

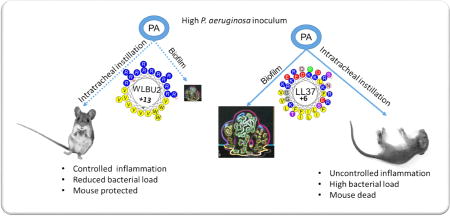
Abstract
Objectives
P. aeruginosa is a common cause of pneumonia in cystic fibrosis patients with the property to generate multidrug resistance against clinically used antibiotics. Antimicrobial peptides (AMPs) are a diverse group of effector molecules of the innate immunity that protect the host against pathogens. However, the lack of activity in common biological matrices has hampered efforts towards clinical development. In this study, we evaluated the therapeutic potential of engineered antimicrobial peptide WLBU2 via direct airway delivery in a murine model of P. aeruginosa infection.
Methods
The human AMP LL37 and WLBU2 were compared for (1) antibiofilm activity using P. aeruginosa on polarized human bronchial epithelial cells; (2) efficacy in P. aeruginosa pneumonia in mice using intra-tracheal (i.t.) instillation of bacteria and AMPs.
Results
WLBU2 (16µM) prevents biofilm formation by up to 3-log compared to 1-log reduction by LL37. With a single dose of 1µg (0.05mg/kg) delivered i.t., the initial effect of LL37 was moderate and transitory, as bacterial load and inflammatory cytokines increased at 24h with observed signs of disease such as lethargy and hypothermia (L&H), consistent with moribund state requiring euthanasia. In sharp contrast, WLBU2 reduced bacterial burden (by 2 logs) and bacteria-induced inflammation (leucocytic infiltrates, cytokine and chemokine gene expression) at 6h and 24h post-exposure, with no observed signs of disease or host toxicity.
Conclusion
These promising results now establish a much lower minimum therapeutic dose (mTd) of WLBU2 (a net gain of 80-fold) compared to the previously reported 4mg/kg systemic mTd, with significant implications for clinical development.
Keywords: LL37, pseudomonas aeruginosa, resistance, antimicrobial peptides, cationic peptides, respiratory infection, antibiotic resistance, pneumonia, WLBU2
Graphical abstract
Introduction
P. aeruginosa is one of the most important opportunistic bacterial pathogens(1). Although it is less frequent in community-acquired pneumonia (CAP), this organism is a common cause of multidrug resistance (MDR)-associated bacterial infections in patients with cystic fibrosis (CF), ultimately resulting in CF-related fatalities(2). It can cause severe hospital-acquired pneumonia (HAP) in immune-compromised patients(3). P. aeruginosa is a member of a group of organisms known as ESKAPE pathogens(4), which typically have a high propensity to develop antibiotic resistance and cause hard-to-treat bacterial infections. In addition, P. aeruginosa has the property to attach to tissues and form sessile communities known as biofilm, which tends to be inherently resistant to standard antibiotic treatment(5). Thus, novel antimicrobial agents with the property to overcome current mechanisms of resistance are urgently needed.
Antimicrobial peptides (AMPs) are a class of agents that may be effective against MDR pathogens, with amphipathicity as a common feature of active AMPs(6). However, natural AMPs have some shortcomings including inhibition of activity in biological matrices and lack of evidence for efficacy in animal models(7), which have influenced clinical development efforts toward topical applications(8). We have postulated that natural AMPs are typically not dedicated antibiotics because, as multifunctional effector molecules of the immune system (e.g., the human AMP LL37)(9), they are mainly responsible for protecting (more preventive than therapeutic) the host from local invasion of pathogens. Therefore, clinical development of AMPs as dedicated antibiotics requires structural optimization or de novo design.
We previously demonstrated that a de novo-engineered AMP, modeled to form an idealized amphipathic helix, requires only two or three different amino acids either to be as effective as LL37 (made of 14 different amino acids) or to display activity in biological matrices and in vivo efficacy in animal models(7, 10, 11). Hence, composed of only Arg, Val, and Trp, the engineered AMP WLBU2 displays broad-spectrum antibacterial activity in saline and divalent cation concentrations. In addition, it demonstrates antiviral and antibiofilm properties as well as systemic efficacy in murine models of P. aeruginosa septicemia(5, 10, 12). What remains unclear is whether local delivery of WLBU2 into the airway could attenuate a respiratory infection. As LL37 is one of the AMPs that normally protect the host against airway infections, we reasoned that local airway delivery of AMPs would represent an effective and appropriate model for comparing WLBU2 and LL37. Thus, we report herein the comparative study of efficacy of WLBU2 and LL37 directly delivered into the airway in a murine model of P. aeruginosa pneumonia.
Materials and Methods
Materials
Eagle's minimum essential medium (EMEM), heat-inactivated fetal bovine serum, and penicillin-streptomycin, protease XIV and DNase were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were reagent grade.
Human bronchial epithelial (HBE) cells
Fully differentiated primary human bronchial epithelial (HBE) cells were derived from lungs removed at the time of lung transplantation from the Center for Organ Recovery and Education (Pittsburgh, PA). Cells were prepared using previously described methods approved by the University of Pittsburgh Institutional Review Board (IRB) (13–17) (13–17) (13–17) (13–17). Briefly, bronchi from the 2nd to 6th generations were digested(16, 17) in MEM containing protease XIV and DNase. The epithelial cells were removed and collected by centrifugation prior to resuspension in BronchialLife Epithelial growth medium (Lifeline Technology; Frederick, MD) and plating onto collagen-treated tissue culture flasks. The cultures were then maintained at air-liquid interface (ALI), with basolateral media changed twice weekly.
Peptide synthesis
WLBU2 (RRWVRRVRRVWRRVVRVVRRWVRR) and LL-37 (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES) were synthesized at the University of Pittsburgh Peptide Synthesis Facility using the previously described standard Fmoc (9-fluorenylmethoxy carbonyl) synthesis protocols(18). For comparison purposes, 1mM peptide represents a concentration of 3.4 mg/mL for WLBU2 and 4.5mg/mL for LL37.
Biotic biofilm assay
Biotic biofilm assay was performed using co-cultures of polarized HBE cells and P. aeruginosa PAO1 with a starting multiplicity of infection (MOI) of 30 and 500µL MEM on the basolateral side as previously described(14). HBE cells were inoculated with PAO1 in 50µL MEM at the apical side for 1 h to allow attachment of the bacteria to the HBE cells in ALI culture. Each AMP (final 16µM concentration), diluted in 50µL PBS, was added for 5 h. After a total of 6h, the biofilms were disrupted by sonication for 30sec (PRO DPS-20 sonicator), with subsequent serial dilution and enumeration on LB agar plates to determine colony-forming units (CFU).
Transepithelial Electrical Resistance (TEER) assay
TEER was examined as a measure of Lung epithelial integrity in differentiated primary HBE cells by using an electronic resistance system (Millicell ERS-2, EMD Millipore). HBE cells were grown on an ALI culture system in 24-transwell plates (Corning, Tewksbury, MA) using BronchiaLife Epithelial Airway Medium (Lifeline, Frederick, MD) in the basolateral compartment. Peptides dissolved in PBS at different concentrations were added onto the apical surface of the HBE cells in a final volume of 100µl. A sterile electrode was applied onto the apical side of the transwell insert containing the HBE cells with/without peptide treatments, and TEER was measured three times for comparison at different time intervals.
Murine infection model
All animal experiments were carried out according to a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pittsburgh based on the National Institutes of Health guide for the care and use of Laboratory animals. 8-week old female wild-type C57BL/6J mice were anesthetized by isoflurane inhalation and instilled intra-tracheally (i.t.) with ~3×106 CFU PAO1 (susceptible to WLBU2 and LL37) in 50µl PBS. One hour post-exposure, each peptide was i.t. administered at 0.05 mg/kg (predetermined in a pilot experiment as minimum dose WLBU2 required to rescue mice from a lethal infection) in 50µl of PBS. Control mice were instilled with 50µl of phosphate buffered saline (PBS) without peptide. Mice were monitored for signs of morbidity and euthanized at 6h or 24h after bacterial exposure.
Lung histopathology
Lung tissues were harvested at 6h or 24 h after bacterial exposure and examined for histopathology. Tissues were fixed in situ with 4% paraformaldehyde for 10 minutes with open chest cavity. The right lobe was embedded in paraffin, and 5µm sections were prepared by staining in hematoxylin and eosin and analyzed for pathological severity.
Bronchoalveolar lavage (BAL) and cell differential counts
Wild-type 8-week old female C57BL/6J mice were anesthetized by isoflurane inhalation prior to peptide administration i.t. in 50 µl of PBS at 0.05mg/kg or 0.35 mg/kg. Control mice were instilled with 50 µl of PBS without peptide. After 24h of treatment, mice were anesthetized with 2.5 % tribromoethanol (Avertin). The trachea was cannulated to allow the lungs to be lavaged twice using 1mL saline, and the BAL samples collected as previously described (19). The number of live immune cells in the BAL was determined using a Vision Cell Analyzer automatic cell counter (Nexcelom, Lawrence, MA). cell differential was determined microscopically using additional aliquots placed on glass microscope slides (Shanon Cytospin; ThermoFisher, Pittsburgh, PA), stained with Diff-Quick. A total of 400 cells per slide were counted at least twice for inflammatory cell differential counts.
Gene expression analysis (Real-time PCR)
Expression levels of different inflammation-related genes were determined at 6h or 24 h from peptide administration (0.05–0.35 mg/kg) in infected or non-infected mice, as previously described(19–22). Briefly, total mRNA was isolated from the right lung using Trizol reagent. Quantitative PCR was performed using ABI7900HT (Applied Biosystems, Foster City, CA) and primers for all genes specified in Figures 3, 4, and 5. Test and calibrator lung RNAs were reverse transcribed using a High-Capacity cDNA reverse transcription kit (Life Technologies). The cDNA was amplified as follows: 50 °C for 2 min, 95 °C for 10 min, 40 cycles; 95 °C for 1 5s; 60 °C for 1min. Three replicates were used to calculate average cycle threshold for each transcript of interest using (β-glucuronidase for normalization (Assays on Demand; Applied Biosystems). Relative mRNA abundance was calculated using the ΔΔCt (cycle threshold) method.
Figure 3. WLBU2 was more effective than LL37 at reducing inflammation induced by P. aeruginosa at 6h after bacterial exposure.

Different inflammatory mediators in the lungs of P. aeruginosa-infected mice after peptide administration (at 0.05 mg/kg) were examined for mRNA levels within 6h (A) or 24h (B) after bacterial exposure. Histologic analysis of lung tissues are shown at the corresponding times of 6h (C) or 24h (D) post-exposure for PBS-, WLBU2-, or LL37-treated mice. Peptides were administered at 0.05 mg/kg 1h after bacterial exposure. Results are mean ± SEM from three independent experiments; n = 4–6 mice for each treatment group; *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 4. Levels of mRNA of different sets of genes in WLBU2- and LL37-treated mice after PAO1 exposure.

Age-matched wild-type C57BL/6J mice were intra-tracheally (i.t.) instilled with 3×106 CFU PAO1 per mouse. Expression of inflammation-related genes showed WLBU2 significantly reduced PAO1-induced inflammation-related mRNA levels compared to LL37 at 6h (A, cytokines and C, chemokines) or 24h (B, cytokines and D, chemokines) post-infection. Results are mean ± SEM from three independent experiments; n = 4–6 mice for each treatment group. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 5. Instillation of AMPs into mouse lungs does not cause pulmonary toxicity.

Wild-type C57BL/6J mice were intra-tracheally (i.t.) instilled with digested AMPs at a concentration of 0.35 mg/kg (WLBU2, LL37 treatment) or PBS (control). AMP treatment did not result in any noticeable difference in total inflammatory cells and differential cell counts (A) as well as expression of inflammation-associated genes (B). The results indicate that WLBU2, similar to LL37, does not cause pulmonary toxicity. Results are mean ± SEM from two independent experiments; n = 4–6 mice for each group.
Data analysis
Data are expressed as mean ± SEM. Statistical comparisons between groups of mice were made using ANOVA, followed by Dunnett’s multiple comparison test (one way ANOVA) or the Bonferroni multiple comparison test (two way ANOVA). A p value <0.05 was considered to be statistically significant.
Results
WLBU2 has been extensively characterized in vitro in comparison to LL37 (23), and presented in Table 1 are various MICs of these two AMPs against diverse strains of multidrug-resistant (MDR) P. aeruginosa. Importantly, both peptides are active against PAO1 with MICs of 2µM (WLBU2) and 4µM (LL37), as previously shown (18).
Table 1.
Activity of WLBU2 and LL37 against MDR strains isolated from Cystic fibrosis patients; PAO1 is included as a control strain susceptible to antibiotics and AMPs
| P. aeruginosa | MIC (µM) | |
|---|---|---|
|
| ||
| LL37 | WLBU2 | |
|
|
||
| PAO1 | 4 | 2 |
| TRPA108 | 2 | 1 |
| TRPA111 | 2 | 1 |
| P001 | 32 | 6 |
| P002 | 32 | 4 |
| P003 | 32 | 6 |
| P004 | 8 | 2 |
| P005 | 8 | 2 |
| TRPA321 | 32 | 2 |
WLBU2 displays antibiofilm activity with no permanent effects on bronchial epithelial integrity
Because our goal was to test the in vivo efficacy of WLBU2 in a respiratory infection model, it was important to first determine in vitro activity of AMPs using a biological matrix that closely mimics the airway epithelium. Hence, we used a well-developed model of maintaining polarized HBE cells in ALI culture to compare the two AMPs of distinct origins (WLBU2 and LL37) but of similar structural properties. After allowing the bacterial cells to attach to HBE cells for 1h, AMPs were added at 16µM (WLBU2 concentration not toxic to HBE cells) on the apical side of the ALI culture. LL37 was able to slightly reduce the formation of bacterial biofilm (Figure 1A). However, the antibiofilm activity of WLBU2 was almost one order of magnitude 10-fold) better than that of LL37 (P<0.05). Furthermore, we determined the influence of these AMPs on epithelial integrity by assessing TEER. Surprisingly, LL37 substantially affected epithelial TEER. Although the initially sharp decline in TEER by AMP treatments was expected, LL37-treated HBE cells never completely recovered at 24 hours after administration (Figure 1C and 1D), with a final reduction in TEER by nearly 1/2 at 32µM or 2/3 at 64µM. By contrast, WLBU2-treated HBE cells completely recovered from the 24h treatment to baseline levels at both concentrations.
Figure 1. Effects of AMPs on Pseudomonas aeruginosa biofilm growth on polarized human airway epithelial (HAE) cells and transepithelial electrical resistance (TEER).

Using a biotic biofilm assay, P. aeruginosa was co-cultured with primary human bronchial epithelial (HBE) cells to examine the ability of WLBU2, compared to LL37, to prevent biofilm formation. Bacteria were allowed to attach to HBE cells for 1h prior to addition of 16µM peptide (A). Peptides, at 32µM (B) and 64µM (C) in PBS, were added to the apical compartment of the cultured HBE cells maintained under ALI condition. TEER was measured at multiple time points to assess recovery time of the ex-vivo epithelium. Results are mean ± SEM from three independent experiments; *p < 0.05, **p < 0.01 for comparison between groups.
WLBU2 significantly reduced bacterial burden and inflammation in PAO1-induced pneumonia
C57BL/6 mice were infected i.t. with PAO1 and treated with AMPs by i.t. instillation (1µg or 0.05mg/kg, based on a pilot experiment in determining minimum WLBU2 dosage protecting mice from a lethal infection) at 1h post-exposure, as described in Materials and Methods. We monitored the animals for either 5h or 23h post-treatment (a total of 6h and 24h post-exposure) with no observed signs of morbidity (e.g., impaired mobility) during the first 6h post-exposure. However, at 24h post-exposure, it became evident that the PBS- and LL37-treated mice were lethargic with only a minor difference in mobility between those two groups, progressing towards moribund state or L&H. By contrast, the WLBU2-treated mice appeared unaffected, with similar mobility to uninfected mice. On necropsies, both LL37 and WLBU2 significantly (P<0.001) reduced bacterial burden by to 2 to 10-fold (LL37) and up to 10 to 100-fold for WLBU2 at 6h post-exposure (Figure 2). By 24h post-exposure, the reduction in bacterial counts due to LL37 treatment was dissipated to less than 2-fold compared to mock-treated mice in the lung. Remarkably, WLBU2 maintained its efficacy after 23h post-treatment, with a 2-log CFU reduction in the lungs compared to mock-treated animals (Figure 2). This substantial reduction in bacterial burden by WLBU2 is consistent with no observed lethargy in these mice compared to LL37- and the PBS-treated animals at 24h post-exposure.
Figure 2. WLBU2-treated mice are protected against PAO1-induced respiratory infection.

Age- and gender-matched wild-type C57BL/6J mice were intra-tracheally (i.t.) instilled with 3×106 CFU PAO1 per mouse. Total lung burden (A) was represented by combining bacterial CFU in BAL (B) and lung homogenate (C) after necropsies at 6h and 24h after PAO1 infection. WLBU2-treated mice showed a significant reduction in their lung bacterial burden compared to LL37- and mock-treated mice. Results are mean ± SEM from two independent experiments; n = 4–6 mice for each treatment group in each experiment; *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we assessed the effects of AMPs on the levels of PAO1-associated inflammation by examining gene expression of major inflammatory cytokines (IL-6 and IL1-β) and chemokines (Cxcl1 and MIP-1α) in the infected mice treated with PBS or 0.05mg/kg AMPs. At 6h post-exposure, LL37 significantly (P<0.05) decreased the expression level of IL1β and MIP-1α, whereas WLBU2 had a much more significant effect (P<0.01) on these two inflammatory mediators (Figure 3A). Similar to the effects on bacterial burden after 24h post-exposure (Figure 3B), WLBU2 had a broad effect (40 to 70% reduction, P<0.05) on the expression of both cytokines (IL-6 and IL-1β) and chemokines (Cxcl1 and MIP). LL37- and PBS-treated animals displayed similar levels (exception, lower IL-1β level in LL37-treated mice compared to control) of these inflammatory cytokines and chemokines. WLBU2-treated mice also displayed substantially lower levels of inflammatory infiltrates compared to lung tissues of PBS- and LL37-treated mice, which display moderate (PBS) to mild (LL37) levels of leukocyte infiltrates at 6h (Figure 3C) post-exposure. By 24h post-exposure (Figure 3D), these leucocyte infiltrates already became substantially more prominent in both PBS- and LL37-treated mice, while reaching baseline level in lung tissues of WLBU2-treated mice. A more extensive examination of inflammatory mediators (NF-kb, TNF-α, IL-10, CxCl1, Cxcl2, and Cxcl3) revealed a similar relationship between LL37 and WLBU2 (Figure 4). Finally, we demonstrated at 7µg (7 times the minimum effective dose) delivered i.t., both LL37 and WLBU2 had no effect on cell differential and inflammatory mediators in the lungs of uninfected mice (Figure 5). All of these findings indicate that WLBU2 displayed superiority over LL37 in efficacy via direct delivery into the airway.
DISCUSSION
The current antibiotic armamentarium is revealed insufficient to treat respiratory infections, one of the leading causes of death worldwide(24, 25). To address this urgency, our strategy has been to develop de novo-designed AMPs (eCAPs) to overcome the limitations of natural AMPs(26, 27). These eCAPs demonstrate broad-spectrum activity in biologically relevant environments and remain effective against ESKAPE pathogens that are resistant to LL37 and colistin(23). In addition to the previously demonstrated systemic efficacy of WLBU2(10), we showed in this study that: (i) WLBU2 displayed significantly higher antibiofilm activity than LL37 without any permanent effect on the integrity of the epithelial cells; (ii) WLBU2 reduced bacterial burden and P. aeruginosa-associated inflammation. Importantly, these mice showed no physical signs of disease, in sharp contrast to LL37-treated mice. Remarkably, WLBU2 achieved this level of efficacy with just a single dose of 1µg (0.05mg/kg). The significance of the current study is the discovery of a substantial gain (~80-fold) in minimum dose required for therapeutic efficacy over the previously reported minimum systemic dose of 4 mg/kg.
Considering the protective role of LL37 in preventing bacterial colonization of human tissues (e.g., skin, reproductive tract, gastrointestinal tract, respiratory tract), it is not surprising that it displays broad antimicrobial activity. Indeed, LL37 reduced biofilm formation and bacterial load in our mice. However, it is important to underscore that our study tested activity in the presence of high bacterial load (infection) consistent with established colonization, which must be distinguished from the typically lower inoculum of bacteria interacting with human epithelia prior to eradication by AMPs (homeostasis), thereby preventing colonization and infection in most cases. As a result, the effects of LL37 were only transitory in our study. By contrast, the effects of WLBU2 on bacterial burden and PAO1-induced inflammation were sustained throughout the 24h post-treatment, with multiple-log reduction in bacterial burden, leucocytic infiltrates, and inflammatory cytokines at both 6h and 24h. Unlike the PBS- and LL37-treated mice, the WLBU2-treated mice never displayed any sign of L&H suggesting that the substantial decrease in bacterial burden and inflammation due to WLBU2 treatment was sufficient to reverse the lethality of the disease. Surprisingly, the LL37-treated HAE cells never fully recovered toward their initial TEER compared to PBS- or WLBU2-treated cells. This does not necessarily mean that LL37 is more toxic to mammalian cells than WLBU2, as we learned from previous toxicity assays(18). Because LL37 displays multiple functional properties in the context of the host immunity, it is possible that the lower TEER of HAE cells is related to the property of LL37 to facilitate the infiltration of other cellular components of the immune system.
With these promising data, an important question is why WLBU2 displays in vivo efficacy while most natural AMPs fail as therapeutics in animal models of infection. The answer lies in the design concept of WLBU2(11). Most mammalian AMPs have evolved in the context of the host innate immunity to perform multiple functions, which is reflective of the diversity in amino acid composition with various degrees of amphipathicity(28). We previously demonstrated that this primary sequence diversity is not required for antimicrobial function(10). In fact, it may explain the lack of structural optimization of natural AMPs necessary to remain active in in vitro models of biological matrices and in animal models. Hence, the design strategy resulting in the structure of WLBU2 is based on the principle of optimal amphipathicity while minimizing the diversity of amino acid composition using only hydrophobic (Val and trp) and cationic (Arg) amino acids. Thus, in contrast to the 14 different amino acids found in LL37, we excluded from the primary sequence the amino acids that may potentially interfere with the structural determinants of optimal antimicrobial function. Other investigators also use such a design principle to develop anticancer AMPs(29, 30). Together, these studies provide strong rationale for further AMP development to help mitigate the problem of drug resistance.
The data highlight the advantage of AMP optimization based on the principle of amphipathicity. The current state of the AMP field combined with the evolutionary capacity of the microbial world requires continuous efforts for enhanced structural optimization. Lessons from previous failures will allow progression towards further increasing safety as well as enhancing therapeutic efficacy and pharmacokinetic properties.
Acknowledgments
ACKNOWLEDGMENTS AND FUNDING
This research is supported by NIH awards R01 HL-091938, HL-125128, AI-133351, and a grant (CIA-123062) from Flight Attendant Medical Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
R.C.M.holds stock in Peptilogics and serves on an advisory board for Peptilogics. Although a potential financial conflict of interest was identified based on the authors' relationship with Peptilogics, the research findings included in this publication may not necessarily be related to the interests of Peptilogics. The other authors declare no conflict of interest.
AUTHORS’ CONTRIBUTIONS TO THE MANUSCRIPT
C.C. performed the experiments and analyzed data; R.C.M. discussed data and reviewed the manuscript; B.D. and Y.P.D. designed the experiments, analyzed data, and wrote the manuscript.
References
- 1.Speert DP. Molecular epidemiology of Pseudomonas aeruginosa. Front Biosci. 2002;7:e354–61. doi: 10.2741/A929. [DOI] [PubMed] [Google Scholar]
- 2.Jelsbak L, Johansen HK, Frost AL, Thogersen R, Thomsen LE, Ciofu O, et al. Molecular epidemiology and dynamics of Pseudomonas aeruginosa populations in lungs of cystic fibrosis patients. Infection and immunity. 2007;75(5):2214–24. doi: 10.1128/IAI.01282-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biedenbach DJ, Giao PT, Hung Van P, Su Minh Tuyet N, Thi Thanh Nga T, Phuong DM, et al. Antimicrobial-resistant Pseudomonas aeruginosa and Acinetobacter baumannii From Patients With Hospital-acquired or Ventilator-associated Pneumonia in Vietnam. Clin Ther. 2016;38(9):2098–105. doi: 10.1016/j.clinthera.2016.07.172. [DOI] [PubMed] [Google Scholar]
- 4.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48(1):1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 5.Lashua LP, Melvin JA, Deslouches B, Pilewski JM, Montelaro RC, Bomberger JM. Engineered cationic antimicrobial peptide (eCAP) prevents Pseudomonas aeruginosa biofilm growth on airway epithelial cells. J Antimicrob Chemother. 2016;71(8):2200–7. doi: 10.1093/jac/dkw143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hiemstra PS, Amatngalim GD, van der Does AM, Taube C. Antimicrobial Peptides and Innate Lung Defenses: Role in Infectious and Noninfectious Lung Diseases and Therapeutic Applications. Chest. 2016;149(2):545–51. doi: 10.1378/chest.15-1353. [DOI] [PubMed] [Google Scholar]
- 7.Deslouches B, Islam K, Craigo JK, Paranjape SM, Montelaro RC, Mietzner TA. Activity of the de novo engineered antimicrobial peptide WLBU2 against Pseudomonas aeruginosa in human serum and whole blood: implications for systemic applications. Antimicrob Agents Chemother. 2005;49(8):3208–16. doi: 10.1128/AAC.49.8.3208-3216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lipsky BA, Holroyd KJ, Zasloff M. Topical versus systemic antimicrobial therapy for treating mildly infected diabetic foot ulcers: a randomized, controlled, double-blinded, multicenter trial of pexiganan cream. Clin Infect Dis. 2008;47(12):1537–45. doi: 10.1086/593185. [DOI] [PubMed] [Google Scholar]
- 9.Hell E, Giske CG, Nelson A, Romling U, Marchini G. Human cathelicidin peptide LL37 inhibits both attachment capability and biofilm formation of Staphylococcus epidermidis. Lett Appl Microbiol. 2010;50(2):211–5. doi: 10.1111/j.1472-765X.2009.02778.x. [DOI] [PubMed] [Google Scholar]
- 10.Deslouches B, Gonzalez IA, DeAlmeida D, Islam K, Steele C, Montelaro RC, et al. De novo-derived cationic antimicrobial peptide activity in a murine model of Pseudomonas aeruginosa bacteraemia. J Antimicrob Chemother. 2007;60(3):669–72. doi: 10.1093/jac/dkm253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deslouches B, Phadke SM, Lazarevic V, Cascio M, Islam K, Montelaro RC, et al. De novo generation of cationic antimicrobial peptides: influence of length and tryptophan substitution on antimicrobial activity. Antimicrob Agents Chemother. 2005;49(1):316–22. doi: 10.1128/AAC.49.1.316-322.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melvin JA, Lashua LP, Kiedrowski MR, Yang G, Deslouches B, Montelaro RC, et al. Simultaneous Antibiofilm and Antiviral Activities of an Engineered Antimicrobial Peptide during Virus-Bacterium Coinfection. mSphere. 2016;1(3) doi: 10.1128/mSphere.00083-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cappiello F, Di Grazia A, Li-Av SZ, Scali S, Ferrera L, Galietta L, et al. Esculentin-1a-derived peptides promote clearance of P. aeruginosa internalized in cystic fibrosis bronchial cells as well as lung cells migration: Biochemical properties and a plausible mode of action. Antimicrob Agents Chemother. 2016 doi: 10.1128/AAC.00904-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Bartlett JA, Di ME, Bomberger JM, Chan YR, Gakhar L, et al. SPLUNC1/BPIFA1 contributes to pulmonary host defense against Klebsiella pneumoniae respiratory infection. Am J Pathol. 2013;182(5):1519–31. doi: 10.1016/j.ajpath.2013.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y, Di ME, Chu HW, Liu X, Wang L, Wenzel S, et al. Increased susceptibility to pulmonary Pseudomonas infection in Splunc1 knockout mice. J Immunol. 2013;191(8):4259–68. doi: 10.4049/jimmunol.1202340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cappiello F, Di Grazia A, Segev-Zarko LA, Scali S, Ferrera L, Galietta L, et al. Esculentin-1a-Derived Peptides Promote Clearance of Pseudomonas aeruginosa Internalized in Bronchial Cells of Cystic Fibrosis Patients and Lung Cell Migration: Biochemical Properties and a Plausible Mode of Action. Antimicrobial agents and chemotherapy. 2016;60(12):7252–62. doi: 10.1128/AAC.00904-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Grazia A, Cappiello F, Cohen H, Casciaro B, Luca V, Pini A, et al. D-Amino acids incorporation in the frog skin-derived peptide esculentin-1a(1–21)NH2 is beneficial for its multiple functions. Amino acids. 2015;47(12):2505–19. doi: 10.1007/s00726-015-2041-y. [DOI] [PubMed] [Google Scholar]
- 18.Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Mietzner TA, Montelaro RC. Rational design of engineered cationic antimicrobial peptides consisting exclusively of arginine and tryptophan, and their activity against multidrug-resistant pathogens. Antimicrob Agents Chemother. 2013;57(6):2511–21. doi: 10.1128/AAC.02218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di YP. Assessment of pathological and physiological changes in mouse lung through bronchoalveolar lavage. Methods in molecular biology. 2014;1105:33–42. doi: 10.1007/978-1-62703-739-6_3. [DOI] [PubMed] [Google Scholar]
- 20.Lukinskiene L, Liu Y, Reynolds SD, Steele C, Stripp BR, Leikauf GD, et al. Antimicrobial activity of PLUNC protects against Pseudomonas aeruginosa infection. J Immunol. 2011;187(1):382–90. doi: 10.4049/jimmunol.1001769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leikauf GD, Concel VJ, Bein K, Liu P, Berndt A, Martin TM, et al. Functional genomic assessment of phosgene-induced acute lung injury in mice. Am J Respir Cell Mol Biol. 2013;49(3):368–83. doi: 10.1165/rcmb.2012-0337OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Travanty E, Zhou B, Zhang H, Di YP, Alcorn JF, Wentworth DE, et al. Differential Susceptibilities of Human Lung Primary Cells to H1N1 Influenza Viruses. J Virol. 2015;89(23):11935–44. doi: 10.1128/JVI.01792-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Burns JL, Montelaro RC. Engineered cationic antimicrobial peptides to overcome multidrug resistance by ESKAPE pathogens. Antimicrob Agents Chemother. 2015;59(2):1329–33. doi: 10.1128/AAC.03937-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Keefe GE, Caldwell E, Cuschieri J, Wurfel MM, Evans HL. Ventilator-associated pneumonia: bacteremia and death after traumatic injury. J Trauma Acute Care Surg. 2012;72(3):713–9. doi: 10.1097/TA.0b013e3182349d14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim C, Takahashi E, Hongsuwan M, Wuthiekanun V, Thamlikitkul V, Hinjoy S, et al. Epidemiology and burden of multidrug-resistant bacterial infection in a developing country. Elife. 2016;5 doi: 10.7554/eLife.18082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deslouches B, Hasek ML, Craigo JK, Steckbeck JD, Montelaro RC. Comparative functional properties of engineered cationic antimicrobial peptides consisting exclusively of tryptophan and either lysine or arginine. J Med Microbiol. 2016;65(6):554–65. doi: 10.1099/jmm.0.000258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steckbeck JD, Deslouches B, Montelaro RC. Antimicrobial peptides: new drugs for bad bugs? Expert Opin Biol Ther. 2014;14(1):11–4. doi: 10.1517/14712598.2013.844227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott MG, Davidson DJ, Gold MR, Bowdish D, Hancock RE. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J Immunol. 2002;169(7):3883–91. doi: 10.4049/jimmunol.169.7.3883. [DOI] [PubMed] [Google Scholar]
- 29.Papo N, Seger D, Makovitzki A, Kalchenko V, Eshhar Z, Degani H, et al. Inhibition of tumor growth and elimination of multiple metastases in human prostate and breast xenografts by systemic inoculation of a host defense-like lytic peptide. Cancer Res. 2006;66(10):5371–8. doi: 10.1158/0008-5472.CAN-05-4569. [DOI] [PubMed] [Google Scholar]
- 30.Deslouches B, Di YP. Antimicrobial peptides with selective antitumor mechanisms: prospect for anticancer applications. Oncotarget. 2017 doi: 10.18632/oncotarget.16743. [DOI] [PMC free article] [PubMed] [Google Scholar]