

Abstract
In previous studies, it was shown that post-conditioning, a transient period of brief ischemia following prolonged severe ischemia in the retina, could provide significant improvement in post-ischemic recovery, attenuation of cell loss, and decreased apoptosis. However, the mechanisms of post-conditioning in the retina have not been elucidated. We hypothesized that two kinases, mitogen-activated protein kinase p38α and protein kinase B (Akt), were involved in the mechanism of post-conditioning. Ischemia was induced in rat retina in vivo. Recovery after ischemia followed by 8 min of post-conditioning early in the reperfusion period after prolonged ischemia was assessed functionally (electroretinography) and histologically at 7 days after ischemia. We examined the role of p38α and Akt subtypes 1–3 in post-conditioning by intravitreal injection of interfering RNA 6 h prior to ischemia and post-conditioning and compared the results to injection of non-silencing interfering RNA sequence. The blockade of p38α significantly decreased the recovery after ischemia and post-conditioning, and enhanced cell loss and disorganization of the retina. Blockade of Akt1, and to a lesser degree, Akt2, significantly decreased the recovery after ischemia and enhanced cell loss and disorganization. These differences in the effects of blockade of Akt subtypes were not explainable by distribution of Akt subtypes in the retina, which were similar. In conclusion, both p38 and Akt are essential components of the neuroprotection induced by post-ischemic conditioning in the retina.
Keywords: Retinal ischemia, Post-conditioning, p38, Akt
Introduction
Retinal ischemia, associated with glaucoma, diabetic retinopathy, central retinal artery occlusion, and other vascular diseases, causes visual loss. Our previous studies demonstrated that the detrimental effects of retinal ischemia can be attenuated by subjecting the retina in vivo to brief periods of non-damaging ischemia, known as retinal ischemic preconditioning (IPC) (Roth et al. 1998). The involvement of a number of signaling molecules in IPC has been reported, including, among others, Akt/protein kinase B subtypes, protein kinase C, erythropoietin, mitogen-activated protein kinase p38 (MAPK p38), and HSP27 (Whitlock et al. 2005; Dreixler et al. 2008, 2009a, b, c). Although preconditioning confers robust endogenous neuroprotection, translational relevance is limited by the necessity to apply the preconditioning stimulus prior to the harmful ischemia, which is usually unknown in its onset.
More recently, it has been shown that applying a brief ischemic stimulus after prolonged ischemia (post-conditioning, “post-C”) also provided significant neuroprotection, with improved post-ischemic functional recovery, decreased histological damage, and attenuation of apoptosis (Fernandez et al. 2009; Dreixler et al. 2010). Further, it was shown that new protein synthesis was required for post-C (Fernandez et al. 2009). These results with post-C are very exciting and directly clinically translatable to patients who have experienced retinal ischemia, but the mechanisms of post-C in the retina have not been determined.
A potential signaling protein in post-C is Akt/protein kinase B. The cytoplasmic 57-kDa protein serine/threonine kinase is recognized as one of the most important protein kinases at the core of numerous and diverse physiological functions. The Akt pathway has also been identified as a cellular survival signal in several neuronal cell types and in protection from oxidative stress (Manning and Cantley 2007). By phosphorylating Ser136 on Bad, it interferes with mitochondrial cytochrome c release and inhibits caspase 9 (Kamada et al. 2007), thereby attenuating apoptosis. It has been established that there are distinct distribution patterns and functional roles of the three isoforms of Akt, suggesting that there may be a similar difference in the role of subtypes in disease states (Tschopp et al. 2005). In earlier studies, using interfering RNA (siRNA) in the retinal ischemia preconditioning model in rats in vivo, we showed the specific involvement of Akt subtypes 2 and 3 in IPC (Dreixler et al. 2009c). However, inhibition of any of the Akt subtypes 1–3 attenuated the mimicking of IPC produced by opening of the mitochondrial KATP channel (Dreixler et al. 2009c). Since all Akt subtypes appear to influence preconditioning in the retina, we hypothesize that post-conditioning alters the post-ischemic molecular environment of retinal cells similarly, and that post-C is significantly regulated by Akt in the reperfusion period.
Another possible signaling intermediary in post-C is the mitogen-activated protein kinase (MAPK) p38α. MAPKs couple cell-surface receptors to critical regulatory targets and gene transcription, respond to chemical and physical stressors, and play a key role in cell survival and adaptation (Coulthard et al. 2009). MAPKs are activated by adenosine receptors and their expression is influenced by PKC and PKA, receptor tyrosine kinases, stress molecules, and cytokines (Hazzalin and Mahadevan 2001). We previously found distinct temporal, cell-specific expression patterns of MAPKs ERK, JNK, and p38 after retinal ischemia or IPC. In particular, phosphorylated p38 increased significantly after ischemia in a specific time course, beginning at 1 h, persisting up to 1 week later. Blocking p38 or ERK, but not JNK, completely attenuated ischemic injury and dramatically reduced apoptosis-related gene expression (Roth et al. 2003). However, among the MAPKs, p38 blockade also prevented IPC neuroprotection (Dreixler et al. 2009a). Thus, p38 appears to be a critical link in retinal cell survival/death, where its activation contributes to injury following ischemia, but its earlier activation produces changes that contribute to subsequent protection from ischemia, through its involvement in ischemic preconditioning. We hypothesize a similarly essential role for p38α in ischemic post-conditioning.
Materials and Methods
Retinal Ischemia and Preconditioning
Wistar rats (200–250 g) purchased from Harlan (Indianapolis, IN, USA) were maintained on a 12 h on/12 h off light cycle. Procedures (Roth et al. 2006; Dreixler et al. 2008) conformed to the ARVO Resolution on the Use of Animals in Research and were approved by our Animal Care Committee. For retinal ischemia, rats were anesthetized with chloral hydrate, 275 mg/kg i.p., and intraocular pressure (IOP) increased to 130–135 mmHg for 55 min using a pressurized bag of sterile ocular irrigating solution (BSS; Alcon, Fort Worth, TX, USA) connected to a 27-g needle positioned in the center of the anterior chamber. IOP was measured continuously using the needle and pressure tubing via a three-way stopcock interposed into the line, with an electronic pressure transducer (Hewlett-Packard) zeroed to the level of the eye. IOP exceeded the systolic blood pressure, monitored non-invasively using a tail blood pressure cuff (IITC Life Science, Woodland Hills, CA, USA). Post-conditioning was accomplished by allowing reperfusion for 5 min after ischemia ended, confirmed by direct observation under the operating microscope, and then increasing the IOP to 160–165 mmHg for 8 min, after which the anterior chamber needle was removed.
Body temperature was maintained at 36–37°C by a servo-controlled heating blanket (Harvard Apparatus, Natick, MA, USA) to prevent a protective effect on ischemia of hypothermia (Faberowski et al. 1989). To preclude ischemic tolerance due to hypoxia (Zhu et al. 2007), O2 saturation was measured with a pulse oximeter (Ohmeda, Louisville, CO, USA) on the rat’s tail. Supplemental oxygen was administered when necessary to maintain O2 saturation >94%.
Electroretinography before and after Retina Ischemia
Procedures were similar to those we reported previously (Dreixler et al. 2009a, 2010). Animals were dark-adapted for at least 2 h before recordings. For baseline and post- ischemic (i.e., after 7 days) follow-up ERG, rats were injected i.p. with ketamine (35 mg/kg) and xylazine (5 mg/ kg). Corneal analgesia was with one to two drops of 0.5% proparacaine (Alcon). Pupils were dilated with 0.5% tropicamide (Alcon), and cyclomydril 0.2% cyclopentolate HCl and 1% phenylephrine HCl (Alcon).
The ERG was recorded at baseline (prior to experiments) and 7 days after ischemia and post-conditioning by placing platinum needle electroencephalogram electrodes (Grass, Providence, RI, USA) in contact with the corneal surfaces of both eyes and a reference electrode on the tongue, as we reported previously (Dreixler et al. 2009a, 2010). Stimulus-intensity ERG analysis was achieved on a UTAS-E4000 using a full-field Ganzfeld stimulator (LKC Technologies, Gaithersburg, MD, USA), with the rat’s head centered 7 in. from the stimulator. The low-pass filter was 0.05 Hz and the high-pass 500 Hz. Flash intensity varied electronically from −3.39 log cd s/m2 to 1.89 log cd s/m2. Settings were confirmed by photometry (EG & G Model 550 photometer; Electro-Optics, Boulder, CO, USA). Responses were averaged for three to 10 flashes delivered 4 to 60 s apart depending upon flash intensity, with number of flashes decreasing and time between them increasing with flash intensity. Flashes were progressively delivered from the lowest intensity to the highest to prevent possible effect upon dark adaptation, and at least 1 min elapsed between the series of flashes for the three highest intensity settings.
Recorded time, intensity, and amplitude were exported and analyzed in Matlab (Math Works, Natick, MA). Recordings were first baseline-corrected for drift and low-frequency noise. Peak a-wave amplitude was calculated as the negative minimum following the light stimulus, and the b-wave amplitude as difference between the a-wave and the maximum value recorded thereafter. Oscillatory potentials (OPs) were measured by extracting OP wavelet components using fast Fourier transformation. The sum of the root mean squares (sum RMS) of the amplitudes of the OP wavelets was calculated. Scotopic P2 for the rod bipolar response was calculated using the Lamb and Pugh model (Bui et al. 2005).
Histology
Eyes enucleated on the seventh day after ischemia were immediately placed in Davidson’s fixative (11% glacial acetic acid, 2% neutral buffered formalin, and 32% ethanol in H2O) for 24 h, then transferred to 70% ethanol for 24 h and stored in PBS at 4 C. Eyes were embedded in paraffin, sectioned to 4 μm, and stained with hematoxylin and eosin (H&E). Sections were examined by light microscopy and cell counts in the retinal ganglion cell (RGC) layer quantified as described earlier (Roth et al. 1998; Junk et al. 2002; Dreixler et al. 2009b). Briefly, the average numbers of cells in the retinal ganglion cell layer were counted over a distance of 150 μm approximately 1,500 μm from both sides of the optic nerve head using ×40 optics. Number of cells is reported as cells per region of interest.
RNA Interference
Target sequences for the interfering RNA (siRNA; Qiagen, Valencia, CA, USA) for p38α, the three Akt subtypes, and the non-silencing siRNA are shown in Table 1. The siRNAs were designed using neural-network technology as described previously (Huesken et al. 2005). Using BLAST, we found the siRNAs were 100% homologous to the mRNA sequence of the targets. siRNA design was then checked for homology to all other sequences of the genome, 3′ UTR/seed analysis, single-nucleotide polymorphisms, and interferon motif avoidance (Farh et al. 2005; Hornung et al. 2005; Judge et al. 2005) (http://www1.qiagen.com/Products/GeneSilencing/HPOnGuardsiRNADesign.aspx). Sequences of the introduced siRNA are uniquely specific to the targeted gene (see reviews for more details) (Dorsett and Tuschl 2004; Genc et al. 2004; Lehner et al. 2004; Mello and Conte 2004). A 2-μl mixture of four siRNAs to the targets, or a single negative control, non-silencing siRNA not corresponding to any known rat gene (Qiagen) in RPMI media (Invitrogen, Carlsbad, CA, USA) and RNAiFect transfection reagent (Qiagen) at a final concentration of 3 μM was injected into the mid-vitreous of both eyes of the rats with a microsyringe (Hamilton, Reno, NV, USA) as previously described (Roth et al. 2006). Penetration of the siRNA into the inner retinal layers was confirmed previously (Dreixler et al. 2008). The specificity for the targeted siRNA has been shown previously for p38α siRNA (Dreixler et al. 2009a) and for all three Akt subtype siRNAs (Dreixler et al. 2009c).
Table 1.
Target sequences for siRNA
| siRNA target sequence(s) | |
|---|---|
| Non-silencing siRNA p38α siRNA |
ATT TCT CCG AAC GTG TCA CGT AAGCTCTTGCGCATGCCTACT AAGGTCCCTGGAAGAATTCAA AAGAAGCTGTCGAGACCGTTT AAGAGCTGACCTACGATGAA |
| Akt1 siRNA | ATG GAG TGT GTG GAC AGT GAA ACG CTA CTT CCT CCT CAA GAA TGG GAA GGT GAT CCT GGT GAA CCG CCT CTG CTT TGT CAT GGA |
| Akt2 siRNA | AAC GAC TTC GAT TAT CTC AAA TCG GTT CTT CCT CAG CAT CAA ACA AGG TAC TTT GAT GAT GAA AAC AAT TTC TCT GTA GCA GAA |
| Akt3 siRNA | AGC GAT GTT ACC ATC GTT AAA CAG GAT CAT GAG AAA CTC TTT CGG CTC ATT CAT AGG CTA TAA CAG GAA GAC TTG CAT TAT CTT |
Immunohistochemistry
To examine cellular localization of Akt subtypes, eyes were removed from euthanized rats and evaluated with immunohistochemistry as previously described (Roth et al. 2003, 2006). Primary antibodies (1:50 concentration) included rabbit polyclonal anti-Akt1 (Calbiochem, La Jolla, CA, USA), rabbit polyclonal anti-Akt2 (Cell Signaling, Beverly, MA, USA), rabbit polyclonal anti-Akt3 (Cell Signaling), biotinylated anti-rat Thy-1 (BD Pharmingen, San Diego, CA, USA), mouse monoclonal anti-PKCα (Transduction Laboratories, Lexington, KY, USA), mouse monoclonal anti-syntaxin (Sigma, St. Louis, MO, USA), mouse monoclonal anti-calbindin (Sigma), mouse monoclonal anti-vimentin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and mouse monoclonal anti-GFAP (Sigma). Control sections for primary antibodies were incubated with non- immune serum. Sections (10 μm) were exposed to the appropriate secondary antibodies: fluorescein-conjugated avidin (1:500, Jackson ImmunoResearch, West Grove, PA, USA), goat anti-mouse IgG FITC-conjugate (1:500; Southern Biotechnology, Birmingham, AL), or goat anti-rabbit IgG rhodamine-conjugate (1:500; Jackson ImmunoResearch). Antifade mounting media containing DAPI (EMC Biosciences, La Jolla, CA, USA) was applied and sections were cover-slipped.
Studies
The roles of p38α and the Akt subtypes in Post-C were specifically examined using RNA interference. Then 3 μM siRNAs for the specific targets or a negative control siRNA was injected into the vitreous of both eyes 6 h before ischemia. Follow-up ERGs were done 7 days later. The eyes were then removed for histological examination.
Data Handling and Statistical Analysis
ERG data were corrected for daily variation in the normal eye and for simultaneous reference to the baseline in the ischemic eye, as previously described (Roth et al. 2006; Dreixler et al. 2008, 2009b, c). The a- and b-waves, SUM RMS of the OPs, and P2 from ischemic eyes 7 days after ischemia in the groups for comparison were expressed as stimulus-intensity plots (intensity, log cd s/m2 on the x- axis, and % recovery of baseline on the y-axis) (Dreixler et al. 2008). The data were normally distributed as confirmed using normality plots and skewness/kurtosis in Stata version 10.0 (College Station, TX, USA). Statistical analysis of the ERG data was achieved with an unpaired t test. For histopathological data analysis, we used the Wilcoxon non-parametric test.
Results
Akt Subtype Cellular Localization in the Retina
In a previous study, we defined the cellular localization of p38 in the retina (Roth et al. 2003). To define the cellular localization of the Akt subtypes in the retina, frozen sections of rat retina were studied using immuno-histochemistry. We co-stained with the following anti-bodies to specific protein markers for co-localization of the Akt subtypes in retinal cells: (1) Thy1 for retinal ganglion cells (Dreixler et al. 2008), (2) vimentin (Verardo et al. 2008) and GFAP (Roth et al. 2003; Dreixler et al. 2008) for Müller cells and astrocytes, (3) syntaxin for amacrine cells (Roth et al. 2003), (4) PKCα for bipolar cells (Roth et al. 2003), and (5) calbindin for horizontal cells (Hirano et al. 2007; Dreixler et al. 2008). Control sections incubated with non-immune serum demonstrated no staining (not shown).
There were differences in the distribution of Akt subtypes. Figure 1 shows co-localization of Akt1 with cellular markers in RGCs (a) and amacrine and displaced amacrine cells (c). Figure 2 shows co-localization of Akt2 with cellular markers in bipolar cells (b), amacrine cells (c) and horizontal cells (d). We note that some apparent Akt2 co-localization with astrocytic markers may be attributed to non-specific staining. Figure 3 shows co-localization of Akt3 with cellular markers in RGCs (a), bipolar cells (b), amacrine cells (c), horizontal cells (d), and astrocytes/ Müller cells (e and f). We also note co-localization of Akt3 with Thy1 and calbindin in other retinal layers that may be attributed to non-specific staining.
Fig. 1.
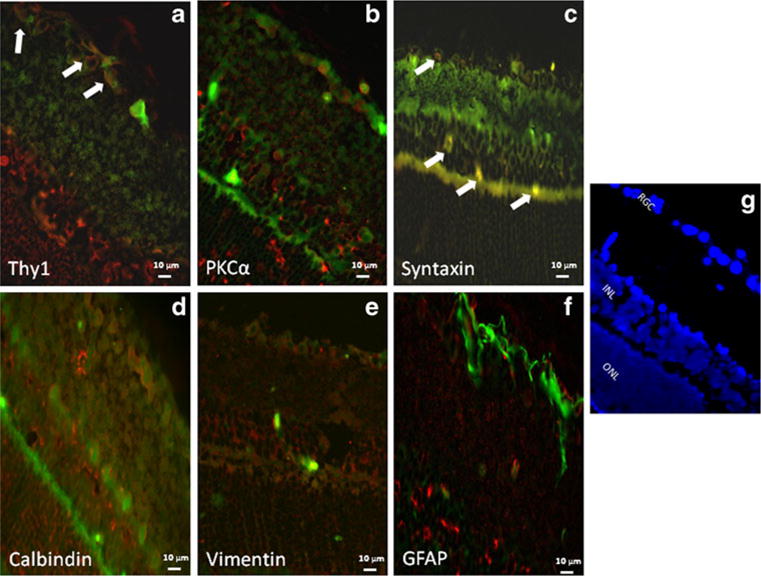
Co-localization of Akt1 (red) and retinal cell markers (green). Co-localization (as depicted by an orange/yellow color) of Akt1with the retinal ganglion cell marker, anti-Thy1 (a) and with amacrine cell marker, anti-syntaxin (c) is indicated by white arrows. The fluorescent images utilized ×40 oil magnification. Control sections for primary antibodies incubated with non-immune serum demonstrated no staining (not shown). Scale bars are shown at the bottom of each figure. Orientation of the retinal layers are shown in (g) by the nuclear staining by DAPI. The figure shows that Akt1 is present primarily in cells in the retinal ganglion cell layer
Fig. 2.
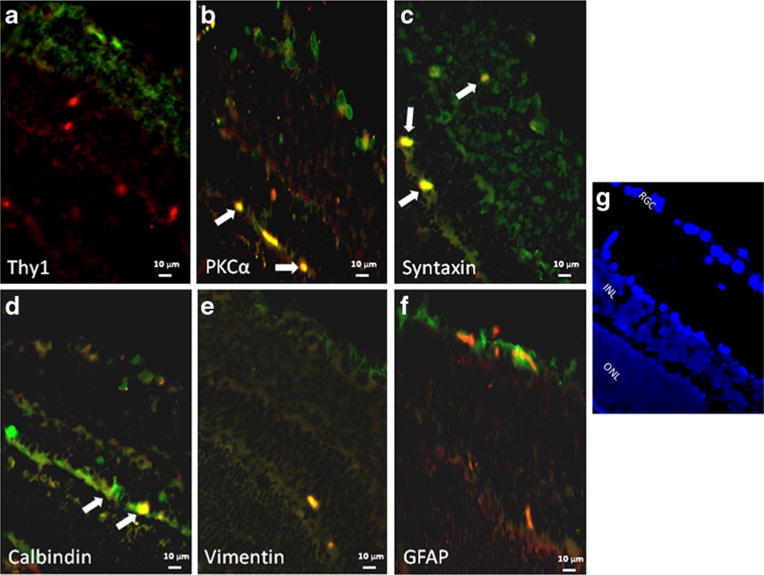
Co-localization of Akt2 (red) and retinal cell markers (green). Co-localization (as depicted by an orange/yellow color) of Akt2 with bipolar cell marker, anti-PKCα (b), with amacrine cell marker, anti- syntaxin (c), and with horizontal cell marker, anti-calbindin (d), is indicated by white arrows. The fluorescent images utilized ×40 oil magnification. Control sections for primary antibodies incubated with non-immune serum demonstrated no staining (not shown). Scale bars are shown at the bottom of each figure. Orientation of the retinal layers are shown in (g) by the nuclear staining by DAPI. The figure shows that Akt2 is present in amacrine cells (including displaced amacrine cells in the RGC layer), bipolar cells, and horizontal cells. We also note that some apparent Akt2 co-localization with astrocytic markers may be attributed to non-specific staining
Fig. 3.
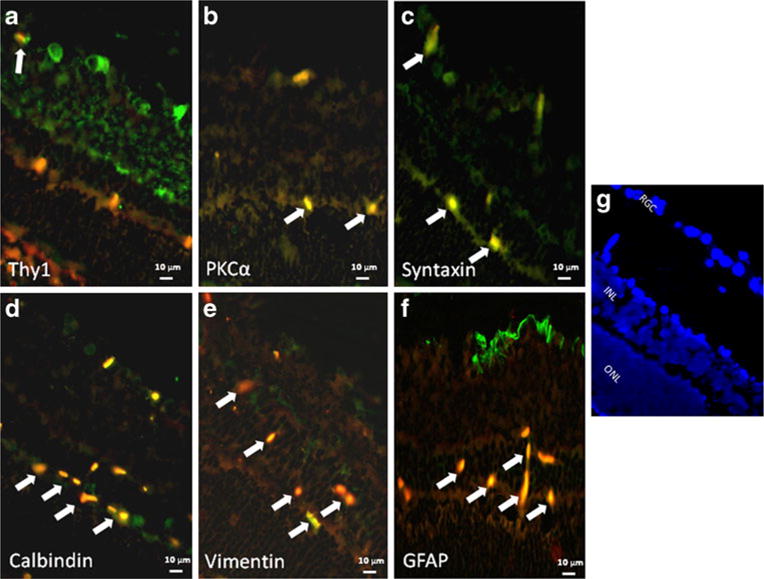
Co-localization of Akt3 (red) and retinal cell markers (green). Co-localization (as depicted by an orange/yellow color) of Akt3 with the retinal ganglion cell marker, anti-Thy1 (a), with bipolar cell marker, anti-PKCα (b), with amacrine cell marker, anti-syntaxin (c), with horizontal cell marker, anti-calbindin (d), and with astrocyte/ Müller cell marker, anti-vimentin (e) and anti-GFAP (f), is indicated by white arrows. The fluorescent images utilized ×40 oil magnification. Control sections for primary antibodies incubated with non-immune serum demonstrated no staining (not shown). Scale bars are shown at the bottom of each figure. Orientation of the retinal layers are shown in (g) by the nuclear staining by DAPI. The figure shows that Akt3 is present in nuclei throughout the inner retina and RGCs, amacrine cells, bipolar cells, horizontal cells, and in astrocytes and Müller cells. We also note co-localization of Akt3 with Thy1 and calbindin in unexpected retinal layers that may be attributed to non-specific staining
Effects of Silencing p38α on Post-conditioning
Intravitreal injection of p38α siRNA 6 h prior to ischemia (n=6) significantly decreased the a-wave, b-wave, OP RMS, and P2 recovery at 7 days after ischemia+post-C when compared to the non-silencing RNA injected (n=5) control group (Fig. 4) over a range of flash intensities from −1.02 to 1.4 log cd s/m2.
Fig. 4.
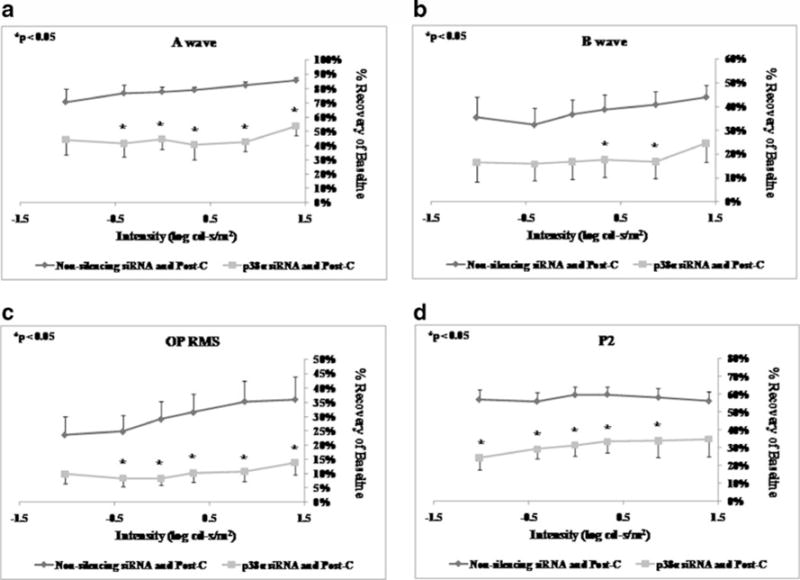
ERG stimulus-intensity response in ischemic post-conditioned retinae treated with siRNA to p38α vs. non-silencing sequence. The recovery relative to baseline at 7 days after ischemia is shown on the y- axis. Double normalized (for baseline in ischemic eye and for variation in the non-ischemic eye) ERG data for a-wave (a), b-wave (b), OP RMS (c), and P2 (d) over a range of flash intensities suggest that p38α is essential in post-conditioning. Solid lines with diamonds=non- silencing siRNA+ischemia+Post-C (n=5). Solid lines with squares= p38α siRNA+ischemia+Post-C (n = 6). Statistical analysis was achieved using unpaired t tests
Histological examination of the retinas 7 days after ischemia+post-C showed that the number of cells in the RGC layer in the ischemic retinae significantly decreased from 11.8±0.6 for the paired control non-ischemic retina to 7.9±0.8 (p=0.03; n=6) in the p38α siRNA group (Table 2; Fig. 5). By comparison, the number of cells in the RGC layer in the ischemic retinae was relatively unchanged from 10.4±0.9 for the paired control non-ischemic retinae to 9.8±0.9 (p=0.40; n=5) in the ischemic retinae for the non-silencing siRNA group, showing that the preservation of histological structure of the retina after ischemia by post-C was attenuated by the blockade of p38α (Table 2; Fig. 5).
Table 2.
Number of retinal ganglion cells per region of interest in the normal and ischemic retinae 7 days after ischemia+Post-C for all the siRNA treatments
| Normal retina | Ischemic retina | p value | |
|---|---|---|---|
| Non-silencing siRNA/post-conditioning | 10.4±0.9 | 9.8±0.9 | 0.40 |
| p38α siRNA/post-conditioning | 11.8±0.6 | 7.9±0.8 | 0.03 |
| Akt1 siRNA/post-conditioning | 11.6±0.4 | 6.7±0.5 | 0.03 |
| Akt2 siRNA/post-conditioning | 11.3 ±0.9 | 7.9±0.9 | 0.04 |
| Akt3 siRNA/post-conditioning | 10.6±0.2 | 7.1±0.4 | 0.07 |
Post-conditioning (top row) prevented the loss of retinal ganglion cells; this effect was attenuated by p38α, Akt1, and Akt2 siRNA. Akt3 siRNA caused a decrease that was not significant. Statistical analysis was achieved using the non-parametric Wilcoxon test
Fig. 5.
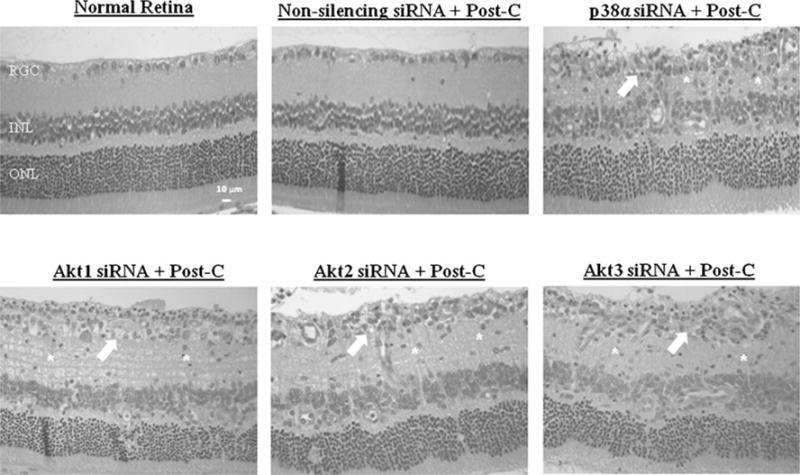
Representative histopathological images of hematoxylin-and- eosin-stained retinae in 4-μm-thick sections for each of the experimental groups. These sections were prepared from retinae removed from the rats at 7 days following ischemia. Arrows indicate layers demonstrating cell loss and asterisks denote regions of inflammatory cell infiltration. The retinal layers and scale bars are depicted in the normal retina image
Effects of Akt Subtype siRNA on Post-conditioning
Intravitreal injection of Akt1 siRNA 6 h prior to ischemia (n=6) significantly decreased the a-wave, b-wave, OP RMS, and P2 recovery at 7 days after ischemia+post-C when compared to the non-silencing RNA injected (n=5) control group (Fig. 6) over a range of flash intensities from −1.02 to 1.4 log cd s/m2. Injection of Akt2 siRNA 6 h prior to ischemia (n=5) significantly decreased only the a-wave. The b-wave, OP RMS, and P2 recovery at 7 days after ischemia+ post-C were not significantly different when compared to the non-silencing RNA injected (n=5) control group (Fig. 7). Injection of Akt3 siRNA 6 h prior to ischemia (n=9) significantly decreased the a-wave at 7 days after ischemia+ post-C, when compared to the non-silencing RNA injected (n=5) control group (Fig. 8) over a range of flash intensities from −1.02 to 1.4 log cd s/m2. The b-wave and OP RMS recoveries were unchanged, and at only one intensity was there a significant effect upon P2.
Fig. 6.
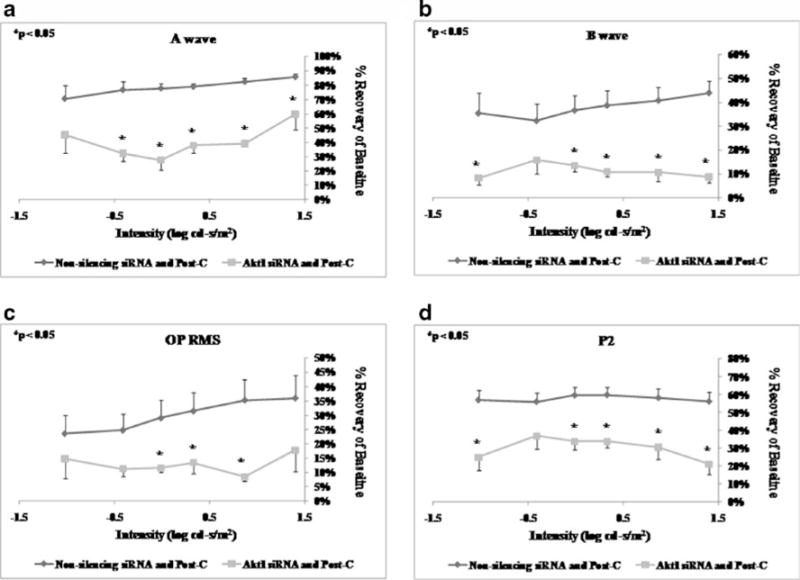
ERG stimulus-intensity response in ischemic post-conditioned retinae treated with siRNA to Akt1 vs. non-silencing sequence. The recovery relative to baseline at 7 days after ischemia is shown on the y-axis. Double normalized (for baseline in ischemic eye and for variation in the non-ischemic eye) ERG data for a-wave (a), b-wave (b), OP RMS (c), and P2 (d) over a range of flash intensities suggest that Akt1 is essential in post-conditioning. Solid lines with diamonds= non-silencing siRNA+ischemia+Post-C (n=5). Solid lines with squares=Akt1 siRNA+ischemia+Post-C (n=6). Statistical analysis was achieved using unpaired t tests
Fig. 7.
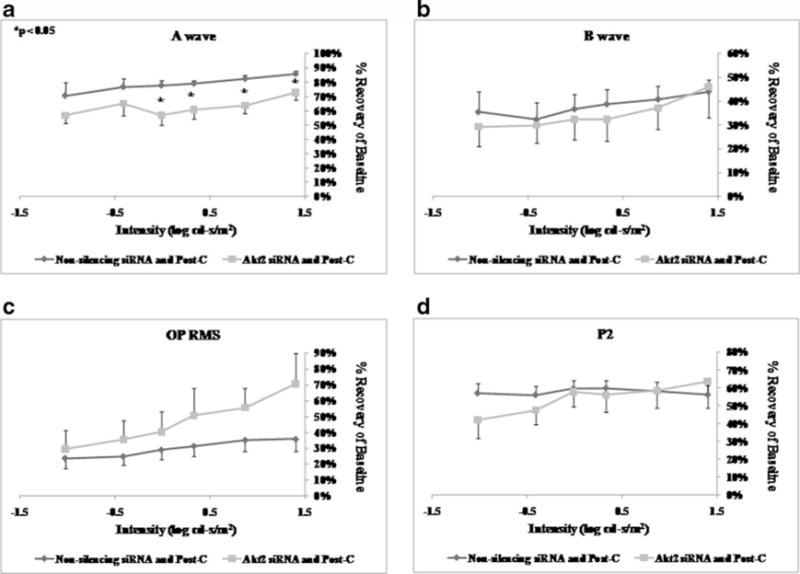
ERG stimulus-intensity response in ischemic post-conditioned retinae treated with siRNA to Akt2 vs. non-silencing sequence. The recovery relative to baseline at 7 days after ischemia is shown on the y-axis. Double normalized (for baseline in ischemic eye and for variation in the non-ischemic eye) ERG data for a-wave (a), b-wave (b), OP RMS (c), and P2 (d) over a range of flash intensities suggest that Akt2 has a minor role in post-conditioning. Solid lines with diamonds=non-silencing siRNA+ischemia+Post-C (n=5). Solid lines with squares=Akt2 siRNA+ischemia+Post-C (n=5). Statistical analysis was achieved using unpaired t tests
Fig. 8.
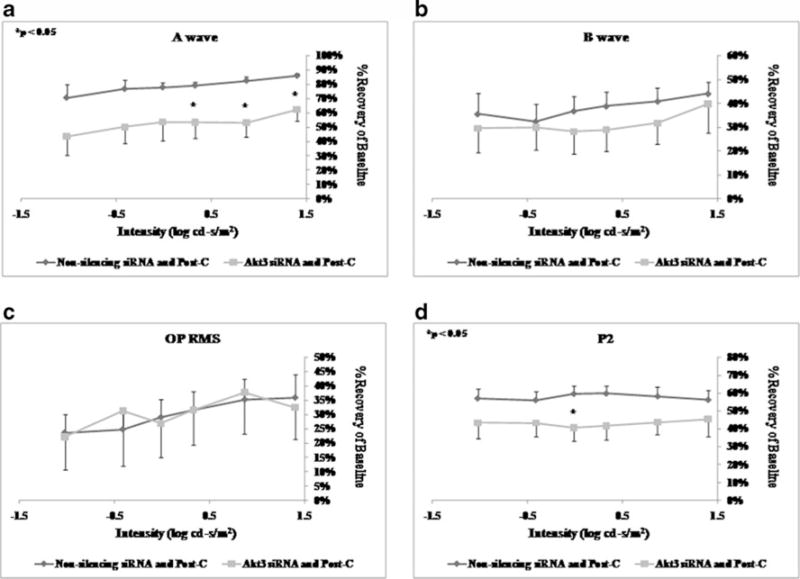
ERG stimulus-intensity response in ischemic post-conditioned retinae treated with siRNA to Akt3 vs. non-silencing sequence. The recovery relative to baseline at 7 days after ischemia is shown on the y-axis. Double normalized (for baseline in ischemic eye and for variation in the non-ischemic eye) ERG data for a-wave (a), b-wave (b), OP RMS (c), and P2 (d) over a range of flash intensities suggest that Akt3 is not involved in post-conditioning. Solid lines with diamonds=non-silencing siRNA+ischemia+Post-C (n = 5). Solid lines with squares=Akt3 siRNA+ischemia+Post-C (n=9). Statistical analysis was achieved using unpaired t tests
Histological examination of the retinas 7 days after ischemia+ post-C showed that the number of cells in the RGC layer in the ischemic retinae significantly decreased from 11.6±0.4 for the paired control non-ischemic retina to 6.7±0.5 (p=0.03; n=6) in the Akt1 siRNA group (Table 2; Fig. 5). In the Akt2 siRNA-treated group, the retinas 7 days after ischemia+ post-C showed that the number of cells in the RGC layer in the ischemic retinae significantly decreased from 11.3±0.9 for the paired control nonischemic retina to 6.7±0.5 (p=0.03; n = 5; Table 2; Fig. 5). Finally, the retinas 7 days after ischemia+post-C showed that the number of cells in the RGC layer in the ischemic retinae showed a non-significant decrease from 10.6±0.2 for the paired control non-ischemic retina to 7.1 ± 0.4 (p=0.07; n=5) in the Akt3 siRNA group (Table 2; Fig. 5).
Discussion
Our results confirmed the hypothesis that p38α and Akt/ protein kinase B are essential in retinal ischemic postconditioning, and that knockdown of these two kinases attenuates the enhanced functional recovery after ischemia that is induced by post-C. We demonstrated that (1) silencing p38α using p38α-directed siRNA significantly reduced the ameliorative effects of post-conditioning on retinal function as demonstrated by a reduction in recovery ERG waves after ischemia; (2) inhibition of specific Akt subtypes also attenuated the protective effects of post-conditioning after ischemia; and that (3) histological protection of the retina was also compromised when the rats were exposed to the target siRNAs, as seen with the disrupted retinal structure and significantly reduced cell count in the retinal ganglion cell layer.
Our previous studies have shown that p38α expression increases following ischemia, with increased activation lasting for up to 1 week after ischemia, particularly in the inner retina. Since p38 functions as a pro-apoptotic protein by, among other effects, activating p53, it is not surprising that blockade of p38 activity using the specific inhibitor SB203580 decreased ischemic injury and apoptosis gene related expression after retinal ischemia (Roth et al. 2003). However, challenging the view that p38 is exclusively a cell death inducing kinase, it is increasingly being shown to be associated with neuroprotection. We recently demonstrated that p38α is also involved in the neuroprotective effects of ischemic preconditioning. Blockade of p38α using interfering RNA attenuated the effect of ischemic preconditioning to decrease damage after retinal ischemia. Moreover, transient activation of p38α by systemic administration of anisomycin mimicked the neuroprotective effect of ischemic preconditioning (Dreixler et al. 2009a). We also demonstrated that blockade of MKP-1, a phosphatase that specifically deactivates p38 in retina, blocked the neuro-protective effect of ischemic preconditioning (Dreixler et al. 2011). We theorized that MKP-1 precisely controls the level of p38 activity in the retina, and excessive p38 activation, as in ischemia, is damaging rather than neuroprotective.
Post-conditioning represents a very different approach from IPC in that the transient ischemic stimulus is placed after the damaging ischemia has ended. Nonetheless, the current study shows that post-conditioning and preconditioning share similar mechanisms, i.e., the involvement of p38α and Akt (Dreixler et al. 2009a, c).
An interfering RNA directed specifically towards p38α was used to inhibit the translation of the protein. It has been shown previously that the predominant p38 isoform in the retina is p38α, while p38β was undetectable (Kikuchi et al. 2000) justifying our specific targeting of p38α. ERG data clearly showed that p38α has a significant role in the post-conditioning effect. All waveforms of the ERG were significantly reduced, implying that multiple cell types of the retina may be involved in p38’s post-conditioning role. These results are also consistent with our earlier demonstration of p38 localization in the inner retina in rats (Roth et al. 2003).
Another signaling pathway involved in conferring significant neuroprotective properties in the retina is protein kinase B, or Akt. Inactivation of the Akt pathway was associated with photoreceptor cell death in mice (Jomary et al. 2006), and exogenous administration of BDNF prevented the dephosphorylation of Akt in rat RGCs in vivo and significantly suppressed NMDA-induced RGC death (Nakazawa et al. 2005). There are three specific subtypes of Akt, and recently it has been increasingly recognized that they subserve different functions (Manning and Cantley 2007). In retina, rod photoreceptor cells in Akt2 knock-out mice had significantly greater sensitivity to photic injury than rods in heterozygous or wild-type mice, while Akt1 deletion had no effect (Li et al. 2007). Further supporting a role of specific subtypes, we have recently shown that interfering RNA directed against Akt subtypes 2 and 3 prevented the ameliorative effect of IPC on post-ischemic functional recovery (Dreixler et al. 2009c). However, all three Akt subtypes were involved in the mimicking of IPC produced by administration of diazoxide, a mitochondrial KATP channel opener (Dreixler et al. 2009c).
Post-C activated Akt, and its neuroprotective effect in rat brain, was attenuated by blocking PI-3-kinase (Gao et al. 2008; Scartabelli et al. 2008). These studies suggested involvement of Akt in post-conditioning. Accordingly, we tested the hypothesis that specific Akt subtypes are responsible for post-conditioning in the retina. Interestingly, only blockade of Akt1 significantly altered functional recovery across all functional measurements, although Akt2, but not Akt3 blockade, also reduced the number of surviving cells in the RGC layer after ischemia followed by post-C, and Akt2 and Akt3 affected only recovery of the a- wave. Moreover, the co-localization of Akt1 primarily to the retinal ganglion cell layer suggests a link to the functional attenuation afforded to the Akt1-directed siRNA. This suggests that in post-conditioning, there is no redundancy in the effects of Akt subtypes. Our results do not appear to correlate to the localization of Akt subtypes in the retina, which was similar for all Akt subtypes. Therefore, differences in subcellular localization and function or in downstream mediators are possible reasons for the differential effects of the Akt subtypes.
As a novel strategy that could be potentially used to protect against retinal ischemia, post-C shifts the focus of neuroprotection away from the paradigm of pre-ischemic intervention to manipulations that alter the post-ischemic state in a manner that is favorable to survival of retinal cells. Results from this study have shown the involvement of two important cell signaling kinases, p38 and Akt, in retinal post-C. Further investigation of the mechanisms of post-C may enable this novel neuroprotective strategy to be applied clinically to patients that have sustained ischemic events in the retina.
Acknowledgments
Supported by National Institutes of Health (Rockville, MD) grants RO1 EY10343 and RO1 EY10343-16S2 (American Recovery and Reinvestment Act) to Dr. Roth, AG029795-02 for the Medical Student Summer Research Program at the Pritzker School of Medicine, UL1RR024999 to the University of Chicago Institute for Translational Medicine; the Illinois Society for the Prevention of Blindness (Chicago, IL); and the Dean’s Research Advisory Committee of the Division of Biological Sciences of the University of Chicago. Ajay Sampat was the recipient of a Medical Student Research Fellowship Award from the American Academy of Neurology, St Paul, MN.
Footnotes
There is no conflict of interest or commercial interest for any of the authors.
Contributor Information
John C. Dreixler, Department of Anesthesia and Critical Care, University of Chicago Medical Center, 5841 South Maryland Avenue, Box MC 4028, Chicago, IL 60637, USA
Ajay Sampat, Pritzker School of Medicine, University of Chicago Medical Center, 5841 South Maryland Avenue, Box MC 4028, Chicago, IL 60637, USA.
Afzhal R. Shaikh, Department of Anesthesia and Critical Care, University of Chicago Medical Center, 5841 South Maryland Avenue, Box MC 4028, Chicago, IL 60637, USA
Michael Alexander, Department of Anesthesia and Critical Care, University of Chicago Medical Center, 5841 South Maryland Avenue, Box MC 4028, Chicago, IL 60637, USA.
Marcus M. Marcet, Department of Surgery (Ophthalmology) and Department of Pathology, University of Chicago Medical Center, 5841 South Maryland Avenue, Box MC 4028, Chicago, IL 60637, USA
Steven Roth, Department of Anesthesia and Critical Care, University of Chicago Medical Center, 5841 South Maryland Avenue, Box MC 4028, Chicago, IL 60637, USA.
References
- Bui BV, Edmunds B, Cioffi GA, Fortune B. The gradient of retinal functional changes during acute intraocular pressure elevation. Invest Ophthalmol Vis Sci. 2005;46:202–213. doi: 10.1167/iovs.04-0421. [DOI] [PubMed] [Google Scholar]
- Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med. 2009;15:369–379. doi: 10.1016/j.molmed.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett Y, Tuschl T. siRNAs: applications in functional genomics and potential as therapeutics. Nat Rev. 2004;3:318–329. doi: 10.1038/nrd1345. [DOI] [PubMed] [Google Scholar]
- Dreixler JC, Shaikh AR, Shenoy SK, Shen Y, Roth S. Protein kinase C subtypes and retinal ischemic preconditioning. Exp Eye Res. 2008;87:300–311. doi: 10.1016/j.exer.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreixler JC, Barone FC, Shaikh AR, Du E, Roth S. Mitogen- activated protein kinase p38alpha and retinal ischemic preconditioning. Exp Eye Res. 2009a;89:782–790. doi: 10.1016/j.exer.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreixler JC, Hagevik S, Hemmert JW, Shaikh AR, Rosenbaum DM, Roth S. Involvement of erythropoietin in retinal ischemic preconditioning. Anesthesiology. 2009b;110:774–780. doi: 10.1097/ALN.0b013e31819c4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreixler JC, Hemmert JW, Shenoy SK, Shen Y, Lee HT, Shaikh AR, Rosenbaum DM, Roth S. The role of Akt/protein kinase B subtypes in retinal ischemic preconditioning. Exp Eye Res. 2009c;88:512–521. doi: 10.1016/j.exer.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreixler J, Shaikh A, Savoie B, Alexander M, Marcet M, Roth S. Retinal ischemic postconditioning is not additive with ischemic preconditioning. Exp Eye Res. 2010;91:844–852. doi: 10.1016/j.exer.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreixler JC, Bratton A, Du E, Shaikh AR, Marcet MM, Roth S. Mitogen activated protein kinase phosphatase-1 (MKP-1) in retinal ischemic preconditioning. Experimental Eye Research. 2011 doi: 10.1016/j.exer.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faberowski N, Stefansson E, Davidson RC. Local hypothermia protects the retina from ischemia. A quantitative study in the rat. Invest Ophthalmol Vis Sci. 1989;30:2309–2313. [PubMed] [Google Scholar]
- Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- Fernandez DC, Bordone MP, Chianelli MS, Rosenstein RE. Retinal neuroprotection against ischemia-reperfusion damage induced by postconditioning. Investig Ophthalmol Vis Sci. 2009;50:3922–3930. doi: 10.1167/iovs.08-3344. [DOI] [PubMed] [Google Scholar]
- Gao X, Zhang H, Takahashi T, Hsieh J, Liao J, Steinberg GK, Zhao H. The Akt signaling pathway contributes to postconditioning’s protection against stroke; the protection is associated with the MAPK and PKC pathways. J Neurochem. 2008;105:943–955. doi: 10.1111/j.1471-4159.2008.05218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genc S, Koroglu TF, Genc K. RNA interference in neuroscience. Mol Brain Res. 2004;132:260–270. doi: 10.1016/j.molbrainres.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Hazzalin CA, Mahadevan LC. MAPK-regulated transcription: a continuously variable gene switch. Nat Rev. 2001;3:30–40. doi: 10.1038/nrm715. [DOI] [PubMed] [Google Scholar]
- Hirano AA, Brandstatter JH, Vila A, Brecha NC. Robust syntaxin-4 immunoreactivity in mammalian horizontal cell processes. Vis Neurosci. 2007;24:489–502. doi: 10.1017/S0952523807070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G. Sequence- specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005;11:263–270. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- Huesken D, Lange J, Mickanin C, Weiler J, Asselbergs F, Warner J, Meloon B, Engel S, Rosenberg A, Cohen D, Labow M, Reinhardt M, Natt F, Hall J. Design of a genome-wide siRNA library using an artificial neural network. Nat Biotechnol. 2005;23:995–1001. doi: 10.1038/nbt1118. [DOI] [PubMed] [Google Scholar]
- Jomary C, Cullen J, Jones SE. Inactivation of the Akt survival pathway during photoreceptor apoptosis in the retinal degeneration mouse. Invest Ophthalmol Vis Sci. 2006;47:1620–1629. doi: 10.1167/iovs.05-1176. [DOI] [PubMed] [Google Scholar]
- Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. 2005;23:457–462. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- Junk AK, Mammis A, Savitz SI, Singh M, Roth S, Malhotra S, Rosenbaum PS, Cerami A, Brines M, Rosenbaum DM. Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2002;99:10659–10664. doi: 10.1073/pnas.152321399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada H, Nito C, Endo H, Chan PH. Bad as a converging signaling molecule between survival PI3-K/Akt and death JNK in neurons after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2007;27:521–533. doi: 10.1038/sj.jcbfm.9600367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi M, Tenneti L, Lipton SA. Role of p38 mitogen-activated protein kinase in axotomy-induced apoptosis of rat retinal ganglion cells. J Neurosci. 2000;20:5037–5044. doi: 10.1523/JNEUROSCI.20-13-05037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner B, Fraser AG, Sanderson CM. How to use RNA interference. Brief Funct Genomics Proteomics. 2004;3:68–83. doi: 10.1093/bfgp/3.1.68. [DOI] [PubMed] [Google Scholar]
- Li G, Anderson RE, Tomita H, Adler R, Liu X, Zack DJ, Rajala RVS. Nonredundant role of Akt2 for neuroprotection of rod photoreceptor cells from light-induced cell death. J Neurosci. 2007;27:203–211. doi: 10.1523/JNEUROSCI.0445-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. Akt/Pkb signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Conte D. Revealing the world of RNA interference. Nature. 2004;431:338–342. doi: 10.1038/nature02872. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Shimura M, Endo S, Takahashi H, Mori N, Tamai M. N-Methyl-D-aspartic acid suppresses Akt activity through protein phosphatase in retinal ganglion cells. Mol Vis. 2005;11:1173–1182. [PubMed] [Google Scholar]
- Roth S, Li B, Rosenbaum PS, Gupta H, Goldstein IM, Maxwell KM, Gidday JM. Preconditioning provides complete protection against retinal ischemic injury in rats. Invest Ophthalmol Vis Sci. 1998;39:775–785. [PubMed] [Google Scholar]
- Roth S, Shaikh AR, Hennelly MM, Li Q, Bindokas V, Graham CE. Mitogen-activated protein kinases and retinal ischemia. Invest Ophthalmol Vis Sci. 2003;44:5383–5395. doi: 10.1167/iovs.03-0451. [DOI] [PubMed] [Google Scholar]
- Roth S, Dreixler JC, Shaikh AR, Lee KH, Bindokas V. Mitochondrial potassium ATP channels and retinal ischemic preconditioning. Invest Ophthalmol Vis Sci. 2006;47:2114–2124. doi: 10.1167/iovs.05-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scartabelli T, Gerace E, Landucci E, Moroni F, Pellegrini-Giampietro DE. Neuroprotection by group I mGlu receptors in a rat hippocampal slice model of cerebral ischemia is associated with the PI3K-Akt signaling pathway: a novel postconditioning strategy? Neuropharmacology. 2008;55:509–516. doi: 10.1016/j.neuropharm.2008.06.019. [DOI] [PubMed] [Google Scholar]
- Tschopp O, Yang Z-Z, Brodbeck D, Dummler BA, Hemmings-Mieszczak M, Watanabe T, Michaelis T, Frahm J, Hemmings BA. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development. 2005;132:2943–2954. doi: 10.1242/dev.01864. [DOI] [PubMed] [Google Scholar]
- Verardo MR, Lewis GP, Takeda M, Linberg KA, Byun J, Luna G, Wilhelmsson U, Pekny M, Chen D-F, Fisher SK. Abnormal reactivity of Muller cells after retinal detachment in mice deficient in GFAP and vimentin. Invest Ophthalmol Vis Sci. 2008;49:3659–3665. doi: 10.1167/iovs.07-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock NA, Agarwal N, Ma J-X, Crosson CE. Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Invest Ophthalmol Vis Sci. 2005;46:1092–1098. doi: 10.1167/iovs.04-0043. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Zhang Y, Ojwang BA, Brantley MA, Jr, Gidday JM. Long-term tolerance to retinal ischemia by repetitive hypoxic preconditioning: role of HIF-1alpha and heme oxygenase-1. Invest Ophthalmol Vis Sci. 2007;48:1735–1743. doi: 10.1167/iovs.06-1037. [DOI] [PubMed] [Google Scholar]