

Abstract
Southwest China is one of the major global biodiversity hotspots. The Tanaka line, extending within southwestern China from its northwest to its southeast, is an important biogeographical boundary between the Sino-Japanese and Sino-Himalayan floristic regions. Understanding the evolutionary history of the regional keystone species would assist with both reconstructing historical vegetation dynamics and ongoing biodiversity management. In this research, we combined phylogeographic methodologies and species distribution models (SDMs) to investigate the spatial genetic patterns and distribution dynamics of Quercus kerrii, a dominant evergreen oak inhabiting southwest China lowland evergreen-broadleaved forests (EBLFs). A total of 403 individuals were sampled from 44 populations throughout southwest China. SDMs and mismatch distribution analysis indicated that Q. kerrii has undergone northward expansion since the Last Glacial Maximum (LGM). Quantitative analysis revealed that the range expansion of Q. kerrii since the LGM exceeded that of the sympatric mid-elevation species Quercus schottkyana, likely owing to their contrasting distribution elevations and habitat availabilities. The historical climate change since the LGM and the latitude gradient of the region played an important role in shaping the genetic diversity of Q. kerrii. The genetic differentiation index and genetic distance surface of Q. kerrii populations east of the Tanaka line exceeded those to its west. The long-term geographic isolation and environmental heterogeneity between the two sides of the Tanaka line might increase species divergence patterns and local adaptation. This study provides new insights into the historical dynamics of subtropical EBLFs and the changing biota of southwest China.
Introduction
Ancient geological events (e.g. the uplift of mountains and volcanic eruptions) and climate change (e.g. glacier/inter-glacier cycles) shape the divergence of species at the intraspecific and interspecific levels (Favre et al. 2015; Hewitt 2004). Meanwhile, long-term environmental heterogeneity along altitudinal or latitudinal gradients also contributes to genetic differentiation among species (Gugger et al. 2013). The evolutionary processes accompanying this heterogeneity can be reconstructed based on contemporary patterns of genetic diversity and genetic differentiation of organisms (Qiu et al. 2011). For example, phylogeographic studies have indicated that many species in northern Europe migrated from three southern peninsulas—the Iberian Peninsula, the Italian Peninsula and the Balkan Peninsula—since the last glacial maximum (LGM; Comes and Kadereit 1998). Understanding the effects of orogeny, climatic oscillations and environmental heterogeneity on the spatial genetic patterns of species, especially for the dominant species of vegetation or endangered species, is informative not only for disentangling the complex evolutionary histories of species, but also for predicting the responses of organisms to future climate change.
Southwest China is a global biodiversity hotspot owing to its complex topographic and drainage systems (Yang et al. 2004). It is rich in vascular plant species and harbours remarkably high rates of endemic and relict species (López-Pujol et al. 2011). The timing and geological mechanisms underlying the establishment of this biodiversity hotspot in southwest China has fascinated numerous botanists and evolutionary biologists for some time (Meng et al. 2017; Zhang et al. 2010; Zhao and Gong 2015). A growing number of studies have indicated that topographic and drainage diversity in this region has contributed greatly to the pattern of genetic diversity in tropical–subtropical plants (Yue et al. 2012; Zhang et al. 2010). However, these studies have mainly focused on shrubs (Liu et al. 2009; Yue et al. 2012), temperate deciduous trees (Tian et al. 2015; Zhao and Gong 2015) and conifers (Ma et al. 2006; Wang et al. 2013). Upper canopy trees play a crucial role in the ecological functions and biodiversity of evergreen-broadleaved forests (EBLFs; Fang et al. 2014). However, the dynamics and genetic structures of these key tree species in tropical–subtropical Asia have received scarce attention (Jiang et al. 2016).
Subtropical EBLFs constitute the primary native vegetation types of southwest China and thus play important ecological and economic roles in the area (Harrison et al. 2001). Strong macrofossil and palynological evidence has indicated that EBLFs were widely established in southern China no later than the Middle Miocene (Jacques et al. 2011; Sun et al. 2011). Molecular data and species distribution models (SDMs) have indicated that the EBLFs were stable and widespread in East Asia since the LGM (Jiang et al. 2016; Xu et al. 2015), although biome reconstructions based on palynological data have indicated that this vegetation type retreated south of 24°N during the LGM (Harrison et al. 2001). Southwest China contains an extensive transitional zone between two floristic regions, the Sino-Japanese and Sino-Himalayan floristic sub-regions. The boundary between these two sub-regions has been referred to as the ‘Tanaka–Kaiyong line’ (Li and Li 1992; Tanaka 1954), which is comprised of the ‘Tanaka line of Citrus Distribution’ and the ‘Kaiyong line of Orchid Distribution’ (Li and Li 1997). The Tanaka line is also the distribution boundary of a series of closely related plant species and genera pairs in subtropical Asia (Li and Li 1997; Tanaka 1954). However, the biogeographical significance of this line is still poorly understood. Based on comparisons of the regional flora, the substitution distribution pattern between closely related species and genera on the two sides of the Tanaka line have been hypothesized to be an outcome of long-term geographical isolation and environmental heterogeneity of the biota (Li and Li 1997). The plant species found east of the Tanaka line have been considered to be relic species, while those to its west are considered to be more recently derived (Sun 2001). Recent phylogeographic studies in southwest China have revealed prominent genetic diversification patterns across the east and west sides of the Tanaka line as well (Tian et al. 2015; Zhang et al. 2006; Zhao and Gong 2015). These studies have mainly attributed this striking pattern to the geographical barriers and isolation associated with the rapid uplifting of the Yunnan–Guizhou Plateau and the intensification of Asian monsoons since the late Miocene along the Tanaka line and throughout adjacent regions (Fan et al. 2013; Tian et al. 2015; Zhao and Gong 2015). The contribution of long-term environmental heterogeneity between the two sides of the Tanaka line to local differentiation and adaptation has been largely overlooked by current phylogeographic studies.
Quercus kerrii, which belongs to the Group Cyclobalanopsis (Fagaceae), is a widespread species in southwest China and north Indo-China. Its distribution spans across the Tanaka line. This region is also known as the species diversity centre for the Eurasian oaks, especially for the Group Cyclobalanopsis (Luo and Zhou 2000). Quercus kerrii is a keystone species in semi-dry to humid EBLFs at low elevations (50–1300 m asl).
Frequent introgression between closely related species of oaks (Eaton et al. 2015) can result in biased estimates of intraspecific genetic structure. Quercus kerrii and Q. austrocochinchinensis are a pair of sister species as inferred by molecular phylogenetic reconstruction using ITS sequences (Deng et al. 2013) and RAD-seq (Hipp et al. 2015). Quercus austrocochinchinensis is an endangered species consisting of only four small populations. Previous investigations into their morphologies and molecular markers have demonstrated that ongoing hybridization and introgression occurs between the two species (An et al. 2017). However, the observed introgression of SSR alleles was asymmetric, occurring only from Q. kerrii to Q. austrocochinchinensis (An et al. 2017). When hybridization occurs between a dominant species and a rare species, genetic assimilation of the rare species is more likely to occur (Levin et al. 1996). Therefore, introgression between Q. kerrii and Q. austrocochinchinensis has contributed little to the genetic diversity and differentiation of Q. kerrii. Furthermore, Q. kerrii is a keystone species in local low-elevation EBLFs, and it plays a critical role in biological conservation and regional carbon storage and cycling. Quercus kerrii provides a unique opportunity to investigate the roles of environmental factors in genetic diversification of species in tropical-subtropical China.
In this study, we examined population samples of Q. kerrii from southwest China to (1) reveal spatial genetic patterns in Q. kerrii; (2) investigate the historical dynamics of Q. kerrii populations in southwest China; (3) explore the influence of environmental factors on the genetic diversity of Q. kerrii and genetic divergence between Q. kerrii populations. This study provides new insight into historical vegetation dynamics and the flora evolution of southwest China EBLFs.
Materials and methods
Potential distribution and environment factors analysis
Maximum entropy models performed by MaxEnt v3.3.3k (Phillips et al. 2006) were used to compare geographic distributions of Q. kerrii in the present and LGM periods. Asian subtropical–tropical lowland ecosystems are severely impacted by human disturbances (e.g. the expansion of crop farming lands). As a result, large numbers of Q. kerrii populations previously recorded in herbaria no longer exist, and most herbarium records lack specific GPS coordinates. Therefore, when recorded specimen localities corresponded to villages, locations have been converted to imprecise GPS coordinates. Additionally, Asian oak specimens are commonly misidentified in the main herbaria. We found quite a few Asian oak specimens were misidentified as Q. kerrii (e.g. Q. franchetii, Q. aliena, Q. griffithii and even species of Lithocarpus and Castanopsis). In order to ensure the accuracy of current species localities, we only used the 44 GPS records collected from this study to estimate the potential species distributions. Nineteen bioclimatic variables characterizing recent (1950–2000) temperature and precipitation patterns and LGM (CCSM) periods at a 2.5-arc-min resolution were downloaded from WorldClim (available online at http://www.worldclim.org/). Multicollinearity among variables was examined using a Pearson correlation matrix estimated in R v3.3.3 (https://www.r-project.org/). We reduced the subsets of variables with high correlations (r > 0.8) to single variables.
According to the geographic distribution of Q. kerrii in China, we divided the 44 populations of Q. kerrii into four groups, including (1) 20 populations from the Lancang River and adjacent regions (group LCJ); (2) 10 populations located at the Red River drainage and adjacent regions (group HH); (3) 11 populations from the Nan-pan-jiang River basin (group NPJ) and from the east side of the Tanaka line and (4) 3 populations from Hainan Island in the South China Sea (group HN). The detailed distribution and collection information are summarized in Table 1 and Fig. 1b. As groups HH and LCJ are only separated by a short geographic distance and are located on the west side of the Tanaka line, we refer to the two groups collectively as HH–LCJ. We also estimated the potential distribution range of Q. kerrii at the present period for HH–LCJ and NPJ subpopulations, respectively. For all sites and HH–LCJ sites, respectively, we used 75% of the sites for training the model and 25% for testing. The mean value of ten replicate results was used for all estimations. The Jackknife procedure (Pearson et al. 2007) was applied to model the NPJ subpopulations because the sites in this group numbered <25. By removing sites one-by-one from the number of total sites n, the model was built using the remaining n−1 localities. Therefore, n time models were built for a species with n sites. The mean values of n time models were used as the potential distribution of the species. Model performance was evaluated by the area under the curve (AUC) value. Species distribution maps of Q. kerrii in the present and LGM periods were visualized in ArcGIS 10.2 (Environmental Systems Research Institute, Inc., ESRI; Redlands, CA, USA).
Table 1.
Sampling information and genetic diversity of Quercus kerrii populations
| cpDNA | SSR | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Location | Long/lat | N | Haplotypes | h | π×103 | N | A r | He | C A | C B |
| Group LCJ | |||||||||||
| 1 | Longling, YN | 99.01/24.31 | 10 | H1(10) | 0 | 0 | 10 | 2.88 | 0.61 | 0.76 | 0.24 |
| 2 | Zhenkang, YN | 98.89/23.87 | 8 | H2(8) | 0 | 0 | 8 | 2.80 | 0.61 | 0.77 | 0.23 |
| 3 | Cangyuan, YN | 98.94/23.44 | 3 | H2(3) | 0 | 0 | 3 | — | — | 0.87 | 0.13 |
| 4 | Gengma, YN | 99.28/23.66 | 10 | H3(10) | 0 | 0 | 10 | 2.69 | 0.58 | 0.58 | 0.42 |
| 5 | Yongde, YN | 99.48/23.85 | 9 | H3(9) | 0 | 0 | 9 | 2.73 | 0.58 | 0.85 | 0.15 |
| 6 | Lincang, YN | 99.81/24.11 | 10 | H3(10) | 0 | 0 | 10 | 2.74 | 0.59 | 0.87 | 0.13 |
| 7 | Lincang, YN | 100.07/24.10 | 10 | H3(10) | 0 | 0 | 9 | 2.58 | 0.56 | 0.69 | 0.31 |
| 8 | Cangyuan, YN | 99.34/23.36 | 10 | H3(10) | 0 | 0 | 10 | 2.89 | 0.62 | 0.81 | 0.19 |
| 9 | Lincang, YN | 100.26/23.82 | 10 | H2(10) | 0 | 0 | 10 | 2.76 | 0.58 | 0.71 | 0.29 |
| 10 | Lincang, YN | 100.32/23.66 | 10 | H2(5),H3(5) | 0.56 | 0.53 | 10 | 2.90 | 0.62 | 0.85 | 0.15 |
| 11 | Lincang, YN | 100.16/23.53 | 10 | H2(10) | 0 | 0 | 10 | 2.79 | 0.60 | 0.75 | 0.25 |
| 12 | Shuangjiang, YN | 100.03/23.34 | 10 | H2(10) | 0 | 0 | 10 | 2.79 | 0.60 | 0.78 | 0.22 |
| 13 | Ximeng, YN | 99.61/22.81 | 9 | H3(9) | 0 | 0 | 10 | 2.57 | 0.54 | 0.52 | 0.48 |
| 14 | Lancang, YN | 100.14/22.94 | 10 | H2(9),H4(1) | 0.20 | 0.57 | 10 | 2.87 | 0.62 | 0.77 | 0.23 |
| 15 | Pu'er, YN | 100.53/23.00 | 10 | H2(10) | 0 | 0 | 10 | 2.69 | 0.56 | 0.86 | 0.14 |
| 16 | Ximeng, YN | 99.59/22.35 | 10 | H1(9),H5(1) | 0.20 | 0.19 | 10 | 2.88 | 0.62 | 0.72 | 0.28 |
| 17 | Lancang, YN | 100.18/22.66 | 10 | H1(10) | 0 | 0 | 10 | 2.80 | 0.61 | 0.87 | 0.13 |
| 18 | Lancang, YN | 100.23/22.60 | 10 | H1(8),H2(1),H4(1) | 0.38 | 0.90 | 10 | 2.76 | 0.60 | 0.75 | 0.25 |
| 19 | Simao, YN | 100.79/22.68 | 10 | H2(10) | 0 | 0 | 10 | 2.77 | 0.58 | 0.86 | 0.14 |
| 20 | Lincang, YN | 100.32/23.62 | 9 | H2(1),H3(8) | 0.22 | 0.21 | 20 | 2.73 | 0.57 | 0.77 | 0.23 |
| Group HH | |||||||||||
| 21 | Jingdong, YN | 100.83/24.54 | 10 | H2(10) | 0 | 0 | 9 | 2.89 | 0.62 | 0.63 | 0.37 |
| 22 | Pu'er, YN | 100.90/24.45 | 5 | H2(5) | 0 | 0 | 5 | 2.82 | 0.64 | 0.69 | 0.31 |
| 23 | Pu'er, YN | 101.25/24.71 | 10 | H6(10) | 0 | 0 | 10 | 2.76 | 0.56 | 0.68 | 0.32 |
| 24 | Jingdong, YN | 101.16/24.23 | 9 | H1(9) | 0 | 0 | 10 | 2.76 | 0.59 | 0.84 | 0.16 |
| 25 | Zhenhuan, YN | 101.21/24.06 | 10 | H1(6),H4(4) | 0.53 | 2.02 | 10 | 2.81 | 0.60 | 0.72 | 0.28 |
| 26 | Xinping, YN | 101.85/24.03 | 10 | H2(10) | 0 | 0 | 10 | 2.79 | 0.59 | 0.28 | 0.72 |
| 27 | Eshan, YN | 102.15/24.18 | 9 | H2(9) | 0 | 0 | 10 | 2.96 | 0.63 | 0.40 | 0.60 |
| 28 | Jiangcheng, YN | 102.02/23.02 | 10 | H2(10) | 0 | 0 | 10 | 2.65 | 0.54 | 0.86 | 0.14 |
| 29 | Jiangcheng, YN | 101.87/22.65 | 10 | H4(1),H7(9) | 0.20 | 1.32 | 10 | 2.73 | 0.59 | 0.78 | 0.22 |
| 30 | Yuanjiang, YN | 102.85/23.03 | 2 | H4(2) | 0 | 0 | 1 | — | — | 0.95 | 0.05 |
| Group NPJ | |||||||||||
| 31 | Xilin, GX | 104.79/24.48 | 9 | H8(9) | 0 | 0 | 9 | 2.92 | 0.63 | 0.61 | 0.39 |
| 32 | Longlin, GX | 104.88/24.67 | 7 | H9(7) | 0 | 0 | 9 | 2.85 | 0.62 | 0.33 | 0.67 |
| 33 | Xilin, GX | 105.04/24.49 | 7 | H10(7) | 0 | 0 | 7 | 2.69 | 0.56 | 0.45 | 0.55 |
| 34 | Longlin, GX | 105.65/24.73 | 8 | H8(8) | 0 | 0 | 13 | 3.2 | 0.68 | 0.39 | 0.61 |
| 35 | Heng, GZ | 105.92/25.02 | 5 | H2(5) | 0 | 0 | 8 | 3.03 | 0.64 | 0.32 | 0.68 |
| 36 | Tianlin, GX | 105.81/24.65 | 2 | H11(2) | 0 | 0 | 1 | — | — | 0.12 | 0.88 |
| 37 | Luotuo, GZ | 106.64/25.28 | 6 | H2(6) | 0 | 0 | 12 | 3.17 | 0.67 | 0.38 | 0.62 |
| 38 | Tianlin, GX | 106.22/24.31 | 7 | H2(7) | 0 | 0 | 5 | 3.14 | 0.69 | 0.22 | 0.78 |
| 39 | Tianlin, GX | 106.37/24.16 | 6 | H12(6) | 0 | 0 | 5 | 3.06 | 0.64 | 0.27 | 0.73 |
| 40 | Funing, YN | 105.84/23.72 | 10 | H2(9),H6(1) | 0.20 | 0.19 | 9 | 2.99 | 0.64 | 0.57 | 0.43 |
| 41 | Chihe, GX | 107.34/24.83 | 8 | H2(8) | 0 | 0 | 11 | 2.94 | 0.63 | 0.03 | 0.97 |
| Group HN | |||||||||||
| 42 | Nanchahe, HN | 109.20/19.14 | 2 | H2(2) | 0 | 0 | — | — | — | — | — |
| 43 | Sanya, HN | 109.09/18.47 | 10 | H2(10) | 0 | 0 | 10 | 2.52 | 0.50 | 0.06 | 0.94 |
| 44 | Jianfengling, HN | 108.88/18.72 | 10 | H2(10) | 0 | 0 | 10 | 2.27 | 0.46 | 0.03 | 0.97 |
YN Yunnan Province, GX Guangxi Province, GZ Guizhou Province, HN Hainan Province, N number of sampled individuals, h haplotype diversity, π nucleotide diversity, A r standardized allelic richness, He expected heterozygosity, C A probability of population belong to group A, C B, probability of population belong to group B
Fig. 1.

a The distribution area of this studies. b Geographic distribution and c network of 12 cpDNA haplotypes of Quercus kerrii. b The pie charts reflect the occurrence frequency of each haplotype in each population. Haplotype colours correspond to those shown in the lower-left panel. The two dotted lines represent the Tanaka line and species genetic barriers, respectively. c The circle sizes are in proportion to the number of individuals of each haplotype. Haplotypes are coloured yellow, red, green and violet to represent the populations belonging to groups LCJ, HH, NPJ and HN, respectively
Principal component analysis (PCA) for the climate variables of sample sites was performed using R v3.3.3 (https://www.r-project.org/). The estimated principal components summarized the overall pattern of variation in 19 climate variables among populations during the present and LGM periods. To compare the environmental changes for populations since the LGM (E change), the absolute values of standardized PC1 scores from the present period (E pre) minus scores from the LGM period (E LGM) were calculated. After standardization, values nearer to 0 indicate that the environment has been relatively stable since the LGM, while values near to 1 indicate that the environmental changes are significant. The present and LGM period environmental conditions as well as environmental changes since the LGM at the sampling sites were visualized using ArcGis 10.2 (ESRI). The t test was performed in R v3.3.3 (http://www.r-project.org/) to compare the differences in climatic factors among groups.
Population sampling and genotyping
A total of 403 individuals were sampled from 44 natural populations of Q. kerrii in China from October 2009 to August 2014 (Table 1). For each population, individuals that were at least 10 m away from each other were sampled. Fresh leaf samples were cleaned and stored in silica gel immediately until DNA extraction was conducted. Voucher specimens for each individual were collected and are stored at the Herbarium of the Shanghai Chenshan Botanical Garden (CSH).
Total genomic DNA was extracted using the modified CTAB method from silica-dried leaf tissue (Doyle 1987). After screening universal primer sets published previously, two chloroplast DNA (cpDNA) intergenic spacers, psbA-trnH (Shaw et al. 2005) and ycf1 (Dong et al. 2015), and 10 nuclear microsatellite loci (nSSR; Table S1) with the highest level of polymorphism were selected to examine the genetic diversity and population structure of these 44 Q. kerrii populations. The PCR reaction protocols for cpDNA and nSSR loci followed the procedures described by Jiang et al. (2016). Successfully amplified PCR products of cpDNA and nSSR markers were sequenced or genotyped by Shanghai Majorbio Bio-pharm Technology Co., Ltd (Shanghai, China). The cpDNA sequence chromatograms were checked and assembled using Sequencher version 4.1.4 (Gene Codes Corporation, Ann Arbor, MI, USA). Then the sequences were aligned using CLUSTAL_W implemented in MEGA 6.0 (Tamura et al. 2013) and subsequently adjusted manually. The allele sizes of each microsatellite marker were genotyped using GENEMARKER version 2.2.0 (Soft Genetics LLC, State Collage, PA, USA). MicroChecker version 2.2.3 (Van Oosterhout et al. 2004) was used to check for genotyping errors and null alleles.
cpDNA analysis
The haplotypes of two combined cpDNA sequences were extracted using DnaSP 5 (Librado and Rozas 2009) and deposited into GenBank (accession number given in the ‘Data Archiving’ section). Spatial distribution maps of haplotypes were drawn using ArcGis 10.2 (ESRI). Haplotype diversity and nucleotide diversity were calculated using ARLEQUIN v3.5 (Excoffier and Lischer 2010). Total gene diversity (H T), within-population gene diversity (H S), G ST and N ST (based solely on haplotype frequencies and consideration of similarities among different haplotypes, respectively) for all populations and each group were calculated using HAPLONST (Pons and Petit 1996). A G ST value significantly lower than N ST according to a U-statistic indicates that there is phylogeographic structure within the populations. A median-joining network (Bandelt et al. 1999) of cpDNA was constructed using the programme Network 4.6.1.0 (available at http://www.fluxus-engineering.com/sharenet_rn.htm).
Tajima’s D statistic (Tajima 1989) and Fu’s F S test were calculated (Fu 1997) in ARLEQUIN v3.5 (Excoffier and Lischer 2010) to test the population expansion hypothesis. Significant negative values of these statistics is a signature of recent population expansion. Mismatch distributions were computed by using ARLEQUIN v3.5 (Excoffier and Lischer 2010) for the cpDNA dataset, and significance was tested by comparison of the empirical estimates to the distribution from 1000 permutations of the data. A multimodal distribution indicates population equilibrium, whereas a unimodal distribution indicates a population expansion (Rogers and Harpending 1992). Sum-of-squared deviations, Harpending’s raggedness index and their p-values were computed in order to assess deviations from the sudden expansion model.
nSSR analysis
We calculated the H e (expected heterozygosity) and A r (allelic richness) values for populations with five or more individuals. The pairwise F ST, A r, pA r (private allelic richness), H e (expected heterozygosity) and H o (observed heterozygosity) values were also calculated at the group level. The A r and pA r values were calculated by rarifying the allele data to be consistent with 6 (at population level) or 38 (at group level) gene copies using HP-RARE (Kalinowski 2005). Pairwise F ST, H o and H e values were calculated using ARLEQUIN v3.5 (Excoffier and Lischer 2010).
Both Bayesian clustering and principal coordinate analyses (PCoA) were used to estimate the genetic structure of Q. kerrii. Bayesian clustering was performed with InStruct (Gao et al. 2007) using the admixture model. The number of clusters (K) was set to vary from 1 to 10. Each cluster was repeated 10 times. We performed a run length of 200,000 MCMC (Markov chain Monte Carlo) iterations with a burn-in of 50% of the chain length for each cluster (K). The best fit for the number of clusters was determined using the ΔK method (Evanno et al. 2005). Ten runs of InStruct with the optimum K value were aligned using CLUMPP (Jakobsson and Rosenberg 2007) based on a greedy algorithm. PCoA can be used to visualize similarities or dissimilarities of microsatellite data based on a genetic distance matrix without any population genetic model assumptions. We performed the PCoA using GenAlEx 6.5 (Peakall and Smouse 2006) based on the genetic distances among populations and among groups. The first three (Gp1, Gp2 and Gp3) and two (Gg1 and Gg2) principal coordinates at population level and group level were visualized respectively.
Landscape pattern of Q. kerrii
A general linear model (GLM) analysis performed in R v3.3.3 (http://www.r-project.org/) was used to estimate the correlation of genetic diversity (A r and H e) with environment and geographic factors at the population level. Five variables—present (E pre) and LGM (E LGM) period environment, environmental change since LGM (E change), latitude and longitude—were used as explanatory covariates. The initial model included all explanatory covariates, and the most suitable models were selected based on a backward elimination procedure. The genetic significant difference level among the Q. kerrii groups were tested using the Gp1 and C A (Cluster A from the Bayesian clustering) values.
Alleles in Space (Miller 2005) was used to explore the possible genetic discontinuities and barriers among populations according to cpDNA and nSSR polymorphism. To avoid the effects of genetic distances among populations caused by geographical distances, residual genetic distances were used to construct the possible genetic discontinuities and barriers among populations. The residual genetic distances were derived from the linear regression of all pairwise genetic distances across geographical distances (Manni et al. 2004). First, a connectivity network of sample locations was generated using the Delaunay triangulation rule (Brouns et al. 2003). Then, residual genetic distances between observations connected in the network were placed at the midpoints of each connection to form a three-dimensional surface plot, where x and y axes were equivalent to the appropriate population’s geographical coordinates and the z axis represented the residual genetic distance. The possible genetic barriers were estimated using Monmonier’s maximum difference algorithm (Monmonier 1973). First, the initial barrier segment was identified based on the greatest residual genetic distance between any two locations joined in the Delaunay triangulation connectivity network. Second, the initial barrier was identified with respect to one direction until an external edge of the connectivity network was encountered. Finally, the second step was repeated in the opposite direction.
Results
Past and present distribution modelling
After excluding the highly autocorrelated climate variables, a total of 10 variables were used for the modelling analysis. The AUC values for the climate modelling exceeded 0.99, indicating a good performance of the models. Among all sites, habitat suitability values of populations were 0.14–0.78 and 0.12–0.72 for the present and LGM periods, respectively. The range of Q. kerrii populations in the present period was larger than that of the LGM period (Fig. 4). To quantitatively analyse the extent of species expansion, we calculated the ratio of the LGM and present potential distribution areas based on the maximum sensitivity plus specificity value as the species presence/absence threshold. The ratio of the present range to the LGM range was 1.27. The environmental factor of total precipitation in the warmest quarter (bio_18) explained more than half of all variation (54.85%, SD = 1.23) in the distribution of Q. kerrii, and the remaining nine environmental factors contributed about 45.15% of the variation. The modelling result indicated that there are non-overlapping distributions of Q. kerrii in the present period between the HH–LCJ and NPJ regions (Fig. 5).
Fig. 4.

Potential species distributions in the a Last Glacial Maximum and b present periods. The colours of the rectangles in the lower left refers to different ranges of habitat suitability. Dots represent the points of sampled populations in our study, and dot colours represent the different ranges of PCA1 scores estimated from 19 environmental variables
Fig. 5.

Potential species distribution areas of Q. kerrii on the west side of the Tanaka line (represented in yellow) and east of the Tanaka line (represented in green). The maximum training sensitivity plus specificity threshold was used to determine the species presence threshold. The sizes of red dots represent the extent of climate change since the Last Glacial Maximum
PCA 1 score values of environment variables were between −2.3 and 2.3 for populations in the LGM period (E LGM) and between −2.0 and 1.4 for populations in the present period (E pre; Fig. 4). Groups LCJ and HH had similar habitats during the present (E pre) and LGM (E LGM) periods. The environmental changes since the LGM (E change) between groups LCJ and HH showed no significant differences (Table 5). On the contrary, these three environmental factors significantly differed between the groups located east (i.e. group NPJ) and west (i.e. groups LCJ, HH and HH–LCJ; Table 5) of the Tanaka line.
Table 5.
The P-values of t test among groups. Below diagonal were genetic structure (in turn, Gp1 and CA) and above were environment factors (in turn, present and LGM period environment and environmental change since LGM)
| LCJ | HH | NPJ | HN | LCJ–HH | |
|---|---|---|---|---|---|
| LCJ | — | 0.60/0.26/0.35 | 0.00/0.00/0.00 | 0.12/0.07/0.87 | — |
| HH | 0.60/0.23 | — | 0.01/0.02/0.00 | 0.16/0.03/0.62 | — |
| NPJ | 0.00/0.00 | 0.00/0.00 | — | 0.50/0.01/0.00 | 0.00/0.00/0.00 |
| HN | 0.00/0.00 | 0.00/0.00 | 0.00/0.00 | — | — |
| LCJ–HH | — | — | 0.00/0.00 | — | — |
cpDNA results
The two cpDNA regions were successfully sequenced from 370 Q. kerrii individuals from 44 populations. After alignment, the total 1058 bp of cpDNA sequences contained 17 polymorphisms, including 2 indels and 15 nucleotide substitutions. The indels were treated as single-base mutations in the following analyses. In total, 12 haplotypes were identified (Fig. 1b), of which, H2 was the most common haplotype shared by all groups. H1 and H3 were less common haplotypes. H1 existed both in groups LCJ and HH (i.e. HH–LCJ). H3 existed only in group LCJ. H4 and H6 were rare haplotypes, but shared by various groups. H8 was shared by two populations within group NPJ. Four unique haplotypes, H9, H10, H11 and H12, were each found in all individuals in four populations from group NPJ (32, 33, 36 and 39, respectively). The haplotype network indicates that the haplotypes were arranged in a star-like structure (Fig. 1c). Haplotype diversity and nucleotide diversity were zero in more than 80% of the populations (Table 1). The highest haplotype diversity estimates were observed in populations 10 (h = 0.56) and 25 (h = 0.53), and the highest nucleotide diversity estimates were found in populations 25 (π = 2.02 × 10−3) and 29 (π = 0.32 × 10−3; Table 1). Groups NPJ and HN exhibited the highest and lowest degrees of genetic differentiation, respectively (Table 2). The degree of differentiation within groups LCJ and HH were similar. Within each group and among all the populations of Q. kerrii, no phylogeographic structure was detected.
Table 2.
Measures of genetic diversity of the Quercus kerrii at group level based on cpDNA and nSSR data
| Population ID | cpDNA | SSR | ||||||
|---|---|---|---|---|---|---|---|---|
| H T (se) | H S (se) | G ST(se) | N ST (se) | H O | H E | A r | pA r | |
| LCJ | 0.67(0.04) | 0.08(0.04) | 0.89(0.05) | 0.86(0.06) | 0.52 | 0.61 | 5.43 | 0.36 |
| HH | 0.69(0.14) | 0.06(0.06) | 0.92(0.08) | 0.83(0.16) | 0.54 | 0.61 | 5.55 | 0.38 |
| NPJ | 0.81(0.10) | 0.02(0.02) | 0.98(0.02) | 0.98(0.02) | 0.59 | 0.67 | 6.55 | 1.05 |
| HN | N/A | N/A | N/A | N/A | 0.46 | 0.51 | 4.65 | 0.68 |
| All | 0.71(0.06) | 0.05(0.02) | 0.93(0.03) | 0.92(0.04) | — | — | — | — |
Mismatch distribution analysis for cpDNA was performed to assess the demographic dynamics. The observed unimodal distribution for all samples suggested a recent history of population expansion (Fig. S1). The two test statistics for selective neutrality were negative but non-significantly (Tajima’s D = −1.142, p = 0.131; Fu’s F S = −2.088, p = 0.241), which weakly suggests a sudden population expansion hypothesis.
nSSR results
A total of 105 alleles were obtained based on 10 nSSR loci, with the allele number varying from 5 to 22 per locus (Table S1). The genetic diversity index A r and H e values of Q. kerrii at the population level were 2.27–3.20 and 0.46–0.69, respectively (Table 1). The genetic diversity indices (H o, H e, A r and pA r) at the group level indicated that group NPJ had the highest genetic diversity, and groups LCJ and HH had similarly low genetic diversity (Table 2). The values of pairwise F ST among groups were 0.004–0.185 (Table 3). The genetic differentiation between group HN and the other groups were higher. Groups LCJ and HH showed the lowest levels of genetic differentiation.
Table 3.
Pairwise F ST-values between the groups of Quercus kerrii. Below diagonal were F ST-values and above were p-values
| LCJ | HH | NPJ | HN | |
|---|---|---|---|---|
| LCJ | — | 0.018 | 0.000 | 0.000 |
| HH | 0.004 | — | 0.000 | 0.000 |
| NPJ | 0.030 | 0.026 | — | 0.000 |
| HN | 0.185 | 0.176 | 0.116 | — |
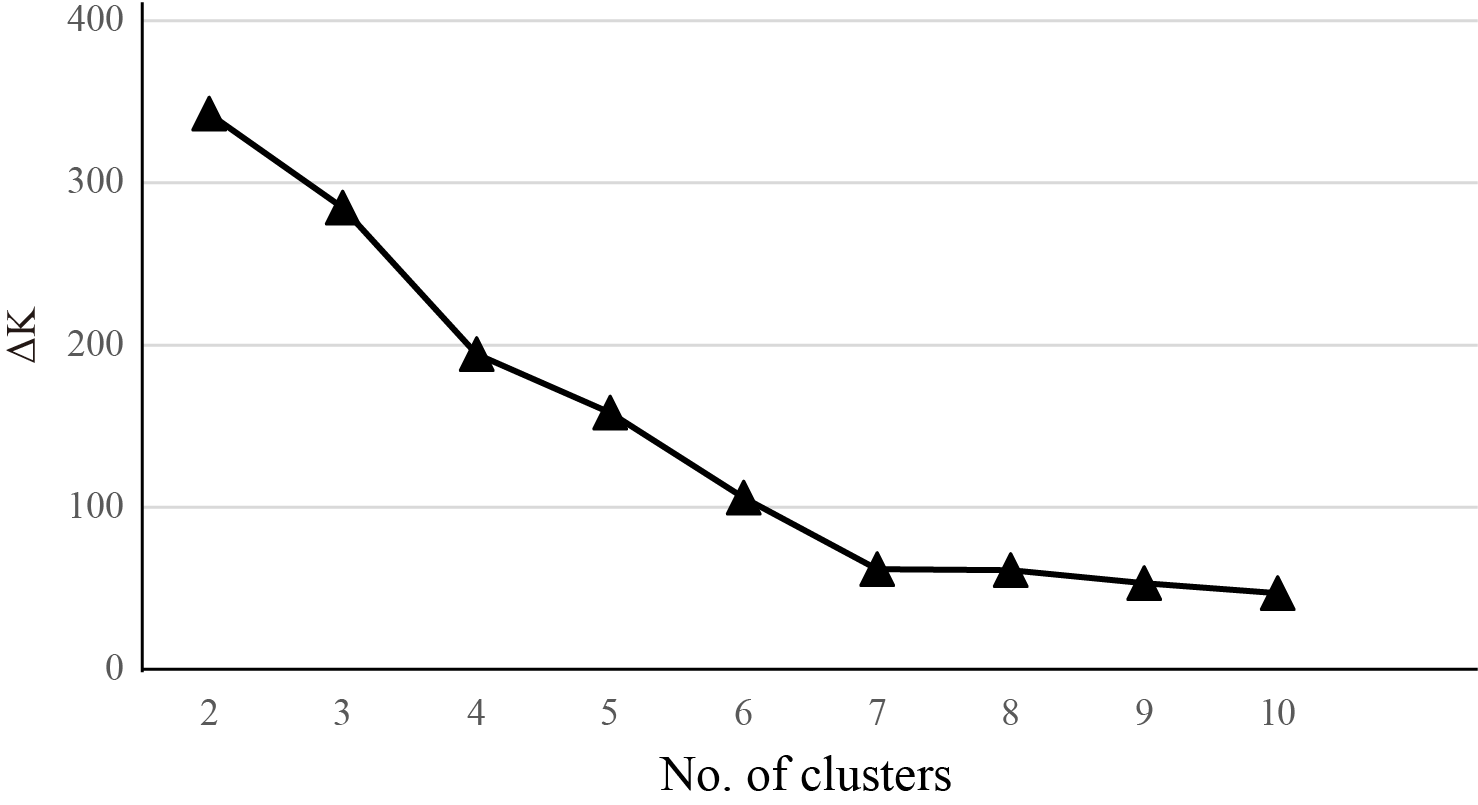
In the Bayesian clustering analysis, the highest ΔK was obtained when K = 2 (Fig. S2). Genetic admixture was widespread in Q. kerrii populations (Table 1). PCoA for the nSSR data at population and group levels revealed consistent patterns (Fig. 2). Group HN was clearly distinct from the other groups. There was no genetic differentiation between groups LCJ and HH. However group NPJ was separated from HH–LCJ.
Fig. 2.

Plots of the first three and two coordinates of the principal coordinates analysis (PCoA) at population level a and group level b based on the nSSR pairwise differentiation matrix for Quercus kerrii
GLM analysis indicated that the model that best explains the genetic diversity (A r and H e) of Q. kerrii contained the variables E change, latitude and E LGM, but only E change and latitude were significant correlated with A r and H e (p < 0.01; Table 4). The strength of the influence of E change on the Q. kerrii genetic diversity was greater than that of latitude. Except for differentiation between groups LCJ and HH, the genetic structure among the other groups exhibited significant differences (Table 5).
Table 4.
Values from best model for regression analysis between the genetic diversity of Quercus kerrii nSSR with environment and geography factors based on the backward eliminating procedure
| A r | H e | |||||||
|---|---|---|---|---|---|---|---|---|
| Est | SEa | t | p | Est | SEa | t | p | |
| (Intercept) | 0.50 | 0.47 | 1.06 | 0.29 | −0.26 | 0.17 | −1.48 | 0.15 |
| E Pre | −0.09 | 0.05 | −2.01 | 0.05 | — | — | — | — |
| E LGM | 0.33 | 0.19 | 1.76 | 0.09 | 0.23 | 0.09 | 2.60 | 0.01 |
| E change | 0.42 | 0.11 | 3.99 | 0.00 | 0.20 | 0.06 | 3.35 | 0.00 |
| Longitude | — | — | — | — | — | — | — | — |
| Latitude | 0.09 | 0.02 | 5.08 | 0.00 | 0.03 | 0.01 | 5.17 | 0.00 |
Est estimate, — eliminating variables. Significant p-values are in bold (p < 0.01), and marginally significant values (0.01 < p < 0.10) are italicized
aStandard error
The genetic landscape shape analysis also produced somewhat similar patterns of population differentiation among cpDNA and nSSR markers (Fig. 3). The surface plots of cpDNA and nSSR genetic distances showed a drastic drop in group HN, which was consistent with the lower genetic diversity indices observed in this group. The nSSR and cpDNA genetic distances increased from groups HH–LCJ to NPJ. Meanwhile, a genetic barrier for cpDNA was detected between group HH–LCJ and the other two groups based on Monmonier’s maximum difference algorithm (Fig. 1b).
Fig. 3.
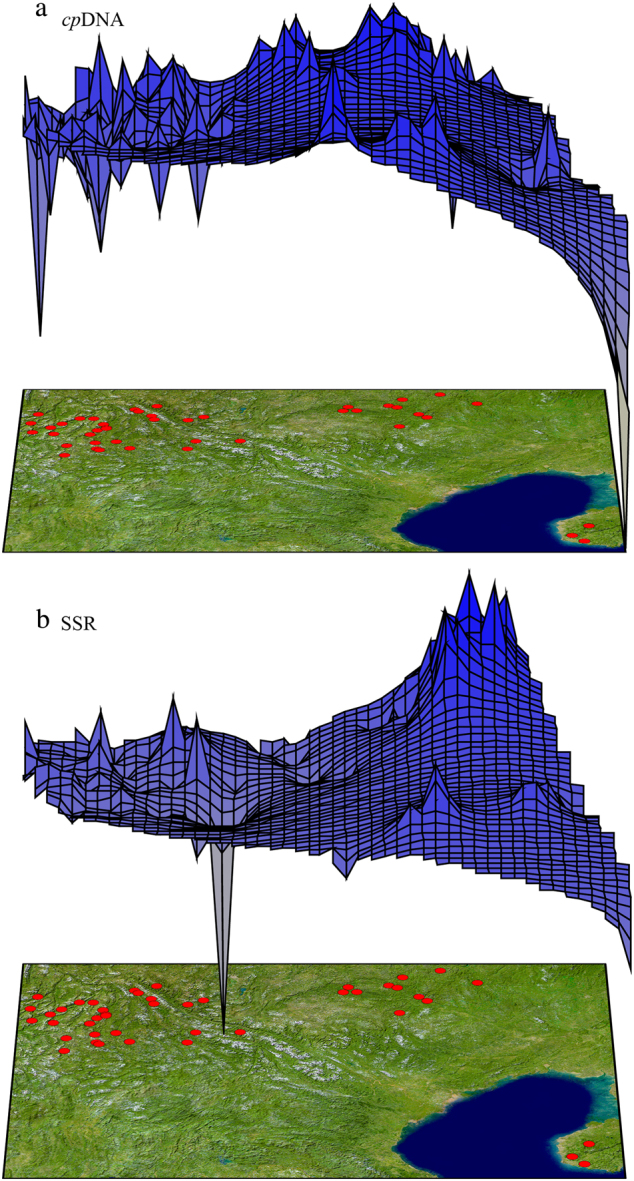
Genetic landscape shape analysis for Quercus kerrii populations based on a cpDNA variation and on b nSSR variation at ten microsatellite loci. The x and y axes show the geographical locations within a Delaunay triangulation network constructed among the sampled populations. Surface heights reflect genetic distances among populations
Discussion
Demographics and distribution dynamics
East Asia was not directly impacted by extensive and unified ice-sheets during the Quaternary period (Liu 1988), but the climatic fluctuations of this region, especially since the LGM period, played an important role in shaping present species distributions and spatial genetic patterns (Qiu et al. 2011). We observed the potential distribution area of Q. kerrii in the present period is 1.27 times greater than that of the LGM period, suggesting that the distribution range of Q. kerrii underwent northward expansion since the LGM (Fig. 4). The neutral test for the cpDNA sequences suggested that the species did not undergo significant demographic expansion, but the star-shaped haplotype network (Fig. 1b) as well as the unimodal distribution revealed by the mismatch distribution analysis (Fig. S1) implied a demographic expansion occurred in Q. kerrii. The consistent SDMs and molecular results indicated that the range expansion of Q. kerrii could have been coupled with demographic expansion. This dynamic pattern fits with the general pattern of organisms responding to climatic change, as species experienced southward retreated and northward colonization during the glacial/interglacial period (Hewitt 2004). However, the fluctuation of the distribution range of Q. kerrii was far less than that indicated by previous reconstructions of the East Asia biome using palynological data. These analyses have indicated that the EBLFs of East Asia obviously retreated to the south region, to a latitude of about 24°N during the LGM (Harrison et al. 2001; Yu et al. 2000). The distributions of other dominant tree species of EBLFs in south China also suggest that subtropical tree species have had relatively stable distributions since the LGM. For example, the phylogeographic studies on Quercus glauca in southeast China (Xu et al. 2015) and Quercus schottkyana (Jiang et al. 2016) in southwest China indicate that their distributions have been stable and widespread since the LGM. Diversified topography and low amplitude fluctuations of climate during the Quaternary period in southwest China have determined that this region has acted as refugia for many plants and have provided relatively stable and suitable habitats.
The distributions of Q. kerrii and Q. schottkyana (another evergreen oak species in the Group Cyclobalanopsis) mostly overlap in southwest China. However, quantitative analysis for the SDM results indicated that Q. kerrii has undergone a larger expansion of potential distribution range since the LGM than Q. schottkyana, with potential distribution expansions of 1.27 and 1.03 times, respectively (Jiang et al. 2016). Except for dispersal distances of seed and pollen gains, the propensity for species to shift in altitudinal range at the regional scale also affects the range dynamics of species in response to climatic fluctuations. Species are expected to track warming and cooling climates by shifting their ranges to respectively higher and lower latitudes or elevations (Chen et al. 2011; Zhu et al. 2012). Owing to the climate diversity along the vertical gradient of mountains, species that are distributed along middle elevations of montane areas can easily migrate up or down elevations as the climate warms or cools, thus tracking suitable habitats. Quercus kerrii and Q. schottkyana have similar biological traits, including the limited dispersal ability of their seeds, wind pollination and substantial longevity. Both are local dominant species of EBLFs in southwest China as well; therefore, the two species might exhibit similar local adaptability, though this is limited by their distinct distribution elevations. Quercus schottkyana is commonly found at elevations of 1500–2500 m asl, the middle mountain region; these species may be able to migrate upward or downward along mountains in response to warming or cooling climate patterns. The main distribution of Q. kerrii in southwest China is restricted to low elevations (50–1300 m asl) in the river gorges and the basins scattered throughout the mountains. Therefore, as the climate cools, its suitable habitats become more limited, with a tendency for the species distribution to retreat to southern low-land refugia but expand northward when climate warms, for example, during the Quaternary glacial/interglacial cycles. The species’ distinct levels of resistance to low temperatures in their habitats may underlie the different demographic dynamics of the two species during the Quaternary glacial cycles.
Genetic isolation and environmental heterogeneity
We determined that the genetic diversity and genetic structure of Q. kerrii populations differed between the NPJ and HH–LCJ groups, which is consistent with the genetic barriers also occurring between the two groups (Fig. 1b). The SDM analysis for HH–LCJ and NPJ regions respectively indicated the existence of unsuitable habitat zones between the two regions as well (Fig. 5). Overall, this genetic differentiation and geographical isolation zones are geographically consistent with the Tanaka line, a straight line running approximately between 98°E, 28°N and 108°E, 19°N (Fig. 1b). Although it is difficult to estimate the precise divergence times between HH–LCJ and NPJ groups based on our data, our results suggested that the isolation between NPJ and HH–LCJ Q. kerrii groups might have coincided with the establishment of the Tanaka line.
The Tanaka line is an important biogeographic boundary describing the biota of the East Asian tropical-subtropical regions. There are a series of prominent species or genus pairs that are separated by the Tanaka line (Li and Li 1992). Previous phylogeographic studies on perennial herbs (Zhang et al. 2006), shrubs (Fan et al. 2013) and deciduous trees (Tian et al. 2015) found obviously divergent intraspecific spatial genetic pattern across the two sides of the Tanaka line. The complexity of the topography and climate of tropical–subtropical Yunnan are closely related to the uplift of the Himalayas and the consequent establishment of modern Asian monsoon systems in the Neogene (Shi et al. 1999). The fossil evidence suggests that the rapid uplift of the Yunnan-Guizhou Plateau occurred at the late Miocene and Pliocene, which was simultaneous to the key establishing stage of the modern Asian monsoon systems (Favre et al. 2015; Jacques et al. 2014). The paleoclimate reconstruction based on fossil assemblages demonstrated that the Southeast Asian and East Asian monsoon co-occurred in the Yunnan during the Late Miocene (Jacques et al. 2011) with a weaker monsoon intensity than is typical today (Li et al. 2015). The modern East Asian monsoons were gradually established and grew stronger between the Pliocene and Quaternary (An et al. 2001). East of the Tanaka line is mainly affected by the East Asian monsoon, while the west is under the strong influence of the Southeast Asian monsoon (i.e. the Indian Ocean monsoon; Li and Li 1992). The evidence from fossil assemblage, palaeogeography and paleoclimate data suggests that the biogeographic significance of the Tanaka line is related to historical tectonic movements and/or long-term environment differentiation, which has acted as a barrier to plant dispersal and gene flow between populations at the east and west sides of the Tanaka line (Li and Li 1997; Zhao and Gong 2015).
A large number of phylogeographic studies have indicated that the formation and establishment of drainage systems (which are also related to the uplift of the Yunnan–Guizhou Plateau) played a key role in disrupting gene flow between populations distributed across valleys or adjacent rivers (Yue et al. 2012). However, our study did not reveal these relationships between contemporary drainage systems and spatial genetic patterns of Q. kerrii. Within groups, the haplotype distributions of Q. kerrii populations on either side of rivers showed no obvious differences (Fig. 1b). The populations comprising the two groups HH and LCJ are separated by the Lan-cang River and Red River, respectively, but each group of populations exhibits similar genetic diversity and differentiation levels (Tables 2, 3 and 5). This phenomenon indicated that regardless of the poor dispersal ability of Q. kerrii acorns, the contemporary drainage patterns in southwest China are not the main factor that has shaped the current genetic patterns of the species.
The genetic pattern present in Q. kerrii suggests that the recent environmental heterogeneity to the east and west of the Tanaka line might play a key role in shaping genetic differentiation patterns within species. High genetic differentiation and the higher genetic distances of populations to the east of the Tanaka line (group NPJ) relative to those west of the Tanaka line (i.e. group HH–LCJ; Fig. 3) were detected. Correspondingly, the climatic factors of the east and west sides of the Tanaka line significantly differ in the present and at the LGM as well (Fig. 4 and Table 5). The east area of the Tanaka line experienced more severe climatic fluctuations since the LGM than the west side did (Fig. 5). Meanwhile, our niche modelling demonstrated there were no overlaps in potential distributions for NPJ and HH–LCJ groups, which further implicated the obvious niche differentiation that might occur between the populations east and west of the Tanaka line (Fig. 5). Environmental heterogeneity across a landscape can create genetic heterogeneity through evolutionary processes, e.g. natural selection, genetic drift and the founder effect (Antonovics 1971; Mitton 2000). The populations from group NPJ with the highest genetic diversity and genetic differentiation indices are located around the peripheral distribution area of Q. kerrii. These populations may therefore undergo more severe and very divergent environmental conditions compared to those in the core distribution area, which may, in turn, enhance divergence driven by local adaptation.
Generally, regions with long-term habitat stability have higher genetic diversity. On the contrary, we detected that environmental change since the LGM is significantly positively correlated with Q. kerrii genetic diversity (Table 4). Both interspecies hybridization and the admixture of individuals from different isolated populations within unstable habitat areas can boost species genetic diversity (Ortego et al. 2014; Ortego et al. 2012). There is ongoing hybridization and introgression between Q. kerrii and Q. austrocochinchinensis; however, the introgression from Q. kerrii to Q. austrocochinchinensis is predominant (An et al. 2017). Moreover, introgression at low to moderate levels only has a limited contribution to increasing genetic diversity in oaks (Ortego et al. 2014). Therefore, the admixture of populations with different genealogical origins in an unstable habitat during climate fluctuation seems a better explanation for the positive correlation observed between environmental change and genetic diversity in Q. kerrii.
Our study revealed some of the factors that influence key EBLF oak species through climatic change. The lowland EBLF species Q. kerrii is more sensitive to climatic change than the mid-elevation montane species Q. schottkyana. The EBLFs at low elevations in Indo-China might be more vulnerable to climatic change than those at mid-elevation montane areas; in turn, more conservation and management attention are needed for species occupying these areas. The regional genetic differentiation and genetic distances of Q. kerrii were uneven. Environmental heterogeneity between the east and west of the Tanaka line plays an important role in shaping genetic diversity and differentiation. Our study supports the hypothesis that long-term isolation and environmental heterogeneity between populations east and west of the Tanaka line drive inter- and intraspecific divergence of plants in the tropical and subtropical regions of Southeast Asia, which supports the biogeographic significance of the Tanaka line. However, the contributions of isolation and local adaptation to the divergence between the populations locates at the east and west of the Tanaka line still requires further investigation using more taxa and high-throughput genotyping of genome-wide markers.
Data archiving
The cpDNA sequences reported in this study were deposited in GenBank under accession numbers MF138014–MF138030. The nuclear microsatellite loci data (nSSR) is available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.0r20b
Electronic supplementary material
{kind=link}
{kind=link}
Acknowledgements
We are grateful to Yan-Shi Xiong and Yi-Gang Song of Chenshan Botanical Garden for their help with sampling. This work was supported by grants from the National Natural Science Foundation of China (31700174 and 31270267), the Shanghai Municipal Administration of Forestation and City Appearances (G152428, G162404, G162405 and G172406), and Southeast Asia Biodiversity Research Institute, Chinese Academy of Sciences (Y4ZK111B01).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Electronic supplementary material
The online version of this article (10.1038/s41437-017-0012-7) contains supplementary material, which is available to authorized users.
References
- An M, Deng M, Zheng SS, Jiang XL, Song YG. Introgression threatens the genetic diversity of Quercus austrocochinchinensis (Fagaceae), an endangered oak: a case inferred by molecular markers. Front Plant Sci. 2017;8(229):299. doi: 10.3389/fpls.2017.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Z, Kutzbach JE, Prell WL, Porter SC. Evolution of Asian monsoons and phased uplift of the Himalayan-Tibetan Plateau since Late Miocene times. Nature. 2001;411(6833):62–66. doi: 10.1038/35075035. [DOI] [PubMed] [Google Scholar]
- Antonovics J. The effects of a heterogeneous environment on the genetics of natural populations: The realization that environments differ has had a profound effect on our views of the origin and role of genetic variability in populations. Am Sci. 1971;59(5):593–599. [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Brouns G, De Wulf A, Constales D. Delaunay triangulation algorithms useful for multibeam echosounding. J Surv Eng ASCE. 2003;129(2):79–84. doi: 10.1061/(ASCE)0733-9453(2003)129:2(79). [DOI] [Google Scholar]
- Chen IC, Hill JK, Ohlemüller R, Roy DB, Thomas CD. Rapid range shifts of species associated with high levels of climate warming. Science. 2011;333(6045):1024–1026. doi: 10.1126/science.1206432. [DOI] [PubMed] [Google Scholar]
- Comes HP, Kadereit JW. The effect of quaternary climatic changes on plant distribution and evolution. Trends Plant Sci. 1998;3(11):432–438. doi: 10.1016/S1360-1385(98)01327-2. [DOI] [Google Scholar]
- Deng M, Zhou ZK, Li QS (2013) Taxonomy and systematics of Quercus subgenus Cyclobalanopsis. Int Oaks 24:48–60
- Dong WP, Xu C, Li CH, Sun JH, Zuo YJ, Shi S, et al. ycf1, the most promising plastid DNA barcode of land plants. Sci Rep. 2015;5:8348. doi: 10.1038/srep08348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JJ. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–15. [Google Scholar]
- Eaton DA, Hipp AL, González‐Rodríguez A, Cavender‐Bares J. Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution. 2015;69(10):2587–2601. doi: 10.1111/evo.12758. [DOI] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005;14(8):2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10(3):564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- Fan DM, Yue JP, Nie ZL, Li ZM, Comes HP, Sun H. Phylogeography of Sophora davidii (Leguminosae) across the ‘Tanaka‐Kaiyong Line’, an important phytogeographic boundary in Southwest China. Mol Ecol. 2013;22(16):4270–4288. doi: 10.1111/mec.12388. [DOI] [PubMed] [Google Scholar]
- Fang J, Guo Z, Hu H, Kato T, Muraoka H, Son Y. Forest biomass carbon sinks in East Asia, with special reference to the relative contributions of forest expansion and forest growth. Glob Chang Biol. 2014;20(6):2019–2030. doi: 10.1111/gcb.12512. [DOI] [PubMed] [Google Scholar]
- Favre A, Päckert M, Pauls SU, Jähnig SC, Uhl D, Michalak I, et al. The role of the uplift of the Qinghai‐Tibetan Plateau for the evolution of Tibetan biotas. Biol Rev. 2015;90(1):236–253. doi: 10.1111/brv.12107. [DOI] [PubMed] [Google Scholar]
- Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147(2):915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Williamson S, Bustamante CD. A Markov chain Monte Carlo approach for joint inference of population structure and inbreeding rates from multilocus genotype data. Genetics. 2007;176(3):1635–1651. doi: 10.1534/genetics.107.072371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gugger PF, Ikegami M, Sork VL. Influence of late quaternary climate change on present patterns of genetic variation in valley oak, Quercus lobata Née. Mol Ecol. 2013;22(13):3598–3612. doi: 10.1111/mec.12317. [DOI] [PubMed] [Google Scholar]
- Harrison SP, Yu G, Takahara H, Prentice IC. Palaeovegetation (Communications arising): diversity of temperate plants in east Asia. Nature. 2001;413(6852):129–130. doi: 10.1038/35093166. [DOI] [PubMed] [Google Scholar]
- Hewitt GM. Genetic consequences of climatic oscillations in the Quaternary. Philos T R Soc B. 2004;359(1442):183–195. doi: 10.1098/rstb.2003.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp AL, Manos P, McVay JD, Cavender-Bares J, Gonzalez Rodriguez A, Romero-Severson J et al (2015) A phylogeny of the world’s oaks Botanical Society of America Conference 2016, Edmonton, Canada
- Jacques FM, Guo SX, Su T, Xing YW, Huang YJ, Liu YSC, et al. Quantitative reconstruction of the Late Miocene monsoon climates of southwest China: a case study of the Lincang flora from Yunnan Province. Palaeogeogr Palaeoclimatol Palaeoecol. 2011;304(3):318–327. doi: 10.1016/j.palaeo.2010.04.014. [DOI] [Google Scholar]
- Jacques FMB, Su T, Spicer RA, Xing YW, Huang YJ, Zhou ZK. Late Miocene southwestern Chinese floristic diversity shaped by the southeastern uplift of the Tibetan Plateau. Palaeogeogr Palaeoclimatol Palaeoecol. 2014;411:208–215. doi: 10.1016/j.palaeo.2014.05.041. [DOI] [Google Scholar]
- Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23(14):1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- Jiang XL, Deng M, Li Y. Evolutionary history of subtropical evergreen broad-leaved forest in Yunnan Plateau and adjacent areas: an insight from Quercus schottkyana (Fagaceae) Tree Genet Genomes. 2016;12(6):104. doi: 10.1007/s11295-016-1063-2. [DOI] [Google Scholar]
- Kalinowski ST. hp‐rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes. 2005;5(1):187–189. doi: 10.1111/j.1471-8286.2004.00845.x. [DOI] [Google Scholar]
- Levin DA, Francisco-Ortega J, Jansen RK. Hybridization and the extinction of rare plant species. Conserv Biol. 1996;10(1):10–16. doi: 10.1046/j.1523-1739.1996.10010010.x. [DOI] [Google Scholar]
- Li SF, Mao LM, Spicer RA, Lebreton-Anberree J, Su T, Sun M, et al. Late Miocene vegetation dynamics under monsoonal climate in southwestern China. Palaeogeogr Palaeoclimatol Palaeoecol. 2015;425:14–40. doi: 10.1016/j.palaeo.2015.02.030. [DOI] [Google Scholar]
- Li XW, Li J. On the validity of tanaka line & its significance viewed from the distribution of Eastern Asiatic Genera in Yunnan. Acta Bot Yunnanica. 1992;14(1):1–12. [Google Scholar]
- Li XW, Li J. The Tanaka-Kaiyong line-an important floristic line for the study of the flora of East Asia. Ann Mo Bot Gard. 1997;84(4):888–892. doi: 10.2307/2992033. [DOI] [Google Scholar]
- Librado P, Rozas J. DnaSPv5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Liu Kb. Quaternary history of the temperate forests of China. Quat Sci Rev. 1988;7(1):1–20. doi: 10.1016/0277-3791(88)90089-3. [DOI] [Google Scholar]
- Liu YF, Wang Y, Huang HW. Species‐level phylogeographical history of Myricaria plants in the mountain ranges of western China and the origin of M. laxiflora in the Three Gorges mountain region. Mol Ecol. 2009;18(12):2700–2712. doi: 10.1111/j.1365-294X.2009.04214.x. [DOI] [PubMed] [Google Scholar]
- López‐Pujol J, Zhang FM, Sun HQ, Ying TS, Ge S. Centres of plant endemism in China: places for survival or for speciation? J Biogeogr. 2011;38(7):1267–1280. doi: 10.1111/j.1365-2699.2011.02504.x. [DOI] [Google Scholar]
- Luo Y, Zhou Z. Phytogeography of Quercus subg Cyclobalanopsis. Acta Bot Yunnanica. 2000;23(1):1–16. [Google Scholar]
- Ma XF, Szmidt AE, Wang XR. Genetic structure and evolutionary history of a diploid hybrid pine Pinus densata inferred from the nucleotide variation at seven gene loci. Mol Biol Evol. 2006;23(4):807–816. doi: 10.1093/molbev/msj100. [DOI] [PubMed] [Google Scholar]
- Manni F, Guerard E, Heyer E. Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Hum Biol. 2004;76(2):173–190. doi: 10.1353/hub.2004.0034. [DOI] [PubMed] [Google Scholar]
- Meng HH, Su T, Gao XY, Li J, Jiang XL, Sun H, et al. Warm–cold colonization: response of oaks to uplift of the Himalaya–Hengduan Mountains. Mol Ecol. 2017;26(12):3276–3294. doi: 10.1111/mec.14092. [DOI] [PubMed] [Google Scholar]
- Miller M. Alleles In Space (AIS): computer software for the joint analysis of interindividual spatial and genetic information. J Hered. 2005;96(6):722–724. doi: 10.1093/jhered/esi119. [DOI] [PubMed] [Google Scholar]
- Mitton JB (2000) Selection in natural populations. Oxford University Press, Oxford
- Monmonier MS. Maximum‐difference barriers: An alternative numerical regionalization method. Geogr Anal. 1973;5(3):245–261. doi: 10.1111/j.1538-4632.1973.tb01011.x. [DOI] [Google Scholar]
- Ortego J, Gugger PF, Riordan EC, Sork VL. Influence of climatic niche suitability and geographical overlap on hybridization patterns among southern Californian oaks. J Biogeogr. 2014;41(10):1895–1908. doi: 10.1111/jbi.12334. [DOI] [Google Scholar]
- Ortego J, Riordan EC, Gugger PF, Sork VL. Influence of environmental heterogeneity on genetic diversity and structure in an endemic southern Californian oak. Mol Ecol. 2012;21(13):3210–3223. doi: 10.1111/j.1365-294X.2012.05591.x. [DOI] [PubMed] [Google Scholar]
- Peakall R, Smouse PE. GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes. 2006;6(1):288–295. doi: 10.1111/j.1471-8286.2005.01155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson RG, Raxworthy CJ, Nakamura M, Townsend Peterson A. Predicting species distributions from small numbers of occurrence records: a test case using cryptic geckos in Madagascar. J Biogeogr. 2007;34(1):102–117. doi: 10.1111/j.1365-2699.2006.01594.x. [DOI] [Google Scholar]
- Phillips SJ, Anderson RP, Schapire RE. Maximum entropy modeling of species geographic distributions. Ecol Model. 2006;190(3):231–259. doi: 10.1016/j.ecolmodel.2005.03.026. [DOI] [Google Scholar]
- Pons O, Petit R. Measwring and testing genetic differentiation with ordered versus unordered alleles. Genetics. 1996;144(3):1237–1245. doi: 10.1093/genetics/144.3.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu YX, Fu CX, Comes HP. Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol Phylogenet Evol. 2011;59(1):225–244. doi: 10.1016/j.ympev.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992;9(3):552–569. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- Shaw J, Lickey EB, Beck JT, Farmer SB, Liu W, Miller J, et al. The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am J Bot. 2005;92(1):142–166. doi: 10.3732/ajb.92.1.142. [DOI] [PubMed] [Google Scholar]
- Shi Y, Li J, Li B. Uplift of the Qinghai-Xizang (Tibetan) plateau and east Asia environmental change during late Cenozoic. Acta Geogr Sin. 1999;54:20–28. [Google Scholar]
- Sun BN, Wu JY, Liu YS, Ding ST, Li XC, Xie SP, et al. Reconstructing Neogene vegetation and climates to infer tectonic uplift in western Yunnan, China. Palaeogeogr Palaeoclimatol Palaeoecol. 2011;304(3):328–336. doi: 10.1016/j.palaeo.2010.09.023. [DOI] [Google Scholar]
- Sun H. Evolution of arctic-tertiary flora in Himalayan-Hengduan Mountains. Acta Bot Yunnanica. 2001;24(6):671–688. [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T (1954) Species problem in citrus. Japanese Society for the Promotion of Science, Tokyo
- Tian B, Zhou ZL, Du FK, He CZ, Xin PY, Ma HC. The Tanaka line shaped the phylogeographic pattern of the cotton tree (Bombax ceiba) in southwest China. Biochem Syst Ecol. 2015;60:150–157. doi: 10.1016/j.bse.2015.04.014. [DOI] [Google Scholar]
- Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P. MICRO‐CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004;4(3):535–538. doi: 10.1111/j.1471-8286.2004.00684.x. [DOI] [Google Scholar]
- Wang B, Mao JF, Zhao W, Wang XR. Impact of geography and climate on the genetic differentiation of the subtropical pine Pinus yunnanensis. PLoS ONE. 2013;8(6):e67345. doi: 10.1371/journal.pone.0067345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Deng M, Jiang XL, Westwood M, Song YG, Turkington R. Phylogeography of Quercus glauca (Fagaceae), a dominant tree of East Asian subtropical evergreen forests, based on three chloroplast DNA interspace sequences. Tree Genet Genomes. 2015;11(1):1–17. doi: 10.1007/s11295-014-0804-3. [DOI] [Google Scholar]
- Yang YM, Tian K, Hao JM, Pei SJ, Yang YX. Biodiversity and biodiversity conservation in Yunnan, China. Biodivers Conserv. 2004;13(4):813–826. doi: 10.1023/B:BIOC.0000011728.46362.3c. [DOI] [Google Scholar]
- Yu G, Chen XD, Ni J, Cheddadi R, Guiot J, Han H, et al. Palaeovegetation of China: A pollen data‐based synthesis for the mid‐Holocene and last glacial maximum. J Biogeogr. 2000;27(3):635–664. doi: 10.1046/j.1365-2699.2000.00431.x. [DOI] [Google Scholar]
- Yue LL, Chen G, Sun WB, Sun H. Phylogeography of Buddleja crispa (Buddlejaceae) and its correlation with drainage system evolution in southwestern China. Am J Bot. 2012;99(10):1726–1735. doi: 10.3732/ajb.1100506. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li QJ, Li HT, Chen J, Li DZ. Genetic diversity and geographic differentiation in Tacca chantrieri (Taccaceae): an autonomous selfing plant with showy floral display. Ann Bot. 2006;98(2):449–457. doi: 10.1093/aob/mcl123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MW, Rao DQ, Yang JX, Yu GH, Wilkinson JA. Molecular phylogeography and population structure of a mid-elevation montane frog Leptobrachium ailaonicum in a fragmented habitat of southwest China. Mol Phylogenet Evo. 2010;54(1):47–58. doi: 10.1016/j.ympev.2009.10.019. [DOI] [PubMed] [Google Scholar]
- Zhao YJ, Gong X. Genetic divergence and phylogeographic history of two closely related species (Leucomeris decora and Nouelia insignis) across the ’Tanaka Line’ in Southwest China. BMC Evol Biol. 2015;15(1):1. doi: 10.1186/s12862-014-0274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Woodall CW, Clark JS. Failure to migrate: lack of tree range expansion in response to climate change. Glob Chang Biol. 2012;18(3):1042–1052. doi: 10.1111/j.1365-2486.2011.02571.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.