

Abstract
Development of an effective treatment against advanced tumors remains a major challenge for cancer immunotherapy. Approximately 50% of human melanoma is driven by B-Raf proto-oncogene mutation (BRAF-mutant). Tumors with such mutation are desmoplastic, highly immunosuppressive, and often resistant to immune checkpoint therapies. We have shown that immunotherapy mediated by low dose doxorubicin-induced immunogenic cell death was only partially effective for this type of tumor, and not effective in long-term inhibition of tumor progression. Wnt family member 5A (Wnt5a), a signaling protein highly-produced by BRAF-mutant melanoma cells, has been implicated in inducing dendritic cell tolerance and tumor fibrosis, thus hindering effective antigen presentation and T cell infiltration. We hypothesized that Wnt5a is a key molecule controlling the immunosuppressive tumor microenvironment in metastatic melanoma. Accordingly, we have designed and generated a trimeric trap protein, containing the extracellular domain of Fizzled 7 receptor that binds Wnt5a with a Kd ~278 nM. Plasmid DNA encoding for the Wnt5a trap was delivered to the tumor by using cationic lipid-protamine-DNA (LPD) nanoparticles. Expression of Wnt5a trap in the tumor, although transient, was greater than in that of any other major organs including liver, resulting in a significant reduction of the Wnt5a level in the tumor microenvironment without systematic toxicity. Significantly, combination of Wnt5a trapping and low dose doxorubicin showed great tumor growth inhibition and host survival prolongation. Our findings indicated that efficient local Wnt5a trapping significantly remodeled the immunosuppressive tumor microenvironment to facilitate immunogenic cell death-mediated immunotherapy.
Keywords: B-Raf proto-oncogene-mutant melanoma, Wnt family member 5A, immunogenic cell death, immune-trap, nanoparticle, tumor microenvironment
Graphical Table of Contents
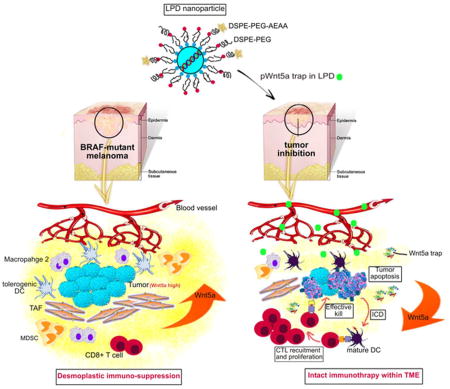
Wnt5a, Wnt family member 5A; BRAF, B-Raf proto-oncogene; LPD, lipid-protamine-DNA; DC, dendritic cell; TAF, tumor associated fibroblast; MDSC, myeloid-derived suppressor cell; ICD, immunogenic cell death; CTL, cytotoxic T lymphocyte; TME, tumor microenvironment.
Melanoma derived from melanocytes is the most aggressive genre of skin cancer.1 Among which, approximately 50% is driven by the B-Raf proto-oncogene mutations. These mutations sustainably activate the mitogen-activated protein-kinase (MAPK) kinase (MEK) - extracellular signal regulated kinase (ERK) - MAP kinase pathway, leading to uncontrolled tumor cell proliferation and invasiveness.2 Despite considerable advancement in the study of cancer biology and drug discovery, it is still a major undertaking to cure most advanced melanoma patients. Even with conventional anticancer chemotherapy, the first line response rate remains low.3, 4
Based on clinical evidence of chemotherapy, the definition of immunogenic cell death (ICD) was derived, revealing how tumor-specific immune responses shape the therapeutic outcome.5 ICD refers to the apoptosis of tumor cell that does release tumor associated antigens, and sequentially stimulate a specific immune response against such antigens. The ICD, in turn, will improve the immunogenic potential as a form of dendritic cell (DC) vaccines. Therapeutic vaccination can be clinically successful as a monotherapy; however, in BRAF-mutant melanoma, the desmoplastic immunosuppressive tumor microenvironment (TME) placed a major hurdle for treatment efficacy.6, 7 Herein, ICD-mediated vaccination should be combined with a co-treatment that overcomes immune evasion in order to achieve desired therapeutic efficacy.
It has been reported that in advanced melanoma patients, expression of the soluble Wnt family member 5A (Wnt5a) ligand induces a paracrine signaling pathway that drives local DC tolerization and fibrotic TME.8, 9 Interestingly, melanoma-derived Wnt5a promotes tumor growth and metastasis, which correlates with the inhibition of antitumor adaptive T-cell responses.10 We hypothesize that compared with wild-type tumor, BRAF-mutant melanoma correlates with increased Wnt5a release. Furthermore, Wnt5a represents a critical mediator of tumor immune evasion and immunotherapy resistance, and the inhibition of this soluble mediator will augment the efficacy of vaccination.
In this work, we designed and generated a fusion protein that specifically and potently binds to and disrupts the biological functions of Wnt5a. For the purpose of local TME ‘trapping’, plasmid containing the Wnt5a trap cDNA was formulated and specifically delivered to the BRAF-mutant melanoma in a syngeneic host by using a cationic nanocarrier, the lipid-protamine-DNA (LPD) nanoparticles (see Graphical Table of Contents). We found that the administration of the trap along with immunogenic cell death-mediated vaccination generated robust innate and adaptive immune responses, resulting in significant tumor regression in the murine model. Moreover, this combination therapy delayed tumor metastasis and improved long-term survival, providing a strong rationale for pursuing this strategy in clinical studies.
RESULTS AND DISCUSSION
Immunogenic cell death induced by low dose Doxorubicin (DOX)
For the purpose of effective cytotoxic T cell killing, we introduced low dose DOX to induce immunogenic cell death within the tumor, thus aiding in activating immune microenvironment. DOX is a small molecule drug that has been used as a first line chemotherapy for the treatment of melanoma. It has been reported that low dose of Dox induced ICD in various cancer cell lines including wild-type melanoma (B16F10).11 To determine the best dosing strategy of DOX-induced ICD, MTT assay was performed (Figure 1A). Followed by calreticulin (CRT) exposure and high mobility group box 1 (HMGB1) release as standard markers for drug-induced (0.2 mM DOX) tumor cell immunogenicity,12 ICD was detected on BRAF-mutant BPD6 cell line in vitro (Figure 1B).
Figure 1. ICD induced by low dose DOX.

(A) MTT result on BPD6 cell line in vitro. n = 5. (B) Fluorescence imaging detecting ICD markers: CRT and HMGB1 on BPD6 cells treated with low dose of DOX. Cell nuclei were stained with Hoechst 33432. Scale bar indicates 10 µm. (C) ELISpot test depicting IFN-γ secreted by re-stimulated splenocytes of mice treated with or without low dose DOX. n = 3. (D) Flow cytometry analysis shows ICD induced increase in: intra-tumoral inflammatory cells (CD8+CD45+) and activation of CD8+ T cell within TME. n = 3. (E) HMGB1 and CRT immunofluorescence staining in tumors slide sections, treated with or without low dose of DOX. n = 3. Scale bar indicates 300 µm. (F) Mean days of mouse survival in PBS and low dose DOX treated groups. n = 8–10. Data present mean ± SE. n.s.: p > 0.05, **: p < 0. 01.
Low dose DOX efficiently induced IFN-γ production under BRAF-mutant antigen re-stimulation in vitro, analyzed by ELISpot assay, indicating strong systemic immune response against the tumor-specific antigen mimicking in vivo settings (Figure 1C). Thus, low dose DOX (0.544 mg/kg) was given i.p. to mice, at an early point of desmoplastic tumor growth, as to induce ICD and the release of tumor antigens for effective DC presentation and cytotoxic T cell recruitment. After DOX treatment, we found induced intra-tumoral inflammatory cells and activated CD8+ T cell greatly increased within whole tumor (Figure 1D). DOX therapy significantly increased HMGB1 and CRT in vivo, as stained in tumor sections (Figure 1E), which further confirmed ICD production in vivo, and demonstrated an effective way of boosting systemic immunity. Although the DOX treatment showed modest tumor growth inhibition, we found no significant prolongation of host survival, necessitating further investigation into remodeling the immunosuppressive TME.
Wnt5a is a key molecule controlling the immunosuppressive desmoplastic TME
Wnt5a is a member of the Wnt family that plays pivotal roles in activating several noncanonical Wnt signaling pathways. In normal tissues, such pathways mainly regulate major developmental processes, including stem cell self-renewal, proliferation, differentiation, and polarity.13 It has been reported that homozygous Wnt5a-ko-mice died postnatally, associated with significant lower dermal lymphatics and multiple defects including skeletal and internal organs.14 Wnt5a-defient mice resulted in reduction of Wnt/β-catenin signaling, which would impair osteoblast differentiation and enhance adipocyte differentiation.15, 16 Moreover, the abnormal activation or inhibition of Wnt5a signaling has been demonstrated in controlling tumor progression, more specifically, in an immunosuppressive manner.16, 17 In melanoma, previous studies have demonstrated that Wnt5a promotes differentiation of monocytes into tolerogenic DCs, thus hindering antigen presentation and effective T-cell mediated killing.18 Furthermore, tolerogenic DCs mediate Treg cell differentiation in the presence of TGF-β.19 More recently, Wnt/TGF-β signaling pathway has been further investigated, in the present of upregulating Yes-Associated Protein 1 (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) signaling, thus directly linked to physiological morphology of fibrosis (Figure 2A).9
Figure 2. Wnt5a is a key molecule controlling the immunosuppressive desmoplastic TME.

(A) Figure legend depicting Wnt5a functions on both DCs and Fibroblasts within TME. Wnt5a is highly expressed among BRAF-mutant melanoma, compared to BRAF-wild-type, in both clinical (panel B, TCGA database, n = 368) and murine samples (panel C, Western blot, n = 3). (D) High level of Wnt5a correlates with poorer patient overall survival. n = 29. (E) Masson’s trichrome staining illustrating BRAF-mutant melanoma with desmoplastic collagen-rich TME (black arrows), compared to wild-type in both human and mouse specimens. Scale bar indicates 300 µm. n = 3. *: p < 0.05, **: p < 0. 01.
We first confirmed that high expression level of Wnt5a was present in melanoma tissue of both patients and murine models (Figure 2B and C), with significantly difference (p = 0.0097 from TCGA analysis) between BRAF-mutant and wild-type tumors. Western blot analysis further confirmed this difference in murine melanoma cell lines, i.e. B16F10 (BRAF-wild type): BPD6 (BRAF-mutant). In melanoma patients, excessive Wnt/β-catenin level has been associated with poorer response to immune checkpoint therapies.20 Our analysis based on the TCGA database further confirmed a strong correlation (p = 0.005) between higher Wnt5a expression with shorter patient overall survival (Figure 2D and Figure S1).
In this work, we used the BPD6 murine BRAF-mutant (BRAFV600E, PTEN−/−) model that highly resembles aggressive clinical melanoma for therapeutic studies.21 Compard to the wildtype, BRAF-mutant melanoma demonstrated a desmoplastic collagen-rich TME in both murine and human specimens (Figure 2E). Since Wnt5a plays an important role in mediating immunosuppressive desmoplastic morphology, we hypothesized that effective local and transient inhibition of Wnt5a would remodel the suppressive TME and facilitate immunotherapy without systemic disruption of the multifaceted roles of Wnt5a in normal organ functions.
Local distribution and transient expression of Wnt5a trap
To efficiently compete with endogenous Wnt5a receptors and ‘trap’ Wnt5a locally within TME, we developed a trimeric trap that binds to mouse Wnt5a with a Kd in the range of medium nanomolars (Figure 3B). Wnt proteins are secreted glycoproteins that bind to the N-terminal extracellular cysteine-rich domain (CRD) of the Frizzled (FZD) receptor family. Quantitative measurement of the interactions between different isoforms of Wnt ligand and FZD receptor is challenging due to the lipid modification of Wnt that makes expression, purification and crystallization of active Wnt ligands difficult. The problem is further complicated by the significant promiscuity where a certain FZD receptor binds to multiple Wnt ligands.
Figure 3. Local distribution and expression of Wnt5a trap.

(A and B) Development and characterization of Wnt5a Trap protein. (A) SDS-PAGE of the Wnt5a trap in the presence (lane 2) and absence (lane 1) of reducing agent DTT. (B) The binding affinity between Wnt5a and FZD7-based trap measured by MST. (C) Bio-distribution of DiI-loaded LPD NPs among tumor and organs. n = 3. (D) Expressions of His-tagged Wnt5a trap in different organs were quantified by ELISA and compared with the injection of free trap protein. n = 5. (E) Fluorescent imaging depicting effective local Wnt5a trapping in tumor slide sections. Numbers indicate Wnt5a expression (yellow). Scale bar indicates 300 µm. (F) Mean days of mouse survival. n = 8–10. Data present mean ± SE. n.s.: p > 0.05, **: p < 0. 01.
We chose the CRD of FZD7 to develop a Wnt5a trap for the following considerations. First, FZD7 is one of the highest expressing FZD receptors in DCs, and tolerogenic DCs are the major immune cells we want to target in the work.22 Second, in the process of tumor progression, FZD7 is found most commonly upregulated among the whole FZD family. This finding is confirmed in various types of cancer, include colorectal cancer, triple negative breast cancer, etc.23 Third, FZD7 plays a vital role in the interaction between cancer stem cell and tumorigenesis.24 Therefore, an affinity molecule based on the CRD of FZD7 has the potential to serve as a decoy to trap Wnt5a while at the same time competitively reduce the numerous biological functions of this highly expressing FZD receptor. Since the oligomeric status of Wnt ligands Wnt5a was implicated in the literature, we designed a trimeric Wnt5a trap with multi-valency and avidity feature by genetically fusing the CRD of FZD7 with a robust trimerization domain from cartilage matrix protein (CMP-1) that is very abundant in mouse and human cartilage.25 The strong hydrophobic and ionic interactions among this trimerization domain result in a parallel, disulfide-linked, and rod-shaped trimeric structure with high stability.26, 27 Since the trimeric trap is formed through self-assembly of three identical monomers, it only requires a relatively small gene to encode the monomeric trap, making the gene to be delivered much shorter and easier to encapsulate.
To construct such an original Wnt5a trap, the optimized coding sequence for the monomeric trap was cloned into the expression vector pcDNA3.1, driven by a CMV promoter (Figure S2A). To facilitate trap secretion after expression, a strong signaling peptide from human serum albumin preproprotein was incorporated at the N-terminus, whereas an FLAG/His (6×) tag was also brought in at the C-terminus, thus to facilitate protein purification and in vivo expression analysis. The amino acid sequence of the monomeric version of Wnt5a trap was listed in Figure S2B. The recombinant Wnt5a trap was expressed in and purified from 293 T cells. The theoretic MW of the monomeric trap should be around 26 kDa but significant glycosylation is expected, as shown in Figure 3A with higher MW. We characterized the binding strength and specificity of the FZD7 CRD and Wnt5a. As shown in Figure 3B, the resulting FZD7-based trap should bind to Wnt5a with a Kd in the range of medium nanomolars.
The LPD NP formulation, which preferentially delivers macromolecules including plasmid DNA for tumor immunotherapy, has been well established in the Huang lab.28 To prepare LPD, plasmid DNA encoding for Wnt5a trap protein was condensed with cationic protamine to form a slightly anionic complex core. The core was further coated with the preformulated cationic liposomes (DOTAP, Cholesterol and DSPE-PEG), and modified with tumor targeting ligand DSPE-PEG-AEAA. The size (~100 nm), the spherical shape, as well as homogenous distribution of LPD NPs were confirmed by TEM images (Figure S2C), consistent with DLS size distribution (Figure S3). Through IVIS imaging, DiI-loaded NPs were found mainly distributed in the tumor 24 h after i.v. injection into mice (Figure 3C). Although liver may take up NPs, it was significantly lower than tumor (p < 0.01). Within the liver, NPs of size ~100 nm mainly internalized by Kupffer cells under phagocytosis, but PEGylated NPs are taken up less efficiently than more charged NPs.29 Kupffer cells are non-parenchymal cells within the liver that are very difficult to transfect, even with Lipofectin® (superior to other lipids for transfection), the transfection efficiency remains low.30 Our data indicated that although liver (as of NP size) and lung (as of NP slightly positive charge) took up NPs,31, 32 the transfection and expression of plasmid were mainly within the tumor (Figure S2D, 14.1 ± 3.9 versus 35.2 ± 8.6). The Wnt5a trap expression within other major organs was minor and also transient. This is presumably due to the efficient targeting effect of AEAA against sigma receptor 1 that is highly expressed on the surface of melanoma cells.
By introducing His-tag into the C-terminus of plasmid map, the expression of the Wnt5a trap against Wnt5a was assessed through ELISA, and further compared with the injection of purified trap protein (Figure 3D). His-tag ELISA showed the expression of trap was transient within one week. Consistent with IVIS imaging, tumor is the major trap producing organ with the help of AEAA targeted local NP delivery. Compared to direct injection of trap protein, the half-life of plasmid delivery was significantly prolonged. As illustrated in Figure 3D, free protein trap was cleared rapidly, with significantly lower concentration among all organs at each timepoint being monitored. Within the tumor, the AUC value for trap expression using NP delivery of plasmid DNA was 2.3-fold higher than that of the free protein trap. Presumably, the trap binding with Wnt5a prevents the Wnt5a binding with anti-Wnt5a antibody, thus the local concentration of Wnt5a measured in the tumor was significantly reduced (Figure 3E), indicating that the locally expressed and secreted Wnt5a trap neutralized Wnt5a in situ. Nevertheless, we found no significant elongation in the host survival, compared to the PBS control (Figure 3F), suggesting that remodeling of TME by trapping Wnt5a alone was insufficient for effective tumor therapy.
Combination of DOX-induced ICD and Wnt5a trapping significantly inhibited tumor progression
Effective tumor immunotherapy is often achieved by combination with the stimulation of the immune system, i.e. with a vaccine, or by remodeling the TME.33 As demonstrated above, neither ICD induced by low dose DOX (a form of vaccine) nor Wnt5a trap (a TME modulator) could produce significant host survival prolongation in this aggressive melanoma model. Therefore, we explored whether a combination of both therapies could bring in a synergistic therapeutic effect.
Indeed, with the help of low dose DOX, the local Wnt5a trapping strategy greatly inhibited tumor growth (Figure 4A and Figure S4). Significantly, the median survival of the mice treated with ICD/trap combo therapy was prolonged from approximately 45 days to 65 days (Figure 4B). To address the action mechanism, we performed treatment by pre-depletion of CD4+ or CD8+ T cells. As shown in Figure 4C, the therapeutic effect was partially abolished by the pre-treatment with either anti-CD8 or anti-CD4 antibody, whereas it was not affected when an isotype matched IgG control was used in the pre-treatment. TUNEL assay demonstrated a large increase of apoptotic cells within TME, indicating local tumor cell death (Figure 4D). There was also a significant (p < 0.01) decrease in collagen content (by Masson Trichrome staining) of the tumor with the combination therapy (Figure 4E). In either case, DOX treatment alone or Wnt5a trap alone had brought only a partial effect.
Figure 4. Combination therapy significantly inhibited tumor progression.

(A) Tumor inhibition of combination therapy, compared with untreated or monotherapy groups. n = 8. (B) Long term survival monitored over two months. n = 8–10. (C) Tumor bearing mice were pretreated with 3 daily injections of anti-CD8 and/or anti-CD4 antibody (300 µg/mice) to deplete the CD8+ and/or CD4+ T cells in vivo. Isotype IgG was used as control. The efficacy of combination therapy with or without different T cell depletion was compared by monitoring tumor burden after treatment cycles. n = 5. (D) TUNEL assay depicting apoptotic region within tumor slide sections. n = 3. (E) Masson’s trichrome staining depicting collagen distribution within TME. n = 3. Scale bars indicate 300 µm. *: p < 0.05, **: p < 0. 01, ***: p < 0. 001.
Treatment-induced remodeling of the tumor microenvironment
To investigate whether the immunosuppressive microenvironment of the tumor was indeed remodeled after the combination therapy, we analyzed the immune cells in the tumor by flow cytometry. As illustrated in Figure 5A, the efficient tumor inhibition could be attributed to the remodeling of the immunosuppression of TME,34 including significant increased CD103+ DCs (required for antigen transportation, T cell priming, as well as induction of intact anti-tumor immunity35), increased DC maturation (CD8+CD11c+), enhanced effector T cells infiltration (CD45+CD8+) and activation (CD8+CD62Llow), and the reduction in the suppressive immune cells such as MDSCs (CD11b+Gr-1+), M2 macrophages (F4/80+CD206+), and PD-L1+ cells (CD274+). Meanwhile, immune cytokines such as IL-12α, TNF-α and IFN-γ were dramatically increased at detected transcriptional level (Figure 5C), indicating a Th-2 to Th-1 phenotype switch to an immuno-stimulatory TME.36 The switch would greatly facilitate tumor antigen presentation (with a specific increase in IL-12α), and result in an intensified cytotoxic T cell mediated tumor-specific killing,37 as demonstrated by extensive IFN-γ production in ELISpot assay (Figure 5B). IFN-γ is produced mainly by CTLs mediating adaptive immune responses. The ELISpot assay used in our study detected and enumerated antigen-reactive T cells that secrete IFN-γ in vitro upon re-stimulation by tumor-specific antigen, mimicking systemic immune response in vivo.
Figure 5. Remodeling of tumor microenvironment.

(A) Flow cytometry analysis of immune functioning cells within TME. n = 3. (B) ELIspot assay depicting IFN-γ production under different treatments. n = 3. (C) RT-PCR analysis of both pro-inflammatory and anti-inflammatory cytokines within TME. n = 6. *: p < 0.05, **: p < 0. 01, ***: p < 0. 001.
Combination therapy demonstrated long-lasting suppressed tumor metastasis
Over two months of survival study, we kept monitoring tumor metastasis in major organs. Consistent with the clinical observation, we found that liver and lung were the major metastatic sites in the untreated murine model.38 As demonstrated in Figure S5, the combination therapy suppressed metastasis in these major organs. The observed long-term efficacy could be due to many factors. For one, innate immunity alone would be sufficient, particularly natural killer (NK) cells, has been reported to prevent metastasis in nude mice which are immune deficient.39 Secondly, the significant tumor inhibition would restrict cancer cell spreading, possibly via suppressing the epithelial-mesenchymal transition (EMT) by down-regulating pro-metastatic markers, such as CCL7/CCR3 or CCL21/CCR7 crosstalks.40, 41
Nevertheless, at late stage of tumor inhibition, the recruitment of both CD8+ and CD4+ T lymphocytes for an efficient systemic immune response would help in tumor restriction and reducing metastasis. CD8 T cells are critical in direct anti-tumoral activities. In our study, effective DC activation (CD11c+MHCII+) was observed, CD8+ T cells and CD69+CD8+ T cells were also found increased within LNs (Figure 6C), as well as within TME (Figure 5A, CD8+ and CD62LlowCD8+ T cells). Memory CD8 T cells (CD8+CD44+) were also found significantly increased within TME (Figure 6D). For CD4 T cells, we found a significant increase in memory CD4 T cells (CD4+CD62L+) within TME (Figure 6A) and LNs (Figure 6B). Furthermore, CD4+ T cells were sorted (at the endpoint of tumor inhibition study) from spleens of mice subjected to either Combo therapy or PBS as control. Among which, antigen-specific CD4 cells undergo in vitro proliferation when re-stimulated with tumor cell lysate containing tumor antigens (Figure 6E). The division of these antigen-reactive cells (Q1 population) was significantly increased, indicating a generation of memory CD4 immunity in vitro mimicking in vivo settings.
Figure 6. Combination therapy demonstrated long-lasting overall immune response.

(A) Immunofluorescent staining for memory CD4 T cells within TME. n = 3. Scale bar indicates 100 µm. (B) and (C) Flow cytometry analysis of T cells and DCs functions within draining LNs. n = 3. (D) Immunofluorescent staining for memory CD8 T cells within TME. n = 3. Scale bar indicates 100 µm. (E) In vitro CD4+ T cell proliferation assay depicting division of antigen-reactive cells (Q1) between groups. n = 3. *: p < 0.05, **: p < 0. 01.
To explore whether the observed therapeutic efficacy could be reproduced in other models of melanoma, we established a second BRAF-mutant melanoma model using D4M cells (BRAFV600E, PTEN−/−, syngeneic with C57BL/6 mice).42 As shown in Figure S6A, D4M tumor also demonstrated a desmoplastic tumor morphology as the BPD6 tumor. Under the same treatment protocols, combination of low dose DOX and Wnt5a trap resulted in superior tumor growth inhibition as compared to either treatment alone (Figure S6B), implicating that our therapeutic strategy could have great potential for translational applications.
Safety evaluation of the combination therapy
We evaluated the safety and side toxicity of the therapies by performing extensive toxicological pathology analysis. It has been reported that Wnt5a plays an important role in the liver. Wnt5a, also participates in hepatic stellate cell activation through Wnt/Ca2+ pathway and may serve as a therapeutic target in the treatment of liver fibrosis.43 Thus the homeostasis of Wnt5a is directly associated with proper liver functions and worth further investigation. Minor and transient Wnt5a trapping might be beneficial. Throughout the trapping therapeutic window and tumor inhibition study, as shown in Figure S7, we found no significant morphological damage was caused in the liver, neither in other major organs, including kidney, lung, spleen, and lung. Compared with non-tumored control group, mice under different treatment of therapies demonstrated none noticeable systematic toxicities (Figure S7). No significant body weight changes were found in any of the treatment groups (Figure 7A). The serum biochemical parameter analysis and the whole blood cell counts remained within the normal ranges for all the groups, suggesting no systemic anemia or inflammation occurred after treatments, nor major liver disfunctions noticed (Figure 7B).
Figure 7. Toxicity evaluation of therapies.

(A) Mice body weight changes under tumor inhibition study. (B) Whole cell counts and serum biochemical marker analysis of tumor bearing mice. Fresh whole blood and serum were collected at endpoint of study. Non-tumored mice were also examined as control. n = 5. n.s.: p > 0.05.
CONCLUSIONS
Inspiring progress has been achieved in cancer immunotherapy against various solid tumors including melanoma.44, 45 Compared to conventional chemotherapy, immunotherapy can elicit significant durable overall survival. However, such response rate remains low, as of only poor portion of patients may benefit from current treatments.46 This is presumably due to the high complexity of TME that plays crucial roles in either dampening or enhancing the immune responses.47 In this study, we focused on the dynamic interaction between metastatic melanoma cells and their critical immunomodulator, Wnt5a, within the TME. We demonstrated the synergistic effect of a combination therapy of clinically feasible chemotherapy-induced immunogenic cell death with the blockade of Wnt5a-mediated immunosuppression.
Among human melanoma, the activation of tumor cell intrinsic Wnt/β-catenin pathway mediates the deficiency in T cell priming tumor-associated antigens in vivo, followed by resistant to anti-PD-L1 and anti-CTLA4 blockage therapy.20, 48 Owing to its diverse functions, Wnt5a signaling in immunosuppression and cancer progression is varied and complex with mechanism still elusive, and whether it plays a role in cancer promotion or suppression depends on specific cancer types. The persistent activation of Wnt5a and its downstream signaling pathways in BRAF-mutant type is particularly mortal.49 So far, systemically applied monoclonal antibodies (mAbs) against Wnt5a are not yet available for patients.10 Moreover, without specific targeting, a disruption in the functioning of Wnt5a molecule among normal tissues may lead to imbalance in immunologic tolerance and result in severe inflammatory diseases.50 In our established Wnt5ahigh BRAF-mutant melanoma mouse models, the advantage of our approach is to locally normalize, rather than systemically deplete, the expression of Wnt5a within TME under a transient therapeutic window. Compared to systemic large-size (~150 kDa) mAb therapies which have been reported with subsequent multiple autoimmune diseases,51 the local and transient expression of a small-size (~26 kDa) Wnt5a ‘trap’ by tumor-specific NP delivery offers great advantages in both therapeutic efficacy and safety.25 Basically, what we have established is a “Wnt5a KD” tumor model in a transient way. The Wnt5a trap used in this work is based on the CRD of FZD7. Since FZD7 is also implicated in the interaction with other Wnt ligands, it is possible that levels of Wnt ligands other than Wnt5a (i.e. Wnt3a) were also reduced in TME.52
Conclusively, the local and transient Wnt5a trapping efficiently remodels the fibrotic immunosuppressive TME, recovers DC functions, and facilitates T cell infiltration, providing a promising platform for the treatment of desmoplastic BRAF-mutant melanoma. This is especially true when combined with a commonly accepted chemotherapy that can further stimulate immune responses, or other forms of tumor-specific vaccination.7, 53 For the future study of the proposed approach, the modified TME may also allow for a second-wave chemotherapeutic nano-therapy, or personalized neoantigens incorporated into therapeutic vaccines to further enhance treatment specificity.54, 55 For the potential translation of the present work, since low dose DOX is clinically acceptable, our approach shows a high potential for clinical translation, especially for patients suffering from chemo-toxicities.56, 57 Currently, we are further investigating into formulation scale-up, prolonged safety evaluation, as well as any potential side effects associated with change in dosing strategy.
MATERIALS AND METHODS
Materials
The 1,2-distearoryl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol-2000)] ammonium salt (DSPE-PEG), dioleoyl phosphatidic acid (DOPA) and 1,2-dioleoyl-3-trimethylammonium-propane chloride salt (DOTAP) were purchased from Avanti Polar Lipids (Alabaster, AL). The DSPE-PEG-AEAA was synthesized according to previous publication of our lab.58 Doxorubicin, cholesterol and protamine were purchased from Sigma-Aldrich (St. Louis, MO). DiI was purchased from ThermoFisher Scientific (Waltham, MA).
Cell lines and animals
Murine BRAF-mutant melanoma cell lines BPD6 and D4M (BRAFV600E, PTEN−/−, syngeneic with C57BL/6) was kindly provided by Brent Hanks (Duke Cancer Institute) and cultivated in RPMI-1640 Medium added with 10% FBS and 1% Penicillin/Streptomycin (Invitrogen, Carlsbad, CA) at 37 °C and 5% CO2. Six-week-old female C57BL/6 mice were purchased from Charles River Laboratories. All animal regulations and procedures were accepted by Institutional Animal Care and Use Committee, University of North Carolina at Chapel Hill.
Antibodies
InVivoMAb anti-mouse CD8α of clone 53-6.7 and anti-mouse CD4 of clone GK1.5 were purchased from BioXcell (West Lebanon, NH). Primary antibodies, fluorescent conjugated primary and secondary antibodies used for immunostainings (IF), western blots (WB), and flow cytometry (flow cytr) were listed in Table S1.
Wnt5a expression in TCGA human patients and survival analysis
The TCGA SKCM (Skin Cutaneous Melanoma)) cohort data was downloaded from the Broad Institute Genome Data Analysis Centers (GDAC) by using its fbget tool to directly access the GDAC FireBrowse API interface. All the samples are either “TP” or “TM” type. For Wnt5a expression analysis, we noticed a significant difference in Wnt5a expression levels between “TP” and “TM” types. The results shown are based on all “TM” type samples, as the majority of the samples are “TM”. The same trend was observed for the “TP” samples. The p-value was calculated with a t-test. For the survival analysis, we chose to use the “TP” samples, since these patients were diagnosed at earlier stages than the “TM” patient and their survival data is thus relatively more accurate. The survival time was calculated as days to death for deceased patients, and the days to last follow-up for alive patients. Data were represented as scatterplot of Wnt5a mRNA expression level vs days of survival for human primary solid SKCM tumor samples. Kaplan-Meier curves and Median Survival were quantified and calculated using Prism 5.0 Software. P-value less than 0.05 was considered significantly difference. *: p < 0.05, **: p < 0. 01, ***: p < 0. 001.
Construction of Wnt5a trap gene
To construct the Wnt5a trap plasmid (pWnt5a trap), the codon-optimized coding sequences of the N-terminal extracellular cysteine-rich domain (CRD) of mouse FZD7 (residues 33–180) and the C-terminal trimerization domain of cartilage matrix protein (residues 458–500) were used for assembling the trap gene. A flexible hinge region with optimized length was introduced between the Wnt5a-binding CRD and the trimerization domain. The final sequence for the monomeric Wnt5a trap codes for a secretion signaling peptide, Wnt5a-binding FZD7 CRD, hinge peptide, trimerization domain, FLAG tag, and His (6×) tag, respectively. The complete cDNA was cloned into pcDNA3.1 between Nhe I and Xho I sites and the accuracy was confirmed by DNA sequencing. The pWnt5a trap map and the amino acid sequence are available in Figure S2.
Expression and purification of recombinant trap protein
293T cells were cultured until 70–80% confluence. To transfect the cells, 24 µg pTrap (or pcDNA3.1 negative control) and 40 µL lipofectamine were added to each 10-cm plate. The serum concentration was reduced after transfection. The 293T cells were monitored each day to ascertain their survival. Ten mL supernatant was harvested after 24, 48, and 72 h, and kept at 4°C for further purification. The supernatants were concentrated with 10 kDa MWCO spin filters to 200 µL and subjected to His-Mag-Ni-Sepharose beads to purify His (6×)-tagged trap protein. The purified proteins were analyzed on 10% SDS-PAGE gel.
Binding kinetics
The binding affinities of FZD-CDR-based proteins to Wnt5a were accessed with Microscale Thermophoresis (MST).59 In brief, Wnt5a-binding protein was first fluorescently labeled by using RED-tris-NTA dye. Ten µL of the labeled protein was then supplied to 10 µL of serially 2-fold diluted mouse Wnt5a using a PBST buffer (PBS with 0.05% Tween 20). The resulting samples were subsequently loaded into capillaries, and the thermophoresis of each sample was measured using Auto Red laser power and medium MST power on Monolith NT.115 (NanoTemper Technologie, Munich, Germany).
Preparation and Characterization of LPD
LPD NPs were synthesized according to published procedures from Huang lab.60 Briefly, DOTAP and cholesterol (1:1, mol/mol) liposomes were prepared by a hydration-extrusion method. LPD cores were self-assembled as adding 100 µL of 20 µg protamine in DI water to 100 µL of 50 µg Wnt5a trap plasmid. After incubation for 10 min at room temperature, 60 µL of the liposomes were added. We then introduced 10 µL DSPE-PEG and 10 µL DSPE-PEG-AEAA at 60 °C for 15 min. Finally, 20 µL 20% glucose solution was added to adjust the osmotic pressure. NP size and NP surface charge were measured by a Malvern ZetaSizer Nano series (Westborough, MA). Followed by negatively staining, NPs were imaged with a JEOL 100 CX II TEM (JEOL, Japan).
Tumor growth inhibition, metastasis suppression and survival analysis
On day 0, mice were inoculated subcutaneously with 1 × 106 BPD6 cells on lower flank area. Once tumor volume reached ~200 mm3 (0.5 × length × width × height), mice were then randomized into 5 groups (n = 5~8) as follows: Untreated group (the PBS group), Control plasmid trap (the pGFP group), Low dose Doxorubicin (the DOX group), Wnt5a trap (the Wnt5a trap group), and Combination of low dose Doxorubicin with Wnt5a trap (the Combo group). DOX group was i.p. treated (0.544 mg/kg) on day 10 and 12. Control or Wnt5a trap was i.v. treated (50 µg plasmid/mouse) on day 10, 12, 14 and 16. Tumor size (digital caliper) and animal weight were monitored every 2–3 days. Mice were sacrificed before tumors reached 20 mm in one dimension. At the endpoint, tumors, major organs and blood samples were harvested and tested. Long-term survival was also monitored for over two months. Kaplan-Meier curves and Median Survival were quantified and calculated using Image J. At the endpoint of survival monitor, metastasis study was performed as major organs were harvested, fixed and processed with H&E staining for pathology observation.
Bio-distribution of LPD NPs
DiI-labeled LPD NPs were synthesized with liposomes containing approximately 0.05% of hydrophobic dye DiI. Mice were intravenously injected with DiI-labeled LPD NPs, and sacrificed after 24 h. Tumor and major organs were collected accordingly and subject to IVIS® Kinetics Optical System (Perkin Elmer, CA) (excitation/emission = 520/560 nm) for imaging and quantifications.
Expression of Wnt5a trap
Mice bearing BPD6 allografts were intravenously injected with LPD NPs containing 50 µg plasmid. Mice were humanely sacrificed at 1, 2, 4, 8 days post final dosage. Tumor and major organs were harvested, and total protein were purified and quantified with BCA Protein Assay Kit (Pierce, Rockford, IL). Expression of Wnt5a trap was quantified with ELISA (Cell biolabs, INC., n = 5) using anti-His antibody against the His-Tag engineered at the C-terminus of Wnt5a. Trap protein was also directly intravenously injected into mice and compared with the plasmid counterpart.
Tumor bearing mice treated with pGFP were sacrificed two days after final dose. Liver, lung, and tumor were further sectioned by a cryostat (H/I Hacker Instruments & Industries, Winnsboro, SC) to quantify the distribution of LPD NPs within these tissues. Accumulation and distribution of NPs among these tissues were quantified to compare (n = 3).
Flow cytometry assay
Immune cell populations were analyzed by flow cytometry.61 Briefly, Tumor tissues or LNs were collected by collagenase A at 37 °C for 40–50 min. Single cells were harvested in PBS and stained with fluorescein-conjugated antibodies. Penetration buffer (BD, Franklin Lakes, NJ) were added for any intracellular cytokine staining.
In vitro CD4+ T cell purification and proliferation
At the endpoint of tumor inhibition study, the CD4 positive T cells were sorted from whole spleen of mice among PBS or Combo groups.62 Sorted cells were stained with 5 µM CFSE, pulsed with DOX-treated cell lysate and cultured with RBC-depleted splenocytes as well as 25 U/mL IL-2 + 5 µg/mL soluble CD28 in vitro (0.5 × 105 cells/well in 96-well plate, with 200 µL compete culture medium) for 3–4 days. All cells before/after culture were subject to flow cytometry for quantification of CD4+CFSE+ cell population. The percentage of CD4+ cell proliferation (Q1 population) was quantified by CFSE staining via flow cytometry, and the divisions of antigen-reactive cells were statistically compared between groups, quantified by Flowjo software.
Immunofluorescence staining
Staining was performed on paraffin-embedded sections from tumor tissues. Briefly, all tissues for paraffin-embedding were resected, rinsed in PBS, and placed in 4% PFA for over 48 h at 4 °C. Immunofluorescence staining was performed by deparaffinization, antigen retrieval, permeabilization, and blocking in 1% bovine serum albumin. All antibodies conjugated with fluorophores were incubated overnight at 4 °C, followed by nuclei counterstained with Prolong® Diamond Antifade Mountant with DAPI (ThermoFisher Scientific). Stained slides were imaged with Zeiss 880 Confocal microscopy (Germany). Three randomly microscopic fields were selected and quantified by Image J software.
ICD determination
ICD dosing was determined by in vitro CRT exposure and HMGB1 release.63 Briefly, BPD6 cells treated with low dose of Doxorubicin were harvested, PBS washed and fixed in 0.25% PFA. Primary and secondary antibodies were diluted in blocking buffer, and added to collected samples for 30 min each. Cells were then mounted, and nuclei were stained with Hoechst 33432 (ThermoFisher Scientific), followed by Confocal imaging. For intracellular staining of HMGB1, cells were permeabilized with 0.1% Triton X-100 for 10min before blocking.
Quantitative real-time PCR (RT-PCR) assay
Total RNA was extracted from the tumor tissues following protocol of RNeasy® Microarray Tissue Mini Kit (Qiagen, Hilden, Germany). We then reverse-transcribed cDNA with iScript™ cDNA Synthesis Kit and amplify cDNA with iScript™ Reverse Transcription Supermix for RT-PCR (Bio-Rad, Hercules, CA). RT-PCR primers, all mouse specific, are listed in Table S2. RT-PCR reactions were performed with 7500 Real-Time PCR System and subject to analysis with 7500 Software, compared to and normalized by GAPDH endogenous control.
ELISPOT assay for IFN-γ production
Mice were subject to different treatment according to groups. At the endpoint day of tumor inhibition study, spleen and draining LNs of mice were collected into single cells under sterile condition, and seeded on capture antibody-coated 96-well plate. The single-cell suspensions were then co-cultured with 5 µM of BRAFV600E peptide at 37 °C for 18 – 20 h. The BRAFV600E peptide is specific to BRAFV600EPten−/− tumor model as a strong stimulator of an antigen-specific response.7 OVA peptide and BRAFWT peptide were used as controls. Cells were subsequently removed by three rounds of washing. IFN-γ production was measured with BD™ ELISPOT assay system (BD Pharmingen, San Diego, CA) and compared between treatment groups.
TUNEL assay
Assay performed following DeadEnd Fluorometric TUNEL System (Promega, Madison, WI) instruction and imaged with fluorescence microscopy. Fragmented DNAs of apoptotic cells were fluorescently stained with FITC and defined as TUNEL-positive nuclei. Slides were mounted, and nuclei were stained with Prolong® Diamond Antifade Mountant with DAPI (ThermoFisher Scientific), followed by imaging under Confocal microscopy. Three fields were randomly selected and quantified.
H&E staining and blood toxicity analysis
At the endpoint day of tumor inhibition study, tumor bearing mice under different treatments were humanely sacrificed, whole blood, serum, and major organs were harvested. Organs were collected for H&E staining by UNC histology facility. Indicators of renal and liver function such as creatinine, blood urea nitrogen (BUN), serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were tested based on blood and serum.
Patient tumor samples
H&E sections from paraffin-embedded biopsies of BRAF-mutant melanoma patients were obtained from Department of pathology, Xinhua Hospital, China according to an approved patient sample management protocol. Informed consent was obtained from patient before evaluation.
Statistical analysis
One-way ANOVA and a two tailed Student's t-test were performed in Prism 5.0 Software. Data were compared with PBS control group. P-value less than 0.05 was considered significantly difference.
Supplementary Material
Acknowledgments
The work was supported by NIH grants CA198999 (to L.H.), CA157738 (to R.L.), R15GM122006 (to D.W.). We appreciate Wenbin Guan from Department of Pathology, Xinhua Hospital, China for providing the human melanoma samples. L.H. was a Senior Visiting Scholar of the State Key Laboratory of Molecular Engineering of Polymers, Fudan University, China.
Footnotes
Conflict of Interest: The trap technology has been licensed to OncoTrap Inc. L.H. and R.L. are co-founders.
Supporting Information Available: Figure S1–S7, the Antibody list and the Primer list for real-time PCR are described. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gloster HM, Jr, Brodland DG. The Epidemiology of Skin Cancer. Dermatol. Surg. 1996;22:217–226. doi: 10.1111/j.1524-4725.1996.tb00312.x. [DOI] [PubMed] [Google Scholar]
- 2.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, et al. Mutations of the Braf Gene in Human Cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.Crosby T, Fish R, Coles B, Mason MD. Systemic Treatments for Metastatic Cutaneous Melanoma. Cochrane Database Syst Rev. 2000:CD001215. doi: 10.1002/14651858.CD001215. [DOI] [PubMed] [Google Scholar]
- 4.Liu Q, Das M, Liu Y, Huang L. Targeted Drug Delivery to Melanoma. Adv. Drug Deliv Rev. 2017 doi: 10.1016/j.addr.2017.09.016. [DOI] [PubMed] [Google Scholar]
- 5.Garg AD, Agostinis P. Editorial: Immunogenic Cell Death in Cancer: From Benchside Research to Bedside Reality. Front. Immunol. 2016;7:110. doi: 10.3389/fimmu.2016.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cintolo JA, Datta J, Xu S, Gupta M, Somasundaram R, Czerniecki BJ. Type I-Polarized Braf-Pulsed Dendritic Cells Induce Antigen-Specific Cd8+ T Cells That Impact Braf-Mutant Murine Melanoma. Melanoma Res. 2016;26:1–11. doi: 10.1097/CMR.0000000000000203. [DOI] [PubMed] [Google Scholar]
- 7.Liu Q, Zhu H, Liu Y, Musetti S, Huang L. Braf Peptide Vaccine Facilitates Therapy of Murine Braf-Mutant Melanoma. Cancer Immunol. Immunother. 2017 doi: 10.1007/s00262-017-2079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holtzhausen A, Zhao F, Evans KS, Tsutsui M, Orabona C, Tyler DS, Hanks BA. Melanoma-Derived Wnt5a Promotes Local Dendritic-Cell Expression of Ido and Immunotolerance: Opportunities for Pharmacologic Enhancement of Immunotherapy. Cancer Immunol. Res. 2015;3:1082–1095. doi: 10.1158/2326-6066.CIR-14-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piersma B, Bank RA, Boersema M. Signaling in Fibrosis: Tgf-Beta, Wnt, and Yap/Taz Converge. Front. Med. 2015;2:59. doi: 10.3389/fmed.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shojima K, Sato A, Hanaki H, Tsujimoto I, Nakamura M, Hattori K, Sato Y, Dohi K, Hirata M, Yamamoto H, Kikuchi A. Wnt5a Promotes Cancer Cell Invasion and Proliferation by Receptor-Mediated Endocytosis-Dependent and -Independent Mechanisms, Respectively. Sci. Rep. 2015;5:8042. doi: 10.1038/srep08042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inoue S, Setoyama Y, Odaka A. Doxorubicin Treatment Induces Tumor Cell Death Followed by Immunomodulation in a Murine Neuroblastoma Model. Exp. Ther. Med. 2014;7:703–708. doi: 10.3892/etm.2014.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Angelova AL, Grekova SP, Heller A, Kuhlmann O, Soyka E, Giese T, Aprahamian M, Bour G, Ruffer S, Cziepluch C, Daeffler L, Rommelaere J, Werner J, Raykov Z, Giese NA. Complementary Induction of Immunogenic Cell Death by Oncolytic Parvovirus H-1pv and Gemcitabine in Pancreatic Cancer. J. Virol. 2014;88:5263–5276. doi: 10.1128/JVI.03688-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumawat K, Gosens R. Wnt-5a: Signaling and Functions in Health and Disease. Cell. Mol. Life Sci. 2016;73:567–587. doi: 10.1007/s00018-015-2076-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buttler K, Becker J, Pukrop T, Wilting J. Maldevelopment of Dermal Lymphatics in Wnt5a-Knockout-Mice. Dev. Biol. 2013;381:365–376. doi: 10.1016/j.ydbio.2013.06.028. [DOI] [PubMed] [Google Scholar]
- 15.Okamoto M, Udagawa N, Uehara S, Maeda K, Yamashita T, Nakamichi Y, Kato H, Saito N, Minami Y, Takahashi N, Kobayashi Y. Noncanonical Wnt5a Enhances Wnt/Beta-Catenin Signaling During Osteoblastogenesis. Sci. Rep. 2014;4 doi: 10.1038/srep04493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Y, Kipps TJ, Zhang S. Wnt5a Signaling in Normal and Cancer Stem Cells. Stem Cells Int. 2017;2017:5295286. doi: 10.1155/2017/5295286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asem MS, Buechler S, Wates RB, Miller DL, Stack MS. Wnt5a Signaling in Cancer. Cancers. 2016;8 doi: 10.3390/cancers8090079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valencia J, Hernandez-Lopez C, Martinez VG, Hidalgo L, Zapata AG, Vicente A, Varas A, Sacedon R. Wnt5a Skews Dendritic Cell Differentiation to an Unconventional Phenotype with Tolerogenic Features. J. Immunol. 2011;187:4129–4139. doi: 10.4049/jimmunol.1101243. [DOI] [PubMed] [Google Scholar]
- 19.Kushwah R, Hu J. Role of Dendritic Cells in the Induction of Regulatory T Cells. Cell Biosci. 2011;1:20. doi: 10.1186/2045-3701-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spranger S, Bao R, Gajewski TF. Melanoma-Intrinsic Beta-Catenin Signalling Prevents Anti-Tumour Immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 21.Madhunapantula SV, Robertson GP. The Pten-Akt3 Signaling Cascade as a Therapeutic Target in Melanoma. Pigm. Cell Melanoma Res. 2009;22:400–419. doi: 10.1111/j.1755-148X.2009.00585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou J, Cheng P, Youn JI, Cotter MJ, Gabrilovich DI. Notch and Wingless Signaling Cooperate in Regulation of Dendritic Cell Differentiation. Immunity. 2009;30:845–859. doi: 10.1016/j.immuni.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King TD, Zhang W, Suto MJ, Li YH. Frizzled7 as an Emerging Target for Cancer Therapy. Cell Signal. 2012;24:846–851. doi: 10.1016/j.cellsig.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu X, Jiao JG, Li YD, Tian T. Overexpression of Fzd7 Promotes Glioma Cell Proliferation by Upregulating Taz. Oncotarget. 2016;7:85987–85999. doi: 10.18632/oncotarget.13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miao L, Li J, Liu Q, Feng R, Das M, Lin CM, Goodwin TJ, Dorosheva O, Liu R, Huang L. Transient and Local Expression of Chemokine and Immune Checkpoint Traps to Treat Pancreatic Cancer. ACS Nano. 2017;11:8690–8706. doi: 10.1021/acsnano.7b01786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dames SA, Kammerer RA, Wiltscheck R, Engel J, Alexandrescu AT. Nmr Structure of a Parallel Homotrimeric Coiled Coil. Nat. Struct. Biol. 1998;5:687–691. doi: 10.1038/90444. [DOI] [PubMed] [Google Scholar]
- 27.Kim D, Kim SK, Vaencia CA, Liu RH. Tribody: Robust Self-Assembled Trimeric Targeting Ligands with High Stability and Significantly Improved Target-Binding Strength. Biochemistry. 2013;52:7283–7294. doi: 10.1021/bi400716w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miao L, Liu Q, Lin CM, Luo C, Wang Y, Liu L, Yin W, Hu S, Kim WY, Huang L. Targeting Tumor-Associated Fibroblasts for Therapeutic Delivery in Desmoplastic Tumors. Cancer Res. 2017;77:719–731. doi: 10.1158/0008-5472.CAN-16-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-Liver Interactions: Cellular Uptake and Hepatobiliary Elimination. J. Control Release. 2016;240:332–348. doi: 10.1016/j.jconrel.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 30.Jarnagin WR, Debs RJ, Wang SS, Bissell DM. Cationic Lipid-Mediated Transfection of Liver Cells in Primary Culture. Nucleic Acids Res. 1992;20:4205–4211. doi: 10.1093/nar/20.16.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frohlich E. The Role of Surface Charge in Cellular Uptake and Cytotoxicity of Medical Nanoparticles. Int. J. Nanomed. 2012;7:5577–5591. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shang L, Nienhaus K, Nienhaus GU. Engineered Nanoparticles Interacting with Cells: Size Matters. J. Nanobiotechnol. 2014;12 doi: 10.1186/1477-3155-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hodge JW, Ardiani A, Farsaci B, Kwilas AR, Gameiro SR. The Tipping Point for Combination Therapy: Cancer Vaccines with Radiation, Chemotherapy, or Targeted Small Molecule Inhibitors. Semin. Oncol. 2012;39:323–339. doi: 10.1053/j.seminoncol.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joyce JA, Fearon DT. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science. 2015;348:74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- 35.Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, Chakarov S, Rivera C, Hogstad B, Bosenberg M, Hashimoto D, Gnjatic S, Bhardwaj N, Palucka AK, Brown BD, Brody J, et al. Expansion and Activation of Cd103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic Pd-L1 and Braf Inhibition. Immunity. 2016;44:924–938. doi: 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaiko GE, Horvat JC, Beagley KW, Hansbro PM. Immunological Decision-Making: How Does the Immune System Decide to Mount a Helper T-Cell Response? Immunology. 2008;123:326–338. doi: 10.1111/j.1365-2567.2007.02719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovarik J, Siegrist CA. Optimization of Vaccine Responses in Early Life: The Role of Delivery Systems and Immunomodulators. Immunol. Cell Biol. 1998;76:222–236. doi: 10.1046/j.1440-1711.1998.00746.x. [DOI] [PubMed] [Google Scholar]
- 38.Tas F. Metastatic Behavior in Melanoma: Timing, Pattern, Survival, and Influencing Factors. J. Oncol. 2012;2012:647684. doi: 10.1155/2012/647684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Habu S, Fukui H, Shimamura K, Kasai M, Nagai Y, Okumura K, Tamaoki N. In Vivo Effects of Anti-Asialo Gm1. I. Reduction of Nk Activity and Enhancement of Transplanted Tumor Growth in Nude Mice. J. Immunol. 1981;127:34–38. [PubMed] [Google Scholar]
- 40.Lee YS, Kim SY, Song SJ, Hong HK, Lee Y, Oh BY, Lee WY, Cho YB. Crosstalk between Ccl7 and Ccr3 Promotes Metastasis of Colon Cancer Cells Via Erk-Jnk Signaling Pathways. Oncotarget. 2016;7:36842–36853. doi: 10.18632/oncotarget.9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang L, Wang D, Li Y, Liu Y, Xie X, Wu Y, Zhou Y, Ren J, Zhang J, Zhu H, Su Z. Ccl21/Ccr7 Axis Contributed to Cd133+ Pancreatic Cancer Stem-Like Cell Metastasis Via Emt and Erk/Nf-Kappab Pathway. PLoS One. 2016;11:e0158529. doi: 10.1371/journal.pone.0158529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jenkins MH, Steinberg SM, Alexander MP, Fisher JL, Ernstoff MS, Turk MJ, Mullins DW, Brinckerhoff CE. Multiple Murine Braf(V600e) Melanoma Cell Lines with Sensitivity to Plx4032. Pigm. Cell Melanoma Res. 2014;27:495–501. doi: 10.1111/pcmr.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiong WJ, Hu LJ, Jian YC, Wang LJ, Jiang M, Li W, He Y. Wnt5a Participates in Hepatic Stellate Cell Activation Observed by Gene Expression Profile and Functional Assays. World J. Gastroenterol. 2012;18:1745–1752. doi: 10.3748/wjg.v18.i15.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ye Y, Wang C, Zhang X, Hu Q, Zhang Y, Liu Q, Wen D, Milligan J, Bellotti A, Huang L, Dotti G, Gu Z. A Melanin-Mediated Cancer Immunotherapy Patch. Sci. Immunol. 2017;2 doi: 10.1126/sciimmunol.aan5692. [DOI] [PubMed] [Google Scholar]
- 45.Fearon D. Combination Immunotherapy for Cancer. J. Exp. Med. 2016;213:1115. doi: 10.1084/jem.2137insight2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Menon S, Shin S, Dy G. Advances in Cancer Immunotherapy in Solid Tumors. Cancers. 2016;8 doi: 10.3390/cancers8120106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Church SE, Galon J. Tumor Microenvironment and Immunotherapy: The Whole Picture Is Better Than a Glimpse. Immunity. 2015;43:631–633. doi: 10.1016/j.immuni.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 48.Kaur A, Webster MR, Weeraratna AT. In the Wnt-Er of Life: Wnt Signalling in Melanoma and Ageing. Br. J. Cancer. 2016;115:1273–1279. doi: 10.1038/bjc.2016.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Webster MR, Xu M, Kinzler KA, Kaur A, Appleton J, O'Connell MP, Marchbank K, Valiga A, Dang VM, Perego M, Zhang G, Slipicevic A, Keeney F, Lehrmann E, Wood W, Becker KG, Kossenkov AV, Frederick DT, Flaherty KT, Xu XW, et al. Wnt5a Promotes an Adaptive, Senescent-Like Stress Response, While Continuing to Drive Invasion in Melanoma Cells. Pigm. Cell Melanoma Res. 2015;28:184–195. doi: 10.1111/pcmr.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pashirzad M, Shafiee M, Rahmani F, Behnam-Rassouli R, Hoseinkhani F, Ryzhikov M, Moradi Binabaj M, Parizadeh MR, Avan A, Hassanian SM. Role of Wnt5a in the Pathogenesis of Inflammatory Diseases. J. Cell. Physiol. 2017;232:1611–1616. doi: 10.1002/jcp.25687. [DOI] [PubMed] [Google Scholar]
- 51.Nagasaka M, Alhasan RS, Crosby M, Thummala N, Kim S, Abrams J, Sukari A. Toxicities Associated with Checkpoint Inhibitor Immunotherapy: The Karmanos Cancer Center Experience. J. Clin. Oncol. 2017;35 [Google Scholar]
- 52.Wei W, Chua MS, Grepper S, So SK. Soluble Frizzled-7 Receptor Inhibits Wnt Signaling and Sensitizes Hepatocellular Carcinoma Cells Towards Doxorubicin. Mol. Cancer. 2011;10:16. doi: 10.1186/1476-4598-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu L, Wang Y, Miao L, Liu Q, Musetti S, Li J, Huang L. Combination Immunotherapy of Muc1 Mrna Nano-Vaccine and Ctla-4 Blockade Effectively Inhibits Growth of Triple Negative Breast Cancer. Mol. Ther. 2018;26:45–55. doi: 10.1016/j.ymthe.2017.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu K, Miao L, Goodwin TJ, Li J, Liu Q, Huang L. Quercetin Remodels the Tumor Microenvironment to Improve the Permeation, Retention, and Antitumor Effects of Nanoparticles. ACS Nano. 2017;11:4916–4925. doi: 10.1021/acsnano.7b01522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The Future of Cancer Treatment: Immunomodulation, Cars and Combination Immunotherapy. Nat. Rev. Clin. Oncol. 2016;13:394. doi: 10.1038/nrclinonc.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of Pegylated Liposomal Doxorubicin: Review of Animal and Human Studies. Clin. Pharmacokinet. 2003;42:419–436. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 57.Rafiyath SM, Rasul M, Lee B, Wei G, Lamba G, Liu D. Comparison of Safety and Toxicity of Liposomal Doxorubicin Vs. Conventional Anthracyclines: A Meta-Analysis. Exp. Hematol. Oncol. 2012;1:10. doi: 10.1186/2162-3619-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li SD, Huang L. Nanoparticles Evading the Reticuloendothelial System: Role of the Supported Bilayer. Biochim. Biophys. Acta. 2009;1788:2259–2266. doi: 10.1016/j.bbamem.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jerabek-Willemsen M, Andre T, Wanner R, Roth HM, Duhr S, Baaske P, Breitsprecher D. Microscale Thermophoresis: Interaction Analysis and Beyond. J. Mol. Struct. 2014;1077:101–113. [Google Scholar]
- 60.Miao L, Guo S, Lin CM, Liu Q, Huang L. Nanoformulations for Combination or Cascade Anticancer Therapy. Adv. Drug Deliv Rev. 2017 doi: 10.1016/j.addr.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Young YK, Bolt AM, Ahn R, Mann KK. Analyzing the Tumor Microenvironment by Flow Cytometry. Methods Mol. Biol. (N. Y.) 2016;1458:95–110. doi: 10.1007/978-1-4939-3801-8_8. [DOI] [PubMed] [Google Scholar]
- 62.Wang LX, Plautz GE. Tumor-Primed, in Vitro-Activated Cd4(+) Effector T Cells Establish Long-Term Memory without Exogenous Cytokine Support or Ongoing Antigen Exposure. J. Immunol. 2010;184:5612–5618. doi: 10.4049/jimmunol.0903549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bianchi ME. Killing Cancer Cells, Twice with One Shot. Cell Death Differ. 2014;21:1–2. doi: 10.1038/cdd.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.