIRCRESAHA.117.311876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marian AJ, Zhao G, Seta Y, Roberts R, Yu QT. Expression of a mutant (arg92gln) human cardiac troponin t, known to cause hypertrophic cardiomyopathy, impairs adult cardiac myocyte contractility. Circulation research. 1997;81:76–85. doi: 10.1161/01.res.81.1.76. [DOI] [PubMed] [Google Scholar]
- 31.Marian AJ, Yu QT, Mann DL, Graham FL, Roberts R. Expression of a mutation causing hypertrophic cardiomyopathy disrupts sarcomere assembly in adult feline cardiac myocytes. Circulation research. 1995;77:98–106. doi: 10.1161/01.res.77.1.98. [DOI] [PubMed] [Google Scholar]
- 32.Tsybouleva N, Zhang L, Chen S, Patel R, Lutucuta S, Nemoto S, DeFreitas G, Entman M, Carabello BA, Roberts R, Marian AJ. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation. 2004;109:1284–1291. doi: 10.1161/01.CIR.0000121426.43044.2B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ. Suppression of canonical wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. The Journal of clinical investigation. 2006;116:2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lombardi R, Bell A, Senthil V, Sidhu J, Noseda M, Roberts R, Marian AJ. Differential interactions of thin filament proteins in two cardiac troponin t mouse models of hypertrophic and dilated cardiomyopathies. Cardiovascular research. 2008;79:109–117. doi: 10.1093/cvr/cvn078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. Tophat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by rna-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ utrs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (msigdb) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (msigdb) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. Consensuspathdb: Toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39:D712–717. doi: 10.1093/nar/gkq1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamburov A, Stelzl U, Lehrach H, Herwig R. The consensuspathdb interaction database: 2013 update. Nucleic Acids Res. 2013;41:D793–800. doi: 10.1093/nar/gks1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herwig R, Hardt C, Lienhard M, Kamburov A. Analyzing and interpreting genome data at the network level with consensuspathdb. Nat Protoc. 2016;11:1889–1907. doi: 10.1038/nprot.2016.117. [DOI] [PubMed] [Google Scholar]
- 44.Supek F, Bosnjak M, Skunca N, Smuc T. Revigo summarizes and visualizes long lists of gene ontology terms. PloS one. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hribal ML, Nakae J, Kitamura T, Shutter JR, Accili D. Regulation of insulin-like growth factor-dependent myoblast differentiation by foxo forkhead transcription factors. The Journal of cell biology. 2003;162:535–541. doi: 10.1083/jcb.200212107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Renault VM, Thekkat PU, Hoang KL, White JL, Brady CA, Kenzelmann Broz D, Venturelli OS, Johnson TM, Oskoui PR, Xuan Z, Santo EE, Zhang MQ, Vogel H, Attardi LD, Brunet A. The pro-longevity gene foxo3 is a direct target of the p53 tumor suppressor. Oncogene. 2011;30:3207–3221. doi: 10.1038/onc.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alkass K, Panula J, Westman M, Wu TD, Guerquin-Kern JL, Bergmann O. No evidence for cardiomyocyte number expansion in preadolescent mice. Cell. 2015;163:1026–1036. doi: 10.1016/j.cell.2015.10.035. [DOI] [PubMed] [Google Scholar]
- 48.Schobesberger S, Wright P, Tokar S, Bhargava A, Mansfield C, Glukhov AV, Poulet C, Buzuk A, Monszpart A, Sikkel M, Harding SE, Nikolaev VO, Lyon AR, Gorelik J. T-tubule remodelling disturbs localized beta2-adrenergic signalling in rat ventricular myocytes during the progression of heart failure. Cardiovascular research. 2017;113:770–782. doi: 10.1093/cvr/cvx074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo Y, VanDusen NJ, Zhang L, Gu W, Sethi I, Guatimosim S, Ma Q, Jardin BD, Ai Y, Zhang D, Chen B, Guo A, Yuan GC, Song LS, Pu WT. Analysis of cardiac myocyte maturation using casaav, a platform for rapid dissection of cardiac myocyte gene function in vivo. Circulation research. 2017 doi: 10.1161/CIRCRESAHA.116.310283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reynolds JO, Quick AP, Wang Q, Beavers DL, Philippen LE, Showell J, Barreto-Torres G, Thuerauf DJ, Doroudgar S, Glembotski CC, Wehrens XH. Junctophilin-2 gene therapy rescues heart failure by normalizing ryr2-mediated ca2+ release. International journal of cardiology. 2016;225:371–380. doi: 10.1016/j.ijcard.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Donti TR, Stromberger C, Ge M, Eldin KW, Craigen WJ, Graham BH. Screen for abnormal mitochondrial phenotypes in mouse embryonic stem cells identifies a model for succinyl-coa ligase deficiency and mtdna depletion. Disease models & mechanisms. 2014;7:271–280. doi: 10.1242/dmm.013466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liao CY, Anderson SS, Chicoine NH, Mayfield JR, Academia EC, Wilson JA, Pongkietisak C, Thompson MA, Lagmay EP, Miller DM, Hsu YM, McCormick MA, O’Leary MN, Kennedy BK. Rapamycin reverses metabolic deficits in lamin a/c-deficient mice. Cell Rep. 2016;17:2542–2552. doi: 10.1016/j.celrep.2016.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calnan DR, Brunet A. The foxo code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 54.Yang L, Jiang J, Drouin LM, Agbandje-McKenna M, Chen C, Qiao C, Pu D, Hu X, Wang DZ, Li J, Xiao X. A myocardium tropic adeno-associated virus (aav) evolved by DNA shuffling and in vivo selection. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3946–3951. doi: 10.1073/pnas.0813207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prasad KM, Xu Y, Yang Z, Acton ST, French BA. Robust cardiomyocyte-specific gene expression following systemic injection of aav: In vivo gene delivery follows a poisson distribution. Gene therapy. 2011;18:43–52. doi: 10.1038/gt.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reay DP, Niizawa GA, Watchko JF, Daood M, Reay JC, Raggi E, Clemens PR. Effect of nuclear factor kappab inhibition on serotype 9 adeno-associated viral (aav9) minidystrophin gene transfer to the mdx mouse. Mol Med. 2012;18:466–476. doi: 10.2119/molmed.2011.00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Falk DJ, Mah CS, Soustek MS, Lee KZ, Elmallah MK, Cloutier DA, Fuller DD, Byrne BJ. Intrapleural administration of aav9 improves neural and cardiorespiratory function in pompe disease. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:1661–1667. doi: 10.1038/mt.2013.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smuder AJ, Sollanek KJ, Min K, Nelson WB, Powers SK. Inhibition of forkhead boxo-specific transcription prevents mechanical ventilation-induced diaphragm dysfunction. Crit Care Med. 2015;43:e133–142. doi: 10.1097/CCM.0000000000000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, Blaauw B, DePinho RA, Sandri M. Regulation of autophagy and the ubiquitin-proteasome system by the foxo transcriptional network during muscle atrophy. Nature communications. 2015;6:6670. doi: 10.1038/ncomms7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cao DJ, Jiang N, Blagg A, Johnstone JL, Gondalia R, Oh M, Luo X, Yang KC, Shelton JM, Rothermel BA, Gillette TG, Dorn GW, Hill JA. Mechanical unloading activates foxo3 to trigger bnip3-dependent cardiomyocyte atrophy. J Am Heart Assoc. 2013;2:e000016. doi: 10.1161/JAHA.113.000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sengupta A, Molkentin JD, Paik JH, DePinho RA, Yutzey KE. Foxo transcription factors promote cardiomyocyte survival upon induction of oxidative stress. The Journal of biological chemistry. 2011;286:7468–7478. doi: 10.1074/jbc.M110.179242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blice-Baum AC, Zambon AC, Kaushik G, Viswanathan MC, Engler AJ, Bodmer R, Cammarato A. Modest overexpression of foxo maintains cardiac proteostasis and ameliorates age-associated functional decline. Aging cell. 2017;16:93–103. doi: 10.1111/acel.12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.