

Abstract
We reported a preclinical cancer vaccine that simultaneously introduced an mRNA antigen and an immune checkpoint blocking siRNA into the antigen-presenting cells. This was achieved by formulating both nucleic acid-based immunotherapeutics into a lipid-coated calcium phosphate (LCP) nanoparticle (NP) as a carrier to address the delivery challenge. The PEGylated lipid NPs were functionalized with mannose as the targeting ligand to facilitate the preferential uptake by the dendritic cells (DCs) in the lymph nodes after subcutaneous administration. The calcium phosphate core allowed acid-mediated dissolution in the endo-lysosomal compartment, which prompted rapid release of cargoes after cellular internalization of NP. LCP mRNA vaccine encoding TRP2 elicited a robust antigen-specific cytotoxic T cell response and a humoral immune response in a C57BL/6 mouse model of B16F10 melanoma. The immune responses efficaciously inhibited the melanoma growth. Moreover, co-delivery of PD-L1 siRNA and mRNA vaccine resulted in the downregulation of PD-L1 in the DCs that presented tumor antigens, significantly prompting T cell activation and proliferation. The enhanced T cell response had a profound inhibitory effect on tumor growth and metastasis. Generally, the work provided a paradigm for the development of an mRNA vaccine carrier to boost the anticancer immune response.
Keywords: mRNA vaccine, melanoma, nanoparticle, immune checkpoint blockade
Graphical Abstract
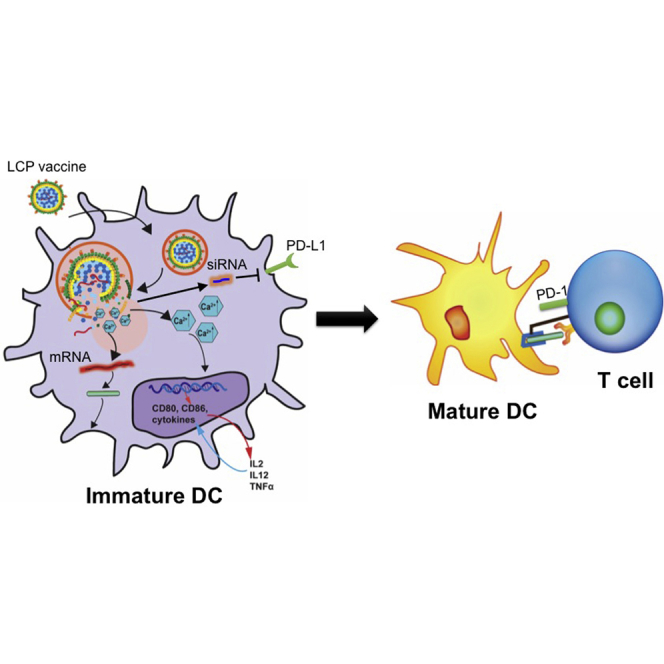
Co-delivery of mRNA encoding a cancer antigen and siRNA against the checkpoint molecule PD-L1 to the dendritic cells in the lymph node has been successfully achieved using a lipid calcium phosphate nanoparticle. The blockade of the immune checkpoint in dendritic cells enhances the activation and proliferation of antigen-specific CD4 and CD8 T cells.
Introduction
Cancer vaccines are designed to target professional antigen-presenting cells (APCs) to induce a potent, antigen-specific immune response as a form of active immunotherapy. The process and presentation of tumor-associated antigens by APCs can break the immune tolerance and elicit antigen-specific effector and memory T cells to reduce tumor burden and prevent tumor relapse. In the past two decades, mRNA has gained attention in an anticancer vaccination approach.1 Intracellularly delivered mRNA could be used as a source for any protein antigens without the possibility of genomic integration. Non-dividing cells, including the dendritic cells (DCs), could be readily transfected by mRNA, but not by DNA.2 Although clinical studies on ex vivo mRNA-modified DCs were launched in the late 1990s,3 the direct use of a cell-free mRNA vaccine was not clinically evaluated until a decade later.4 Weide et al.4 confirmed this in the first clinical trial, when autologous total tumor mRNA was intradermally injected to treat metastatic melanoma. Although granulocyte-macrophage colony stimulating factor (GM-CSF) was administered in situ to recruit more DCs for transfection, no clinical response was observed.4 The disappointing clinical results indicated that lack of efficient cellular uptake and improved mRNA stability were the major obstacles.4, 5, 6, 7
Vaccine-induced T cell immune response involves multiple steps. They include antigen processing and presentation by the APCs, which subsequently lead to activation and proliferation of a specific clone of T cells in the secondary lymphoid tissues. Among the sophisticated regulatory mechanisms, immune checkpoint pathways that either diminish or boost the amplitude of immune responses are extensively investigated. This mechanism, however, is hijacked by the tumor through upregulation of inhibitory checkpoint signals, leading to T cell anergy and immune tolerance of the tumor cells. Although clinical successes of antibody therapies targeting inhibitory signaling receptors such as cytotoxic T lymphocyte-associated antigen 4 (CTLA4)8 or programmed cell death protein 1 (PD-1)9 indicated that the antitumor immunity could be boosted at multiple regulatory levels to achieve therapeutic goals,10 systemic administration of the antibodies risks breaking peripheral tolerance and causing autoimmune diseases, especially because PD-1 is ubiquitously expressed in the lymphoid cells, such as DCs and macrophages. As an alternative strategy, we proposed to block the checkpoint signal pathway between the APCs and the T cells and thus boost T cell activation and proliferation in an antigen-specific manner.
We have previously established a lipid-coated calcium phosphate formulation called lipid calcium phosphate (LCP) nanoparticles (NPs). This formulation has been used for the delivery of nucleic acids,11, 12 peptides,11, 13 and chemodrugs.14 In this study, we investigated the use of LCP NPs as vaccine carriers to deliver antigen mRNA alone or with small interfering RNA (siRNA) targeting an immune checkpoint to DCs in vivo. This LCP-based mRNA vaccine was evaluated in an immune competent B16F10 melanoma model. mRNA encoding a melanoma-associated antigen, tyrosinase-related protein 2 (TRP2), and siRNA targeting programmed cell death protein ligand 1 (PD-L1) were packaged in the LCP NPs. The immunogenic potency was evaluated by the antigen-specific T cell response and the inhibition of tumor growth. Moreover, the adjuvant properties of the LCP NPs were investigated to rationalize the development of an ideal mRNA vaccine carrier.
Results
Chemically Modified mRNA Was Readily Packaged in LCP NPs
Single-stranded RNAs with certain structural features can interact with RNA sensors in the host cells such as Toll-like receptors,15 retinoic acid-inducible protein I,16 and protein kinase R (PKR),17 the activation of which shuts down the translation system and results in low transgene expression. Incorporation of chemically modified nucleosides in RNA preparation has been demonstrated to substantially reduce the activation of RNA sensors and enhance protein expression.18 Alternatively, the RNA sequence could be engineered to minimally elicit immune response and achieve potent protein expression.19 In this study, we adapted the method reported by Warren et al.20 by incorporating 5-methylcytidine (m5C) triphosphate and pseudouridine (φ) triphosphate in the in vitro synthesis of mRNA to ensure a minimal innate immune response and a higher expression level of the antigen.21 In addition, mRNA transcripts were constructed in such a way that they contained a fragment of the 3′ UTR of human α-globin, as well as a 80–100 poly(A) tail for improved stability and translatability.20, 22
Our lab has developed an LCP NP system that has successfully delivered nucleic acid-based therapeutics such as siRNA and plasmid DNA to the target cell in vivo for anticancer therapy. In this study, single-stranded mRNA was loaded in the LCP NPs in the same manner as siRNA. Basically, mRNA was co-precipitated with calcium phosphate by mixing two reverse microemulsions containing mRNA with calcium ions and phosphate ions. The formed NP, which was known as the calcium phosphate (CaP) core, was stabilized by dioleoyl phosphatydic acid (DOPA) and suspended in the oil phase. The final particle was prepared by coating the core particle with 1,2-dioleoyl-3-trimethylammonium-propane chloride salt (DOTAP) and 1,2-distearoryl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol-2000)] ammonium salt (DSPE-PEG) so that it could be stably suspended in the aqueous phase. The hydrodynamic size of the LCP NP was ∼45 nm (Figure S1A) as determined by dynamic light scattering (DLS). The zeta potential was approximately 0 mV (Figure S1B), which is indicative of full PEGylation of the LCP NP. Transmission electron microscopy (TEM) images were taken to investigate the NP morphology and to confirm the size (Figures S1C and S1D). The LCP loaded with mRNA was spherical with a diameter of around 25 nm after uranium acetate staining (Figure S1D). The discrepancy of sizes determined by DLS, and TEM arises because DLS measures the hydrodynamic diameter, taking hydration due to PEG into account, while TEM does not. To determine the encapsulation efficiency, a trace amount of 3H-labeled cytosine triphosphate was incorporated into the in vitro transcription for labeling mRNA. The encapsulation efficiency of mRNA loading was about 60% after optimization.
mRNAs Loaded into LCP NP Were Rapidly Released after Cellular Uptake
As shown in previous studies, the dissolution of CaP core in the acidic endo-lysosomal compartment causes high osmotic pressure, which ruptures the endosomal membrane.23 This mechanism accounts for the efficient endosomal escape and cargo release after cellular uptake of LCP NPs. To track the intracellular trafficking of mRNA cargoes under confocal microscopy, Cy3-labeled cytidine triphosphate (CTP) was incorporated into RNA molecules during the in vitro transcription. After incubation of mannose functionalized LCP NPs with murine JAWSII DCs on ice for 30 min, LCP primarily adhered to the surface of cells (Figure 1Ai). The 37°C heat pulse induced energy-dependent receptor-mediated endocytosis. Within the first 20 min of internalization, Cy3-labeled mRNA mostly co-localized within the endosomal compartment, which was stained with LysoTracker (Figure 1Aii). Longer incubation (40 and 60 min) led to the spread of the Cy3-labeled mRNA throughout the cells (Figures 1Aiii and 1Aiv). The fluorescence intensity of Cy3 in the treated cells also increased with time. The condensation of mRNA with CaP resulted in Cy3 fluorescence quenching, which could be reversed by dissolving the CaP in acid (Figure S1E). The enhancement of the fluorescence intensity in the 60 min image compared with that in the 10 min image indicated the release and de-condensation of mRNA after endosomal escape.
Figure 1.

LCP NP Efficiently Delivered mRNA to Murine Dendritic Cell JAWSII and Resulted in Prompt Transgene Expression
(A) Intracellular trafficking studies of Cy3-labeled mRNA cargoes delivered by LCP NP using confocal microscopy. The images taken 0, 20, 40, and 60 min after heat pulse were presented to demonstrate endosomal escape and the release of mRNA cargoes. The bottom panel displays the zoomed-in view of single cells in the bottom-right corner of the top panels. (B) Flow cytometric analysis of the transfection efficiency of the DCs by modified and unmodified mRNA delivered by LCP NPS. UM-mRNA, unmodified mRNA; M-mRNA, modified mRNA. (C) Quantitation of the percentage of transfected DCs by LCP containing modified and unmodified mRNA. Data points represent group mean ± SD (n = 4 for all groups, Student’s t test). (D) Real-time monitoring of transgene expression using luciferase as a model gene. Data points represent group mean ± SD (n = 4 for all time points).
The high release efficiency led to high transfection efficiency, as demonstrated by model gene-encoding EGFPs. Twenty-four hours after transfection, up to 68% of the DCs expressed EGFP, as determined by flow cytometry (Figure 1B). As a comparison, LCP loaded with unmodified mRNA only transfected about 41% of the DCs in vitro (Figure 1C). The discrepancy was probably because unmodified RNA carried 5′-triphosphate, which could activate PKR, a global repressor of protein translation, and result in the suppression of the transgene expression.24 In the kinetics study, chemically modified mRNA encoding firefly luciferase was synthesized and used to transfect the murine DCs. Luciferase is a short-lived enzyme with a half-life of approximately 2 hr.25 The high turnover rate allows a more accurate estimation of available mRNA for translation than using transcripts for EGFP. Although the release of mRNA started within 1 hr of transfection (Figure 1A), the translation product was not detectable until hour 2 (Figure 1D). The expression level peaked around 4 hr post-transfection and plateaued thereafter. The mRNA co-precipitated with CaP during packaging; therefore, the dissolution of CaP accommodated the rapid expression of mRNA.
The Adjuvant Activity of LCP NPs Prompts the Maturation of DCs
LCP NP is an artificial nanosized Ca2+ reservoir, which is advantageous for the maturation of DCs, because Ca2+ ionophores can stimulate immature DCs to acquire many of the morphological and functional properties of the mature DCs.26, 27, 28 To visualize the real-time change of intracellular Ca2+ concentration after LCP NP administration, immature JAWSII DCs were pretreated with Fluo-4 AM, a cell permeant calcium indicator. The cells show a minimal fluorescence baseline at rest, as expected; however, binding free Ca2+ to Fluo-4 increases fluorescence emission intensity up to 100-fold. Fluo-4 fluorescence intensity began to increase around 20 min after transfection and continued until 60 min after transfection (Figure 2A). Because the dissociation constant (KD) for Fluo-4 calcium binding is 345 nM, the increased fluorescence intensity reflected a significant increase in the intracellular Ca2+ level above the resting status (10–50 nM).
Figure 2.

The Administration of LCP NP Triggered Cytokine Expression, Increased Intracellular Ca2+ Concentration, and Induced DC Maturation
(A) Real-time monitoring of intracellular Ca2+ using Fluo-4 AM as a Ca2+ indicator. Intracellular Ca2+ in the JAWSII cells increased within 60 min of transfection of LCP. (B) Flow cytometry analysis of co-stimulation marker expression levels 24 hr after transfection. DOTAP-LCP, LCP with an outer leaflet of DOTAP; DOPC-LCP, LCP with an outer leaflet of DOPC; DOTAP-lipo, lipoplex formed between cationic liposomes (DOTAP/cholesterol/DSPE-PEG/DSPE-PEG-mannose) and mRNA. (C–E) RT-PCR analysis of cytokine transcripts stimulated by either empty LCP or LCP loaded with chemically modified mRNA. The y axis indicated the fold change of the cytokine expression. Data points represent group mean ± SD (n = 3 for all groups).
The effects of the LCP on the maturation of JAWSII cells were evaluated based on the expression level of CD80 and CD86 using flow cytometry. LCP NP-treated cells exhibited the greatest upregulation of CD80 and CD86 (Figure 2B). In contrast, lipoplex formulated by mixing DOTAP/cholesterol/DSPE-PEG liposomes and mRNA showed only limited upregulation of both surface markers compared with untreated cells. Given that all particles used identical targeting ligands, this difference in maturation activity most likely stemmed from the presence of CaP, which was the major component of LCP NPs but was absent from lipoplex. The contribution of cationic lipids to DC maturation was also investigated, because they were frequently used as a vaccine adjuvant.29 However, our results demonstrated that LCP NPs could induce the expression of co-stimulating molecules regardless of the type of outer leaflet lipid coating (Figure 2B). This suggests that Ca2+ plays a more important role in DC maturation than the cationic lipid adjuvant in LCP NP-based vaccine.
The cytokine expression profiles of DCs treated with LCP were characterized as well. A panel of cytokines, including interleukin (IL)-2, tumor necrosis factor alpha (TNF-α), IL-12, IL-6, and IL-10, was examined using RT-PCR analysis of total RNA extracted from DCs treated with mRNA-loaded or empty LCP NPs. The results revealed a pulse of pro-inflammatory cytokines with up to 5-fold increase of TNF-α (Figure 2C), 10-fold increase of IL-12 (Figure 2D), and 3-fold increase of IL-2 by LCP NPs with or without chemically modified mRNA (Figure 2E). The cytokine release peaked 2 hr post-transfection and was restored to the normal level within 6 hr. No expression was detected in other tested cytokines (data not shown). The data indicated that the presence of mRNA cargo did not induce more pro-inflammatory cytokines than were induced by the adjuvant activity of the vectors, which showed that immuno-stimulant properties of in vitro-transcribed mRNA were mostly abolished because of chemical modification.
LCP NP Efficiently Delivered mRNA to DCs in the Lymph Nodes after Subcutaneous Administration
In addition to its potent adjuvant property, LCP was evaluated as a gene delivery carrier. 3H-CTP was used to label mRNA to track the biodistribution of the mRNA delivered by LCP. Results were detected mRNA in the lymph nodes as early as 4 hr after subcutaneous injection (Figure 3A). The accumulation of mRNA peaked at 8 hr, with 170% injected dose per gram of tissue. The accumulation of LCP in the lymph nodes plateaued until 24 hr after injection. By then, only 10% of the injected LCP NPs were retained at the injection site (data not shown).
Figure 3.

LCP Efficiently Deliver mRNA to DCs in the Lymph Nodes for Transgene Expression
(A) Time course biodistribution of 3H-labeled mRNA delivered by LCP NP. Data points represent group mean ± SD (n = 3 for all groups). (B) Imaging of lymph nodes harvested 24 hr post-administration of LCP vaccine. (C) Flow cytometry analysis of the cell population that took up LCP NP in the lymph nodes and spleen. Data points represent group mean (n = 4 for all groups). (D) Visualization of EGFP expression on the cryosection of the lymph node using confocal microscopy. (E–G) Quantitation of cytokine expression levels using ELISA to determine the induction of cytokine by LCP after subcutaneous inoculation. ND, not detected. Data points represent group mean ± SD (n = 3 for all groups).
The distribution of mRNA was also visualized through an in vivo imaging system using Cy3-labeled mRNA. High levels of fluorescence intensity were observed in the major draining lymph nodes (axillary, brachial, inguinal, and sciatic) 24 hr after injection (Figure 3B). These lymph nodes were subjected to cryosectioning for microscopic analysis.
Further analyses were performed to explore the cell types in the secondary lymphoid tissues that took up the NPs using DiI-labeled LCP NPs. A single-cell suspension was prepared from the collection of axillary, brachial, inguinal, and sciatic lymph nodes harvested from both ipsilateral and contralateral sides of the injection site. Fluorophore-conjugated antibodies against murine CD11b/F4/80 and CD11c/CD123 were used to stain and define cell populations. The data demonstrated that although the absolute percentage of cells that took up the NPs in the lymph node was below 5%, up to 50% of these DiI-positive cells were CD11c+/CD123+ DCs (Figure 3C). In contrast, only 20% of the cells that took up LCP in the spleen were DCs. Such a different distribution pattern in different organs may be related to the location and abundance of different cell populations, as well as the size and surface modification of the LCP. More investigation is needed to elucidate this phenomenon. Because mannose receptor (CD206) is primarily expressed on the surface of macrophages and immature DCs, both macrophages and DCs were the potential targets of the LCP NPs. Having characterized the recipients of LCP NPs in the lymph nodes, chemically modified mRNA encoding EGFP was delivered using LCP NPs to visualize in vivo expression. As was shown under the confocal microscope, EGFP was observed in a fraction of cells in the lymph nodes 24 hr post-injection (Figure 3D). The local cytokine expression levels in the lymph nodes were evaluated with ELISA (Figures 3E–3G) after vaccination. Although the LCP remained in the lymph nodes 24 hr post-injection, the cytokine expression peaked 2 hr post-injection and dropped significantly within 4 hr, which was in consistent with in vitro cytokine induction profiles.
LCP-Based Vaccine Delivered mRNA Vaccine Encoding TRP2 and Induced an Antigen-Specific Immune Response
We then proceeded to evaluate therapeutic efficacy of mRNA vaccine in the murine melanoma syngeneic tumor model. The TRP2 protein, one of the most widely tested tumor-associated antigens for B16F10 cells, was chosen. The expression longevity of TRP2 was also investigated in vivo. Western blot analysis indicated that a single injection, equivalent to 10 μg of modified mRNA, resulted in a sustained expression of TRP2 in the lymph nodes for up to 5 days (Figure 4A). Under the same dose, an in vivo cytotoxic T lymphocyte (CTL) assay was performed one week later to examine the antigen-specific T cell response. As shown in Figure 4B, mice vaccinated with chemically modified mRNA encoding TRP2 specifically killed 64% of TRP2 peptide-pulsed versus ovalbumin (OVA) peptide-pulsed splenocytes. To determine whether the mRNA vaccine was as potent as the antigenic TRP2 peptide (SVYDFFVWL) vaccine, we vaccinated the animals with a previously published formulation of LCP loaded with 50 μg of peptide and 25 μg of CpG oligonucleotide.13 The results showed that animals vaccinated with only 10 μg of mRNA encoding the full-length TRP2 could induce antigen-specific T cell response as potent as that induced by the peptide-based vaccine (Figure 4B). The study also investigated the immune response elicited by chemically modified mRNA versus regular unmodified mRNA. The higher antigen expression because of higher stability of the modified mRNA benefited the application of the vaccine. The modified mRNA resulted in higher CTL response (64%) than that of regular mRNA (43%) (p < 0.001) (Figure 4B).
Figure 4.

LCP Delivered mRNA Vaccine Elicited a Potent Antigen-Specific T Cell Response and Inhibited Tumor Growth In Vivo
(A) Western blot analysis showing the longevity of TRP2 expression in the lymph nodes after vaccination. UN, untreated, D1–D7, 1–7 days post-injection. (B) Killing efficiency mediated by antigen-specific cytotoxic T cells, which were vaccinated with various formulations. Data points represent group mean ± SD (n = 3 for all groups, ***p < 0.001, one-way ANOVA with post-Turkey test). (C) Growth inhibition of B16F10 melanoma on C57BL/6 syngeneic mice after various vaccine treatments. Data points represent group mean ± SEM (n = 5–7, ***p < 0.001, one-way ANOVA). (D) Examination of IgG titers against TRP2 protein after vaccination. Data points represent group mean ± SD (n = 5–7, *p < 0.05, **p < 0.01, one-way ANOVA with post-Turkey test).
The TRP2-specific immune response induced by the mRNA vaccine showed potent tumor inhibition activity even with an established tumor (∼150 mm3 at the time of vaccination). As shown in Figure 4C, mRNA vaccine significantly inhibited tumor growth, compared with the untreated animals (p < 0.001). In addition, animals vaccinated with chemically modified mRNA demonstrated a more effective growth inhibition compared with that of regular mRNA (p < 0.001), which was consistent with the results of the in vivo CTL assay. Naked mRNA only showed moderate tumor growth inhibition activity. Although the modified mRNA vaccine resulted in a comparable killing efficiency to the peptide vaccine in the CTL assay, the mRNA vaccine demonstrated significantly higher tumor growth inhibition activity than the peptide vaccine. The discrepancy was most likely because the full-length antigen could generate a T cell response for multiple epitopes of the same antigen. Therefore, a combinatory array of TRP2-specific T cells should result in a more potent anticancer activity.
Although the induction of a T cell immune response was the major focus for this anticancer vaccine, we also examined the potential humoral immune response elicited by the mRNA vaccine. An ELISA was performed to detect serum immunoglobulin G (IgG) titers against the full-length TRP2 protein at the endpoint of the tumor growth inhibition study. The results indicated that a relatively high titer could be induced by the modified or unmodified mRNA vaccine delivered by LCP (Figure 4D). The TRP2 peptide was unable to induce any humoral immune response, because the peptide can only bind to mouse major histocompatibility complex class I (MHC class I) molecules. The generation of an antibody against TRP2 antigen provided another potential mechanism for tumor growth inhibition, i.e., antibody-dependent cell-mediated cytotoxicity, which is mediated by natural killer cells, macrophages, or neutrophils.
Knockdown of Checkpoint PD-L1 with siRNA Enhanced the Potency of Trp2 mRNA Vaccine
We then proceeded to include siRNA therapeutics to block the inhibitory checkpoint signal PD-1:PD-L1 pathway regarding its role in T cell priming. Because of the flexibility of the LCP NPs, both siRNA and mRNA could be simultaneously packaged in the same NP with minor optimization. 3H-labeled double-stranded oligonucleotide was used as a substitute for siRNA in the formulation study. The results indicated that LCP preferred to encapsulate lower molecular weight double-stranded oligonucleotides (∼20 bp) compared with higher molecular weight mRNAs (∼2,200 nt). By adjusting the input ratio of oligonucleotides to mRNA from 1:9 (w/w) to 5:5 (w/w), the encapsulation efficiency of mRNA significantly dropped from 77% to 50%, whereas siRNA encapsulation remained unchanged (Figure S1E). We used a 1:9 (siRNA:mRNA, w/w) ratio for the following study. Despite the seemingly low mass of siRNA loaded in the LCP NPs, the molarity of siRNA was still more than 50 times higher than that of mRNA because of the lower molecular weight.
The LCP vaccines loaded with 9 μg of Trp2 or control mRNA and 1 μg of PD-L1 or control siRNA were subcutaneously injected into the mice when the tumor reached 300 mm3. The inclusion of PD-L1 siRNA in the vaccine formulation enhanced the potency of the Trp2 mRNA vaccine compared with the mRNA vaccine alone (p < 0.001), demonstrating significant tumor growth inhibition up to 23 days after tumor inoculation (Figure 5A). Using PD-L1 siRNA alone in the LCP vaccine showed modest tumor growth inhibition (p < 0.001). Western blot analysis also confirmed the knockdown of PD-L1 expression in the lymph nodes by siRNA 24 hr post-vaccination (Figure 5B). This enhanced antitumor effect by the combo vaccine was accompanied by an increase in the CD8+ T cell population in the lymph nodes, spleen, and tumors as determined by flow cytometry analysis (Figure 5C). The combination of these data suggested that removal of PD-L1 on APCs led to enhanced CD8+ T cell activation and proliferation due to blockade of the PD-1:PD-L1 pathway.
Figure 5.

LCP Loaded with Trp2 mRNA and Anti-PD-L1 siRNA Could Significantly Inhibit Large Tumor Growth and Metastasis
(A) Tumor growth inhibition study of the B16F10 melanoma-bearing mice after vaccination with different LCP NPs when the tumor reached 300 mm3. Data points represent group mean ± SEM (n = 5–7, ***p < 0.001, two-way ANOVA with post-test). (B) Flow cytometry analysis of CD8+ T cells generated in the lymph nodes, spleen, and tumor at the endpoint of the tumor inhibition study. Data points represent group mean ± SD (n = 3 for all groups). (C) Western blot analysis that confirmed the knockdown of PD-L1 in the lymph nodes 24 hr post-vaccination. (D) Representative of lymph nodes harvested from animals at the endpoint of the tumor inhibition study. From left to right: inguinal, axillary, brachial, and sciatic lymph nodes on the ipsilateral side of the tumor and inguinal, axillary, brachial, sciatic and lymph nodes on the contralateral side of the tumor. (E) H&E staining images of representative lymph node sections from each treatment group. (Yellow circle highlights the metastasis in the lymph nodes.)
Because melanoma is notorious for metastasis, we examined the lymph nodes of the mice at the endpoint of the experiment to check whether the vaccine could inhibit metastasis of the cancer cells to the sentinel and distant lymph nodes. Results showed that the mice that receiving the combo vaccine had the lowest extent of melanoma metastasis in the lymph nodes (Figures 5D and 5E). In the control groups, even the distant lymph nodes on the contralateral side of the tumor had severe metastases. Although the combo vaccine showed profound therapeutic effects on the primary tumor and potential metastatic sites, it was unclear whether the small primary tumor size, the high number of antigen-specific T cells, or both contributed to the antimetastasis effect.
PD-L1 siRNA in the LCP mRNA Vaccine Was Superior to Anti-PD-L1 Monoclonal Antibody for Promoting T Cell Priming
Because monoclonal antibody (mAb) against PD-L1 has shown an exceptional antitumor effect in preclinical studies, we attempted to compare the adjuvant effect of PD-L1 mAb and siRNA. In a tumor growth inhibition study, B16F10 tumor-bearing mice received LCP (Trp2 mRNA+ siPD-L1), LCP (Trp2 mRNA+siCon), or LCP (Trp2 mRNA) + PD-L1 mAb, and the tumor volumes and animal weights were monitored (Figure 6A). Pharmacodynamics of the mRNA vaccine treatment with a different adjuvant showed a time-dependent pattern. Anti-PD-L1 mAb with mRNA vaccine showed a more potent tumor inhibition effect than siRNA combo vaccine treatment within 4 days of treatment. The average tumor volume of the mAb combo group was about half of that of siRNA combo group (61 ± 20 mm3 versus 112 ± 32 mm3) (Figure 6A, inset). This phenomenon was overturned 6 days post-injection. The effect of the mAb seemed to diminish, and the tumor growth rate started to pick up. The tumors grew at the same rate of the mRNA vaccine-alone group (Figure 6A, inset). The siRNA combo vaccine group, however, showed a dip in the tumor growth curve day 6 post-injection and maintained a relatively flat growth rate.
Figure 6.

PD-L1 siRNA Delivered with mRNA Vaccine Showed a Higher Immune Boosting Effect Compared with PD-L1 mAb
(A) Tumor growth inhibition assay comparing the efficacy of siRNA and mAb against PD-L1 when used with mRNA vaccines. Data points represent group mean ± SEM (n = 5, ***p < 0.001, two-way ANOVA with post-test). Insert: tumor growth curve of the siRNA and mAb groups with high magnification. The arrow indicates the day of vaccination and administration of PD-L1 mAb. (B) Record of animal weight as a means to evaluate treatment toxicity. (C) Representative flow cytometry analysis of CD8+ INFγ+ cells in the CD3+ T cell population. (D) Quantitation of flow cytometry analysis. Data points represent group mean ± SD (n = 5, ***p < 0.001, one-way ANOVA). (E). Representative flow cytometry analysis of CD4+ INFγ+ cells in the CD3+ T cell population. (F) Quantitation of flow cytometry analysis. Data points represent group mean ± SD (n = 5, ***p < 0.001, one-way ANOVA).
In an attempt to elucidate this phenomenon, splenocytes were collected from the mice at the endpoint of the experiment (20 days post-tumor inoculation and 12 days post-vaccination). The splenocytes were activated with full-length TRP2 protein in vitro, and the expression of interferon (IFN)-γ was analyzed using a flow cytometer after the cells were stained with CD3, CD4, CD8, and IFN-γ. The result showed that siRNA against PD-L1, when delivered to the same APCs with mRNA vaccine, could significantly boost antigen-specific CD8+ cells up to 2-fold compared with mRNA vaccine alone (2.21% versus 1.10%). PD-L1 mAb, however, only showed a minor boosting effect (1.41% versus 1.10%) (Figures 6C and 6D). It thus could be speculated that the initial several days of tumor inhibition in the mAb combo group was attributed to the direct blocking of the PD-1:PD-L1 axis between cancer cells and T cells in the tumor. When the PD-L1 mAb was cleared from the circulation, the inhibitory effect started to diminish and tumor growth took off. The same pattern was observed in antigen-specific CD4+ cells (Figures 6E and 6F).
Discussion
Direct use of mRNA for cancer vaccination has been demonstrated to be effective in the induction of antigen-specific T cell responses in several mouse models, whether it was injected as naked mRNA30 or protected mRNA by carriers.31, 32 Our success of mRNA vaccine results from the rational design of the carrier NP, as well as from exploiting the regulatory mechanism for T cell activation and proliferation. The nanotechnology provides an approach that could address the delivery issue in mRNA vaccine development. LCP NPs were core membrane-structured NPs that showed great stability in vivo due to the high density of PEGylation on the lipid membrane.11 The high stability, coupled with the feature of nanosize, ensures the NPs to efficiently drain via the lymphatics into the lymph node after subcutaneous administration,33 leading to high accumulation of NPs in the lymph nodes in which DCs are abundant (Figures 3A and 3B). Nucleic acid cargoes were co-precipitated with CaP core during preparation, allowing full protection of mRNA before disassembly triggered by the acidic environment.23 This assembly mechanism guaranteed efficient cargo release. Reports showed that single-stranded mRNA demonstrated different condensation and de-condensation kinetics compared with plasmid DNA, especially when complexed with cationic polymers.34 RNA with high flexibility resulted in higher binding affinity toward the cationic polymers than plasmid DNA, making it more stable against competitive release by other charged molecules in the cytoplasm.35 In contrast, the encapsulation mechanism of mRNA in the LCP formulation avoided conventional charge-based condensation and led to efficient release of the mRNA cargo.
As was often observed in the clinical studies of mRNA vaccine, adjuvant such as GM-CSF was administered with mRNA antigen to boost the immune response.4, 7 Although single-stranded RNA with several structure features could activate various RNA sensors, such as Toll-like receptor, RIG-I, and PKR,15, 16, 36 to potentiate immune response, this self-adjuvant activity was always accompanied by a low expression level. Warren et al.20 have reported the use of chemically modified nucleoside triphosphates during in vitro synthesis to abolish the immune-stimulating activity and enhance the protein expression level. However, chemically modified mRNA with a low immune-stimulating property is not always favorable for vaccine therapy. Studies on mRNA vaccines showed some mixed signals. Oberli et al.37 employed lipoplex composed of novel lipids to deliver mRNA vaccine only to find that unmodified mRNA induced a higher antigen-specific T cell response than chemically modified mRNAs. They speculated that the IFN-α induced by unmodified mRNA was necessary for the induction of CD8+ T cells. However, Udhayakumar et al.38 reported a polyplex system using an amphipathic polypeptide for mRNA vaccine therapy. They observed that the polyplex with chemically modified mRNA induced a lower level of cytokines and reduced DC maturation, although they showed a higher antigen expression level. The higher expression by modified mRNA was superior to the high cytokine levels induced by unmodified mRNA, leading to higher T cell induction and vaccine efficacy. These seemingly contradictory results unanimously point to an old rational: inducing a potent immune response requires an abundance of antigen, as well as adjuvant. Therefore, we synthesized the chemically modified mRNA to achieve high and sustained expression of antigens. This approach resulted in the sustained in vivo expression of TRP2 protein in the lymph node for up to 5 days post-injection (Figure 4A). The CTL study and efficacy studies confirmed that the modified mRNA was superior to the unmodified mRNA in the context of LCP NPs, because it caused stronger tumor suppression (Figure 4C), probably mediated by higher CD8+ T cell response (Figure 4B). However, the low immunostimulating property of the chemically modified mRNA was compensated by the calcium ions released from the LCP NPs, as was evidenced by the fluorescent Ca2+ indicator (Figure 2A). Calcium ionophores were claimed to be capable of rapidly and consistently inducing myeloid origin mononuclear cells to acquire mature DC morphological properties.27 The increase of intracellular Ca2+ activates transcription factors such as NFAT or nuclear factor κB (NF-κB),39 which in turn activate the expression of co-stimulating receptors CD80 and CD86 (Figure 2B), as well as the pro-inflammatory cytokines. The LCP NPs could rapidly induce high levels of cytokine expression, regardless of the payload it carried (Figures 2C–2E).
Moreover, mRNA vaccine showed a more potent antitumor effect than peptide vaccine (Figure 4C), although both induced comparable levels of cytotoxic T cells in the CTL assay (Figure 4B). The mRNA vaccine could generate multiple epitopes for antigen presentation and induce CD8+ T cells targeting various epitopes, which might account for the discrepancy. It was also possible that antigen-specific antibody elicited by mRNA vaccine, but not peptide vaccine, contribute to the difference in antitumor effect (Figure 4D).
The LCP NPs conferred a formulation advantage to mRNA vaccine by protecting and targeting the vaccine to the APCs. In addition, LCP served as the calcium ion depot that facilitates the DC maturation, whereas this adjuvant activity was often achieved by co-delivery of molecules with specific pathogen-associated molecular patterns to activate pattern recognition receptors.40 Other than sustained antigen presentation and DC maturation, our study proposed and successfully validated that immune response by a vaccine could be enhanced by blocking PD-1: PD-L1 ligation between APCs and T cells during antigen presentation instead of blocking the ligations between T cells and cancer cells, which are extensively investigated.
PD-L1, a counterreceptor for PD-1, is widely expressed in both lymphoid tissues and non-lymphoid tissues.41 The ligation between PD-L1 and PD-1 leads to T cell exhaustion and negative regulation in T cell activation.42, 43, 44 Blocking the ligation with antibodies against either the ligand or the receptor has shown great promise in clinical trials for late-stage cancers.45 Despite the complex mechanism of action for PD-L1: PD-1 blockade, major preclinical and clinical studies considered the PD-1 (or PD-L1) antibody therapy as tools of interest to inhibit the effector T cell anergy in the tumor microenvironment.10 However, early research by Freeman et al.42 suggested the engagement of PD-1 by PD-L1 led to the inhibition of T cell proliferation and cytokine secretion. In their study, the co-stimulatory signal (CD80/CD86) on APCs was considerably offset by the presence of PD-L1, as was shown by an 80% reduction of proliferation rate.42 The function of PD-1: PD-L1 between APCs and T cells was confirmed in a PD-L1 knockout mouse model, in which both CD4+ and CD8+ T cell response were markedly elevated compared with that of the wild-type mice.46 Another study suggested that the ligation between PD-1: PD-L1 would block the stop signal sent by T cell receptor and shorten the physical contact duration between T cell and DCs.47 These studies undoubtedly implied a novel therapeutic target to enhance the vaccine potency by shifting the balance between stimulatory and inhibitory signals during the early stage of T cell response. The blockade of PD-L1: PD-1 pathway by RNA interference prompted the activation and proliferation of T cells during the antigen presentation of DCs in the secondary lymphoid tissues. The elevated T cell response, reflected by a substantial increase in the population of CD8+ tumor-infiltrating lymphocytes (Figure 5B), effectively sequestered the primary tumor growth (Figure 5A). Moreover, the increased number of CD8+ T cells in the spleen and lymph nodes showed a systemic response that was effective in inhibiting melanoma metastasis (Figures 5D and 5E). In contrast, the knockdown of PD-L1 alone in the absence of mRNA vaccines failed to inhibit the tumor growth due to the lack of antigen presentation, reiterating the importance of coupling a cancer vaccine with immune checkpoint therapy. The immune checkpoint inhibition in the current study is limited only to those DCs that express and present the antigen due to the co-delivery of both mRNA (antigen source) and siRNA (checkpoint inhibitor) with the same NPs. The antigen-specific inhibition of the checkpoint regulation is one of salient points of the study.
The tumor inhibition study, which was designed to compare the widely used anti-PD-L1 antibody (clone B7-H1) with RNAi therapy regarding their vaccine potentiation efficacy, unfolded the mechanisms of actions for these two molecules targeting the same targets. The blockade of the PD-L1/PD-1 axis using either mAb or siRNA could potentially inhibit tumor growth via two possible pathways: (1) blocking the PD-1:PD-L1 axis between DCs and T cells to enhance T cell priming or (2) blocking the PD-1:PD-L1 axis between T cells and cancer cells to enhance the efficacy of effector T cells. In our study, the administration route and targeting ligand on the LCP NP make it difficult to for LCP vaccine to access cancer cells, because they are designed to target APCs in the lymph node. Therefore, the siRNA in LCP primarily accumulated in the lymph nodes after administration. However, the mAb against PD-L1 could access both lymph nodes and tumors; therefore, it could potentiate the vaccine on both pathways. However, the results from the T cell IFN-γ assay (Figures 6C and 6E) suggested that co-delivery of PD-L1 siRNA and mRNA antigen into the same APCs showed significantly higher T cell priming efficiency than that induced by the combination of PD-L1 mAb and mRNA vaccine. The tumor inhibition study (Figure 6A) study also suggested that the effect of PD-L1 mAb-mediated tumor inhibition primarily stemmed from the blockade of PD-1:PD-L1 ligation between the T cells and cancer cells, which explained the diminished tumor inhibitory effect once the mAb was cleared from the circulation system.
Conclusions
The administration of mRNA vaccine in vivo appears to be an insurmountable challenge that often compromises the therapeutic efficacy. To address this problem, we have used a lipid-coated CaP NP as a carrier that efficiently delivered mRNA to the DCs in the lymph nodes for antigen expression. Single vaccination using a LCP-based mRNA vaccine could induce a robust antigen-specific T cell response, as well as a humoral immune response that led to significant tumor growth inhibition. Moreover, siRNA against PD-L1 was encapsulated in the LCP to downregulate the negative regulator for T cell activation in the APCs. Simultaneous manipulation of APCs for T cell priming drastically facilitated the activation and proliferation of cytotoxic T cells, consolidating a new mechanism of action for enhancing vaccine immune response. Overall, the study provided a rational design model for an mRNA vaccine. Not only do multiple aspects such as delivery efficiency, adjuvant activity, stability, and translatability of mRNA molecules need to be considered, but each component of the vaccine also needs to function synergistically in a spatiotemporal manner. Our results also imply that effective subunit vaccine can be similarly constructed for prophylactic and therapeutic intervention for infectious diseases.
Materials and Methods
Chemicals and Reagents
Lipids, namely DOPA, DOTAP, and DSPE-PEG, were purchased from Avanti Polar Lipids (Alabaster, AL). Mannose-PEG-DSPE was synthesized using a previously established protocol.13, 48 Toll-like receptor 9 agonist CpG ODN 1826 (5′-TCCATGACGTTCCTGACGTT-3′) and predesigned siRNA against PD-L1 (siRNA ID: SASI_Mm01_00062675) were obtained from Sigma-Aldrich (St. Louis, MO). Cholesterol, cyclohexane, Igepal CO-520, and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise mentioned. H-2Kb restricted peptides TRP2 (SVYDFFVWL, MW 1175), OVA (SIINFEKL, MW 1773), and modified TRP2 peptide, p-TRP2 (pSpSSSVYDFFVWL, MW 1626) were purchased from Peptide 2.0 (Chantilly, VA). Nucleotides (ATP, guanosine triphosphate [GTP], CTP, and uridine triphosphate [UTP]) were purchased from Affymetrix (Santa Clara, CA), and modified nucleotides (m5C-5′-triphosphate and pseudouridine-5′-triphosphate) were purchased from TriLink BioTechnologies (San Diego, CA).
Cell Lines and Animals
Both murine immature DC line JAWSII (syngeneic with C57BL/6) and murine melanoma cell line B16F10 (syngeneic with C57BL/6) were purchased from ATCC. JAWSII cells were cultured in MEM-α medium supplemented with 20% fetal bovine serum (Life Technologies, Carlsbad, CA) and 5 ng/mL murine recombinant GM-CSF (BD Biosciences, San Jose, CA). B16F10 cells were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). C57BL/6 female mice aged 6–8 weeks were purchased from NCI (Frederick, MD). All animal protocols were approved by the University of North Carolina at Chapel Hill’s Institutional Animal Care and Use Committee.
In Vitro Transcription of mRNAs
The genes of interest (EGFP, firefly luciferase, and TRP2) mRNA were sub-cloned into a modified pcDNA3.0 vector. The open reading frame of the gene was flanked by the Kozak sequence (5′-GCCACC-3′) in the 5′ UTR and the human α-globin sequence in the 3′ UTR. The template for in vitro transcription was prepared by PCR amplification of the gene, followed by agarose gel electrophoresis purification. mRNAs were synthesized with the MEGAscript T7 Transcription kit (Life Technologies). For two reactions in a 40 μL volume, 1.6 μg of PCR amplicon of the gene was incubated with 2.4 μL Anti-Reverse Cap Analog (ARCA) 3′-O-Me-m7G(5′) ppp(5′)G (100 mM) (NEB, Ipswich, MA), 0.6 μL GTP (100 mM), 3 μL m5C-5′-triphosphate (100 mM), 3 μL pseudouridine-5′-triphosphate (100 mM), 3 μL ATP (100 mM), 4 μL buffer, and 4 μL polymerase. The reaction was incubated at 37°C for 4 hr. The resulting mRNA was polyadenylated using a Poly(A) Tailing kit (Life Technologies). The final product was treated with TURBO DNase provided by the MEGAscript kit and purified with a MEGAclear kit (Life Technologies). For synthesis of 3H-labeled mRNA, 5% CTP was replaced with 3H-labeled CTP (PerkinElmer, Akron, OH) in the reaction. For Cy3-labeled mRNA, 5% CTP was replaced with Cy3-CTP (GE Healthcare, Chalfont, Buckinghamshire, UK).
Preparation of LCP NPs
The LCP NPs were synthesized in a water-in-oil reverse microemulsion. Briefly, 50 μg of mRNA was added to 600 μL of 2.5 M CaCl2, which was subsequently dispersed in 20 mL of cyclohexane/Igepal CO-520 (71:29, v/v) oil phase to form the Ca phase. Another 600 μL of 12.5 mM Na2HPO4 (pH 9.0) were dispersed in a second oil phase. The two microemulsions were mixed and stirred at room temperature for 5 min before 400 μL of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE; 20 mM dissolved in chloroform) was added. The microemulsion was stirred for another 25 min before an equal volume of ethanol was added to break the microemulsion. The CaP cores loaded with mRNA were collected with centrifugation at 10,000× g and then washed twice with 20 mL of ethanol to remove residual cyclohexane and surfactant. The pellets were resuspended in 2 mL of chloroform and stored at −20°C for future use. Final LCP NPs were formed by mixing 2 mL of CaP cores with 140 μL of 20 mM DOTAP, 140 μL of 20 mM cholesterol, 100 μL of 20 mM DSPE-PEG2000, and 40 μL of 10 mM DSPE-PEG-mannose in chloroform. After removal of chloroform under reduced pressure, LCP NPs were formed by rehydration with 100 μL of 5% glucose. LCP NPs were observed using TEM (JEOL 100CX II TEM, JEOL, Japan). Particle size and zeta potential were measured with a Malvern Zetasizer Nano ZS in water (Malvern, Worcestershire, UK).
Real-Time Intracellular Trafficking of mRNA in DCs
To observe intracellular trafficking of Cy3-labeled mRNA delivered by LCP, an amount of LCP NP Cy3-labeled mRNA was added to 5 × 106 JAWSII cells seeded in a 33 mm glass-bottom Petri dish (MatTek, Ashland, MA) to reach a 1 μg/mL mRNA concentration. JAWSII cells were treated with Hoechst 33342 (Thermo Fisher Scientific) and LysoTracker (Thermo Fisher Scientific) to stain nuclei, endosomes and lysosomes before treatment per the manufacturer’s instructions. LCP NPs were incubated with cells on ice for 30 min and then washed with ice-cold PBS twice. Cells were later transferred to a 37°C chamber attached to an Olympus FV1000 confocal microscope (Olympus, Center Valley, PA). The images were taken every 5 min for 1 hr total. The images were processed using an FV10-ASW 4.0 Viewer (Olympus).
Transfection Study
To evaluate the transfection efficiency of LCP NPs on JAWSII cells, a volume of LCP NPs equivalent to mRNA encoding EGFP or firefly luciferase was added to 1 × 104 cells in a 96-well plate in Opti-MEM medium so that the final concentration of mRNA was 0.5 μg/mL. The medium was refreshed with full medium 4 hr post-transfection. Images were taken using a Nikon epi-fluorescence microscope (Nikon, Tokyo, Japan). The percentage of transfected cells was determined using flow cytometry (BD Biosciences). Luciferase expression was evaluated using a luciferase assay kit (Promega, Madison, WI). The experiments were performed in triplicate.
Real-Time Monitoring of Intracellular Ca2+ Concentration
2 × 105 JAWSII cell were seeded in a 33 mm glass-bottom Petri dish (MatTek). Cells were incubated with 2.5 mM Fluo-4 AM (Thermo Fisher Scientific) in the presence of 0.02% Pluronic F-127 for 1 hr at 37°C. An amount of LCP NP mRNA was added to the cells to reach a concentration equivalent to 1 μg/mL of mRNA. The cells were incubated on ice for 30 min. Cells were then washed twice with ice-cold PBS before observation with an epifluorescence microscope at room temperature. The quantitation of fluorescence intensity was determined using ImageJ software (NIH).
Biodistribution Study of LCP
6- to 8-week-old female C57BL/6 mice were subcutaneously injected to the flank with LCP loaded with 10 μg 3H-labeled mRNA. The mice were then sacrificed 4, 8, 12, and 24 hr after vaccination, and the major organs were harvested. Inguinal, axillary, brachial, and sciatic lymph nodes were also collected for analysis. Tissues of 100 mg or less were dissolved in 1 mL of Tissue Solubilizer (GE Healthcare) and incubated at 50°C. Next, 200 μL of the preceding samples were mixed with 4 mL of Ultima Gold scintillation cocktail (PerkinElmer) and 300 μL of 30% H2O2. The mixture was then subjected to scintillation counting (BD Biosciences). The experiments were performed in triplicate.
In Vivo CTL Assay
In vivo CTL was conducted per a previously published protocol.13 C57BL/6 mice were vaccinated with LCP mRNA vaccine (equivalent to 10 μg of mRNA) on the flank. Seven days later, the mice were intravenously injected with a mix of 5 × 106 splenocytes, half of which were pulsed by TRP2 peptide (10 μM) while the other half by OVA peptide (10 μM). Both cells were stained with 2 μM of PKH-26 (Sigma-Aldrich, St. Louis, MO). The TRP2-pulsed cells were labeled with 4 μM of carboxyfluorescein succinimidyl ester (CFSE), and OVA-pulsed cells were stained with 0.4 μM CFSE. These two populations were referred to as CFSEhigh (TRP2-pulsed cells) and CFSElow (OVA-pulsed cells). After 18 hr, splenocytes from these treated mice were collected and subjected to flow cytometry analysis. The number of CFSEhigh and CFSElow was calculated, and the in vivo TRP2-specific lysis percentage was enumerated. The experiments were conducted in triplicate.
Tumor Growth Inhibition
For vaccination studies, 6- to 8-week-old female C57BL/6 mice were subcutaneously inoculated with 2 × 105 B16F10 cells on their flanks on day 0. The mice were treated with the LCP vaccine (equivalent to 10 μg of mRNA) when tumor sizes reached 150 mm3. For combo vaccine therapy, 6- to 8-week-old female C57BL/6 mice were inoculated subcutaneously with 2 × 105 B16F10 cells on their lower back on day 0. The animals received vaccines (equivalent to 9 μg of mRNA and 1 μg of siRNA) when the tumor size reached 300 mm3. The tumor size was measured every 2 to 3 days using digital calipers (Thermo Fisher Scientific, Pittsburgh, PA) and calculated using the following equation: . Body weight was also monitored. For the study that compared the efficacy of PD-L1 siRNA and PD-L1 mAb, 6- to 8-week-old female C57BL/6 mice were inoculated subcutaneously with 2 × 105 B16F10 cells on their lower back on day 0. On day 12, animals received one dose of LCP (Trp2 mRNA+ siCon), LCP (Trp2 mRNA+ siPD-L1) (equivalent to 9 μg of mRNA and 1 μg siRNA), or LCP (Trp2 mRNA) (equivalent to 9 μg of mRNA), together with PD-L1 mAb (clone B7-H1, 5 mg/kg, intraperitoneally) (Bioxcell, West Lebanon, NH). The tumor size was measured every two days.
ELISA
The quantitation of serum IgG against full-length TRP2 protein was conducted using an ELISA. A 96-well plate was coated with 20 μg/mL of full-length TRP2 protein, which was expressed and purified from E. coli in the presence of sodium bicarbonate buffer (100 mM) overnight at 4°C. After blocking and washing, the serum was diluted at different titers and incubated in the 96-well plate at room temperature for 2 hr. After washing, horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Santa Cruz) was added to the plate before incubating for another 2 hr. Next, 200 μL of tetramethylbenzidine (TMB) substrate was dispensed into the wells before incubating at room temperature for 30 min. Finally, 100 μL of stopping solution was added. The plate was then read at 450 nm absorbance using a plate reader (PerkinElmer).
For ELISA that determined the cytokine expression in the lymph nodes, animals received LCP NP injection equivalent to 10 μg of mRNA on the flank of the mice. Animals were sacrificed 2 and 4 hr post-injection. The axillary, bronchial, inguinal, and sciatic lymph nodes from the injection sides were collected and homogenized in 100 μL of PBS buffer. The supernatant of the homogenate was used for the assay. The ELISA kits for detecting IL-2, IL-12, and TNF-α were purchased from Thermo Fisher Scientific. The assay was performed according to the manufacturer’s manual. 10 μL of the supernatant was used in the assay.
RT-PCR Analysis
To monitor the real-time cytokine expression profile, an amount of LCP NPs equivalent to 1 μg of mRNA was added to 2 × 105 JAWSII cells seeded in a 24-well plate. Cells were harvested 2, 4, 6, 8, and 24 hr post-transfection. Total mRNA was extracted from the cells using an RNeasy kit (QIAGEN, Germantown, MD). RNA was reverse transcribed to cDNA using the SuperScript First-Strand Synthesis System (Thermo Fisher Scientific). Then, 200 ng of cDNA was mixed with a TaqMan Universal PCR Master Mix (Thermo Fisher Scientific) and the reactions were conducted using a 7500 Real-Time PCR System (Thermo Fisher Scientific). Actin was used as an endogenous control. The data were analyzed with the 7500 software. The RT-PCR was performed in triplicate. Primers for RT-PCR were purchased from Thermo Fisher Scientific: TNF-α (Mm00443258_m1), IL-12 (Mm00434165_m1), IL-2 (Mm00434256_m1), IL-6 (Mm00446190_m1), and IL-10 (Mm01288386_m1).
In Vivo Imaging of LCP NPs
An amount of LCP NPs equivalent to 10 μg of Cy3-labeled mRNA was subcutaneously injected into the flanks of the C57BL/6 mice. Twenty-four hours after injection, mice were sacrificed and the major lymph nodes were collected. The distribution of LCP NPs in the lymph nodes was imaged using Kodak In-Vivo FX PRO (Kodak, Rochester, NY). Images were processed using Carestream software. The lymph nodes were then cryosectioned and counterstained with DAPI. The microscale distribution of LCP NP was imaged using a Nikon epifluorescence microscope. To visualize the EGFP expression in the lymph nodes, the tissues were fixed with 2% paraformaldehyde (in PBS) for 20 min before incubating in 30% sucrose solution overnight at 4°C. The samples were then embedded in OTC (Thermo Fisher Scientific, Rockford, IL) and subjected to frozen section. Sections were then counterstained with DAPI before imaging.
Flow Cytometry Analysis
The evaluation of Ca2+-mediated CD80/CD86 expression was conducted using a flow cytometer. 5 × 105 JAWSII cells were seeded in a 6-well plate and treated with an amount of DOTAP-LCP, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)-LCP, or lipoplex equivalent to 1 μg/mL of mRNA. Twenty-four hours post-transfection, cells were harvested and resuspended at 1 × 106 cells/mL. The cells were then stained with fluorescein isothiocyanate (FITC)-anti-CD80 and FITC-anti-CD86 antibody (BD Biosciences) at a 1:100 dilution and room temperature for 1 hr. Cells were washed with PBS before analyzing with flow cytometry. To study the cell population that took up LCP in the lymph node, mice were subcutaneously injected with DiI-labeled LCP in the flank. Twenty-four hours after injection, lymph nodes were collected. The lymph nodes were then treated with collagenase and DNase I to generate a single-cell suspension (1 × 106 cells/mL). The cells were co-stained with either antibodies against CD11b and F4/80 or antibodies against CD11c and CD123 at a 1:100 dilution. The cells were then subjected to flow cytometry analysis after washing. The experiments were conducted in triplicate. The data were analyzed using FlowJo 9.3.2 (FlowJo, Ashland, OR). For the T cell INF-γ expression assay, the splenocytes from vaccinated mice were collected and resuspended in RPMI-1640 full medium. Then, 50 μg of full-length TRP2 protein was added to 106 cells, and the cells were incubated for 6 hr. Brefeldin A buffer (1,000×) (BD Biosciences) was added to the cells 3 hr after the antigen was added. The cells were then stained with fluorescently labeled anti-CD3, CD8, and CD4 antibodies (BD Biosciences). Later, the cells were fixed and permeabilized with a BD fixation and permeabilization kit (BD Biosciences) according to the manufacturer’s manual. The cells were intracellularly stained with anti-IFN-γ antibody (BD Biosciences) and analyzed with flow cytometry.
Western Blot Analysis
Tumor or lymph nodes were collected and homogenized in radioimmunoprecipitation assay (RIPA) buffer. Protein concentrations of the tumor lysates were measured using the BCA assay reagents (Thermo Fisher Scientific) following the manufacturer’s instructions. Protein samples were resolved on NuPAGE 4%–12% Bis-Tris Gels (Thermo Fisher Scientific), transferred to an Immobilon-P transfer membrane (Thermo Fisher Scientific), and probed with the primary antibodies anti-TRP2 (Santa Cruz, Santa Cruz, CA), anti-PD-L1 (Abcam, Cambridge, MA), and anti-GAPDH mAb (Cell Signaling, Danvers, MA) and the HRP-conjugated secondary antibody. Signals were developed using the Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, Rockford, IL).
Statistical Analysis
Statistical analyses were conducted using Prism 5. Differences were considered statistically significant if the p value was less than 0.05.
Author Contributions
Y.W. and L.H. conceived the presented idea and designed the experiments. Y.W., L.Z., Z.X., and L.M. conducted the experiments. Y.W. and L.H. analyzed the results and wrote the manuscript.
Acknowledgments
The work was supported by NIH grants CA149363, CA151652, and CA149387. We thank Andrew Blair, Sara Musetti, and Wantong Song for manuscript editing.
Footnotes
Supplemental Information includes one figure and can be found with this article online at https://doi.org/10.1016/j.ymthe.2017.11.009.
Supplemental Information
References
- 1.Van Lint S., Renmans D., Broos K., Dewitte H., Lentacker I., Heirman C., Breckpot K., Thielemans K. The ReNAissanCe of mRNA-based cancer therapy. Expert Rev. Vaccines. 2015;14:235–251. doi: 10.1586/14760584.2015.957685. [DOI] [PubMed] [Google Scholar]
- 2.Breckpot K., Heirman C., Neyns B., Thielemans K. Exploiting dendritic cells for cancer immunotherapy: genetic modification of dendritic cells. J. Gene Med. 2004;6:1175–1188. doi: 10.1002/jgm.615. [DOI] [PubMed] [Google Scholar]
- 3.Rains N., Cannan R.J., Chen W., Stubbs R.S. Development of a dendritic cell (DC)-based vaccine for patients with advanced colorectal cancer. Hepatogastroenterology. 2001;48:347–351. [PubMed] [Google Scholar]
- 4.Weide B., Carralot J.P., Reese A., Scheel B., Eigentler T.K., Hoerr I., Rammensee H.G., Garbe C., Pascolo S. Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J. Immunother. 2008;31:180–188. doi: 10.1097/CJI.0b013e31815ce501. [DOI] [PubMed] [Google Scholar]
- 5.Kübler H., Scheel B., Gnad-Vogt U., Miller K., Schultze-Seemann W., Vom Dorp F., Parmiani G., Hampel C., Wedel S., Trojan L. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: a first-in-man phase I/IIa study. J. Immunother. Cancer. 2015;3:26. doi: 10.1186/s40425-015-0068-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rittig S.M., Haentschel M., Weimer K.J., Heine A., Muller M.R., Brugger W., Horger M.S., Maksimovic O., Stenzl A., Hoerr I. Intradermal vaccinations with RNA coding for TAA generate CD8+ and CD4+ immune responses and induce clinical benefit in vaccinated patients. Mol. Ther. 2011;19:990–999. doi: 10.1038/mt.2010.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weide B., Pascolo S., Scheel B., Derhovanessian E., Pflugfelder A., Eigentler T.K., Pawelec G., Hoerr I., Rammensee H.G., Garbe C. Direct injection of protamine-protected mRNA: results of a phase 1/2 vaccination trial in metastatic melanoma patients. J. Immunother. 2009;32:498–507. doi: 10.1097/CJI.0b013e3181a00068. [DOI] [PubMed] [Google Scholar]
- 8.Hodi F.S., Mihm M.C., Soiffer R.J., Haluska F.G., Butler M., Seiden M.V., Davis T., Henry-Spires R., MacRae S., Willman A. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl. Acad. Sci. USA. 2003;100:4712–4717. doi: 10.1073/pnas.0830997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brahmer J.R., Drake C.G., Wollner I., Powderly J.D., Picus J., Sharfman W.H., Stankevich E., Pons A., Salay T.M., McMiller T.L. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J., Yang Y., Huang L. Calcium phosphate nanoparticles with an asymmetric lipid bilayer coating for siRNA delivery to the tumor. J. Control. Release. 2012;158:108–114. doi: 10.1016/j.jconrel.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y., Hu Y., Wang Y., Li J., Liu F., Huang L. Nanoparticle delivery of pooled siRNA for effective treatment of non-small cell lung cancer. Mol. Pharm. 2012;9:2280–2289. doi: 10.1021/mp300152v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Z., Ramishetti S., Tseng Y.C., Guo S., Wang Y., Huang L. Multifunctional nanoparticles co-delivering Trp2 peptide and CpG adjuvant induce potent cytotoxic T-lymphocyte response against melanoma and its lung metastasis. J. Control. Release. 2013;172:259–265. doi: 10.1016/j.jconrel.2013.08.021. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y., Peng L., Mumper R.J., Huang L. Combinational delivery of c-myc siRNA and nucleoside analogs in a single, synthetic nanocarrier for targeted cancer therapy. Biomaterials. 2013;34:8459–8468. doi: 10.1016/j.biomaterials.2013.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diebold S.S., Kaisho T., Hemmi H., Akira S., Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 16.Hornung V., Ellegast J., Kim S., Brzózka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K.K., Schlee M. 5′-triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 17.Nallagatla S.R., Hwang J., Toroney R., Zheng X., Cameron C.E., Bevilacqua P.C. 5′-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science. 2007;318:1455–1458. doi: 10.1126/science.1147347. [DOI] [PubMed] [Google Scholar]
- 18.Anderson B.R., Muramatsu H., Nallagatla S.R., Bevilacqua P.C., Sansing L.H., Weissman D., Karikó K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010;38:5884–5892. doi: 10.1093/nar/gkq347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thess A., Grund S., Mui B.L., Hope M.J., Baumhof P., Fotin-Mleczek M., Schlake T. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 2015;23:1456–1464. doi: 10.1038/mt.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warren L., Manos P.D., Ahfeldt T., Loh Y.H., Li H., Lau F., Ebina W., Mandal P.K., Smith Z.D., Meissner A. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y., Su H.H., Yang Y., Hu Y., Zhang L., Blancafort P., Huang L. Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol. Ther. 2013;21:358–367. doi: 10.1038/mt.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kormann M.S., Hasenpusch G., Aneja M.K., Nica G., Flemmer A.W., Herber-Jonat S., Huppmann M., Mays L.E., Illenyi M., Schams A. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011;29:154–157. doi: 10.1038/nbt.1733. [DOI] [PubMed] [Google Scholar]
- 23.Xie Y., Bai O., Zhang H., Li W., Xiang J. Tumor necrosis factor gene-engineered J558 tumor cell-released exosomes stimulate tumor antigen P1A-specific CD8+ CTL responses and antitumor immunity. Cancer Biother. Radiopharm. 2010;25:21–28. doi: 10.1089/cbr.2009.0714. [DOI] [PubMed] [Google Scholar]
- 24.Nallagatla S.R., Toroney R., Bevilacqua P.C. A brilliant disguise for self RNA: 5′-end and internal modifications of primary transcripts suppress elements of innate immunity. RNA Biol. 2008;5:140–144. doi: 10.4161/rna.5.3.6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ignowski J.M., Schaffer D.V. Kinetic analysis and modeling of firefly luciferase as a quantitative reporter gene in live mammalian cells. Biotechnol. Bioeng. 2004;86:827–834. doi: 10.1002/bit.20059. [DOI] [PubMed] [Google Scholar]
- 26.Bagley K.C., Abdelwahab S.F., Tuskan R.G., Lewis G.K. Calcium signaling through phospholipase C activates dendritic cells to mature and is necessary for the activation and maturation of dendritic cells induced by diverse agonists. Clin. Diagn. Lab. Immunol. 2004;11:77–82. doi: 10.1128/CDLI.11.1.77-82.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Czerniecki B.J., Carter C., Rivoltini L., Koski G.K., Kim H.I., Weng D.E., Roros J.G., Hijazi Y.M., Xu S., Rosenberg S.A., Cohen P.A. Calcium ionophore-treated peripheral blood monocytes and dendritic cells rapidly display characteristics of activated dendritic cells. J. Immunol. 1997;159:3823–3837. [PubMed] [Google Scholar]
- 28.Koski G.K., Schwartz G.N., Weng D.E., Czerniecki B.J., Carter C., Gress R.E., Cohen P.A. Calcium mobilization in human myeloid cells results in acquisition of individual dendritic cell-like characteristics through discrete signaling pathways. J. Immunol. 1999;163:82–92. [PubMed] [Google Scholar]
- 29.Yan W., Chen W., Huang L. Reactive oxygen species play a central role in the activity of cationic liposome based cancer vaccine. J. Control. Release. 2008;130:22–28. doi: 10.1016/j.jconrel.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 30.Carralot J.P., Probst J., Hoerr I., Scheel B., Teufel R., Jung G., Rammensee H.G., Pascolo S. Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell. Mol. Life Sci. 2004;61:2418–2424. doi: 10.1007/s00018-004-4255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoerr I., Obst R., Rammensee H.G., Jung G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J. Immunol. 2000;30:1–7. doi: 10.1002/1521-4141(200001)30:1<1::AID-IMMU1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 32.Fotin-Mleczek M., Duchardt K.M., Lorenz C., Pfeiffer R., Ojkić-Zrna S., Probst J., Kallen K.J. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J. Immunother. 2011;34:1–15. doi: 10.1097/CJI.0b013e3181f7dbe8. [DOI] [PubMed] [Google Scholar]
- 33.Tseng Y.C., Xu Z., Guley K., Yuan H., Huang L. Lipid-calcium phosphate nanoparticles for delivery to the lymphatic system and SPECT/CT imaging of lymph node metastases. Biomaterials. 2014;35:4688–4698. doi: 10.1016/j.biomaterials.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uzgün S., Nica G., Pfeifer C., Bosinco M., Michaelis K., Lutz J.F., Schneider M., Rosenecker J., Rudolph C. PEGylation improves nanoparticle formation and transfection efficiency of messenger RNA. Pharm. Res. 2011;28:2223–2232. doi: 10.1007/s11095-011-0464-z. [DOI] [PubMed] [Google Scholar]
- 35.Cheng C., Convertine A.J., Stayton P.S., Bryers J.D. Multifunctional triblock copolymers for intracellular messenger RNA delivery. Biomaterials. 2012;33:6868–6876. doi: 10.1016/j.biomaterials.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alexopoulou L., Holt A.C., Medzhitov R., Flavell R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 37.Oberli M.A., Reichmuth A.M., Dorkin J.R., Mitchell M.J., Fenton O.S., Jaklenec A., Anderson D.G., Langer R., Blankschtein D. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 2017;17:1326–1335. doi: 10.1021/acs.nanolett.6b03329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Udhayakumar V.K., De Beuckelaer A., McCaffrey J., McCrudden C.M., Kirschman J.L., Vanover D., Van Hoecke L., Roose K., Deswarte K., De Geest B.G. Arginine-rich peptide-based mRNA nanocomplexes efficiently instigate cytotoxic T cell immunity dependent on the amphipathic organization of the peptide. Adv. Healthc. Mater. 2017;6 doi: 10.1002/adhm.201601412. [DOI] [PubMed] [Google Scholar]
- 39.Shumilina E., Huber S.M., Lang F. Ca2+ signaling in the regulation of dendritic cell functions. Am. J. Physiol. Cell Physiol. 2011;300:C1205–C1214. doi: 10.1152/ajpcell.00039.2011. [DOI] [PubMed] [Google Scholar]
- 40.Awate S., Babiuk L.A., Mutwiri G. Mechanisms of action of adjuvants. Front. Immunol. 2013;4:114. doi: 10.3389/fimmu.2013.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakanishi J., Wada Y., Matsumoto K., Azuma M., Kikuchi K., Ueda S. Overexpression of B7-H1 (PD-L1) significantly associates with tumor grade and postoperative prognosis in human urothelial cancers. Cancer Immunol. Immunother. 2007;56:1173–1182. doi: 10.1007/s00262-006-0266-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freeman G.J., Long A.J., Iwai Y., Bourque K., Chernova T., Nishimura H., Fitz L.J., Malenkovich N., Okazaki T., Byrne M.C. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong H., Chen L. B7-H1 pathway and its role in the evasion of tumor immunity. J. Mol. Med. (Berl.) 2003;81:281–287. doi: 10.1007/s00109-003-0430-2. [DOI] [PubMed] [Google Scholar]
- 44.Blank C., Gajewski T.F., Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol. Immunother. 2005;54:307–314. doi: 10.1007/s00262-004-0593-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 46.Latchman Y.E., Liang S.C., Wu Y., Chernova T., Sobel R.A., Klemm M., Kuchroo V.K., Freeman G.J., Sharpe A.H. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc. Natl. Acad. Sci. USA. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fife B.T., Pauken K.E., Eagar T.N., Obu T., Wu J., Tang Q., Azuma M., Krummel M.F., Bluestone J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009;10:1185–1192. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu Z., Wang Y., Zhang L., Huang L. Nanoparticle-delivered transforming growth factor-β siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano. 2014;8:3636–3645. doi: 10.1021/nn500216y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.