

Abstract
Fibrous cross-β aggregates (amyloids) and their transmissible forms (prions) cause diseases in mammals (including humans) and control heritable traits in yeast. Initial nucleation of a yeast prion by transiently overproduced prion-forming protein or its (typically, QN-rich) prion domain is efficient only in the presence of another aggregated (in most cases, QN-rich) protein. Here, we demonstrate that a fusion of the prion domain of yeast protein Sup35 to some non-QN–rich mammalian proteins, associated with amyloid diseases, promotes nucleation of Sup35 prions in the absence of pre-existing aggregates. In contrast, both a fusion of the Sup35 prion domain to a multimeric non-amyloidogenic protein and the expression of a mammalian amyloidogenic protein that is not fused to the Sup35 prion domain failed to promote prion nucleation, further indicating that physical linkage of a mammalian amyloidogenic protein to the prion domain of a yeast protein is required for the nucleation of a yeast prion. Biochemical and cytological approaches confirmed the nucleation of protein aggregates in the yeast cell. Sequence alterations antagonizing or enhancing amyloidogenicity of human amyloid-β (associated with Alzheimer's disease) and mouse prion protein (associated with prion diseases), respectively, antagonized or enhanced nucleation of a yeast prion by these proteins. The yeast-based prion nucleation assay, developed in our work, can be employed for mutational dissection of amyloidogenic proteins. We anticipate that it will aid in the identification of chemicals that influence initial amyloid nucleation and in searching for new amyloidogenic proteins in a variety of proteomes.
Keywords: amyloid, amyloid-beta (AB), neurodegeneration, neurodegenerative disease, prion, protein aggregation, protein folding, yeast prion
Introduction
Cross-β fibrous protein polymers, reproduced and spread via nucleated polymerization and termed amyloids, are associated with a variety of human and animal diseases, including Alzheimer's (AD),8 Parkinson's (PD), and Huntington's (HD) diseases (1). Some evidence points to the connection between amyloids and type II diabetes (2, 3). Infectious amyloidogenic prion protein (PrP) is responsible for transmissible spongiform encephalopathies (TSEs), also called prion diseases (4, 5). Some amyloid and prion diseases (including AD and TSEs) are fatal, and most of them are incurable. AD is typically reported as the 6th most frequent cause of death in the United States (6); however, this is certainly an underestimate, as AD was routinely underdiagnosed in the past, and a significant portion of Americans over the age of 65 are dying from complications caused by AD. Therefore, estimates evaluating AD as the 3rd most frequent cause of death in the United States (7), and possibly in other developed countries with a long life-expectancy, are likely to be more realistic. Moreover, health-care costs related to dementias (i.e. mostly to AD) are estimated at the level of $259 billion in the year 2016 (6), and AD is one of the major factors affecting the quality of life at an advanced age (8). In case of TSEs, the epidemics of “mad cow” disease led to huge losses in the European cattle industry (5). In addition to disease-related amyloids and prions, some amyloids or amyloid-like protein polymers have been described that are related to biologically positive functions, such as formation of structural fibrous materials (e.g. silks), storage of peptide hormones, scaffolding of covalent polymers (e.g. melanin), and long-term memory (9, 10).
Despite such a broad biological impact of amyloids and prions, the mechanism of their initial formation in vivo remains a mystery. Many proteins possess an amyloid-forming potential in the test tube (1); however, it is effectively suppressed in the biological systems, apparently due to its interference with the normal (functional) protein fold. In most cases, it is unclear how disease-related or functional amyloids overcome such a suppression. Some amyloid and prion diseases are caused by mutations that either occur in the gene coding for amyloid-forming protein or influence production of this protein (11–13), although the molecular basis of the impact of these mutations on protein structure remains poorly understood in most cases. However, a majority of amyloid diseases (including the vast majority of AD cases) occur sporadically, due to unknown reasons (14). Problems with structural studies of amyloidogenic proteins (e.g. due to their inability to form crystals) and differences between patterns of amyloid formation in vitro (where most proteins can undergo this process depending on conditions) and in vivo (where only a few of them are capable of doing so) further complicate the understanding of the initial steps of amyloid nucleation in natural conditions. Low rates of in vivo amyloid formation and late onset or long incubation periods, detected for most amyloid and prion diseases, make it difficult or impossible to catch amyloid nucleation in mammalian models. As a variety of unrelated proteins with very different sequences are shown to form amyloids, the sequence requirements for amyloid nucleation also remain elusive.
In yeast and other fungi, endogenous prions (in most cases of amyloid nature) manifest themselves as non-Mendelian elements heritable via cytoplasm (15, 16). Yeast prion proteins contain so-called prion domains (PrDs) that are entirely responsible for the intermolecular interaction leading to the formation of an amyloid axis, and are, at least in some cases, distinct from domains responsible for the major cellular functions of the same proteins. Some yeast prions control easily detectable phenotypic traits, typically resulting from a partial loss of the cellular function of a protein as a result of its incorporation into prion polymers. For example, formation of a prion state (termed [PSI+]) by the Sup35 protein, a yeast counterpart of a eukaryotic translation termination factor eRF3 (17, 18), results in defective termination of translation, leading to a read-through of stop codons (nonsense-suppression), an easily detectable phenotype in specially designed yeast strains (15). This enables researchers to develop phenotypic assays for prion formation that are capable of detecting even rare events.
By using yeast models, it was demonstrated that de novo prion nucleation is induced by the transient overproduction of a prion-forming protein or its PrD (19–22). However, an efficient prion induction by overproduced Sup35 protein or its PrD requires the presence of another prion, usually [PIN+] (or [RNQ+]), a prion form of Rnq1 protein (23–25). It was proposed that Rnq1 prion polymers nucleate the initial assembly of the Sup35 polymers. A transient direct association between Rnq1 polymers and Sup35 appears likely as the prion domains of both these proteins are rich in Gln and Asn residues. This is also true for the majority of other yeast prion proteins, and the presence of the other (in most cases, QN-rich) protein in an aggregated form was indeed shown to reproduce the effect of Rnq1 prion on [PSI+] induction (24, 25). It is also possible to promote de novo nucleation of the Sup35 prion in the absence of Rnq1 prion by expressing a modified PrD of Sup35. For example, an attachment of either an artificial highly hydrophobic extension (23) or a polyQ region of the same length as in the human huntingtin protein in case of HD (26) to the Sup35 PrD-containing derivative promoted [PIN+]-independent prion formation. However, it remained unclear whether known amyloidogenic proteins or domains, which are not QN-rich, can influence prion nucleation by a QN-rich yeast protein.
Here, we show that the initial nucleation of Sup35 protein in the absence of other pre-existing prions can be promoted by the attachment of a mammalian amyloidogenic protein (or domain) to Sup35 PrD. Moreover, we demonstrate that a de novo prion nucleation assay in yeast can be applied to dissecting the role of specific regions or amino acid residues of mammalian proteins in altering the amyloidogenic properties of these proteins
Results
Mammalian amyloidogenic proteins do not promote [PSI+] nucleation in trans
It has previously been shown that co-overproduction of some yeast prionogenic proteins can promote prion formation by another yeast prion protein in the strain lacking pre-existing prions (24, 25, 27, 28). To determine whether mammalian amyloidogenic proteins exhibit such an effect on prion formation by the yeast protein Sup35, we have overproduced the mouse prion protein (PrP), associated with TSEs, and the human β-amyloid peptide (Aβ), associated with AD, in a yeast strain lacking pre-existing prions ([pin− psi−]) either individually or simultaneously with separately expressed Sup35 PrD, Sup35N (Fig. 1). In the case of PrP, we have employed the region between positions 90 and 230, which is sufficient to generate and maintain a prion state in mammals (29). In the case of Aβ, the most amyloidogenic and pathogenic (30) 42-residue variant (Aβ(1–42)) has been employed. We have confirmed that the PrP(90–230) protein is produced in yeast (Fig. S1). However, the levels of Aβ(1–42) were below detection limits (data not shown), possibly due to a low proteolytic stability of this short peptide in yeast cells. Therefore, we have also used the PrP(90–231)-GFP and Aβ(1–42)-GFP fusion proteins that are both produced at high levels in yeast cells (Fig. S1A). The PrP- and Aβ-based constructs produced amyloid-like detergent-resistant aggregates in yeast, as shown previously (31) and confirmed by us (Fig. S1B) using the semi-denaturing detergent agarose gel electrophoresis, SDD-AGE (32). To detect [PSI+] formation, we employed the ade1–14 (UGA) reporter (15). The [psi−] strains bearing this reporter are Ade− (i.e. they do not grow on medium lacking adenine) and only rarely produce spontaneous Ade+ colonies, in part due to reversions or suppressor mutations. The conversion of endogenous Sup35 into a prion form leads to a termination defect and read-through of ade1–14, resulting in an Ade+ phenotype. Therefore, [PSI+] induction can be detected as an increase in the frequency of Ade+ colonies over a low background. None of the mammalian proteins (PrP(90–230), PrP(90–231)-GFP, Aβ(1–42), or Aβ(1–42)-GFP) was able to induce [PSI+] formation, both at normal levels of Sup35 and in the presence of excess Sup35N (Fig. 1, A and B). This was in contrast to the yeast prionogenic QN-rich protein Lsb2 (fused to GFP) that promoted [PSI+] formation in the presence of excess Sup35N (Fig. 1, A and B), as shown previously (23, 33, 34).
Figure 1.
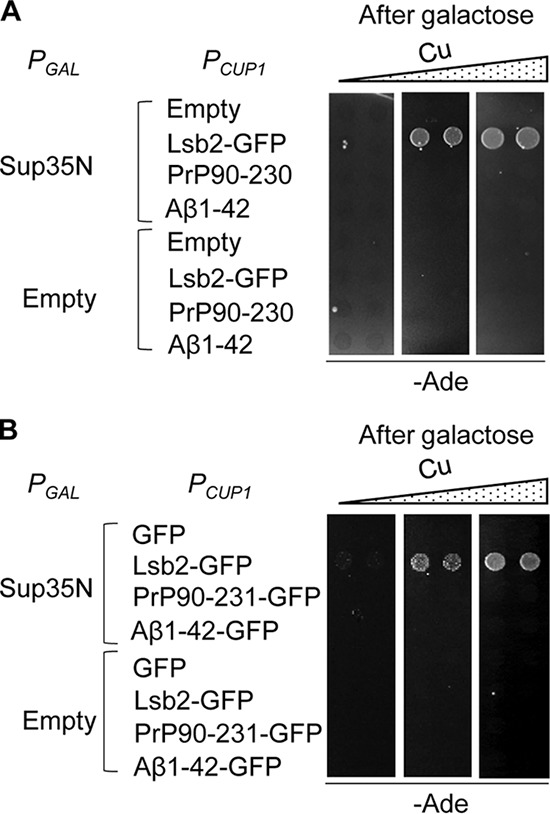
Mammalian amyloidogenic proteins PrP and Aβ(1–42) do not induce formation of the Sup35 prion in trans. Overexpression of PrP(90–230) or Aβ(1–42) (A) or of their respective fusions to GFP (B) from the copper-inducible promoter, PCUP1, induces [PSI+] formation in the [psi− pin−] strain neither on its own nor in the presence of excess Sup35N (produced under the control of the galactose inducible promoter, PGAL). The QN-rich prion-inducing protein Lsb2 fused to GFP (B) is shown as a positive control. Cultures were pre-incubated on the medium containing additional CuSO4 at a concentration of 0, 50, and 150 μm from left to right. The protein levels for PrP(90–230), PrP(90–231)-GFP, and Aβ(1–42)-GFP are shown in Fig. S1A; the formation of detergent-resistant aggregates by PrP(90–231)-GFP and Aβ(1–42)-GFP and shown in Fig. S1B.
Mammalian amyloidogenic proteins promote [PSI+] nucleation when attached to the Sup35 prion domain
Next, we checked what happens if a mammalian amyloidogenic protein is physically attached to the fragment of Sup35 containing the PrD. We prepared a series of such constructs as shown in Fig. 2A. Some of them also contained an HA tag (see “Experimental procedures”), which does not influence the ability of the protein to induce a prion, according to our data (not shown). The Sup35N fragment (roughly equivalent to Sup35 PrD), produced from a copper-inducible (PCUP1) promoter, can slightly induce the formation of Ade+ ([PSI+]) colonies in a [pin−] strain at high concentrations of CuSO4 (e.g. see Table 1), but this effect is weak and is not clearly detectable in plate assays (e.g. see Fig. 2B) unless very high concentrations of CuSO4 and/or very long incubation periods are used.
Figure 2.
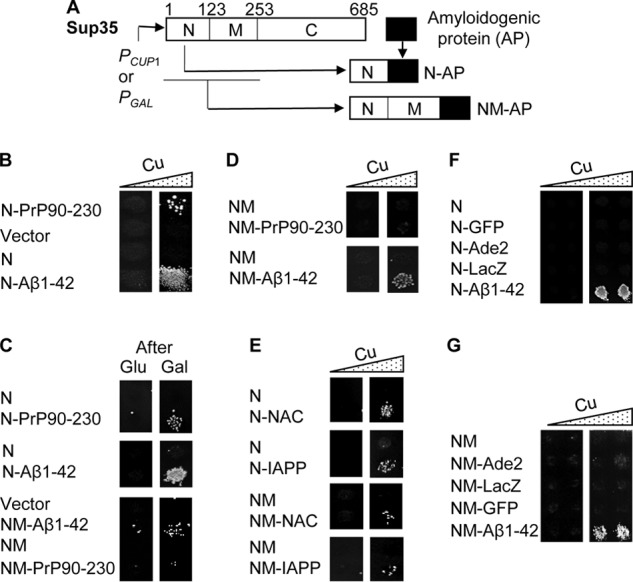
Phenotypic detection of prion nucleation by chimeric constructs containing mammalian amyloidogenic proteins in the yeast model. A, scheme of construction of the chimeric genes that contain regions coding for mammalian amyloidogenic proteins (AP) attached to the C terminus of the region coding for Sup35N or Sup35NM. Numbers indicate amino acid position in the Sup35 sequence. B–D, transient copper-induced (B and D) or galactose-induced (C) overproduction of the chimeric proteins containing Sup35N (B and C) or Sup35NM (D), each fused to PrP(90–230) or Aβ(1–42), promotes the de novo formation of [PSI+] in a [psi− pin−] strain. For frequencies of [PSI+] induction, see Table 1. E, transient overproduction of Sup35N (or NM) each fused to NAC(61–93) or IAPP(41–69) promotes the de novo formation of [PSI+] in a [psi− pin−] strain. F and G, transient overproduction of Sup35N fused to Ade2, LacZ, or GFP fails to promote de novo [PSI+] formation in a [psi− pin−] strain. The Sup35N-Aβ(1–42) (F) or Sup35NM-Aβ(1–42) (G) construct was used as a positive control. B and D–G, images from −Ade plates are shown, obtained without (left column) or with (right column) pre-incubation in the presence of additional (100 μm) CuSO4. C, images from −Ade plates obtained after pre-incubation on the glucose medium (left column) or on the medium with 2% galactose instead of glucose (right column) are shown. A zoomed-in image of the plate used for preparing B, confirming a reproducibility of the effect by showing several transformants for each construct, is included in Fig. S2. Comparison of [PSI+] induction by the PrP(90–230)- and Aβ(1–42)-based chimeric constructs in the [pin−] and [PIN+] backgrounds is shown in Fig. S3, B and C.
Table 1.
Frequencies of [PSI+] induction by chimeric and control plasmids
| Inducer | Frequency (± S.D) of Ade+ colonies per 10,000 cells after 100 μm CuSO4 |
|
|---|---|---|
| 0 h | 24 h | |
| Vector | 0.08 ± 0.02 | 0.07 ± 0.04 |
| Sup35N | 0.07 ± 0.05 | 0.31 ± 0.11 |
| Sup35N-PrP(90–230) | 0.07 ± 0.02 | 8.4 ± 0.7 |
| Sup35N-PrP(120–230) | 0.01 ± 0.02 | 0.25 ± 0.23 |
| Sup35N-PrP(90–144) | 0.11 ± 0.05 | 54 ± 17 |
| Sup35N-PrP(90–159) | 0.06 ± 0.05 | 736 ± 44 |
| Sup35N-PrP(90–171) | 0.07 ± 0.07 | 35 ± 6 |
| Sup35N-Aβ(1–42) | 1.19 ± 0.16 | 1178 ± 208 |
| Sup35N-Aβ(1–40) | 0.09 ± 0.08 | 2.0 ± 0.9 |
| Sup35N-Aβ(1–42)*** (F19S, F20S, I31P) | 0.09 ± 0.05 | 0.36 ± 0.28 |
| Sup35NM | 0.07 ± 0.06 | 0.13 ± 0.04 |
| Sup35NM-Aβ(1–42) | 0.11 ± 0.03 | 30 ± 7 |
| Sup35NM-Aβ(1–42)*** (F19S, F20S, I31P) | 0.01 ± 0.02 | 0.04 ± 0.03 |
No [PSI+] induction occurs when Sup35N alone is expressed from the galactose-inducible PGAL promoter (e.g. see Fig. 1A). The Sup35NM fragment, bearing both PrD and the middle region (Sup35M), which contains a high concentration of charged residues and is responsible for keeping Sup35 in a soluble state, does not induce the formation of Ade+ colonies in the [pin−] strain (e.g. see Fig. 2C and Table 1). Notably, an attachment of the region coding for either mouse PrP(90–230) or human Aβ(1–42) to the C terminus of Sup35N (Fig. 2A) enabled such a chimeric construct to induce Ade+ colonies after incubation in the presence of CuSO4 even in conditions when induction by Sup35N alone was not detectable (see Fig. 2B, Fig. S2, and Table 1). [PSI+] induction by Sup35N-Aβ(1–42) was stronger than that by Sup35N-PrP(90–230), and it could be detected even at background levels of CuSO4 as seen in the quantitative assay (Table 1) and occasionally in the plate assay (e.g. Fig. 2B). More importantly, the ability of these chimeric constructs to induce Ade+ colonies was not promoter-specific and did not depend on the presence of CuSO4 per se, as it was reproduced by using the chimeric constructs expressed from the PGAL promoter (Fig. 2C). Aβ(1–42) also promoted Ade+ formation when fused to the Sup35NM fragment and was expressed from either a PGAL (Fig. 2C) or PCUP1 (Fig. 2D) promoter, albeit at a lower frequency (Table 1), and in the latter case at higher concentrations of CuSO4 when compared with Sup35N-Aβ(1–42). However, we have not detected Ade+ induction by the Sup35NM-PrP(90–230) construct (Fig. 2D).
We have shown that the vast majority of Ade+ colonies induced by the PrP- or Aβ-based chimeric constructs in the [pin−] strain retain suppression after the loss of the inducing plasmid and are curable by serial passages on medium containing an antiprion agent guanidine hydrochloride (GdnHCl) (Table 2). These data confirm that the majority of these colonies arise from the conversion of the endogenous Sup35 protein into a prion form, [PSI+]. Notably, a high expression of the PrP- or Aβ-based constructs (fused to either GFP or Sup35 PrD) did not inhibit the growth of the [pin−] yeast strain (data not shown) and did not increase the levels of the stress-inducible chaperone Hsp104 (Fig. S3A). These results show that prion induction by the PrP or Aβ constructs is not a consequence of proteotoxic stress. As expected, both PrP- or Aβ-based chimeric constructs, as well as control Sup35N protein, efficiently induced [PSI+] formation in a [PIN+] strain (Fig S3, B and C).
Table 2.
Guanidine curability of Ade+ colonies induced by chimeric constructs
| Inducer | Colonies curable by GdnHCl | Total no. of colonies tested |
|---|---|---|
| Sup35N-PrP(90–230) | 35 | 43 |
| Sup35N-PrP(90–144) | 39 | 40 |
| Sup35N-PrP(90–159) | 30 | 33 |
| Sup35N-PrP(90–171) | 27 | 29 |
| Sup35N-Aβ(1–42) | 28 | 29 |
| Sup35N-Aβ(1–40) | 24 | 30 |
The ability to promote [PSI+] nucleation in the absence of [PIN+] upon fusion to Sup35N or NM is not restricted only to Aβ(1–42) or PrP(90–230), as two other human amyloidogenic peptides, namely the aggregation-prone region (“non-amyloid component” or NAC) of α-synuclein, associated with PD (14, 35), and amylin (or IAPP) peptide, associated with type II diabetes (2), also exhibited such an effect (Fig. 2E). In contrast, several proteins without known amyloidogenic properties, specifically yeast AIR-carboxylase (Ade2), bacterial β-galactosidase (LacZ), and jellyfish green fluorescent protein (GFP) did not induce [PSI+] formation at any noticeable level when fused to Sup35N (Fig. 2F) or NM (Fig. 2G). Notably, some of these proteins are known to form multimers, and in a separate experiment employing the ade2 mutant strain, we have specifically shown that the Sup35N-Ade2 and Sup35NM-Ade2 constructs produce functional AIR carboxylase in yeast (Fig. S3D). This confirms that these chimeric proteins form multimeric complexes in yeast, because the functionality of AIR carboxylase depends on its multimerization (36). Therefore, our data show that the ability of a protein to promote prion nucleation in a fusion to a fragment bearing the PrD of Sup35 depends on the amyloidogenic properties of such a protein, rather than with its ability to form multimeric complexes per se.
Mammalian amyloidogenic proteins promote the formation of biochemically and cytologically detectable aggregates in the yeast cells
By using semi-denaturing detergent-agarose gel electrophoresis (SDD-AGE), we have demonstrated that the chimeric proteins containing PrP(90–230) or Aβ(1–42) produce detergent-resistant polymers in the yeast cells lacking pre-existing prions (Fig. 3A), as is typical of yeast prions and amyloids (32), and promote the immobilization of endogenous Sup35 protein into an aggregated fraction (Fig. 3B). Likewise, the chimeric protein, containing the NAC region of α-synuclein, also produced the detergent-resistant aggregates in yeast in the absence of pre-existing prions (Fig. 3A). Thus, phenotypically detectable [PSI+] formation coincides with physical aggregation of the inducer protein and immobilization of the inducee protein into aggregates.
Figure 3.
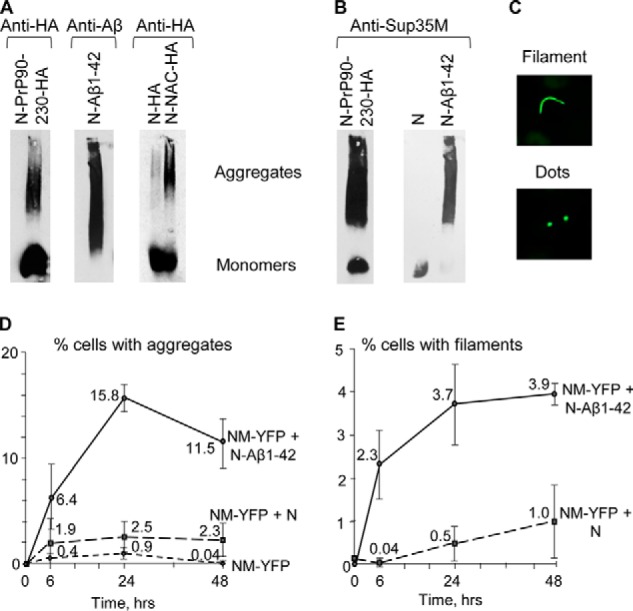
Biochemical and cytological detection of aggregation promoted by chimeric proteins in yeast. A and B, cell lysates of cultures expressing chimeric proteins Sup35N-PrP(90–230)-HA or Sup35N-Aβ(1–42) or Sup35N-NAC-HA in the presence of 100 μm CuSO4 analyzed by SDD-AGE. A, monomers and high molecular weight aggregates of chimeric proteins were detected by either the anti-HA antibody for the HA-tagged Sup35N-PrP(90–230), Sup35N, and Sup35N-NAC constructs or the anti-Aβ 6E10 antibody for the Sup35N-Aβ(1–42) construct. B, immobilization of the endogenous Sup35 protein into an aggregated fraction in the presence of Sup35N-PrP(90–230) or Sup35N-Aβ(1–42) (but not in the presence of control Sup35N) is detected using the anti-Sup35M antibody. C, examples of cells co-expressing the Sup35NM-YFP and Sup35N-Aβ constructs and forming either filamentous (top image) or dot-like (bottom image) fluorescent aggregates. D, kinetics of aggregate formation in the [psi− pin−] strain, bearing various combinations of plasmid-encoded proteins (as indicated), after the addition of CuSO4 up to 50 μm. E, kinetics of the formation of filamentous aggregates in the same experiment that is shown in D. The culture expressing the Sup35NM-YFP construct alone is not shown in E, as it did not produce any filamentous aggregates.
A high level expression of a fluorescently-tagged Sup35NM fragment is known to produce cytologically detectable aggregates efficiently in a strain bearing pre-existing prions (e.g. [PIN+], the prion form of Rnq1) but not in a [pin−] strain (37, 38). These aggregates include both dot-like puncta and filamentous assemblies, seen as lines, ribbons, or rings (see examples in Fig. 3C), and are defined here as “filaments.” It is shown (37, 38) that filaments specifically represent intermediate stages of the prion generation pathway, whereas dots may correspond to other types of intermediates, to mature prions, and possibly to non-prion aggregates. We have confirmed that Sup35NM-YFP produces detectable aggregates only very rarely and transiently in the [pin−] strain (Fig. 3D), and it never produces filaments. In contrast, co-expression of Sup35N-Aβ(1–42) with Sup35NM-YFP in the [pin−] strain resulted in an efficient accumulation of fluorescently-tagged aggregated structures (Fig. 3D), detectable as early as after 6 h of induction, and increased in numbers with prolonged incubation. These structures included filaments (Fig. 3E) similar to those detected previously after the expression of fluorescently-tagged Sup35NM in the [PIN+] cells (37, 38). The co-expression of Sup35N with Sup35NM-YFP produced cytologically detectable aggregates at a significantly lower frequency and with slower kinetics, compared with Sup35N-Aβ(1–42) (Fig. 3D). These data confirm that fusion to Aβ promotes the nucleation of cytologically detectable aggregated structures by Sup35 PrD.
[PSI+] induction by chimeric proteins is not due to the induction of [PIN+]
Both Sup35N-PrP(90–230) and Sup35N-Aβ(1–42) constructs were capable of nucleating the [PSI+] prion in an rnq1Δ strain, lacking the Rnq1 protein (Fig. 4A). This shows that a chimeric protein does not promote formation of [PSI+] indirectly, via inducing [PIN+], a prion form of Rnq1, which would in turn induce [PSI+]. However, it is known that other endogenous yeast QN-rich proteins in an aggregated form can substitute for the [PIN+] in [PSI+] induction (23, 28). To make sure that chimeric PrP- or Aβ-based constructs do not induce [PSI+] by generating other prions that confer a [PIN+]-like effect, we mated a sample of independently obtained Sup35 derivatives, induced by Sup35N-PrP(90–230) or Sup35N-Aβ(1–42) in the [psi− pin−] strain, to the [psi− pin−] strain of opposite mating type, bearing a plasmid with the HSP104 gene under a strong constitutive PGPD promoter and a plasmid with the SUP35 gene under a galactose-inducible PGAL promoter (Fig. 4B). Excess Hsp104 is known to cure [PSI+] (39) but not [PIN+] (23) or the majority of other known yeast prions (40). Therefore, if [PSI+] formation was due to the formation of [PIN+] or another prion with similar [PSI+]-inducing capability, we would expect that the [psi−] derivative of such a [PSI+] isolate, cured of both the inducing plasmid and induced [PSI+], would stay [PIN+] and therefore be reinduced into a [PSI+] state after the overproduction of Sup35. However, the vast majority of [psi−] derivatives, being cured of [PSI+] as well as of the original inducer plasmid and HSP104 plasmid, were unable to turn into a [PSI+] state (Ade+ phenotype) after Sup35 was overproduced on galactose, indicating that they stay [pin−] (Fig. 4C). These data show that [PSI+] nucleation in the presence of chimeric constructs bearing mammalian amyloidogenic proteins is not due to the induction of [PIN+] prion or other yeast non-Sup35 prions with a similar effect.
Figure 4.
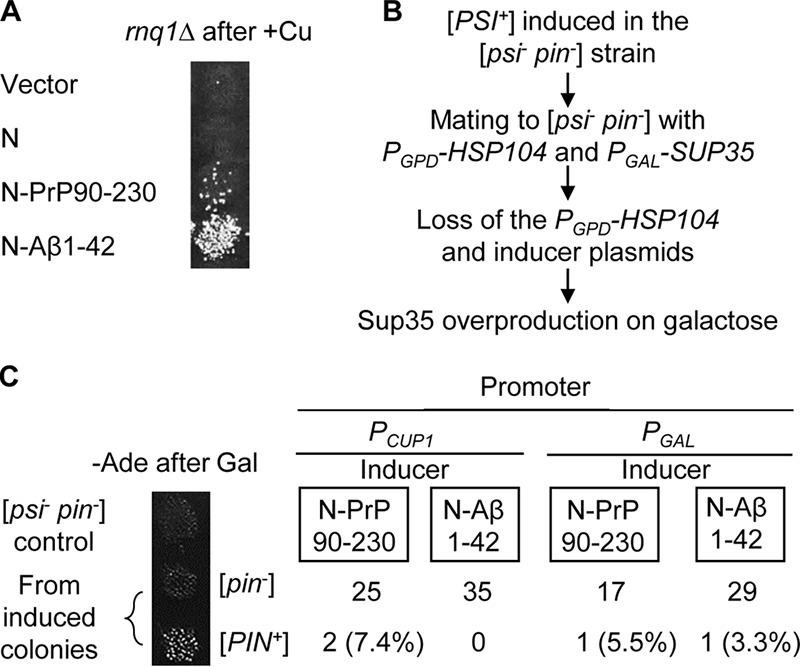
Induction of [PSI+] by chimeric constructs is not due to formation of another prion with a [PSI+]-inducing ([PIN+]) effect. A, induction of [PSI+] by chimeric constructs, expressed in the rnq1Δ strain with the addition of 100 μm CuSO4. B, scheme of the experiment for the detection of the formation [PIN+] or other prions with [PIN+]-like effect in the [PSI+] cells, induced by chimeric constructs. The [PSI+] colonies, induced in the [psi− pin−] strain by plasmids carrying Sup35N-PrP(90–230) or Sup35N-Aβ(1–42) (each colony originated from an independent transformant), were mated to the isogenic [psi− pin−] strain of the opposite mating type, carrying the plasmid with the HSP104 gene under a strong constitutive PGPD promoter and the plasmid with the SUP35 gene under galactose-inducible PGAL promoter. Resulting diploids (cured of [PSI+] by the constitutive overproduction of Hsp104) were then cured of the inducer and PGPD-HSP104 plasmids and placed onto a galactose medium to overexpress Sup35. Following transient induction of Sup35 on galactose, colonies were velveteen replica-plated to the −Ade medium with glucose to check for [PSI+] reinduction. C, results of the experiment are described in B. Only [PIN+] isolates can generate Ade+ (i.e. [PSI+]) colonies in these conditions. Most of the colonies derived from the [PSI+] isolates, which were induced by PrP- or Aβ-containing chimeric constructs, stayed [pin−].
Aβ alterations influence prion nucleation in yeast in the same direction as they do in humans or in vitro
Next, we checked whether alterations in Aβ known to influence prion propagation and disease in mammals have similar effects in yeast. Several variants of Aβ peptide exist in humans, of which Aβ(1–42) and Aβ(1–40) (lacking the last two amino acid residues) are the most abundant ones (41). Of these two, Aβ(1–42) is considered to be the most amyloidogenic and most pathogenic form in humans (30, 42). In yeast, Sup35N-Aβ(1–40) nucleated [PSI+] much less efficiently than did Sup35N-Aβ(1–42) (Fig. 5A), whereas Sup35NM-Aβ(1–40) did not nucleate [PSI+] at all (Fig. 5B). Notably, removal of the two N-terminal amino acid residues of Aβ within the chimeric construct did not inhibit [PSI+] nucleation (Fig. 5C), in agreement with structural models placing the N-terminal region of Aβ outside of the amyloid core (43, 44).
Figure 5.
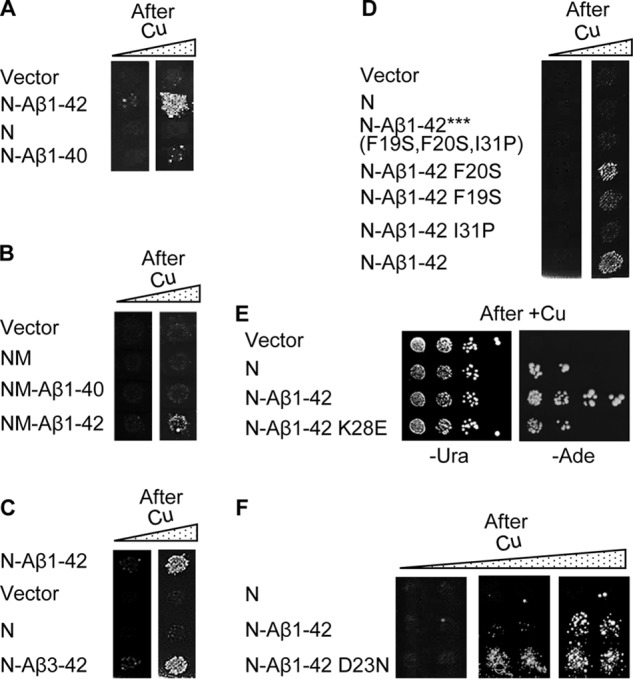
[PSI+] nucleation by chimeric constructs with various Aβ derivatives in yeast. A, Sup35N-Aβ(1–40) construct shows decreased [PSI+] induction in a [psi− pin−] strain, compared with Sup35N-Aβ(1–42). B, Sup35NM-Aβ(1–40) construct does not induce [PSI+] formation in a [psi− pin−] strain. [PSI+] induction by Sup35NM-Aβ(1–42) is shown as a positive control. C, Sup35N-Aβ(3–42) construct induces [PSI+] formation in a [psi− pin−] strain at levels comparable with Sup35N-Aβ(1–42). D, effects of base substitutions at positions 19, 20, and 31 of Aβ and of a combination of these substitutions on [PSI+] induction by the chimeric Sup35N-Aβ(1–42) constructs in a [psi− pin−] strain, compared with wild type Sup35N-Aβ(1–42). A–D, images from −Ade plates are shown, without (left column) or with (right column) pre-incubation on the medium with an additional 100 μm CuSO4. For quantitative data, see Table 1. E, K28E substitution decreases the ability of Sup35N-Aβ(1–42) to induce [PSI+] formation in a [psi− pin−] strain. Serial decimal dilutions of cultures, grown in the presence of 10 μm CuSO4, were spotted onto the −Ura medium selective for the plasmid (left image) and onto the −Ade medium selective for [PSI+] (right image). F, D23N substitution increases the ability of Sup35N-Aβ(1–42) to induce [PSI+] formation in a [psi− pin−] strain. Images from −Ade plates are shown, obtained after pre-incubation on the medium with additional 0, 10, or 50 μm CuSO4, from left to right.
We have also generated several mutations at the positions of Aβ(1–42) known to influence amyloid formation. Previous in vitro experiments and structural data identified positions 19, 20, and 31 as being important for amyloid formation by Aβ (45–47) and located within intramolecular cross-β-sheets of Aβ(1–40) polymers (48, 49). However, according to the most recent structural model of Aβ(1–42) polymers (44), only position 31 is located within one of the β-strands, whereas positions 19 and 20 are involved in hydrophobic interactions. Indeed, the substitution I31P, breaking the proposed β4-strand, greatly decreased [PSI+] nucleation by Sup35N-Aβ42, whereas the substitution F19S caused only a mild decrease, and the substitution F20S had almost no effect (Fig. 5D). Notably, the triple mutation F19S,F20S,I31P entirely abolished [PSI+] nucleation by both Sup35N-Aβ(1–42) (Fig. 5D and Table 1) and Sup35NM-Aβ(1–42) (Table 1). The Lys to Glu substitution, affecting the β3 strand and disrupting a presumable “salt bridge” that involves position Lys-28 (44, 50), also significantly decreased [PSI+] nucleation by Sup35N-Aβ(1–42) in yeast (Fig. 5E). In contrast, the substitution D23N, a so-called “Iowa mutation” associated with the heritable form of AD (11, 13), significantly increased [PSI+] nucleation in yeast (Fig. 5F). These data confirm that the effects of Aβ alterations in the yeast model parallel those detected in vitro or in humans.
Phenotypic characterization of the effects of Aβ alterations was confirmed by aggregation assays in yeast. The Sup35N-Aβ(1–40) construct (Fig. 6A) and Sup35N-Aβ(1–42) triple mutant (F19S,F20S,I31P; Fig. 6B) neither formed detergent-resistant polymers at detectable levels nor immobilized Sup35 into an aggregated state, according to SDD-AGE. Overall, our data show that the effects of Aβ alterations of [PSI+] nucleation in yeast parallel their effects shown in humans or in vitro and/or predicted from structural models.
Figure 6.
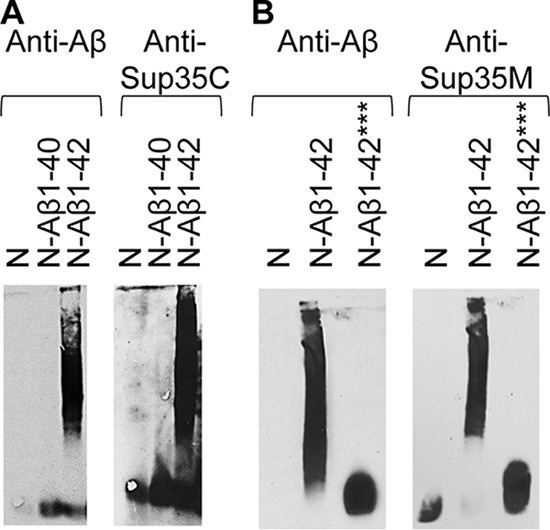
Biochemical detection of the effects of Aβ alterations on protein aggregation in yeast. The SDD-AGE analysis was performed as shown in Fig. 3. Cultures were grown in the presence of 100 μm CuSO4. A, in contrast to Sup35N-Aβ(1–42), the Sup35N-Aβ(1–40) construct does not efficiently aggregate (left image) and does not immobilize endogenous Sup35 into an aggregated fraction (right image). The small Aβ monomers are not seen on the left image as they have run out of the gel. B, Sup35N-Aβ(1–42) protein with triple F19S,F20S,I31P substitution, does not aggregate (left image) and does not immobilize endogenous Sup35 when probed into an aggregated fraction (right image). The image for N-Aβ(1–42) (left) and the images for N and N-Aβ(1–42) (right) in B are the same images that were shown in Fig. 3, A and B, respectively. These images are repeated here as positive (N-Aβ(1–42)) and negative (N) controls. ***, triple mutation F19S,F20S,I31P in amyloid-β peptide.
PrP alterations influence prion nucleation in yeast in the same direction as they influence the formation of PrP prion in mammals
Next, we checked whether correspondence between known effects of sequence alterations on the amyloid formation in other systems and on prion nucleation in yeast also holds true for PrP. Amino acid substitution P101L in mouse PrP (see Fig. 7A) corresponds to the human mutation P102L, associated with a heritable prion disease, and is shown to cause disease accompanied by a production of the infectious PrP protein in mice (10). In contrast, the substitution Q167R is shown to inhibit prion replication in mice (51). In agreement with these data, substitutions P101L and Q167R in the Sup35N-PrP(90–230) construct, respectively, increased or decreased [PSI+] nucleation in the yeast assay (Fig. 7B). The region between amino acid residues 90 and 119 is required for the susceptibility to prion disease in mammals (52). We have shown that a deletion of this region knocks out [PSI+] nucleation by the chimeric Sup35N-PrP protein in yeast (Fig. 7C and Table 1) and essentially eliminates the immobilization of full-size Sup35 protein into aggregates in the yeast cells as detected by SDD-AGE (Fig. 7D). Truncation of human PrP after positions 144 or 159, eliminating the C-terminal region, leads to a heritable disease with symptoms similar to prion disease (53–55), and truncated PrP forms amyloids in vitro (56). We have shown that C-terminal truncations of mouse PrP (at positions 144, 159, or 171) in the Sup35N-PrP chimeras significantly increased both [PSI+] nucleation (Fig. 7C and Table 1) and immobilization of Sup35 into amyloid aggregates (Fig. 7D), and truncation at position 159 of PrP also enabled [PSI+] nucleation in a fusion to Sup35NM (Fig. 7E). These data agree with the notion that C-terminal PrP truncations trigger the formation of disease via nucleating prion-like aggregates, even though transmissibility of such aggregates has not been proven. Notably, the PrP fragment, including only residues from 90 to 119 did not promote [PSI+] nucleation when fused to Sup35 (Fig. S5), indicating that although this region is essential for prion formation (see above), it is not sufficient for this process. The presence of the N-terminal region of PrP(23–89) increased [PSI+] nucleation in yeast, as demonstrated by the ability of the chimeric Sup35NM-PrP(23–230) protein to nucleate [PSI+] (Fig. 7F), in contrast to the Sup35NM-PrP(90–230) construct (see Fig. 2D). Although the 23–89 region of PrP is not necessary for prion formation or propagation in mammals, it contains oligopeptide repeats, whose expansions are known to cause a heritable disease with symptoms similar to a prion disease (57, 58), Overall, our data show that PrP alterations influence its ability to nucleate prions in yeast in the same direction as they influence (or are suggested to influence) prion diseases in mammals and humans.
Figure 7.
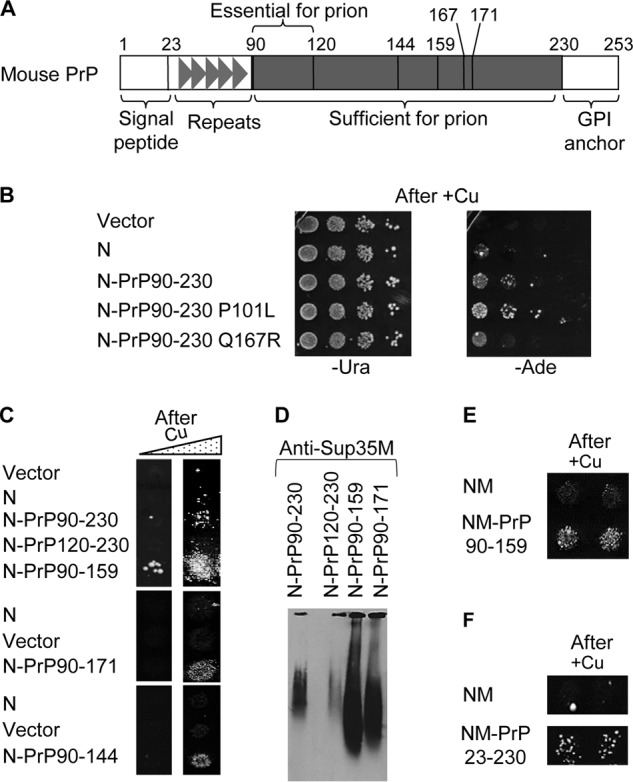
[PSI+] nucleation by chimeric constructs with various PrP derivatives in yeast. A, scheme of construction of the chimeric Sup35N-PrP(90–230) derivatives. Numbers indicate amino acid positions, corresponding to mutations or truncations made in our work. B, phenotypic detection of [PSI+] nucleation by wild type and mutant PrP-based chimeric constructs in yeast. Transient overproduction of the Sup35N-PrP constructs was induced on the medium with additional 100 μm CuSO4, and serial decimal dilutions were spotted onto the −Ura medium selective for the plasmid (left image) and onto the −Ade medium selective for [PSI+] (right image). C, comparison of [PSI+] nucleation by the Sup35N-PrP derivatives with various truncations after growth on the medium with additional 100 μm CuSO4. The Sup35N-PrP(120–230) construct was not able to nucleate [PSI+], whereas the Sup35N-PrP(90–144), Sup35N-PrP(90–159), and Sup35N-PrP(90–171) constructs exhibited increased [PSI+] formation, compared with Sup35N-PrP(120–230). Quantitative data are shown in Table 1. D, SDD-AGE analysis performed in the same way as on Fig. 3B shows that the Sup35N-PrP(120–230) construct cannot promote immobilization of endogenous Sup35 protein into an aggregated fraction, whereas the Sup35N-PrP(90–159) and Sup35N-PrP(90–171) constructs increase immobilization of Sup35 into an aggregated fraction, compared with Sup35N-PrP(90–230). Equal protein amounts were loaded in each case; monomeric fractions are not shown. E and F, Sup35NM fused to PrP(90–159) (D) or to PrP(23–230) (E) can promote formation of [PSI+] in a [psi− pin−] strain after overexpression. B, D, and E, images from −Ade plates are shown, obtained without (B and E, left columns) or with (D, and right columns on B and E) pre-incubation in the presence of additional (100 μm) CuSO4.
Effects of chimeric constructs on prion nucleation are not due to alterations in protein levels
One possible explanation for chimeric constructs, as well as for alterations of PrP or Aβ to influence [PSI+] nucleation, could be through altering the levels of chimeric proteins. To investigate this possibility, we have compared levels of proteins accumulated in yeast cells at the same concentrations of CuSO4. As described previously (59), and confirmed by us (Fig. S4A), Sup35N is accumulated at low levels in yeast, despite the fact that it has a higher prion-inducing activity in comparison with Sup35NM and Sup35. This is probably due to the high misfolding capability and proteolytic instability of Sup35N. The Sup35N-PrP(90–230) chimeric protein was produced at higher levels compared with Sup35N (Fig. S4A). However, this could not explain the increased prion-nucleating activity of Sup35N-PrP(90–230), because the Sup35N-PrP(120–230) derivative, not capable of prion nucleation, was produced at about the same level as Sup35N-PrP(90–230) (Fig. S4B). Moreover, the C-terminal truncated derivatives of Sup35N-PrP, which exhibited increased [PSI+] nucleation, were in fact accumulated at lower levels compared with Sup35N-PrP(90–230) (Fig. S4B). In the Aβ-based series, the prion-inducing Sup35N-Aβ(1–42) construct was accumulated at the same levels as the prion non-inducing Sup35N-Aβ(1–40) and Sup35N-Aβ(1–42) triple F19S,F20S,I31P mutant (Fig. S4, C and D), respectively. The [PSI+]-inducing Sup35NM-Aβ(1–42) construct was accumulated at the same level as the non-inducing Sup35NM-Aβ(1–40) construct, and both were less abundant than the non-inducing control, Sup35NM (Fig. S4E). Overall, our data show that although cellular levels of proteins used in this work could vary in some cases, the differences in prion nucleation cannot be explained by differences in protein abundance.
Different chimeric constructs induce different spectra of prion strains
The Sup35 protein can produce a variety of prion variants or “strains” that presumably correspond to various amyloid structures (20, 37). These strains can be differentiated from each other based on both their phenotypic manifestations and biochemical patterns. “Stronger” strains are characterized by higher levels of nonsense codon read-through (leading to better growth on −Ade medium and lighter color on complete medium in the case of the ade1–14 reporter) and by higher mitotic stability compared with “weaker” strains. This is due to the fact that “stronger” strains are generated by amyloid fibrils with a less rigid amyloid core that are more efficiently fragmented by the yeast chaperone machinery and therefore produce a larger number of oligomeric “seeds,” making immobilization of newly synthesized Sup35 and proliferation of prion state more efficient (60). Once established, the prion strain typically faithfully reproduces its observable characteristics.
To determine whether mammalian amyloidogenic proteins influence the parameters of prion strains produced in yeast, we compared spectra of prion strains generated in the presence of different inducing constructs. For this purpose, [PSI+] isolates were divided into three groups designated as “strong,” “intermediate,” and “weak” strains on the basis of growth on −Ade medium and color on complete (YPD) medium. Strain patterns were scored after elimination of the inducing plasmid, in order to exclude the possibility that the continuous presence of a chimeric construct influences the phenotypic manifestation of a [PSI+] strain. Data are shown in Fig. 8, Fig. S6, and in Table S1. Although Sup35N-PrP(90–230) and Sup35N-Aβ(1–42) induced preferentially or exclusively “strong” strains, the constructs with some deletion PrP derivatives, such as Sup35N-PrP(90–159) and Sup35N-PrP(90–171) induced preferentially or exclusively “intermediate” strains, and Sup35N-Aβ(1–40) produced [PSI+] isolates of all three classes. Notably, some point mutations changed a spectrum of the induced [PSI+] strains. For example, the Sup35N-Aβ(1–42) construct with mutation D23N induced preferentially intermediate [PSI+] isolates, whereas the construct with the mutation K28E construct induced preferentially weak and intermediate [PSI+] isolates. Altogether, these results indicate that the preferable type of a yeast prion strain, at least in part, depends on the mammalian amyloidogenic protein used in the inducing construct.
Figure 8.
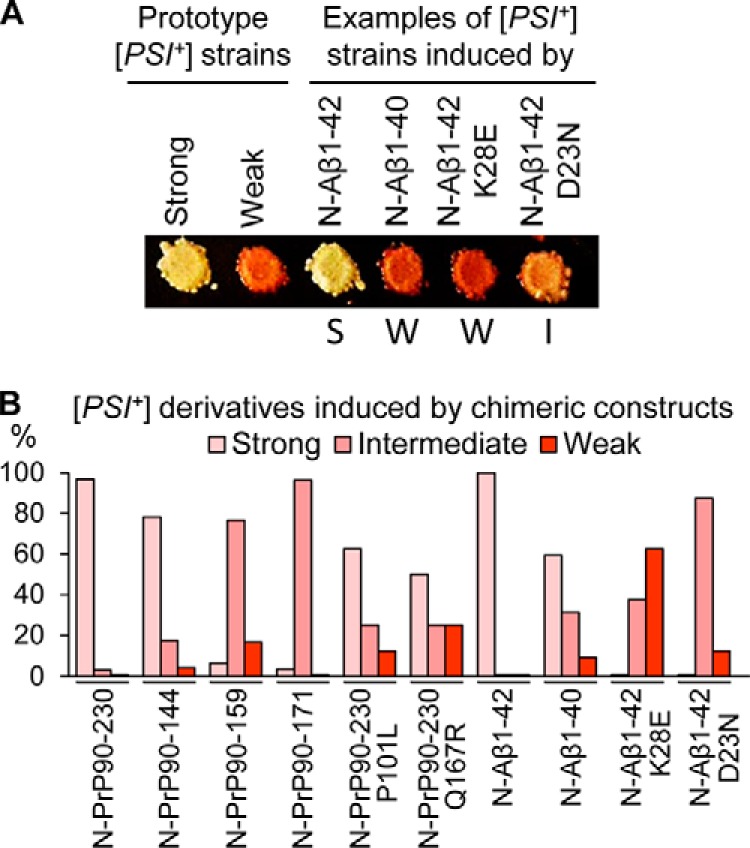
Spectra of prion strains induced by various Sup35N-PrP and Sup35N-Aβ derivatives. A, [PSI+] strains were distinguished by color on YPD and amount of growth on −Ade. Strong [PSI+] appeared white or whitish pink on YPD and grew after 2 days on −Ade; intermediate [PSI+] appeared solid pink on YPD and grew after 4 days on −Ade; weak [PSI+] appeared reddish pink on YPD and grew after 7 days on −Ade. Previously published prototype strains OT56 (for the strong [PSI+] prion) and OT55 (for the weak [PSI+] prion) are shown for comparison with representative strong (S), weak (W), and intermediate (I) isolates, nucleated by the chimeric constructs (as indicated) and tested after the loss of a prion-inducing plasmid. YPD plates were incubated for 4 days at 30 °C, followed by 3 days of refrigeration at 4 °C for the better color development. B, percentages of strong, intermediate, and weak [PSI+] strains induced by wild type and altered Sup35N-PrP and Sup35N-Aβ derivatives in a [psi− pin−] strain. More detailed information, including images for multiple isolates, data for the constructs containing point mutations, actual numbers, and errors (calculated as described in Ref. 94) is presented in Fig. S6 and Table S1.
Discussion
[PIN+]-independent [PSI+] nucleation
Efficient prion nucleation by the overproduced yeast Sup35 protein or its PrD-containing fragments typically requires the presence of another (usually QN-rich) protein in an aggregated form (23–25). A fusion of some Sup35 PrD-containing derivatives to extended polyQ tracts, resembling those associated with HD in humans, or to a yeast prion-forming protein Rnq1 promotes nucleation of the Sup35 prion even in the absence of pre-existing Q/N-rich yeast prions (26, 61). However, expanded polyQ constructs and QN-rich proteins were also reported to promote Sup35 aggregation in trans (62), so that an addition of a polyQ or another QN-rich region to the QN-rich Sup35 PrD could be interpreted as an expansion of Sup35 PrD. Our new data demonstrate (to our knowledge, for the first time) that a fusion of the Sup35 PrD-containing region (Sup35N or Sup35NM) to a non-QN–rich mammalian protein (or protein domain) with proven amyloidogenic properties is sufficient for nucleating the formation of Sup35-based prions in yeast cells lacking known pre-existing prions. An apparent explanation for this result is that mammalian proteins nucleate an amyloid in yeast, thus promoting amyloidization of the attached yeast prion domain (Fig. 9). This leads to immobilization of full-length endogenous yeast protein into prion aggregates, thus allowing for phenotypic detection of a yeast prion. Importantly, a covalent attachment of a mammalian “inducer” to Sup35N (or NM) is required for prion nucleation, as mammalian non–QN-rich amyloidogenic proteins do not promote [PSI+] induction in trans (Fig. 1). As expected, the Sup35N-based chimeric proteins are more efficient in prion nucleation than the Sup35NM-based chimeric proteins, apparently due to an anti-nucleation effect of the M region of Sup35, which contains stretches of potentially repulsive charged residues. This explains why the previous work by Choe et al. (63) failed to detect [PSI+] induction by the Sup35NM-PrP-GFP chimeric protein in the [pin−] cells. Indeed, the Sup35NM-PrP(90–230) chimeric protein also failed to nucleate [PSI+] in our hands (Fig. 2D), although [PSI+] induction was detected for the Sup35N-PrP(90–230) construct (Fig. 2B).
Figure 9.
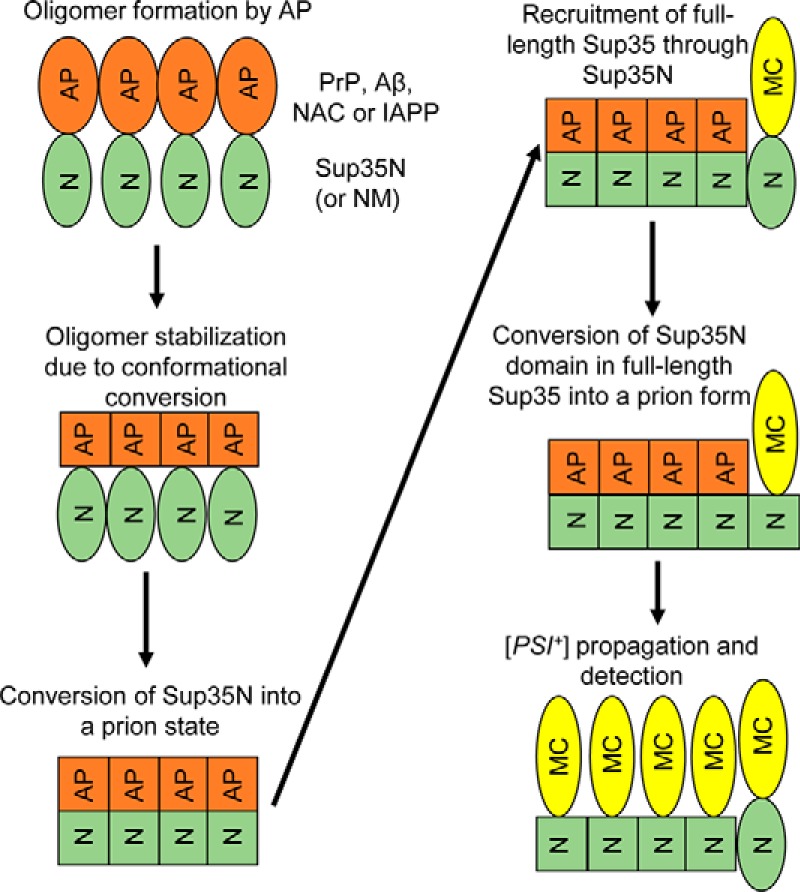
Model for [PSI+] nucleation by mammalian amyloidogenic proteins. N, M, and C - domains of Sup35. AP - mammalian amyloidogenic protein (PrP, Aβ, NAC region of α-synuclein, or IAPP). Non-prion isoforms are designated as ellipses; prion isoforms are shown as squares. For details, see comments in the text.
Role of protein amyloidogenicity in [PSI+] nucleation
Importantly, the formation of amyloid nuclei is not triggered by just any kind of protein multimerization, as shown by the lack of [PSI+] induction in the presence of chimeric constructs, producing non-amyloidogenic multimeric proteins such as such as Ade2 and LacZ (Fig. 2, F and G). Fusions of Sup35N with mammalian amyloidogenic proteins are characterized by higher protein abundance at the same levels of expression, compared with proteolytically unstable Sup35N (Fig. S4A). However, the increased frequency of prion nucleation by Sup35N-based chimeric proteins is not simply due to an increase in the abundance of chimeric constructs. Neither the deletion of the 90–119-region of PrP in Sup35N-PrP nor the triple substitution at positions 19, 20, and 31 of Aβ in Sup35N-Aβ decreased the abundance of chimeric constructs (Fig. S4, B and D); however, they knocked out the ability of a chimeric protein to nucleate [PSI+] (Figs. 5D and 7C). Moreover, the Sup35NM construct is expressed at even higher levels than Sup35NM-based prion-inducing chimeric proteins; however, it cannot nucleate [PSI+] in the [pin−] cells. This shows that the increased prion nucleation by chimeric constructs is a result of their amyloidogenic properties, leading to the initiation of the self-assembly into an amyloid form.
Amino acid residues influencing prion nucleation by Aβ
In the case of Aβ peptide, data from the yeast assay are also in a good agreement with existing results obtained in other systems. For example, the Aβ(1–40) peptide lacking the two C-terminal hydrophobic amino acids, Ile-41 and Ala-42, is considered to be less aggregation-prone and is a typically non-pathogenic Aβ isoform in humans (30, 42). This peptide is drastically inefficient in prion nucleation in the yeast assay, compared with the highly amyloidogenic and presumably pathogenic Aβ(1–42) (Fig. 5A). Although previous structural studies used the in vitro-produced Aβ40 polymers (48, 49), the high-resolution structures of Aβ(1–42) amyloids, mostly based on solid-state NMR, have also been reported recently (43, 44, 50). These structures include two molecules per polymer unit, and five intermolecular β sheets spanning residues 2–6 (β1), 15–18 (β2), 26–28 (β3), 30–32 (β4), and 39–42 (β5) per each “half” of the fibril. The anti-nucleation effects (Fig. 5, D and E) of substitutions I31P (breaking a β4 strand), F19S (disrupting hydrophobic interactions with the β2 strand), and K28E (affecting the β3 strand and disrupting a “salt bridge” potentially involved in stabilization of an amyloid structure) in the yeast assay are in good agreement with the published structural models. Likewise, the pro-nucleation effect (Fig. 5F) of the D23N substitution, corresponding to a so-called “Iowa mutation,” a heritable case of AD (9, 11), is also in good agreement with the models. This substitution removes one of the negatively charged residues presumably facing the solvent that might increase an aggregation propensity.
Sequence requirements for prion nucleation by PrP
The region between residues 90 and 119 of PrP, which is known to be essential for the susceptibility to prion infection in mammals (52), is also required for prion nucleation in yeast, although the N-terminal region of PrP(23–89) is dispensable for both (Fig. 7). Mutation P101L, associated with heritable prion disease in mammals (10), increased, whereas mutation Q167R, inhibiting prion replication in mammals, decreased PrP-dependent prion nucleation in the yeast assay. Increased prion-nucleating ability of the fragments lacking the C-proximal region of PrP (Fig. 7) agrees with previous reports linking C-proximal PrP truncations to a heritable neurological disease in humans (53–55), and it supports a notion that this disease is likely to be prion-like in nature. One possible explanation for this effect is that the α-rich C-proximal domain of PrP stabilizes the native conformation and therefore antagonizes the initiation of the cross-β (prion) conformation (64, 65). Although the structural organization of PrP in a prion form remains a matter of debate (66–68), our data agree with models locating cross-β interactions within the region 90–170, suggesting the retention of the native secondary structure by the C-terminal region of PrP (68–70), and predict that the proposed β-structure at positions 160–164 is dispensable for prion initiation. However, our data do not necessarily contradict a possibility of further expansion of the amyloid core to the C-proximal region as shown for some PrP-based amyloids (71, 72). Most importantly, our yeast assay provides a tool that could be employed to further decipher sequential and structural requirements for initiation of PrP polymerization and conformational conversion.
Impact of a nucleating construct on spectra of induced [PSI+] “strains”
Both yeast (13) and mammalian (73–75) prion and amyloid proteins are known to form various variants or strains that differ from each other by phenotypic and biochemical characteristics and are apparently controlled by distinct protein conformations. Interestingly, we have found out that the spectra of [PSI+] strains induced by different chimeric constructs differ from each other (Fig. 8). One possible explanation for these data is the formation of distinct initial nuclei by different attached regions of chimeric proteins, followed by an expansion of the amyloid region to different regions of the attached Sup35N domain. Such a mechanism would correspond to a “deformed templating” model previously proposed for strain conversions in PrP prions (76). In this scenario, the spectra of [PSI+] strains might corroborate to the differences in the “hybrid” templates formed by the fusion proteins. An alternative explanation is that certain strain conformations formed by Sup35N are more compatible, whereas other strain conformations are less compatible with an amyloid conformation formed by a specific mammalian amyloidogenic protein physically attached to the same molecule.
Potential applications of the yeast prion nucleation assay
As the ability of a chimeric construct to nucleate prion formation clearly depends on the amyloidogenicity of the attached protein, our assay can be used to investigate amyloidogenic and prionogenic properties of mammalian and human proteins. Although several yeast assays for Aβ were proposed previously (31, 77–79), none of them specifically address the initial nucleation of Aβ polymerization. Our approach could be used to map regions and identify amino acid residues specifically important for polymer nucleation, a crucial step triggering the subsequent amyloid formation and pathogenicity of Aβ, PrP, and other disease-related amyloidogenic proteins. Moreover, this assay can be employed to search for chemical factors and conditions specifically modulating the process of initial amyloid nucleation in both a general and a protein-specific manner. This may pave the way for the development of both therapeutic and prophylactic treatments for amyloid diseases that address a triggering mechanism of the disease, initial amyloid nucleation. The major advantage of our system in comparison with previously proposed yeast-based and cell-based assays is that our assay does not require the chimeric fusion protein to propagate a prion state in yeast. Prion detection is achieved by transferring the amyloid state to the endogenous yeast Sup35 protein, so that even transient amyloid formation by a chimeric construct is then fixed and amplified by conversion of an endogenous yeast protein into a prion. Furthermore, non-amyloid multimeric proteins are apparently not capable of nucleating prion formation at high efficiency in our system, making it possible to use this assay for identifying new potentially amyloidogenic proteins or domains, originating from various organisms, including humans. The rapid and easy phenotypic detection of prion nucleation in yeast makes our assay amenable to high-throughput approaches. It is, of course, possible that some sequences identified in such a way would be amyloidogenic only within the context of a chimeric construct containing the prion domain of Sup35, rather than on their own, and/or that some non-canonical assemblies (other than typical amyloids) may facilitate amyloid nucleation. For example, a short artificial extension of highly hydrophobic contents that makes the Sup35N-containing fragment capable of nucleating [PSI+] in the absence of other known prions (23) may fall into this category. However, even such possible “false positives” might in fact turn out to be useful for a better understanding of the mechanism of initial amyloid/prion nucleation in the cell.
Experimental procedures
Yeast strains
The Saccharomyces cerevisiae strains used in this study are listed in Table S2. Haploid [PSI+ PIN+] strains GT81-1C and GT81-1D are meiotic spore clones of the homozygous (except mating type) autodiploid GT81 (80). The [psi− pin−] strains GT409 and GT197 were obtained, respectively, from GT81-1C and GT81-1D via curing them of [PSI+] by guanidinium hydrochloride (GdnHCl), whereas the [psi− PIN+] strain GT159 was obtained via curing GT81-1C of [PSI+] using excess Hsp104. The rnq1Δ strain GT564 was obtained by K. Gokhale in the Chernoff laboratory via replacing the RNQ1 gene with the Schizosaccharomyces pombe ortholog of the HIS3 gene in the strain GT159. To make sure that our results are not strain-specific, we have also checked [PSI+] induction by some chimeric constructs in the [psi− pin−] strain GT17 of the 74-D694 genotype (19, 20, 23). Results obtained with this strain were similar to those seen with GT409. Strain 33G-D373, as described previously (81) and containing a double point mutation in the ADE2 gene, was used for determining the functionality of the Ade2-based chimeric proteins. Prototype “strong” (ψ+1-74-D694 or OT56) and “weak” (ψ+7-74-D694 or OT56) strains, obtained as described earlier (20), and used in our previous work (82), were employed for the phenotypic comparisons with [PSI+] strains, induced by chimeric constructs.
Plasmids and primers
The S. cerevisiae–Escherichia coli shuttle plasmids used in this study and primers used in plasmid constructions are shown in Tables S3 and S4 respectively. The DNA regions coding for Sup35NM (with HA tag) and PrP(90–230) were initially inserted in the pcDNA3.1/Zeo(+) backbone; the chimeric genes coding for Sup35N-PrP(90–230), Sup35NM-PrP(90–230), and Sup35NM-PrP(120–230) were initially generated in pcDNA3.1/Zeo(+) as well (83). Then, respective constructs were excised by using restriction endonucleases BamHI and XbaI or SacI and inserted under the copper-inducible promoter (PCUP1) into a respective centromeric shuttle vector with the URA3 marker. The plasmid pmCUP1-Sup35N-PrP(120–230) was constructed via replacing the EcoRI fragment that contains the PCUP1-SUP35NM fragment from the plasmid pmCUP1-Sup35NM-PrP(120–230) with the EcoRI fragment that contains PCUP1-SUP35N fragment from the plasmid pmCUP1-Sup35N-PrP(90–230). The pmCUP1-Sup35N plasmid was constructed by inserting the PCR-amplified BamHI-SacI fragment that contains the SUP35N region from the plasmid Sup35N-PrP(90–230) into the pmCUP1 vector at the position following the PCUP1 promoter. The genes coding for Sup35N-PrP(90–119), Sup35N-PrP(90–144), Sup35N-PrP(90–159), and Sup35N-PrP(90–171) were constructed by inserting the PCR-amplified BamHI-XbaI fragments that code for the respective PrP domains, from the plasmid pmCUP1-Sup35N-PrP(90–230) into the pmCUP1 vector at the position following the PCUP1 promoter. Constructs coding for the HA-tagged derivatives of the Sup35N and Sup35N-PrP(90–230) proteins were produced by PCR-amplifying the BamHI-SacI fragments, coding for respective proteins, from pmCUP1-Sup35N-PrP(90–230) with primers adding an HA tag coding sequence to a C-terminal end of each fragment, and inserting the resulting constructs into the pmCUP1 vector at the position following the PCUP1 promoter. Both HA-tagged and non-tagged constructs produced the same results in the [PSI+] induction assays. The chimeric gene coding for Sup35NM-PrP(90–159) was constructed by replacing the EcoRI fragment that contains the PCUP1-SUP35N cassette from the plasmid pmCUP1-Sup35N-PrP(90–159), with the EcoRI fragment that contains the PCUP1-SUP35NM cassette from the plasmid pmCUP1-Sup35NM-PrP(90–230). The chimeric gene coding for Sup35N-PrP(23–230) was constructed by inserting the PCR-amplified BamHI-XbaI fragment that codes for the region 23–230 of PrP from plasmid mPrPcyto (83) into the pmCUP1-Sup35NM-PrP(90–230) vector at the position following the Sup35NM-coding sequence, replacing the PrP(90–230)-coding fragment. The gene coding for Sup35NM-Aβ(1–42) was constructed by inserting the PCR-amplified EcoRI-NotI fragment that contains Aβ(1–42) from the plasmid pcDNA3.1(+)-Aβ42 (kindly provided by Dr. K. Ugen, University of South Florida) containing the human Aβ(1–42)-coding sequence (84) into the pmCUP1-Sup35NM-PrP(90–230) vector at the position following the Sup35NM-coding sequence, replacing the PrP(90–230)-coding fragment. The DNA sequence coding for Aβ(1–42) was placed under the PCUP1 promoter by inserting the PCR-amplified BamHI-XbaI fragment that codes for Aβ(1–42), from the plasmid pmCUP1-Sup35NM-Aβ(1–42), into the pmCUP1 vector at the position following the PCUP1 promoter. The genes coding for Sup35N-Aβ(1–42) and Sup35N-Aβ(3–42) were constructed by replacing the EcoRI fragment that contains the PCUP1-SUP35NM cassette from the plasmid pmCUP1-Sup35NM-Aβ42, with the EcoRI fragment that contains PCUP1-SUP35N from the plasmid pmCUP1-Sup35N-PrP(90–230). The digestion of an additional EcoRI site at the 3rd codon of Aβ(1–42) resulted in the generation of pmCUP1-Sup35N-Aβ(3–42), although pmCUP1-Sup35N-Aβ(1–42) was generated by incomplete digestion. To construct the series of plasmids that are more convenient for construction procedures using the EcoRI digestion, the pmCUP1 vector was digested with EcoRI; the resulting 5′-overhang was blunted using mung bean nuclease and religated with the same vector to disrupt the EcoRI site upstream from the sequence coding for PCUP1. This plasmid, named pmCUP1-nERI, was used to construct pmCUP1-nERI-Sup35N-Aβ(3–42) by inserting the PCR-amplified BamHI-XbaI fragment that contains the Sup35N-Aβ(3–42)-coding sequence from the plasmid pmCUP1-Sup35N-Aβ(3–42), into the pmCUP1-nERI vector at the position following the PCUP1 promoter. To disrupt an additional EcoRI recognition site spanning nucleotide positions 7–12 of Aβ(1–42)-coding sequence without changing the amino acid sequence, the 3rd codon of Aβ(1–42) (GAA) that codes for glutamic acid was mutated to the synonymic codon GAG, and the PCR-amplified EcoRI-XbaI fragment containing the Aβ(1–42)-coding sequence with respective change (Aβm(1–42)) was inserted into the plasmid pmCUP1-nERI-Sup35N at the position following the sequence coding for Sup35N. In the [PSI+] induction assays, the Sup35N-Aβm(1–42) construct produced results similar to the unmodified Sup35N-Aβ(1–42) construct. The gene coding for Sup35N-Aβ(1–40) was constructed by inserting the PCR-amplified BamHI-XbaI fragment, which contains Aβ(1–40)-coding sequence from the plasmid pmCUP1-nERI-Sup35N-Aβm(1–42), into the plasmid pmCUP1-nERI-Sup35N-Aβm(1–42) at the position following the sequence coding for Sup35N. The gene coding for Sup35NM-Aβ(1–40) was constructed by replacing the EcoRI fragment, which contains PCUP1-SUP35N cassette, in the plasmid pmCUP1-Sup35N-Aβ(1–40) with the EcoRI fragment that contains the PCUP1-SUP35NM cassette from the plasmid pmCUP1-Sup35NM-PrP(90–230). Individual base substitutions in the pmCUP1-Sup35N-PrP(90-230) and pmCUP1-nERI-Sup35N-Aβm(1–42) plasmids were generated in the Aβ(1–42)-coding sequence using the QuikChange site-directed mutagenesis protocol (Agilent Technologies, Santa Clara, CA). A construct coding for Aβ(1–42) with triple amino acid substitution F19S,F20S,I31P was generated by O. Laur at Emory Custom Cloning Core Facility in the plasmid pmCUP1-Sup35NM-Aβ(1–42). The genes coding for Sup35N-NAC or Sup35N-IAPP were constructed by inserting the PCR-amplified EcoRI-NotI fragment that contains NAC-HA and IAPP regions from the plasmid p106.NAC, containing the human NAC-coding sequence that corresponds to codons 61–95 of α-synuclein gene, and from the plasmid pJ201:66979-IAPP2_optSc, containing the codons 41–70 of human IAPP-coding sequence that corresponds to residues 8–37 in mature amylin, respectively, into the plasmid pmCUP1-nERI-Sup35N-Aβ(1–42) at the position following the sequence coding for Sup35N, replacing the Aβ(1–42)-coding fragment. Original plasmids p106.NAC and pJ201:66979-IAPP2_optSc were kindly provided by Dr. V. Conticello from Emory University. The plasmids pmCUP1-Sup35NM-NAC and pmCUP1-Sup35NM-IAPP were constructed by inserting the PCR-amplified EcoRI-NotI fragments that contain NAC-HA and IAPP regions from pmCUP1-Sup35N-NAC and pmCUP1-Sup35N-IAPP and inserted into the plasmid pcDNA3.1(Zeo)-Sup35NM-PrP(90–230) at the position following the Sup35NM-coding sequence, replacing the PrP(90–230)-coding fragment. Then, respective chimeric genes were cut from plasmids pcDNA3.1(Zeo)-Sup35NM-NAC and pcDNA3.1(Zeo)-Sup35NM-IAPP with BamHI and XbaI and inserted into the pmCUP1 vector at the position following the PCUP1 promoter. The plasmid coding for the C-terminal fusion of Lsb2 with GFP, expressed under the PCUP1 promoter in the pRS316 backbone, was constructed earlier (31). The chimeric gene coding for Sup35N-LacZ was constructed by inserting the PCR-amplified EcoRI-XbaI fragment that contains the lacZ-coding sequence from the plasmid pSVA1 (kindly provided by Dr. M. D. Ter-Avanesyan, Moscow, Russia) into the plasmid pmCUP1-nERI-Sup35N-Aβm(1–42) at the position following the Sup35N-coding sequence, replacing the Aβ(1–42)-coding fragment. The gene coding for Sup35NM-LacZ was constructed by replacing the EcoRI fragment of the plasmid pmCUP1-Sup35N-LacZ that contains the PCUP1-SUP35N cassette with the EcoRI fragment that contains the PCUP1-SUP35NM cassette from the plasmid pmCUP1-Sup35NM-PrP(90–230). The gene coding for Sup35N-GFP was constructed by inserting the PCR-amplified EcoRI-SacII fragment that contains GFP-coding sequence from the plasmid pmCUP1-NM-GFP (85) into pmCUP1-nERI-Sup35N-Aβm(1–42) at the position following the Sup35N-coding sequence, replacing the Aβ(1–42)-coding sequence. The gene coding for Sup35NM-Ade2 was constructed by inserting the Ade2-coding fragment from the plasmid pRS316GAL-Sup35NM-Ade2 into the plasmid pmCUP1-Sup35NM-Aβ(1–42) at the position following the sequence coding for Sup35NM, replacing the Aβ(1–42)-coding sequence. The gene coding for Sup35N-Ade2 was constructed by replacing the EcoRI fragment that contains the PCUP1-SUP35NM cassette from the plasmid pmCUP1-Sup35NM-Ade2, with the EcoRI fragment that contains the PCUP1-SUP35NM cassette from the plasmid pmCUP1-Sup35N-PrP(90–230). Plasmids with constructs under the PGAL promoter were constructed by inserting the BamHI-XbaI fragments with respective chimeric genes from constructs with PCUP1 promoter into the centromeric HIS3 vector pLA1 (82) under the galactose-inducible promoter PGAL. Plasmid pLH105, containing the HSP104 gene under the strong constitutive PGPD promoter, was a gift from S. Lindquist quoted earlier (86). Plasmids pLA1-Sup35N and pLA1-Sup35, containing, respectively, PGAL-SUP35N and PGAL-SUP35N expression cassettes, were described earlier (86, 87). The plasmid pmCUP1-PrP-GFP(URA3), kindly provided by A. P. Galkin and coding for the PrP(90–231)-GFP chimeric protein, was described earlier (88). The plasmid pmCUP1-Aβ(1–42)-GFP(URA3) was constructed via inserting the DNA fragment, encoding Aβ(1–42) and obtained from the human brain mRNA by RT-PCR, with the addition of BamHI and SacII sites into the plasmid pmCUP1-GFP (85), digested with BamHI and SacII. Plasmid pNM-YFP, containing the SUP35NM-YFP construct under the control PCUP1 promoter and kindly provided by I. L. Derkatch, is based on the LEU2 vector pRS315 and was described previously (62). All regions that underwent PCR amplification as well as immediate flanking regions were verified by sequencing, performed at Eurofins MWG Operon (Huntsville, AL). Isolation of plasmid DNA from bacteria was performed according to standard procedures (89).
Enzymes and antibodies
Enzymes used for molecular cloning, PCR, ligation, and site-directed mutagenesis, including restriction endonucleases BamHI, EcoRI, XbaI, NotI, SacI, ClaI, XhoI, mung bean nuclease, Taq and Pfu DNA polymerases, and T4 DNA ligase, were purchased from New England Biolabs. The antibodies to Sup35N and Hsp104 (from S. Lindquist laboratory), Sup35M (4A5), and PrP (4H11) have been described previously (82, 90–92). Antibody to HA was purchased from Covance. Antibody to GFP, Ab-13970, was purchased from Abcam. Antibody to Aβ (6E10, Covance, catalogue number SIG 39320) was a gift of L. Walker (Emory University School of Medicine). Secondary anti-rabbit and anti-rat HRP antibodies were purchased from Sigma. Secondary anti-mouse HRP antibody was purchased from GE Healthcare.
Yeast media and growth conditions
Yeast cultures were grown at 30 °C. Standard yeast media and standard procedures for yeast cultivation and phenotypic and biochemical analyses were used (93). Cell counts were performed using a hemocytometer (Brightline). Optical densities of yeast cultures were measured at 600 nm using Shimadzu UV-2450 spectrophotometer. Standard synthetic medium contains 3 μm copper sulfate (CuSO4); it was supplemented with 10, 50, 100, or 150 μm CuSO4 as indicated to induce higher expression of PCUP1 promoter. Synthetic media lacking adenine, leucine, or uracil are designated as −Ade, −Leu, and −Ura, respectively. In all cases when the carbon source is not specifically indicated, 2% glucose (Glu) was used. The synthetic medium containing 2% galactose (Gal) or 2% galactose and 2% raffinose (Gal + Raf) instead of glucose was used to induce the GAL promoter. Organic complete YPD medium containing yeast extract (1%), peptone (2%), and glucose (2%) was used for color detection. Organic YPG medium containing glycerol (3%) instead of glucose was used to identify respiratory-incompetent (Pet−) transformants that arose due to loss of mitochondrial DNA during transformation and were eliminated from further analysis. Detection assay for [PSI+], based on the read-through of the ade1-14 (UGA) allele, that results in growth on −Ade medium and lighter color on YPD medium is described previously (13) and under the “Results.” Liquid cultures were grown with at least a 1:5 liquid/flask volumetric ratio in a shaking incubator (200–250 rpm). Yeast transformations were performed according to the standard Li+ protocol (92). Curing of [PSI+] by GdnHCl was performed by incubating cultures for three consecutive passages (∼20–40 generations) on YPD plates with 5 mm GdnHCl, followed by streaking out on YPD and checking individual colonies by both color and growth on −Ade medium. The mating assay for the presence of [PIN+] or another prion with a [PSI+]-inducing capability is described in Fig. 4A.
Prion nucleation assays
To check for [PSI+] nucleation, plasmids bearing chimeric and control genes under the PCUP1 or PGAL promoter were transformed into the yeast [psi−] strain. For plate assays, transformants were grown on the media selective for the plasmid (e.g. −Ura) containing 2% glucose as a carbon source and a background concentration (3 μm) of Cu2+, and then velveteen replica-plated onto the same medium with addition of 0, 10, 50, 100, or 150 μm CuSO4 as specified in the figure legends (for PCUP1 constructs) or onto the same medium with 2% galactose instead of glucose (for PGAL constructs) to induce expression of the chimeric genes. After induction (usually for 2 days), plates were replica-plated to −Ade medium with glucose and without additional CuSO4, where overexpression was turned off. [PSI+] formation was scored by growth on −Ade medium, typically after about 10 days of incubation. At least eight (and usually more) independent transformants were checked per each strain/plasmid combination to ensure reproducibility. Transformants carrying the control and experimental plasmids were always compared on one and the same plate. One or two representative transformants for each strain/plasmid combination are shown on the figures. In all cases, there were no differences in growth detected on the completed medium or medium selected for the plasmid (for simplicity, respective images are not shown on most figures).
For semi-quantitative and quantitative measurements of [PSI+] formation, a pre-culture obtained from a fresh transformant colony was grown in the liquid synthetic medium selective for the plasmid up to OD600 = 2.5 and then inoculated into the liquid plasmid-selective media with additional CuSO4 (usually 100 μm) at a starting concentration of 106 cells/ml. Cultures were incubated at 30 °C with shaking, with aliquots taken at desired time points, washed with water, diluted appropriately, and either spotted (as serial decimal dilutions) or plated onto both plasmid-selective medium containing adenine (to count numbers of viable plasmid-containing cells) and plasmid-selective medium lacking adenine (e.g. −Ura−Ade) to detect [PSI+]. Frequency of [PSI+] induction was calculated as a ratio of the number of Ade+ colonies to the total number of viable plasmid-containing cells plated. To ensure accuracy, only dilutions that produced plates with fewer than 500 colonies were counted. For each construct, quantitative assay was repeated with at least three cultures, each originated from an independent transformant to ensure reproducibility. Standard deviations were calculated according to Ref. 94. Cultures with prion-inducing and control plasmids were always run in parallel in the same experiment.
Protein isolation and characterization: SDS-PAGE, SDD-AGE, and Western blotting
For isolation of the total yeast protein, cells grown in the liquid medium were collected by centrifugation at 2000 × g for 5 min at 4 °C, washed with 300 μl of ice-cold lysis buffer (25 mm Tris, pH 7.5, 0.1 m NaCl, 10 mm EDTA, 100 μg/ml cycloheximide, 2 mm benzamidine, 20 μg/ml leupeptin, 4 μg/ml pepstatin A, 1 mm N-ethylmaleimide, 1× protease inhibitor mixture from Roche Diagnostics GmbH, 2 mm phenylmethylsulfonyl fluoride), resuspended in 2 volumes of ice-cold lysis buffer, and mixed to ∼300 μl of acid-washed glass beads. Cells were lysed by vortexing 6 times for 30 s, with at least 1 min on ice in-between each time. Cell debris was removed by centrifugation at 2000 × g for 5 min. The amount of protein in the samples was determined by Bradford reagent (Bio-Rad) and normalized using lysis buffer. The protein samples were boiled for 10 min prior to loading onto SDS polyacrylamide gel. After electrophoresis, proteins were transferred onto Immobilon-P 0.45-μm polyvinylidene difluoride blotting membrane (EMD Millipore) or Amersham Biosciences Protran Premium 0.45-μm nitrocellulose blotting membrane (GE Healthcare) and reacted to appropriate antibodies. Reaction was detected by using the chemiluminescent detection reagents as described in the GE Healthcare protocols. SDD-AGE followed by transfer to the nitrocellulose membrane was performed according to Ref. 32, with modification (addition of 0.1% SDS to the transfer buffer). Protein concentrations were normalized by the Bradford assay.
Fluorescence microscopy
For the microscopic detection of aggregates, the [psi− pin−] yeast strain was co-transformed with the plasmid producing Sup35NM-YFP and either empty control plasmid pRS316, plasmid producing Sup35N-HA, or plasmid producing Sup35N-Aβ(1–42). All the constructs were under the control of the PCUP1 promoter. Cultures were pre-grown overnight in a liquid synthetic medium selective for both plasmids, inoculated at OD600 = 0.5 into fresh medium of the same composition with the addition of CuSO4 up to 50 μm, and grown with shaking. Cells were examined after specified periods of incubation by using a Leica DM6000B microscope and Leica QWin Standard 3.2.0 software (Leica Microsystems GmbH), at ×1000 magnification (×100 objective plus ×10 eye, with NA 1.5 for the objective). Images were captured with a Leica DC 500 color digital camera.
Author contributions
Y. O. C. designed the experimental strategy. P. C., M. S., K. L. C., A. V. R., A. V. G., A. A. R., and J. V. S. performed the experiments. P. C., M. S., A. V. G., A. A. R., I. M. V., and Y. O. C. analyzed the data. A. A. R., C. N.-K., and I. M. V. provided materials. P. C., K. L. C., A. V. G., A. A. R., and Y. O. C. obtained funding. P. C., M. S., and Y. O. C. wrote the initial draft. P. C., I. M. V., and Y. O. C. edited the manuscript. All authors reviewed and approved the final version of the paper.
Supplementary Material
Acknowledgments
We thank T. A. Chernova, V. Conticello, I. L. Derkatch, A. P. Galkin, S. Lindquist, M. D. Ter-Avanesyan, K. Ugen, and L. Walker for plasmids and antibodies; Z. Deckner, K. Gokhale, D. A. Kiktev, O. Laur, B. Parks, N.V. Romanova, the Emory Custom Cloning Core Facility, and the St. Petersburg State University Chromas Core Facility for help in some experiments and constructions; S. G. Inge-Vechtomov, A. Kajava, D. Lynn, A. Mehta, L. Walker, G. A. Zhouravleva, and D. G. Yanchenko for helpful discussions and/or help with manuscript preparation.
This work was supported in part by Emory Alzheimer's Disease Research Center Grant P50AG025688 from the National Institutes of Health, a Creutzfeldt-Jakob Disease Family Foundation (CJD Foundation) grant (to Y. O. C.), Russian Science Foundation (RSF) Grant 14-50-00069 (to Y. O. C.), and Russian Foundation for Basic Research (RFBR) Grant 15-04-08159 (to A. A. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, CJD Foundation, RSF, or RFBR.
This article contains Figs. S1–S6 and Tables S1–S4.
- AD
- Alzheimer's disease
- PD
- Parkinson's disease
- HD
- Huntington's disease
- PrP
- prion protein
- TSE
- transmissible spongiform encephalopathy
- Aβ
- amyloid-β peptide
- PrD
- prion domain
- IAPP
- islet amyloid polypeptide
- SDD-AGE
- semi-denaturing detergent agarose gel electrophoresis
- NAC
- non-amyloid component
- AP
- amyloidogenic protein
- GdnHCl
- guanidine hydrochloride.
References
- 1. Knowles T. P., Vendruscolo M., and Dobson C. M. (2014) The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396 10.1038/nrm3810 [DOI] [PubMed] [Google Scholar]
- 2. Kahn S. E., Andrikopoulos S., and Verchere C. B. (1999) Islet amyloid: a long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 48, 241–253 10.2337/diabetes.48.2.241 [DOI] [PubMed] [Google Scholar]
- 3. Westermark P. (1994) Amyloid and polypeptide hormones: what is their interrelationship? Amyloid 1, 47–60 10.3109/13506129409148624 [DOI] [Google Scholar]
- 4. Cobb N. J., and Surewicz W. K. (2009) Prion diseases and their biochemical mechanisms. Biochemistry 48, 2574–2585 10.1021/bi900108v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prusiner S. B. (1997) Prion diseases and the BSE crisis. Science 278, 245–251 10.1126/science.278.5336.245 [DOI] [PubMed] [Google Scholar]
- 6. Alzheimer's Association (2017) 2017 Alzheimer's disease facts and figures. Alzheimers Dement 13, 325–373 10.1016/j.jalz.2017.02.001 [DOI] [Google Scholar]
- 7. James B. D., Leurgans S. E., Hebert L. E., Scherr P. A., Yaffe K., and Bennett D. A. (2014) Contribution of Alzheimer disease to mortality in the United States. Neurology 82, 1045–1050 10.1212/WNL.0000000000000240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones R. W., Romeo R., Trigg R., Knapp M., Sato A., King D., Niecko T., Lacey L., and DADE Investigator Group (2015) Dependence in Alzheimer's disease and service use costs, quality of life, and caregiver burden: The DADE study. Alzheimer's Dementia 11, 280–290 [DOI] [PubMed] [Google Scholar]
- 9. Fowler D. M., Koulov A. V., Balch W. E., and Kelly J. W. (2007) Functional amyloid from bacteria to humans. Trends Biochem. Sci. 32, 217–224 10.1016/j.tibs.2007.03.003 [DOI] [PubMed] [Google Scholar]
- 10. Nizhnikov A. A., Antonets K. S., Bondarev S. A., Inge-Vechtomov S. G., and Derkatch I. L. (2016) Prions, amyloids, and RNA: pieces of a puzzle. Prion 10, 182–206 10.1080/19336896.2016.1181253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grabowski T. J., Cho H. S., Vonsattel J. P., Rebeck G. W., and Greenberg S. M. (2001) Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 49, 697–705 10.1002/ana.1009 [DOI] [PubMed] [Google Scholar]
- 12. Prusiner S. B. (1993) Genetic and infectious prion diseases. Arch. Neurol. 50, 1129–1153 [DOI] [PubMed] [Google Scholar]
- 13. Van Nostrand W. E., Melchor J. P., Cho H. S., Greenberg S. M., and Rebeck G. W. (2001) Pathogenic effects of D23N Iowa mutant amyloid β protein. J. Biol. Chem. 276, 32860–32866 10.1074/jbc.M104135200 [DOI] [PubMed] [Google Scholar]
- 14. Irvine G. B., El-Agnaf O. M., Shankar G. M., and Walsh D. M. (2008) Protein aggregation in the brain: the molecular basis for Alzheimer's and Parkinson's diseases. Mol. Med. 14, 451–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liebman S. W., and Chernoff Y. O. (2012) Prions in yeast. Genetics 191, 1041–1072 10.1534/genetics.111.137760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wickner R. B., Edskes H. K., Bateman D. A., Kelly A. C., Gorkovskiy A., Dayani Y., and Zhou A. (2013) Amyloids and yeast prion biology. Biochemistry 52, 1514–1527 10.1021/bi301686a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stansfield I., Jones K. M., Kushnirov V. V., Dagkesamanskaya A. R., Poznyakovski A. I., Paushkin S. V., Nierras C. R., Cox B. S., Ter-Avanesyan M. D., and Tuite M. F. (1995) The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 14, 4365–4373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhouravleva G., Frolova L., Le Goff X., Le Guellec R., Inge-Vechtomov S., Kisselev L., and Philippe M. (1995) Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 14, 4065–4072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chernoff Y. O., Derkach I. L., and Inge-Vechtomov S. G. (1993) Multicopy SUP35 gene induces de novo appearance of ψ-like factors in the yeast Saccharomyces cerevisiae. Curr. Genet. 24, 268–270 10.1007/BF00351802 [DOI] [PubMed] [Google Scholar]
- 20. Derkatch I. L., Chernoff Y. O., Kushnirov V. V., Inge-Vechtomov S. G., and Liebman S. W. (1995) Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 144, 1375–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Masison D. C., and Wickner R. B. (1995) Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 270, 93–95 10.1126/science.270.5233.93 [DOI] [PubMed] [Google Scholar]
- 22. Wickner R. B. (1994) [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264, 566–569 10.1126/science.7909170 [DOI] [PubMed] [Google Scholar]
- 23. Derkatch I. L., Bradley M. E., Zhou P., Chernoff Y. O., and Liebman S. W. (1997) Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 147, 507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Derkatch I. L., Bradley M. E., Hong J. Y., and Liebman S. W. (2001) Prions affect the appearance of other prions: the story of [PIN+]. Cell 106, 171–182 10.1016/S0092-8674(01)00427-5 [DOI] [PubMed] [Google Scholar]
- 25. Osherovich L. Z., and Weissman J. S. (2001) Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell 106, 183–194 10.1016/S0092-8674(01)00440-8 [DOI] [PubMed] [Google Scholar]
- 26. Goehler H., Dröge A., Lurz R., Schnoegl S., Chernoff Y. O., and Wanker E. E. (2010) Pathogenic polyglutamine tracts are potent inducers of spontaneous Sup35 and Rnq1 amyloidogenesis. PLoS ONE 5, e9642 10.1371/journal.pone.0009642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suzuki G., Shimazu N., and Tanaka M. (2012) A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 336, 355–359 [DOI] [PubMed] [Google Scholar]
- 28. Du Z., Park K. W., Yu H. Fan Q., and Li L. (2008) Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat. Genet. 40, 460–465 10.1038/ng.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fischer M., Rülicke T., Raeber A., Sailer A., Moser M., Oesch B., Brandner S., Aguzzi A., and Weissmann C. (1996) Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 15, 1255–1264 [PMC free article] [PubMed] [Google Scholar]
- 30. Näslund J., Schierhorn A., Hellman U., Lannfelt L., Roses A. D., Tjernberg L. O., Silberring J., Gandy S. E., Winblad B., and Greengard P. (1994) Relative abundance of Alzheimer Aβ amyloid peptide variants in Alzheimer disease and normal aging. Proc. Natl. Acad. Sci. U.S.A. 91, 8378–8382 10.1073/pnas.91.18.8378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rubel A. A., Ryzhova T. A., Antonets K. S., Chernoff Y. O., and Galkin A. (2013) Identification of PrP sequences essential for the interaction between the PrP polymers and Aβ peptide in a yeast-based assay. Prion 7, 469–476 10.4161/pri.26867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Halfmann R., and Lindquist S. L. (2008) Screening for amyloid aggregation by semi-denaturing detergent-agarose gel electrophoresis. J. Vis. Exp. 2008, 838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chernova T. A., Romanyuk A. V., Karpova T. S., Shanks J. R., Ali M., Moffatt N., Howie R. L., O'Dell A., McNally J. G., Liebman S. W., Chernoff Y. O., and Wilkinson K. D. (2011) Prion induction by the short-lived, stress-induced protein Lsb2 is regulated by ubiquitination and association with the actin cytoskeleton. Mol. Cell 43, 242–252 10.1016/j.molcel.2011.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chernova T. A., Kiktev D. A., Romanyuk A. V., Shanks J. R., Laur O., Ali M., Ghosh A., Kim D., Yang Z., Mang M., Chernoff Y. O., and Wilkinson K. D. (2017) Yeast short-lived actin-associated protein forms a metastable prion in response to thermal stress. Cell Rep. 18, 751–761 10.1016/j.celrep.2016.12.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stefanis L. (2014) α-Synuclein in Parkinson's disease. Cold Spring Harb. Perspect. Med. 2(2): a009399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li S. X., Tong Y. P., Xie X. C., Wang Q. H., Zhou H. N., Han Y., Zhang Z. Y., Gao W., Li S. G., Zhang X. C., and Bi R. C. (2007) Octameric structure of the human bifunctional enzyme PAICS in purine biosynthesis. J. Mol. Biol. 366, 1603–1614 10.1016/j.jmb.2006.12.027 [DOI] [PubMed] [Google Scholar]
- 37. Zhou P., Derkatch I. L., and Liebman S. W. (2001) The relationship between visible intracellular aggregates that appear after overexpression of Sup35 and the yeast prion-like elements [PSI+] and [PIN+]. Mol. Microbiol. 39, 37–46 10.1046/j.1365-2958.2001.02224.x [DOI] [PubMed] [Google Scholar]
- 38. Ganusova E. E., Ozolins L. N., Bhagat S., Newnam G. P., Wegrzyn R. D., Sherman M. Y., and Chernoff Y. O. (2006) Modulation of prion formation, aggregation, and toxicity by the actin cytoskeleton in yeast. Mol. Cell. Biol. 26, 617–629 10.1128/MCB.26.2.617-629.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chernoff Y. O., Lindquist S. L., Ono B., Inge-Vechtomov S. G., and Liebman S. W. (1995) Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268, 880–884 [DOI] [PubMed] [Google Scholar]
- 40. Chernova T. A., Wilkinson K. D., and Chernoff Y. O. (2017) Prions, chaperones and proteostasis in yeast. Cold Spring Harb. Perspect. Biol. 9, a023663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mori H., Takio K., Ogawara M., and Selkoe D. J. (1992) Mass spectrometry of purified amyloid β protein in Alzheimer's disease. J. Biol. Chem. 267, 17082–17086 [PubMed] [Google Scholar]
- 42. Jarrett J. T., Berger E. P., and Lansbury P. T. (1993) The carboxy terminus of the β-amyloid protein is critical for the seeding of amyloid formation–implications for the pathogenesis of Alzheimer's disease. Biochemistry 32, 4693–4697 10.1021/bi00069a001 [DOI] [PubMed] [Google Scholar]
- 43. Colvin M. T., Silvers R., Ni Q. Z., Can T. V., Sergeyev I., Rosay M., Donovan K. J., Michael B., Wall J., Linse S., and Griffin R. G. (2016) Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J. Am. Chem. Soc. 138, 9663–9674 10.1021/jacs.6b05129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Walti M. A., Ravotti F., Arai H., Glabe C. G., Wall J. S., Böckmann A., et al. (2016) Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. U.S.A. 113, E4976–E4984 10.1073/pnas.1600749113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hilbich C., Kisters-Woike B., Reed J., Masters C. L., and Beyreuther K. (1992) Substitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer's disease βA4 peptides. J. Mol. Biol. 228, 460–473 10.1016/0022-2836(92)90835-8 [DOI] [PubMed] [Google Scholar]
- 46. Morimoto A., Irie K., Murakami K., Masuda Y., Ohigashi H., Nagao M., Fukuda H., Shimizu T., and Shirasawa T. (2004) Analysis of the secondary structure of β-amyloid (Aβ42) fibrils by systematic proline replacement. J. Biol. Chem. 279, 52781–52788 10.1074/jbc.M406262200 [DOI] [PubMed] [Google Scholar]
- 47. Williams A. D., Portelius E., Kheterpal I., Guo J. T., Cook K. D., Xu Y., and Wetzel R. (2004) Mapping Aβ amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 335, 833–842 10.1016/j.jmb.2003.11.008 [DOI] [PubMed] [Google Scholar]
- 48. Paravastu A. K., Leapman R. D., Yau W. M., and Tycko R. (2008) Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354 10.1073/pnas.0806270105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tycko R. (2006) Molecular structure of amyloid fibrils: insights from solid-state NMR. Q. Rev. Biophys. 39, 1–55 10.1017/S0033583506004173 [DOI] [PubMed] [Google Scholar]
- 50. Xiao Y., Ma B., McElheny D., Parthasarathy S., Long F., Hoshi M., Nussinov R., and Ishii Y. (2015) Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease. Nat. Struct. Mol. Biol. 22, 499–505 10.1038/nsmb.2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perrier V., Kaneko K., Safar J., Vergara J., Tremblay P., DeArmond S. J., Cohen F. E., Prusiner S. B., and Wallace A. C. (2002) Dominant-negative inhibition of prion replication in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 99, 13079–13084 10.1073/pnas.182425299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Flechsig E., and Weissmann C. (2004) The role of PrP in health and disease. Curr. Mol. Med. 4, 337–353 10.2174/1566524043360645 [DOI] [PubMed] [Google Scholar]
- 53. Ghetti B., Piccardo P., Frangione B., Bugiani O., Giaccone G., Young K., Prelli F., Farlow M. R., Dlouhy S. R., and Tagliavini F. (1996) Prion protein amyloidosis. Brain Pathol. 6, 127–145 10.1111/j.1750-3639.1996.tb00796.x [DOI] [PubMed] [Google Scholar]
- 54. Kitamoto T., Iizuka R., and Tateishi J. (1993) An amber mutation of prion protein in Gerstmann-Sträussler syndrome with mutant PrP plaques. Biochem. Biophys. Res. Commun. 192, 525–531 [DOI] [PubMed] [Google Scholar]
- 55. Lorenz H., Windl O., and Kretzschmar H. A. (2002) Cellular phenotyping of secretory and nuclear prion proteins associated with inherited prion diseases. J. Biol. Chem. 277, 8508–8516 10.1074/jbc.M110197200 [DOI] [PubMed] [Google Scholar]
- 56. Theint T., Nadaud P. S., Surewicz K., Surewicz W. K., and Jaroniec C. P. (2017) 13C and 15N chemical shift assignments of mammalian Y145Stop prion protein amyloid fibrils. Biomol. NMR Assign. 11, 75–80 10.1007/s12104-016-9723-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chiesa R., Piccardo P., Ghetti B., and Harris D. A. (1998) Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 21, 1339–1351 10.1016/S0896-6273(00)80653-4 [DOI] [PubMed] [Google Scholar]
- 58. Prusiner S. B., and Scott M. R. (1997) Genetics of prions. Annu. Rev. Genet. 31, 139–175 10.1146/annurev.genet.31.1.139 [DOI] [PubMed] [Google Scholar]
- 59. Kochneva-Pervukhova N. V., Poznyakovski A. I., Smirnov V. N., and Ter-Avanesyan M. D. (1998) C-terminal truncation of the Sup35 protein increases the frequency of de novo generation of a prion-based [PSI+] determinant in Saccharomyces cerevisiae. Curr. Genet. 34, 146–151 10.1007/s002940050379 [DOI] [PubMed] [Google Scholar]
- 60. Tanaka M., Collins S. R., Toyama B. H., and Weissman J. S. (2006) The physical basis of how prion conformations determine strain phenotypes. Nature 442, 585–589 10.1038/nature04922 [DOI] [PubMed] [Google Scholar]
- 61. Newby G. A., Kiriakov S., Hallacli E., Kayatekin C., Tsvetkov P., Mancuso C. P., Bonner J. M., Hesse W. R., Chakrabortee S., Manogaran A. L., Liebman S. W., Lindquist S., and Khalil A. S. (2017) Genetic tool to track protein aggregates and control protein inheritance. Cell 171, 966–979 10.1016/j.cell.2017.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Derkatch I. L., Uptain S. M., Outeiro T. F., Krishnan R., Lindquist S. L., and Liebman S. W. (2004) Effects of Q/N-rich, polyQ, and non-polyQ amyloids of the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc. Natl. Acad. Sci. U.S.A. 101, 12934–12939 10.1073/pnas.0404968101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Choe Y. J., Ryu Y., Kim H. J., and Seok Y. J. (2009) Increased [PSI+] appearance by fusion of Rnq1 with the prion domain of Sup35 in Saccharomyces cerevisiae. Eukaryot. Cell 8, 968–976 10.1128/EC.00353-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jansen C., Parchi P., Capellari S., Vermeij A. J., Corrado P., Baas F., Strammiello R., van Gool W. A., van Swieten J. C., and Rozemuller A. J. (2010) Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol. 119, 189–197 10.1007/s00401-009-0609-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kovač V., Hafner-Bratkovič I., and Čurin Šerbec V. (2016) Anchorless forms of prion protein-impact of truncation on structure destabilization and prion protein conversion. Biochem. Biophys. Res. Commun. 481, 1–6 10.1016/j.bbrc.2016.11.036 [DOI] [PubMed] [Google Scholar]
- 66. Diaz-Espinoza R., and Soto C. (2012) High-resolution structure of infectious prion protein: the final frontier. Nat. Struct. Mol. Biol. 19, 370–377 10.1038/nsmb.2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Groveman B. R., Kraus A., Raymond L. D., Dolan M. A., Anson K. J., Dorward D. W., and Caughey B. (2015) Charge neutralization of the central lysine cluster in prion protein (PrP) promotes PrP(Sc)-like folding of recombinant PrP amyloids. J. Biol. Chem. 290, 1119–1128 10.1074/jbc.M114.619627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Requena J. R., and Wille H. (2014) The structure of the infectious prion protein: experimental data and molecular models. Prion 8, 60–66 10.4161/pri.28368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Benetti F., and Legname G. (2015) New insights into structural determinants of prion protein folding and stability. Prion 9, 119–124 10.1080/19336896.2015.1022023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Govaerts C., Wille H., Prusiner S. B., and Cohen F. E. (2004) Evidence for assembly of prions with left-handed β-helices into trimers. Proc. Natl. Acad. Sci. U.S.A. 101, 8342–8347 10.1073/pnas.0402254101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cobb N. J., Sönnichsen F. D., McHaourab H., and Surewicz W. K. (2007) Molecular architecture of human prion protein amyloid: a parallel, in-register β-structure. Proc. Natl. Acad. Sci. U.S.A. 104, 18946–18951 10.1073/pnas.0706522104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Saijo E., Hughson A. G., Raymond G. J., Suzuki A., Horiuchi M., and Caughey B. (2016) PrPSc-specific antibody reveals C-terminal conformational differences between prion strains. J. Virol. 90, 4905–4913 10.1128/JVI.00088-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bartz J. C. (2016) Prion strain diversity. Cold Spring Harb. Perspect. Med. 6, a024349 10.1101/cshperspect.a024349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ghaemmaghami S. (2016) Biology and genetics of PrP prion strains. Cold Spring Harb. Perspect. Med. 6, a026922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cohen M., Appleby B., and Safar J. G. (2016) Distinct prion-like strains of amyloid β implicated in phenotypic diversity of Alzheimer's disease. Prion 10, 9–17 10.1080/19336896.2015.1123371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Makarava N., and Baskakov I. V. (2013) The evolution of transmissible prions: the role of deformed templating. PLOS Pathog. 9, e1003759 10.1371/journal.ppat.1003759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bagriantsev S., and Liebman S. (2006) Modulation of Αβ42 low-n oligomerization using a novel yeast reporter system. BMC Biol. 4, 32 10.1186/1741-7007-4-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Porzoor A., and Macreadie I. (2016) Yeast as a model for studies on Aβ aggregation toxicity in Alzheimer's disease, autophagic responses, and drug screening. Methods Mol. Biol. 1303, 217–226 10.1007/978-1-4939-2627-5_12 [DOI] [PubMed] [Google Scholar]
- 79. Treusch S., Hamamichi S., Goodman J. L., Matlack K. E., Chung C. Y., Baru V., Shulman J. M., Parrado A., Bevis B. J., Valastyan J. S., Han H., Lindhagen-Persson M., Reiman E. M., Evans D. A., Bennett D. A., et al. (2011) Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science 334, 1241–1245 10.1126/science.1213210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chernoff Y. O., Galkin A. P., Lewitin E., Chernova T. A., Newnam G. P., and Belenkiy S. M. (2000) Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Mol. Microbiol. 35, 865–876 10.1046/j.1365-2958.2000.01761.x [DOI] [PubMed] [Google Scholar]
- 81. Kushnirov V. V., Ter-Avanesyan M. D., Didichenko S. A., Smirnov V. N., Chernoff Y. O., Derkach I. L., Novikova O. N., Inge-Vechtomov S. G., Neistat M. A., and Tolstorukov I. I. (1990) Divergence and conservation of SUP2 (SUP35) gene of yeast Pichia pinus and Saccharomyces cerevisiae. Yeast 6, 461–472 10.1002/yea.320060603 [DOI] [PubMed] [Google Scholar]
- 82. Newnam G. P., Wegrzyn R. D., Lindquist S. L., and Chernoff Y. O. (1999) Antagonistic interactions between yeast chaperones Hsp104 and Hsp70 in prion curing. Mol. Cell. Biol. 19, 1325–1333 10.1128/MCB.19.2.1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Krammer C., Suhre M. H., Kremmer E., Diemer C., Hess S., Schätzl H. M., Scheibel T., and Vorberg I. (2008) Prion protein/protein interactions: fusion with yeast Sup35p-NM modulates cytosolic PrP aggregation in mammalian cells. FASEB J. 22, 762–773 10.1096/fj.07-8733com [DOI] [PubMed] [Google Scholar]
- 84. Kutzler M. A., Cao C., Bai Y., Dong H., Choe P. Y., Saulino V., McLaughlin L., Whelan A., Choo A. Y., Weiner D. B., and Ugen K. E. (2006) Mapping of immune responses following wild-type and mutant Aβ42 plasmid or peptide vaccination in different mouse haplotypes and HLA Class II transgenic mice. Vaccine 24, 4630–4639 10.1016/j.vaccine.2005.08.036 [DOI] [PubMed] [Google Scholar]
- 85. Serio T. R., and Lindquist S. L. (1999) [PSI+]: an epigenetic modulator of translation termination efficiency. Annu. Rev. Cell Dev. Biol. 15, 661–6703 10.1146/annurev.cellbio.15.1.661 [DOI] [PubMed] [Google Scholar]
- 86. Chernoff Y. O., Newnam G. P., Kumar J., Allen K., and Zink A. D. (1999) Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone Ssb in formation, stability, and toxicity of the [PSI] prion. Mol. Cell. Biol. 19, 8103–8112 10.1128/MCB.19.12.8103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chernova T. A., Allen K. D., Wesoloski L. M., Shanks J. R., Chernoff Y. O., and Wilkinson K. D. (2003) Pleiotropic effects of Ubp6 loss on drug sensitivities and yeast prion are due to depletion of free ubiquitin pool. J. Biol. Chem. 278, 52102–52115 10.1074/jbc.M310283200 [DOI] [PubMed] [Google Scholar]
- 88. Rubel A. A., Saifitdinova A. F., Lada A. G., Nizhnikov A. A., Inge-Vechtomov S. G., and Galkin A. P. (2008) Yeast chaperone Hspl04 regulates gene expression on the posttranscriptional level. Mol. Biol. 42, 123–130 10.1134/S0026893308010184 [DOI] [PubMed] [Google Scholar]
- 89. Sambrook J., and Russell D. W. (2001) Molecular Cloning: A Laboratory Manual. 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 90. Ertmer A., Gilch S., Yun S. W., Flechsig E., Klebl B., Stein-Gerlach M., Klein M. A., and Schätzl H. M. (2004) The tyrosine kinase inhibitor STI571 induces cellular clearance of PrPSc in prion-infected cells. J. Biol. Chem. 279, 41918–41927 10.1074/jbc.M405652200 [DOI] [PubMed] [Google Scholar]
- 91. Parsell D. A., Sanchez Y., Stitzel J. D., and Lindquist S. (1991) Hsp104 is a highly conserved protein with two essential nucleotide-binding sites. Nature 353, 270–273 10.1038/353270a0 [DOI] [PubMed] [Google Scholar]
- 92. Patino M. M., Liu J. J., Glover J. R., and Lindquist S. (1996) Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 273, 622–626 10.1126/science.273.5275.622 [DOI] [PubMed] [Google Scholar]
- 93. Kaiser C., Michaelis S., and Mitchell A. (1994) Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 94. Ghahramani S. (2005) Fundamentals of Probability with Stochastic Processes. 3rd Ed., Pearson Prentice Hall, Upper Saddle River, NJ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.