

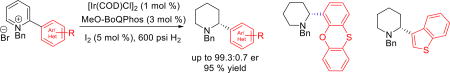
Abstract
Enantioselective synthesis of α-aryl and α-heteroaryl piperidines is reported. The key step is an iridium-catalyzed asymmetric hydrogenation of substituted N-benzyl pyridinium salts. High levels of enantioselectivity up to 99.3:0.7 er were obtained for a range of α-heteroaryl piperidines. DFT calculations support an outer-sphere dissociative mechanism for the pyridinium reduction. Notably, initial protonation of the final enamine intermediate determines the stereochemical outcome of the transformation rather than hydride reduction of the resultant iminium intermediate.
Graphical abstract
N-Heterocyclic piperidine compounds constitute a widespread structural motif in biologically active compounds.1 Enantiomerically pure α-substituted piperidines are also invaluable templates for chiral auxiliaries in asymmetric synthesis.2 As a result, their enantioselective syntheses are highly sought in pharmaceutical and chemical industries.3 Several methods have been reported for the synthesis of chiral N-heterocycle functionalization adjacent to the nitrogen atom,4 including enantioselective α-functionalization. Stereoselective α-lithiation has enjoyed particular success for the generation of enantioenriched 2-arylpyrrolidines,5 but the 6-membered piperidine counterpart has proven to be difficult to functionalize in a similar manner.6
In our ongoing efforts toward the synthesis of enantiomerically pure oxygen-containing α-aryl piperidines, we envisage the development of a direct technology to expedite the synthesis of these valuable biologically relevant subunits. Existing synthetic methods require either cryogenic conditions or long sequences to prepare the requisite molecules. We also sought to access these molecules through an atom-economical enantioselective approach. Recent advances in the asymmetric reduction of the heteroarenes have enabled straightforward synthesis of enantioenriched heterocyclic compounds.7 However, for reduction of the pyridinium salts to prepare enantioenriched α-arylated piperidines, few reports are available,8 and the subtrates are limited to the α-phenyl derivatives without heteroaryl functionality. Herein, we report our findings on a highly efficient and enantioselective synthesis of α-aryl and α-heteroaryl piperidines through an asymmetric hydrogenation strategy applying [Ir(COD)Cl]2/MeO-BoQPhos catalyst system. Furthermore, a detailed mechanistic rationale for the high enantioselectivity was elucidated for the first time by DFT calculations.
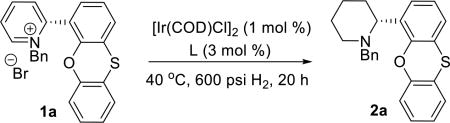
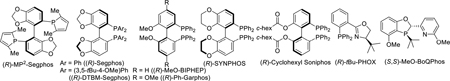
We commenced our investigation with N-benzyl-2-phenoxathiinyl pyridinium salt 1a; which was conveniently prepared by Suzuki coupling of 2-bromopyridine and phenoxathiin-4-boronic acid, followed by treatment with benzyl bromide. A complete ligand screen was conducted with 1 mol % [Ir(COD)Cl]2, 3 mol % ligand at 40 °C and 600 psi H2 pressure (representative examples shown in Table 1). Similar to what has observed for the 2-alkylpyridinium salt reduction,9 atropisomeric C2-symmetric bis(phosphine) ligands showed moderate enantioselectivities in the asymmetric hydrogenation of 1a. Piperidine 2a was produced in 91.8:8.2 enantiomeric ratio with Segphos ligand (entry 3) and 86.3:13.7 er with electron-rich doubly oxygenated MP2-Segphos ligand (entry 1).8b In contrast, the unsymmetrical phosphorous-pyridine based MeO-BoQPhos ligand was discovered to be highly effective for the enantioselective reduction of the α-aryl pyridinium salt; 2a was obtained in the highest enantioselectivity of 99.3:0.7 er (entry 13), with 92% isolated yield (Scheme 1, entry 1).
Table 1.
Ligand Screen for Asymmetric Hydrogenation of 1aa
| ||
|---|---|---|
|
| ||
| entry | ligand | er (2a) |
| 1 | (R)-MP2-Segphos | 86.3:13.7 |
| 2b | (R)-MP2-Segphos | 57.4:42.6 |
| 3 | (R)-Segphos | 91.8:8.2 |
| 4 | (R)-MeO-Biphep | 86.4:13.6 |
| 5 | (R)-Ph-Garphos | 66.5:33.5 |
| 6 | (R)-Synphos | 89.4:10.6 |
| 7 | (R)-DTBM-Segphos | 77.5:22.5 |
| 8 | (R)-Cyclohexyl Soniphos | 84.2:15.8 |
| 9c | (R)-tBuPHOX | 50.2:49.8 |
| 10b,d | (R)-tBuPHOX | 53.2:46.8 |
| 11 | Josiphos-001 | 54.5:45.5 |
| 12 | Josiphos-003 | 50.2:49.8 |
| 13b | (S,S)-MeO-BoQphos | 99.3:0.7 |
| ||
Reaction conditions: a mixture of [Ir(COD)Cl]2 and ligand was stirred in a vial for 15 min in 0.75 mL THF, before addition of 25 mg 1a, 100% conv to the desired product unless specified otherwise.
5 mol % of I2 was added to the catalyst solution before addition of substrate.10
33% conv.
40% conv.
Scheme 1. Asymmetric Hydrogenation of α-(Hetero)aryl-N-benzylpyridinium Saltsa.

a Reactions were run at 40 °C for 24 h. The numbers in parenthesis are isolated yields. b 20 °C. c 50 °C.
Inspired by the high effectiveness of the catalyst system and the importance of chiral aryl piperidines in biological systems, we investigated asymmetric hydrogenation of additional N-benzylpyridinium salts having both α-aryl and α-heteroaryl functionalities (Scheme 1). Enantioenriched 2-benzothiophene piperidine derivative 2b was produced in 96.6:3.4 er and 89% yield. The pyridinium salt bearing a dibenzothiophene substituent was also reduced successfully; piperidine 2c was obtained in 95.1:4.9 er and 93% yield. For 2-thiophene substituted piperidines 2d and 2e, 96.4:3.6 and 96.7:3.3 enantiomeric ratios were obtained, respectively. Similarly, 90.3:9.7 er was produced for both 2-benzofuran and 2-dibenzofuran piperidines 2f and 2g.
N,O-Containing molecules are generally effective chelating ligands for catalysis;11 2-alkoxyaryl pyridinium salts were thus explored. The ortho-methoxy phenyl derivative 2h was obtained in 93.7:6.3 er and 91% yield. The more electron-rich di-tert-butyl analogue 2i was prepared in 95.3:4.7 er and 93% yield. The α-(2,3-dihydrobenzofuran) piperidine 2j was generated in 94.5:5.5 er and 84% yield, and α-diphenylether substituted piperidine 2k was obtained in 93.7:6.3 er and 89% yield. The catalyst system is chemoselective at reducing the pyridine group only and does not cause reduction of other sensitive functional groups such as halides; 2l was produced in 95.5:4.5 er and 92% yield. The reaction conditions are also applicable to electron-deficient aryl substituted pyridinium salts. α-(2,5-Disubstituted fluorophenyl)piperidine 2m was prepared in 94.7:5.3 er and 86% yield. 2-(Trifluorophenyl) derived piperidine 2n was isolated in 93.5:6.5 er and 88% yield. Furthermore, α-(2,4-dimethylphenyl)piperidine 2o was achieved in 97.7:2.3 er and 88% yield. Piperidine derivatives 2p and 2q bearing naphthyl and phenanthrenyl substituents were generated in 94.2:5.8 er and 95.6:4.4 er, respectively. In addition, less sterically demanding α-(2-phenyl)piperidine 2r was also prepared in 92.7:7.3 er and 91% yield.
Since N-alkyl piperidines are present in a number of pharmaceutical active compounds;12 an N-ethyl pyridinium salt 1s was studied yielding piperidine 2s in 99:1 er, which is higher than the N-Bn counterpart 2o. These outcomes represent the highest enantioselectivities in the reduction of 2-(hetero)aryl pyridinium salts. The reduction conditions are applicable to less sterically demanding aryls. Removal of the benzylic N-substituent of the resulting products readily affords enantioenriched α-aryl piperidines.9
To understand the origin of high enantioselectivity, we initiated a DFT study of the mechanism of this transformation. Earlier studies of the reduction of quinolines13 and imines14 using Ir complexes suggest that reaction proceeds via sequential protonation and hydride transfer via an outer sphere mechanism. Although recent work has shown that the active catalyst for imine reductions arising from incorporation of a molecule of starting material into the catalyst via C-H insertion and imine coordination,15 such a species is not possible here with the N-alkylated pyridines. In this system, sequential reduction leads to an enamine, reduction of which determines stereochemical outcome of the entire transformation (Scheme 2).9
Scheme 2. Proposed Catalytic Cycle Through an Outer-Sphere Reaction Mechanism16.

Thus, computational efforts were focused on the reduction of a model enamine (R1 = R2 = Me). Calculations indicate that both protonation and hydride transfer demonstrate significant facial bias that can serve as a basis for enantioselection. However, the two steps give rise to opposite selectivities. Thus, it was imperative to determine which step is selectivity determining. Prior reports on reduction of 2-alkyl pyridinium salts suggested that the second step hydride transfer proceeds through a dissociative mechanism, which controls the stereochemical outcome of the process.9b In this case, the tight ion pair was found to be much lower in energy than the separated ion pair after protonation (Scheme 3). We hypothesize that the tight ion pair causes the catalyst to stay on the same face during both steps. It thus appears that substrate protonation is the enantiodetermining step.
Scheme 3. Energetic Profiles for Protonation and Hydride Transfera.

a Blue and red pathways correspond to reactions on different faces of the substrate. Free energies were computed using PBE-D2/6-311+G(d,p),Ir: LANL2DZ (f),I:2DZ,IEFPCM-THF//PBE/6-31G(d),Ir:LANL2DZ;17 values are in kcal/mol.
Using the corresponding energy gap for the two different facial approaches in the protonation step, the enantioselectivity of the product was calculated (Scheme 4). The selectivity levels obtained in this way matched both the absolute stereochemistry and the selectivity values of the major product obtained experimentally, supporting the idea that protonation is enantiodetermining.18
Scheme 4. Comparison of Experimental and Calculated Enantioselectivities in the Reduction.

Analysis of the protonation transition states using a distortion-interaction approach19 (Scheme 5) indicates that the observed selectivity is primarily due to the distortion of the catalyst structure by the approaching substrate. The apparent lack of specific interactions selectively stabilizing one of the approaches suggests that selectivity relies predominantly on the geometrical fit between catalyst and substrate, providing an example for the well-known ‘lock and key’ concept.
Scheme 5. Distortion-Interaction Analysis of Protonation.

Examination of the transition states and vibrational modes in the distorted structure of the catalyst reveals that shape misfit is primarily localized between the approaching substrate and methoxy group of the catalyst (red arrow on Scheme 6). The methoxy group on the pyridine ring causes interactions such that the substrate orients closer to the other methoxy group indicated in the Scheme 6. In doing so, the substrate incurs a more disfavorable interaction with the indicated methoxy in the higher energy transition state.
Scheme 6. Structures of Protonation Transition States. Van der Waals Radius Spheres are Shown for the Ligand.

In summary, we have discovered the first enantioselective hydrogenation of α-heteroaryl-N-benzylpyridinium salts using an iridium catalyst containing a P,N ligand, MeO-BoQPhos. A variety of 2-(hetero)aryl pyridinium salts were reduced with high levels of enantioselectivity (up to 99.3:0.7 er) which represents the most efficient and practical method for this important transformation. The resultant piperidines can be readily deprotected and transformed into biologically interesting molecules, providing a concise synthesis of chiral piperidine-containing compounds. In addition, detailed DFT computational studies shed light on the reaction mechanism which involves an outer-sphere dissociative mechanism where initial protonation of the final enamine intermediate determines the stereochemical outcome of the transformation.
Supplementary Material
Acknowledgments
We thank the NIH (GM087605 M.C.K.) and Boehringer Ingelheim Pharmaceuticals for financial support. Computational support was provided by XSEDE (TG-CHE120052).
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details and characterization data (PDF)
References
- 1.(a) Feher M, Schmidt JM. J. Chem. Inf. Comput. Sci. 2003;43:218. doi: 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]; (b) Lovering F, Bikker J, Humblet C. J. Med. Chem. 2009;52:6752. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]; (c) Ritchie TJ, MacDonald SJF, Young RJ, Pickett SD. Drug Discovery Today. 2011;16:164. doi: 10.1016/j.drudis.2010.11.014. [DOI] [PubMed] [Google Scholar]; (d) Leeson PD, St-Gallay SA, Wenlock MC. MedChemComm. 2011;2:91. [Google Scholar]
- 2.(a) Andrey O, Alexakis A, Bernardinelli G. Org. Lett. 2003;5:2559. doi: 10.1021/ol0348755. [DOI] [PubMed] [Google Scholar]; (b) Laars M, Kriis K, Kailas T, Müürisepp A-M, Pehk T, Kanger T, Lopp M. Tetrahedron: Asymm. 2008;19:641. [Google Scholar]; (c) Noole A, Lippur K, Metsala A, Lopp M, Kanger T. J. Org. Chem. 2010;75:1313. doi: 10.1021/jo902664v. [DOI] [PubMed] [Google Scholar]
- 3.(a) Amat M, Cantó M, Llor N, Bosch J. Chem. Commun. 2002:526. doi: 10.1039/b200020m. [DOI] [PubMed] [Google Scholar]; (b) Stead D, Carbone G, O’Brien P, Campos KR, Coldham I, Sanderson A. J. Am. Chem. Soc. 2010;132:7260. doi: 10.1021/ja102043e. [DOI] [PubMed] [Google Scholar]; (c) Reddy LR, Das SG, Liu Y, Prashad M. J. Org. Chem. 2010;75:2236. doi: 10.1021/jo902710s. [DOI] [PubMed] [Google Scholar]; (d) Cui Z, Yu H-J, Yang R-F, Gao W-Y, Feng C-G, Lin G-Q. J. Am. Chem. Soc. 2011;133:12394. doi: 10.1021/ja2046217. [DOI] [PubMed] [Google Scholar]; (e) Beng TK, Gawley RE. Org. Lett. 2011;13:394. doi: 10.1021/ol102682r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Vo C-VT, Bode JW. J. Org. Chem. 2014;79:2809. doi: 10.1021/jo5001252. [DOI] [PubMed] [Google Scholar]
- 4.(a) McNally A, Prier CK, MacMillan DWC. Science. 2011;334:1114. doi: 10.1126/science.1213920. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hesp KD, Fernando DP, Jiao W, Londregan AT. Org. Lett. 2014;16:413. doi: 10.1021/ol403367b. [DOI] [PubMed] [Google Scholar]
- 5.(a) Campos KR, Klapars A, Waldman JH, Dormer PG, Chen C-Y. J. Am. Chem. Soc. 2006;128:3538. doi: 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]; (b) O’Brien P, Bilke JL. Angew. Chem. Int. Ed. 2008;47:2734. doi: 10.1002/anie.200704539. [DOI] [PubMed] [Google Scholar]; (c) Klapars A, Campos KR, Waldman JH, Zewge D, Dormer PG, Chen C-Y. J. Org. Chem. 2008;73:4986. doi: 10.1021/jo8006804. [DOI] [PubMed] [Google Scholar]; (d) Stead D, Carbone G, O’Brien P, Campos KR, Coldham I, Sanderson A. J. Am. Chem. Soc. 2010;132:7260. doi: 10.1021/ja102043e. [DOI] [PubMed] [Google Scholar]
- 6.(a) Dearden MJ, Firkin CR, Hermet J-PR, O’Brien P. J. Am. Chem. Soc. 2002;124:11870. doi: 10.1021/ja027774v. [DOI] [PubMed] [Google Scholar]; (b) Coldham I, Leonori D. Org. Lett. 2008;10:3923. doi: 10.1021/ol801579r. [DOI] [PubMed] [Google Scholar]; (c) Coldham I, Raimbault S, Whittaker DTE, Chovatia PT, Leonori D, Patel JJ, Sheikh NS. Chem. Eur. J. 2010;16:4082. doi: 10.1002/chem.200903059. [DOI] [PubMed] [Google Scholar]; (d) Beng TK, Gawley RE. J. Am. Chem. Soc. 2010;132:12216. doi: 10.1021/ja105772z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Legault CY, Charette AB. J. Am. Chem. Soc. 2005;127:8966. doi: 10.1021/ja0525298. [DOI] [PubMed] [Google Scholar]; (b) Wang D-S, Chen Q-A, Li W, Yu C-B, Zhou Y-G, Zhang X. J. Am. Chem. Soc. 2010;132:8909. doi: 10.1021/ja103668q. [DOI] [PubMed] [Google Scholar]; (c) Woodmansee DH, Pfaltz A. Top. Organomet. Chem. 2011;34:31. [Google Scholar]; (d) Wang D-S, Chen Q-A, Lu S-M, Zhou Y-G. Chem. Rev. 2012;112:2557. doi: 10.1021/cr200328h. [DOI] [PubMed] [Google Scholar]; (e) Liu Y, Du H. J. Am. Chem. Soc. 2013;135:12968. doi: 10.1021/ja406761j. [DOI] [PubMed] [Google Scholar]; (f) Zhao D, Glorius F. Angew. Chem. Int. Ed. 2013;52:9616. doi: 10.1002/anie.201304756. [DOI] [PubMed] [Google Scholar]; (g) Huang W-X, Liu L-J, Wu B, Feng G-S, Wang B, Zhou Y-G. Org. Lett. 2016;18:3082. doi: 10.1021/acs.orglett.6b01190. [DOI] [PubMed] [Google Scholar]; (h) Gualandi A, Savoia D. RSC Adv. 2016;6:18419. [Google Scholar]
- 8.(a) Ye Z-S, Chen M-W, Chen Q-A, Shi L, Duan Y, Zhou Y-G. Angew. Chem. Int. Ed. 2012;51:10181. doi: 10.1002/anie.201205187. [DOI] [PubMed] [Google Scholar]; (b) Chang M, Huang Y, Liu S, Chen Y, Krska SW, Davies IW, Zhang X. Angew. Chem., Int. Ed. 2014;53:12761. doi: 10.1002/anie.201406762. [DOI] [PubMed] [Google Scholar]; (c) Renom-Carrasco M, Gajewski P, Pignataro L, de Vries JG, Piarulli U, Gennari C, Lefort L. Adv. Synth. Catal. 2016;358:2589. doi: 10.1002/chem.201601501. [DOI] [PubMed] [Google Scholar]
- 9.(a) Wei X, Qu B, Zeng X, Savoie J, Fandrick KR, Desrosiers J-N, Tcyrulnikov S, Marsini MA, Buono FG, Li Z, Yang B-S, Tang W, Haddad N, Gutierrez O, Wang J, Lee H, Ma S, Campbell S, Lorenz JC, Eckhardt M, Himmelsbach F, Peters S, Patel ND, Tan Z, Yee NK, Song JJ, Roschangar F, Kozlowski MC, Senanayake CH. J. Am. Chem. Soc. 2016;138:15473. doi: 10.1021/jacs.6b09764. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qu B, Mangunuru HPR, Wei X, Fandrick KR, Desrosiers J-N, Sieber JD, Kurouski D, Haddad N, Samankumara LP, Lee H, Savoie J, Ma S, Grinberg N, Sarvestani M, Yee NK, Song JJ, Senanayake CH. Org. Lett. 2016;18:4920. doi: 10.1021/acs.orglett.6b02401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conversion is <20% for MeO-BoQphos in the absence of I2. For additive effect, see: Wang D-W, Wang X-B, Wang D-S, Lu S-M, Zhou Y-G, Li Y-X. J. Org. Chem. 2009;74:2780. doi: 10.1021/jo900073z.
- 11.(a) Beckmann U, Hägele G, Frank W. Eur. J. Inorg. Chem. 2010:1670. [Google Scholar]; (b) Wang M, Wang Z, Shi Y-H, Shi X-X, Fossey JS, Deng W-P. Angew. Chem. Int. Ed. 2011;50:4897. doi: 10.1002/anie.201007960. [DOI] [PubMed] [Google Scholar]; (c) Liu L, Carroll PJ, Kozlowski MC. Org. Lett. 2015;17:508. doi: 10.1021/ol503521b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Stumpf A, Renolds M, Sutherlin D, Babu S, Bappert E, Spindler F, Welch M, Gaudino J. Adv. Synth. Catal. 2011;353:3367. [Google Scholar]; (b) Peng Z, Wong JW, Hansen EC, Puchlopek-Dermenci ALA, Clarke HJ. Org. Lett. 2014;16:860. doi: 10.1021/ol403630g. [DOI] [PubMed] [Google Scholar]
- 13.(a) Zhou H, Li Z, Wang Z, Wang T, Xu L, He Y, Fan Q-H, Pan J, Gu L, Chan ASC. Angew. Chem. Int. Ed. 2008;47:8464. doi: 10.1002/anie.200802237. [DOI] [PubMed] [Google Scholar]; (b) Dobereiner GE, Nova A, Schley ND, Hazari N, Miller SJ, Eisenstein O, Crabtree RH. J. Am. Chem. Soc. 2011;133:7547. doi: 10.1021/ja2014983. [DOI] [PubMed] [Google Scholar]
- 14.Hopmann KH, Bayer A. Organometallics. 2011;30:2483. [Google Scholar]
- 15.Schramm Y, Barrios-Landeros F, Pfaltz A. Chem. Sci. 2013;4:2760. [Google Scholar]
- 16.Saidi O, Williams JMJ. Iridium-Catalyzed Hydrogen Transfer Reactions. In: Andersson P, editor. Iridium Catalysis. Topics in Organometallic Chemistry. Vol. 34. Springer, Berlin, Heidelberg: pp. 77–106.(b) The proposed generation of the Ir-H species is detailed in the Supporting Information.
- 17.Hopmann KH. Organometallics. 2016;35:3795. [Google Scholar]
- 18.(a) Additional analysis (see the Supporting Information) suggests that proton tunneling has a significantly lower rate than the normal thermal mode and, therefore, does not affect selectivity of the reaction. (b) The major enantiomer is (R)-configuration for 2-(hetero)aryl-N-benzyl-piperidines, which was suggested from calculation studies and was also assigned by analogy from 2-alkyl-N-benzyl-piperidines in reference 9b.
- 19.(a) Bickelhaupt FM. J. Comput. Chem. 1999;20:114. [Google Scholar]; (b) Ess DH, Houk KN. J. Am. Chem. Soc. 2007;129:10646. doi: 10.1021/ja0734086. [DOI] [PubMed] [Google Scholar]; (c) Bickelhaupt FM, Houk KN. Angew. Chem. Int. Ed. 2017;56:10070. doi: 10.1002/anie.201701486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.