

Abstract
Nanobodies can be seen as next‐generation tools for the recognition and modulation of antigens that are inaccessible to conventional antibodies. Due to their compact structure and high stability, nanobodies see frequent usage in basic research, and their chemical functionalization opens the way towards promising diagnostic and therapeutic applications. In this Review, central aspects of nanobody functionalization are presented, together with selected applications. While early conjugation strategies relied on the random modification of natural amino acids, more recent studies have focused on the site‐specific attachment of functional moieties. Such techniques include chemoenzymatic approaches, expressed protein ligation, and amber suppression in combination with bioorthogonal modification strategies. Recent applications range from sophisticated imaging and mass spectrometry to the delivery of nanobodies into living cells for the visualization and manipulation of intracellular antigens.
Keywords: antigen-binding proteins, cellular delivery, molecular biology, nanobodies, site-specific functionalization
1. Introduction
Antibodies are the core molecules of the immune system for identifying, targeting, and clearing pathogens from the infected organism. Immunoglobulin G (IgG), a 150 kDa protein consisting of two heavy and two light chains is the predominant antibody type found in nature.1 Since antibodies were used for the detection of rhesus factor immunization and to quantify the amount of insulin present in blood plasma, uncountable analytical applications have been developed.2 The ability to generate humanized and monoclonal antibodies highly specific to almost any antigen of interest has intensified this development and laid the foundation for the targeted therapeutic use of antibodies.3 While early therapeutic concepts exclusively relied on the function of the antibody itself, more recent development combine the target specificity of antibodies with the effectiveness of small drug molecules in so‐called antibody–drug conjugates (ADCs).4 For this, a drug molecule is covalently linked to a functional group within the antibody, which requires selective chemical methods for attachment without interfering with antibody function.5 The same trend of attaching functionality holds true for analytical and diagnostic antibodies. While many of the established methods rely on indirect detection modes like radioactive labelling of the antigen, oxidation by horseradish peroxidase, or the enzyme alkaline phosphatase (in enzyme‐linked immunosorbent assay, ELISA),6 more recent developments have made use of small fluorescent labels that are covalently bound to the primary antibody.7
The generation, production, functionalization, and intracellular application of full‐length antibodies can be challenging. Antibodies are posttranslationally glycosylated proteins and their function has been shown to be dependent on the attached glycans.8 Furthermore, antibodies have a complex structure involving inter‐ and intramolecular disulfide bonds, which is vulnerable towards environmental changes, the reductive milieu of the intracellular environment, and the attachment of payloads. Moreover, conventional IgGs contain a highly conserved loop length for the antigen‐binding domain (complementary determining regions, CDRs), which evolved to bind convex paratopes, thereby limiting the scope of potential antigens.9 For instance, the receptor‐binding domains of various pathogens have evolved as cavities, which prevents the binding of full length IgGs.10
Consequently, novel classes of recombinant antigen‐binding proteins that lack these limitations are on the rise.11 Besides their reduced size and structural complexity, many recombinant antigen‐binding proteins (binders) can be produced in high amounts using eukaryotic and prokaryotic cells, and based on their increased stability towards reductive conditions, can be applied within cellular environments.12 This opens avenues for live‐cell detection and the manipulation of important intracellular processes with minimal impairment to the cell. In contrast, the use of full‐length antibodies is often limited to extracellular targets and fixed or permeabilized tissues. These promising properties have led to the development of various classes of binders that are either immunoglobulin‐derived or synthetic derivatives of completely different protein classes. Nanobodies are noteworthy examples of recombinant antigen‐binding proteins that are distinguished by unique physical properties and binding specificity.7, 13 They are defined as single‐domain variable fragments of camelid‐derived heavy‐chain antibodies (hcAb). Nanobodies are introduced in Section 2 and discussed in comparison with other formats of recombinant binders. In Section 3, an overview of nanobody generation and selection procedures is given, and in Section 4, selected applications of genetically encoded nanobodies in cellular biology and imaging are depicted. In Section 5, techniques for the chemical functionalization of nanobodies will be highlighted. Recent developments allow the generation of homogenous nanobody conjugates that have increased binding affinity and beneficial in vivo properties compared to their randomly functionalized equivalents.14 Finally, in Section 6, advances in the cellular delivery of nanobodies and other binders will be reviewed.
2. Recombinant Antigen‐Binding Proteins: Nanobodies and Others
IgGs are the predominant isotype of immunoglobulins and consist of two identical heavy and two identical light chains that are covalently linked through disulfide bonds.1 The antigen is recognized through an interplay between the variable N‐terminal domains of the heavy (VH) and the light (VL) chain and six CDRs (Figure 1 a).8 Binders derived from IgGs can be classified as antigen‐binding fragments (Fab, ca. 50 kDa), single‐chain variable fragments (scFv, ca. 25 kDa,) and heavy‐ or light‐chain single domains (VH or VL, ca. 12.5 kDa). Fab and scFv binders consist of both the VH and VL domain of the parental IgG, and retain the size and affinity of the area binding the antigen. Due to their reduced size compared to regular IgGs, they show enhanced pharmacokinetic properties for in vivo applications.10a VH and VL are covalently linked by artificial amino acid linkers or disulfides and associated through strong hydrophobic interactions. Ward et al. were able to demonstrate, that functional single VH domains of mice can be secreted from E. coli, and they hypothesized that their reduced size should enable binding to the cavities of pathogens.15 However, these expectations have not been fulfilled. The antigen‐binding area of isolated VH domains is bisected and their binding affinity significantly reduced compared to the parent antibody. Moreover, the hydrophobic amino acids that are essential for VH/VL interaction in full‐length IgGs are solvent‐exposed, thus leading to aggregation and poor solubility.7
Figure 1.
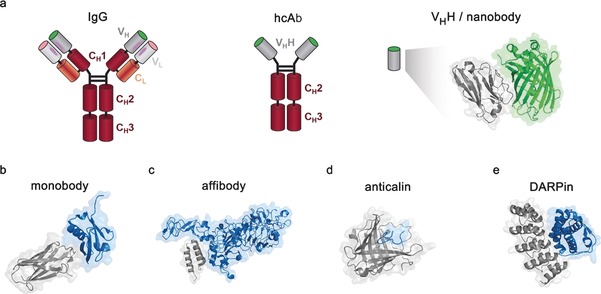
Depiction of IgGs, nanobodies, and other engineered recombinant antigen‐binding proteins. a) Comparison of nanobodies and IgGs. Conventional IgG molecules contain two heavy and two light chains. Light chains contain one constant (CL, orange) and one variable (VL, light gray) domain. The heavy chains contain three constant domains (CH1–3, red) and one variable domain (VH, dark gray). Heavy‐chain antibodies (hcAb) from Camelidae lack the CH1 and CL domain of conventional antibodies. They recognize their antigen through a single variable domain, VHH (dark gray). The X‐Ray structure of a nanobody binding its antigen GFP (green) is shown (PDB ID: 3G9A).17 b) A fibronectin‐based monobody binding the SUMO protein (PDB ID: 3RZW).18 c) An affibody based on Protein A, binding to HER 2 (PDB ID: 3MZW).19 d) A libocalin derived anticalin binding to the Alzheimer's disease (AD)‐relevant amyloid‐β (PDB ID: 4MVI).20 e) A designed ankyrin repeat protein (DARPin) in complex with human interleukin‐4 (PDB ID: 4YDY). Antigens are shown in blue, antigen‐binding proteins in gray.
In the early 1990s, an exceptional class of IgG immunoglobulin was detected in the sera of Camelidae.13a These so called heavy‐chain antibodies (hcAb) are devoid of light chains, with the functional antigen‐binding unit reduced to a single variable domain (VHH, Figure 1 a).7, 16 These properties allow the generation of potent, recombinant VHHs by isolating and engineering the corresponding single domain region of Camelidae B cells after immunization. Based on their small size of around 13–14 kDa, recombinant VHHs are often referred to as nanobodies. Interestingly, similar IgG derivatives have been identified in several cartilaginous fish (Ginglymostoma cirratum, Orectolobidae).21 The structure of nanobodies differs in two ways from VH domains of regular IgGs. First, the CDRs are enlarged to provide a similar antigen‐interacting surface to that of regular IgGs (600–800 Å2, Figure 2).22 These changes result in nanobodies binding their antigen in a convex paratope, thus making them well suited for binding structures that are restricted for full‐length IgGs like immune‐evasive epitopes and cavities.22, 25 Second, hydrophobic amino acids within the conserved framework region (FR) that are responsible for VH/VL interaction are replaced with hydrophilic amino acids.26 In particular, these mutations contribute to the increased solubility and stability of nanobodies, thus allowing a simplified manufacturing process and a wide range of biotechnological applications.27 Moreover, nanobody‐based cancer therapy has revealed that nanobodies possess beneficial biophysical and pharmacological properties for in vivo applications, with indications for a low response from the immune system, which is encouraging for future clinical trials.28 Together, these unique properties of nanobodies have even initiated “camelization” strategies for human derived single VH to increase their binding affinity and stability while maintaining their low immunogenic potential in humans.9b, 29 Alternatively, a humanized nanobody scaffold has been engineered, which facilitates CDR grafting from other nanobodies for the development of nanobody based therapeutics.30 Along these lines, pharmaceutical companies like the Belgian company Ablynx nv have a growing number of pre‐clinical and clinical programs in development that are based on proprietary nanobody technology, thus further emphasizing their potential.31
Figure 2.
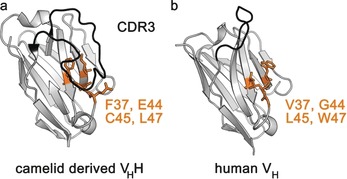
Comparison of the binding regions and surface structures of nanobodies and human‐derived VH domains. a) A VHH (nanobody) from Camelidae with GFP as the antigen (PDB ID: 3G9A).17 b) A human derived variable domain (VH) with vascular endothelial growth factor as the antigen (PDB ID: 2FJF).23 VHHs contain a significantly enlarged CDR3 framework (black), thus ensuring high binding affinity. Several hydrophobic amino acids that are highly conserved in conventional VH domains are mutated within nanobodies, which increases their solubility (orange).24
In addition to immunoglobulin‐derived binders, non‐immunoglobulin‐based proteins have been engineered to specifically bind antigens with similar affinity compared to conventional antibodies. Small proteins that are involved in tight protein–protein interactions serve as scaffolds for the generation of such binders. The specific binding surface of these scaffolds is randomized and high‐affinity binders selected through an in vitro display technique.32 Prominent examples are monobodies,33 anticalins,34 affibodies, and designed ankyrin repeat proteins (DARPins;35 Figure 1 b–e).
Monobodies are recombinant antigen‐binding proteins based on human fibronectin III.33 They are structurally similar to immunoglobulin binders but devoid of intramolecular disulfides, thus making them ideal for intracellular applications.36 The cell wall protein Protein A present in Staphylococcus aureus serves as the basis for affibodies.37 In nature, Protein A binds the fragment crystallizable (Fc) region of immunoglobulins, preventing phagocytosis triggered by an immune response of the host organism.38 Mutagenesis of the binding area resulted in a number of efficient affibodies that bind targets like human insulin or the cytokine TNFα.39 Anticalins are derived from the ß‐barrel‐structured lipocalins, a diverse class of proteins responsible for the transport, storage, synthesis, and sequestration of small hydrophobic molecules.40 Lipocalin‐based libraries have enabled the selection of anticalins against various targets with up to picomolar affinities.34 Finally, DARPins are based on natural ankyrin repeats that are involved in protein–protein interactions. In contrast to other engineered binders, DARPins are characterized by a modular assembly of consecutive repeats engineered to bind a specific target.35 The different classes of engineered recombinant antigen‐binding proteins share many advantageous properties, including high stability and small size. However, the effort needed in generating non‐Ig‐derived recombinant antigen‐binding proteins with sufficient binding affinity is high, thus limiting the applicability of these binders.41
3. Nanobody Generation and Selection Procedures
Nanobodies (and other immunoglobulin‐based recombinant antigen‐binding proteins) can either be generated by immunizing the respective animal with the antigen of interest,27 or by further evolving an existing naïve library.42 In the case of immunization, up to six injections of around 0.5 mg antigen or immobilized antigen (e.g., BSA conjugate) are performed within a time course of several weeks.43 The mRNA is isolated from lymphocytes and its complementary DNA (cDNA) synthesized using reverse transcriptase.16, 43 Next, the specific segment encoding the VHH domain is amplified and potent binders isolated or further engineered using a polypeptide display technique. Phage display is the most common display technique for in vitro binder selection and is capable of screening up to 1011 sequences per library.44 Here, the VHH encoding part is fused to a viral coat protein, leading to the library being displayed on the surface of bacteriophages. Since each phage displays a single VHH variant and contains its genetic information, the most efficient binders can be selected by challenging the library with the immobilized antigen followed by nucleotide sequencing. Alternative screening strategies include yeast and bacterial display, in which binders can be selected by multiparameter and quantitative flow cytometry, as well as mRNA and ribosome display, which are well suited for the selection of large libraries of up to 1015 sequences.44c Once a potent nanobody is selected, it can be readily expressed in high yields of up to several g L−1 in E. coli, S. cerevisiae, P. pastoris, or human cells by using a periplasmic leader sequence.45 Secretion to a non‐reducing environment during expression is advisable, since nanobodies harbor up to two disulfide bridges.16 An overview of the nanobodies presented within this review, together with their targets, functionalization approaches, applications, and known affinity values is given in Table 1.
Table 1.
Overview of the nanobodies discussed in this review.
| Target | Application | Functionalization | Generation | K d [nm] |
|---|---|---|---|---|
| GFP | Protein immobilization46c, 68a,68b | Solid support, randomly attached (NHS‐chemistry),[b] TTL mediated chemoenzymatic biotinylation | Immunization | 0.2327 |
| Detection of PPIs,48 | Lac repressor, genetic fusion | |||
| Protein degradation49 | F‐box domain of Slmb, genetic fusion | |||
| Imaging27, 65, 66, 72 | FPs, genetic fusion; fluorophores and gold nanoparticles randomly attached (NHS‐chemistry), chemoenzymatic TTL[c] mediated site‐specific attachment | |||
| Intracellular protein discovery109 | Polycationic resurfacing, FPs,[d] genetic fusion | 1109 | ||
| Cellular delivery, live cell immunostaining118 | Linear and cyclic cell‐penetrating peptides via EPL[e] | 0.32118 | ||
| l‐Plastin | Trapping of inactive conformation50 | V5‐tag for purification, genetic fusion | Immunization | 40‐8050 |
| P‐glycoprotein | Inhibiting drug‐efflux‐based multidrug resistance51 | – | Immunization | 52051 |
| HypeE | Inhibiting or stimulating AMPylation52 | Fluorophore, biotin, Sortase A mediated | Naïve phage display library | N/A |
| MazE | Crystallization chaperone53a | – | Immunization | N/A |
| β2‐microglobulin | Crystallization chaperone53c | – | Immunization | 1.653c |
| EpsI:EpsJ | Crystallization chaperone53b | – | Immunization | N/A |
| human lysozyme | Studying protein folding using NMR54 | ‐ | Immunization and grafting CDRs to stable nanobodies | 46054 |
| Proclacitonin | High‐throughput assay55b | Chitosane‐graphene nanocomposite, randomly attached (glutaraldehyde) | Immunization | 6.2–24.555b |
| PCNA | Imaging DNA replication27, 60 | FPs, genetic fusion | Immunization | N/A |
| β‐catenin | Imaging of β‐catenin61 | FPs, genetic fusion | Immunization | 1.9‐4461 |
| HIV‐1 capsid protein | Imaging HIV‐162 | FPs, genetic fusion | Immunization | 0.1662 |
| Nuclear lamina | Imaging the cytoskeleton27 | FPs, genetic fusion | Immunization | N/A |
| Target | Application | Functionalization | Generation | Kd [nm] |
| HER2 | Biomarker for breast cancer14a | Radiolabel, engineered C‐terminal cysteine & maleimide chemistry | Immunization | 614a |
| Biomarker for breast cancer86a | Radiolabel, fluorphore, chemoenzymatic attachment by the use of Sortase A | 486a | ||
| Nanobody based activation immunotherapeutic91 | Dinitrophenyl moiety, chemoenzymatic attachment by the use of lipoic acid ligase | 0.51–3.2120 | ||
| Intracellular protein discovery109 | Polycationic resurfacing, FPs, genetic fusion | 1109 | ||
| CAIX | Biomarker for breast cancer75d | Radiolabel, engineered C‐terminal cysteine & maleimide chemistry | Immunization | 1375d |
| PSMA | Biomarker for prostate cancer75a | Radiolabel, engineered C‐terminal cysteine & maleimide chemistry | Immunization | 27.475a |
| TNF | Treatment for autoimmune disorders75e | Linear and branched PEG, C‐terminal engineered cysteine & maleimide chemistry | Unknown | N/A |
| MUC1 | Cancer specific cell killing75c | Polyethyleneimine, engineered C‐terminal cysteine & maleimide chemistry | Naïve phage display library | N/A |
| NPC | STORM imaging14b | Fluorophore, engineered surface cysteine & maleimide chemistry | Immunization | N/A |
| ApoB‐100 | Electrochemical impedance spectroscopy of ApoB‐10077a | Biotin, chemoenzymatic attachment by the use of BirA[f] | Immunization | 5.2–16.7 |
| Testosterone | High‐throughput assay55a | Biotin, chemoenzymatic attachment by the use of BirA | Immunization | 950 |
| Unknown | Immobilization80 | Biotin, chemoenzymatic attachment by the use of transglutaminase | Unknown | N/A |
| Class II MHC | Immune response imaging86b, 87 | Radiolabel, chemoenzymatic attachment by the use of Sortase A. Double functionalization with fluorophore and radiolabel by the use of Sortase A in combination with engineered C‐terminal cysteine & maleimide chemistry | Immunization | N/A |
| Target | Application | Functionalization | Generation | K d [nm] |
| EGFR | Imaging and photoinduced cross‐linking96 | Fluorphore and PEG, amber suppression of AmAzZLys | Immunization | 11.5–14.196 |
| VCAM‐1 | Immobilization98 | Biotin, attachment by EPL | Immunization | N/A |
| PlexinD1 | Tumor targeting100 | Polymersomes, attached by EPL | Naïve phage display library | N/A |
| β‐lactamase | Intracellular protein discovery109 | Polycationic resurfacing, FPs, genetic fusion | Immunization | 1109 |
[a] Protein–protein interactions. [b] N‐Hydroxysuccinimide. [c] Tubulin tyrosine ligase. [d] Fluorescent proteins. [e] Expressed protein ligation. [f] Bacterial biotin ligase.
4. Genetically Encoded Nanobodies in Cellular Biology and Imaging
As already mentioned, nanobodies have advantageous properties for advanced applications in molecular biology.7 They feature high thermal and conformational stability and retain their binding activity after prolonged incubation at elevated temperatures, high salt concentrations, and under different pH conditions.46 Moreover, they are able to efficiently refold and fully restore their antigen affinity after thermal denaturation, thus opening novel opportunities for studying the dynamics of protein folding.41, 47
These robust properties allow them to capture their respective antigens in vitro as well as in vivo, one example being the widely used GFP binder.27 A visual example for antigen capture in living cells is the recently developed fluorescent‐3‐hybrid (F3H) assay to monitor dynamic protein–protein interactions (Figure 3 a).48 Here, a green fluorescent protein (GFP)‐binding nanobody is fused to the Lac repressor, resulting in the recruitment of GFP fusion proteins to artificial LacO DNA repeats. As soon as a second protein labelled with another fluorescent molecule interacts with the GFP fusion, it will be co‐recruited to the anchor site, thereby enabling time‐resolved visualization of protein–protein interactions.
Figure 3.
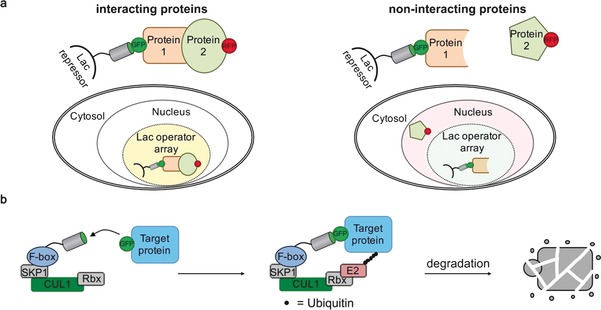
Schematic depiction of nanobody‐based detection of protein–protein interactions in living cells and nanobody‐mediated protein degradation. a) The fluorescent‐3‐hybrid (F3 H)assay is based on a GFP‐binding nanobody fused to the Lac repressor tightly binding to Lac operator DNA repeats stably integrated in the genome, for example, in baby hamster kidney cells. In this way, a GFP‐labelled protein of interest (Protein 1) is recruited to the LacO region within the nucleus. If a second protein (Protein 2) that is labelled with a different fluorescent protein (RFP) interacts with Protein 1, it will be co‐recruited to the LacO region, thereby resulting in strong correlation of the GFP and RFP fluorescence signal. b) Nanobody‐mediated ubiquitin‐dependent protein degradation. A GFP‐binding nanobody is fused to the F‐box domain of the Drosophila melanogaster derived Slmb protein. Together with the S‐phase kinase associated protein 1 (SKP1), Cullin 1 (CUL1), Ring protein (Rbx) subunits, the F‐box forms the E3 enzyme that is responsible for target‐protein recognition and binding of the E2 enzyme. A GFP‐labelled target protein is recruited to the E3 domain upon nanobody binding. Subsequent ubiquitination catalyzed by an E2 enzyme triggers protein degradation of the GFP‐labelled protein.
In recent years, nanobodies have been used to analyze protein function in living cells and organisms. The reversible genetic knockdown of proteins by interfering RNA is a prominent method for elucidating protein function. However, such systems rely on the fast depletion of the target protein. An alternative method to study protein function by reversible knockout makes use of nanobodies that mediate ubiquitin‐dependent protein degradation of the bound protein (Figure 3 b).49 This technology was used against a GFP fusion to show that the myosin II regulatory light chain Sqh is required for dorsal closure in the fruit fly Drosophila. Moreover, nanobodies have been used to sense and trap specific conformations of proteins.17 Nanobodies that bind l‐plastin, an actin‐binding protein involved in immune regulation, that trap the protein in an inactive conformation revealed that l‐plastin plays an important role for immune synapse formation and T‐cell proliferation.50 Moreover, a nanobody that was shown to inactivate the intracellular ATP hydrolysis activity of Pgp, an ABC‐type transporter, could potentially serve as the basis for the development of new therapeutics to overcome drug resistance during cancer therapy (Figure 4).51 Furthermore, nanobodies have been shown to modulate the abundance of posttranslational modifications on proteins in living cells. Truttmann et al. were able to engineer VHHs that either inhibit or stimulate Huntington associated protein E (HYPE)‐mediated AMPylation of proteins and used these tools to identify histones H2, H3, and H4 as new targets for HYPE.52
Figure 4.
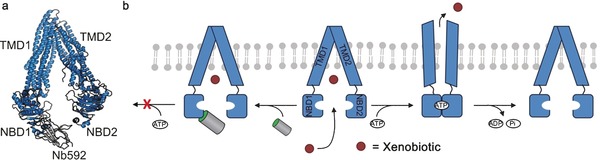
The nanobody Nb592 binds to the ATP‐binding cassette (ABC) transporter P‐glycoprotein (P‐gp) and inhibits its ATPase activity. a) Crystal structure of P‐gp in complex with Nb592. The nanobody specifically binds to the nucleotide‐binding domain 1 (NBD1) of P‐gp. TMD=transmembrane domain. P‐gp is shown in blue, Nb592 in gray (PDB ID: 4KSD).51 b) Upon ATP binding and dimerization of the NBDs, restructuring of the P‐gp and its TMD occurs and the xenobiotic is transported across the cellular membrane and into the extracellular environment. Upon ATP hydrolysis and the release of ADP and Pi release, the P‐gps ground state is restored. In contrast, once Nb592 is bound to NBD1, NBD dimerization, ATP complexation, and transporter activity is inhibited.
Another important application of nanobodies is their use as crystallization chaperones for intrinsically disordered proteins and large molecular complexes. The production of protein crystals with sufficient quality for X‐ray crystallography of such proteins can be highly challenging, and co‐crystallization with nanobodies has been shown to improve the crystallization behavior significantly. In this way, the structures of disordered proteins like the addiction antidote MazE and the amyloidogenic β2‐microglobulin, as well as the structure of a large protein complex of EpsJ and EpsI that is involved in the secretion of proteins, have been solved.53 In addition to their use as crystallization chaperones, nanobodies have been utilized as NMR probes to study protein structure and folding.54
Finally, nanobodies have even been shown to bind challenging epitopes such as small molecules. This has led, for example, to the development of various nanobody‐based high‐throughput assays to identify and quantify clinically relevant biomarkers, including testosterone and proclacitonin.55
The fluorescent labelling of proteins has become one of the most important tools for visualizing and understanding cellular structures and intracellular processes. Fluorescent proteins (FPs), like GFP, are the most frequently used biosensors, and their fusion to a protein of interest enables the dynamic visualization of proteins of interest in living cells.56 Even though genetic fusion to FPs is straightforward, their fluorescence properties, as well as a tendency towards photobleaching, limit their spectroscopic use, and their possible impact to the biological function of the protein of interest is often underestimated.57 To improve the spectroscopic properties of FP‐tagged proteins of interest, Kirchhofer et al. developed GFP‐binding nanobodies that are able to modulate the absorption properties of GFP by inducing structural changes in the environment of the chromophore (Figure 5 a). These changes stabilize GFP fluorescence in living cells and resulted in higher fluorescence sensitivity and spatial resolution.17
Figure 5.
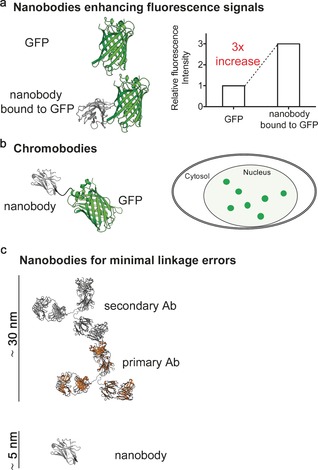
Nanobodies in imaging. a) Upon binding of its antigen, the GFP‐binding nanobody GBP stabilizes and enhances the fluorescence signal of GFP when imaged in living cells. The nanobody (VHH) is depicted in gray, GFP in green (PDB ID: 3K1K17). b) FP‐tagged nanobodies (so called chromobodies) avoid the need for FP fusion to the protein of interest and maintain its endogenous expression level. Various chromobodies have been engineered and used as powerful imaging tools. The nanobody (VHH) is depicted in gray, GFP in green (PDB ID: 3G9A).17 c) Nanobodies have the ability to minimize linkage errors during super‐resolution microscopy experiments. They significantly reduce the spatial distance to the actual specimen compared to classical experimental setups composed of full‐length primary and secondary antibodies. The nanobody is depicted in gray (PDB ID: 3G9A17), the secondary and primary antibodies in gray and orange, respectively (PDB ID: 1IGT58).
Nevertheless, several studies have revealed that the genetic fusion of fluorescent proteins to a protein of interest can result in loss and even alteration of function.59 The co‐expression of an FP‐tagged nanobody (so called chromobody) with high affinity to the protein of interest can serve as a powerful alternative (Figure 5 b). As well as avoiding genetic manipulation of the target protein, its endogenous expression level is also retained, thereby increasing experimental authenticity.27 In exchange, possible functional and structural changes upon antigen binding need to be examined and evaluated. Among others, chromobodies have been developed for the live‐cell imaging of endogenous DNA replication in human cells,60 endogenous β‐catenin,61 HIV‐1 infection,62 cytoskeletal components27 and the progression of apoptosis.63 Moreover, nanobodies have shown beneficial characteristics for super‐resolution microscopy (Figure 5 c). Conventional detection reagents composed of a primary antibody and a fluorescently labelled secondary antibody lead to high linkage errors and loss of resolution since the actual dye is removed from the target structure (up to 30 nm). Nanobodies have a diameter of 2.5 nm and a height of approximately 4 nm, which makes them better suited for high‐resolution imaging of cellular structures.24 Guizetti and co‐workers used a GFP‐binding nanobody to elucidate the abscission stages of human cells and identified contractile filament helices with a diameter of 17 nm to be a central component of intercellular bridges.64 The gain in resolution was demonstrated with super‐resolution imaging of microtubules.65 This concept of using nanobodies for better fluorescence signal and resolution in imaging experiments has subsequently been further developed by labelling nanobodies with small organic fluorophores and gold nanoparticles.65, 66 In recent years, a number of such methods to facilitate the development of binders with advanced properties have been developed. An overview is given in the following section, and applications of the resulting functionalized nanobodies are discussed.
5. Chemical and Enzymatic Functionalization of Nanobodies: Concepts and Applications
The chemical labelling of nanobodies with fluorophores, their immobilization on solid supports, or their functionalization with recognition motifs, delivery agents, and other chemical groups broadly expands their applicability for imaging, proteomics, and novel therapeutic tools. While traditionally, nanobodies used in imaging are expressed fused to FPs like GFP and RFP, the labelling of nanobodies with small organic probes is expanding their utility as tools for biological research, including in super‐resolution imaging.65, 66 Due to their low production costs and long‐term stability, numerous nanobodies have been immobilized on different matrices and been used for immunoaffinity chromatography.67 This is of particular interest for state‐of‐the‐art mass spectrometry (MS)‐based proteomics since such technologies require the efficient enrichment of defined targets from complex protein mixtures. In this context, immobilized nanobodies that specifically bind fluorescently labelled proteins has allowed the combinatorial analysis of protein–protein interactions, DNA methyltransferase activity, and histone‐tail binding by fluorescent microscopy and mass spectrometry (Figure 6).27, 46c, 68
Figure 6.
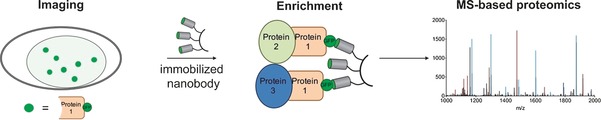
FP‐binding nanobodies like the GFP‐binding GBP enable the combinatorial analysis of proteins and their interaction partners through imaging and mass spectrometry‐based proteomics. A protein of interest (Protein 1) expressed as an FP fusion can be imaged using conventional methods. A nanobody that binds the FP is chemically immobilized on a solid support, which facilitates enrichment of the target protein and any interacting protein (Proteins 2 and 3). Subsequent MS analysis enables the identification and assignment of the co‐enriched interacting proteins.
Lys‐Selective Functionalization
In first proof‐of‐principle studies, nanobodies were randomly labelled at solvent‐exposed lysine residues by N‐hydroxysuccinimide (NHS) ester containing fluorophores to give heterogeneous protein mixtures (Figure 7 a).65, 69 Similarly, NHS‐functionalized matrices have been used to covalently attach and immobilize nanobodies.66, 70 Even though the unselective lysine labelling of nanobodies has proven valuable, it has been shown to affect the CDR loops, leading to a significant reduction in epitope recognition.14b
Figure 7.
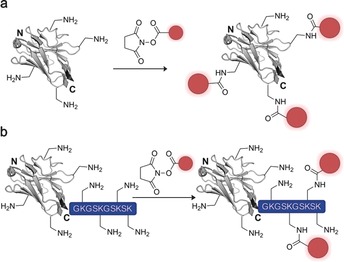
Random labeling of nanobodies. a) NHS‐activated probes/drugs are reacted with the nucleophilic ‐amine of a solvent‐exposed lysine residue, resulting in heterogeneous nanobody conjugate mixtures with partly reduced binding affinities. b) The C‐terminal fusion of a poly‐lysine stretch to nanobodies is intended to prevent unselective NHS‐based labeling of Lys residues within the CDR loops responsible for antigen binding. The nanobody is depicted in gray, the introduced functionality (e.g., fluorophore, drug, tracer) in red. The crystal structure of a GFP‐binding nanobody is used in (a) and (b) [PDB ID: 3G9A].17
Moreover, these conventional bioconjugation technologies have proven unfavorable for in vivo therapy and diagnostics since unselective functionalization of antigen‐binding proteins leads to variations in the number of probes attached, which, in combination with alteration of epitope recognition, can lead to impaired pharmacokinetic properties and stability.4b,4c, 71 Therefore, the site‐specific attachment of tracers and drugs to nanobodies to give homogenous conjugates with a defined number of probes per binder can offer advantageous properties. Platanova and co‐workers gained higher conjugation control by genetically adding a poly‐lysine stretch to the C terminus of GFP‐ and RFP‐binding nanobodies (Figure 7 b).72 Incubating the nanobodies with NHS‐activated fluorophores resulted in a labeling ratio of 1.0–1.5 fluorescent molecules per nanobody. However, whether the poly‐lysine stretch prevented fluorophore conjugation to ‐amino groups within the nanobody sequence was not shown and seems unlikely. In recent years, a number of methods for the site‐specific functionalization of proteins have been applied to homogeneously modify nanobodies. These methods include selective modification of unpaired cysteine residues, chemoenzymatic systems, expansion of the genetic code, and expressed protein ligation (EPL).
Labeling of (Unpaired) Cysteine Residues
Increased homogeneity of protein conjugates can be achieved by addressing cysteine residues, which are less abundant in comparison to lysine.73 In this case, the free thiol group of a reduced cysteine is converted with a cysteine‐selective chemical entity carrying the probe or drug. Among others, maleimides are the most common functional groups used for the labelling of cysteines.74 However, since proteins often contain several cysteine residues that are involved in the formation of interchain disulfide bonds, an additional reduction step is required and the resulting conjugates are once again heterogeneous mixtures. The introduction of an additional cysteine into the protein of interest is a possible way to circumvent this limitation and has found widespread application in nanobody functionalization.14a, 75 In most cases, the cysteine has been introduced at the C terminus of the nanobody, thus ensuring that the conjugation site is most distal from the antigen‐binding interface (Figure 8 a). Massa et al. used an engineered cysteine nanobody to produce homogeneous biomarkers for human epidermal growth factor receptor 2 (HER2)‐expressing cancer cells.14a Along these lines, single cysteine nanobodies against carbonic anhydrase IX (CAIX) and prostate‐specific membrane antigen (PSMA9), labelled with an infrared dye and a radiolabel, respectively, have been applied to the in vivo diagnosis of breast and prostate cancer (Figure 8 b).75a,75d
Figure 8.
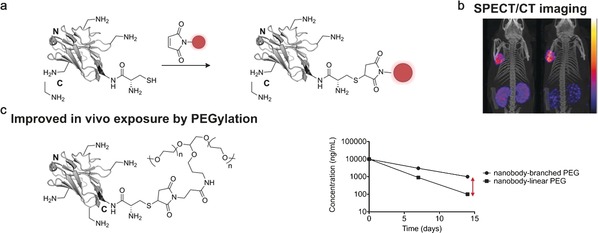
Nanobody functionalization through labelling of unpaired cysteine residues. a) Nanobodies do not possess free cysteine residues. Therefore, incorporating a single cysteine residue into a nanobody (mostly to the C terminus) is an easy way to site‐specifically attach a probe or drug through cysteine‐selective chemistry (shown for a maleimide‐functionalized probe). b) A prostate cancer specific nanobody (antigen PSMA) containing a C‐terminal unpaired cysteine was site‐specifically functionalized with a radiolabel by the use of maleimide chemistry. Single‐photon emission computed tomography (SPECT)/CT images of mice bearing prostate cancer tumors in the left shoulder were taken 3 h (left) and 24 h (right) after injection of the nanobody. Scale from 0 to 0.015 kBq (left) and 0 to 0.005 kBq (right).75a c) A tumor necrosis factor alpha (TNF) nanobody was conjugated to linear and branched polyethylene glycol (PEG) chains through maleimide chemistry. Subsequent pharmacokinetic experiments in mice, rats, and monkeys revealed that the branched PEG conjugates show significantly improved in vivo circulation time compared to linear PEG in all tested species as schematically shown in the concentration–time plot. The nanobody is depicted in gray, the introduced functionality in red. The crystal structure of a GFP‐binding nanobody is used in (a) and (c) [PDB ID: 3G9A].17
In 2012, Vugmeyster and co‐workers covalently attached branched and linear polyethylene glycol (PEG) linkers to the C terminus of single cysteine nanobodies to prolong their in vivo circulation time.75e Pharmacokinetic and biodistribution profiles in three different species showed that the site‐specific attachment of PEG chains successfully protected the nanobody by masking sites related to cellular uptake, proteolysis, and other clearance pathways (Figure 8 c). Moreover, DNA‐binding polyethyleneimine‐maleimide has been conjugated to a single cysteine nanobody against MUC1‐over‐expressing cancer cells to selectively induce apoptosis by polyethylenimine/DNA delivery.75c However, the C‐terminal attachment of cysteine residues often results in dimerization of the nanobodies and glutathione capping of the unpaired cysteine, thus making an additional reduction step prior to functionalization unavoidable.14a Therefore, Pleiner et al. analyzed the tertiary structure of a nanobody binding the Xenopus nuclear pore complex and engineered a cysteine at the surface of the nanobody framework region that is less prone towards capping and protein dimerization.14b In general, however, introducing additional cysteines into a nanobody can result in reduced expression yields.14a, 71
Chemoenzymatic Labelling:
Ever since the bacterial biotin ligase (BirA) was repurposed to site‐specifically biotinylate a protein of interest, different labelling methods have been developed that are built upon the reinterpretation of a naturally occurring enzyme.76 In vivo biotinylation by BirA was one of the first chemoenzymatic methods applied to nanobodies. Here, the C terminus of the nanobody is genetically fused to a short biotin acceptor domain (BAD) and co‐expressed in human cells with BirA. The enzyme activates biotin through monophosphorylation and transfers the biotinyl moiety to the target protein during expression. This system has been applied to generate nanobody‐based ELISA and immunosensors for the rapid detection of influenza and apolipoprotein (Figure 9 a).77 However, being limited to the biotinylation of proteins restricts the applicability of this method.
Figure 9.
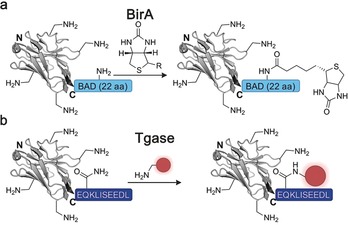
Chemoenzymatic functionalization of nanobodies. a) The biotin ligase BirA enables the in vivo attachment of biotin to nanobodies functionalized with the 22 amino acid biotin acceptor domain (BAD, light blue). b) Transglutaminases have been used to attach a functionality to nanobodies by generating an isopeptide bond between the glutamine of a short C‐terminal recognition sequence (dark blue) and an amine‐carrying probe. The nanobody is depicted in gray, the introduced functionality in red. The crystal structure of a GFP‐binding nanobody is used in (a) and (b) [PDB ID: 3G9A].17
Transglutaminases are another family of enzymes that have been adapted to site‐specifically modify proteins. In nature, they play an important role in the crosslinking of proteins and catalyze the formation of an isopeptide bond between the ‐carbonyl amide group of glutamines and the ‐amine group of lysines.78 In principle, transglutaminases can target any glutamine residue within a protein of interest as long as it is positioned in a disordered or highly flexible region of the biomolecule.79 Since nanobodies do not contain such glutamine residues, transglutaminases were successfully applied for their site‐specific modification by placing a glutamine‐containing c‐myc‐tag (EQKLISEEDL) at the protein C terminus (Figure 9 b).80 Although this was only used to biotinylate nanobodies, Fabs and other antigen‐binding proteins have been functionalized with different entities, including fluorophores and drugs, and in principle, these findings should be applicable to nanobodies too.80, 81
The transpeptidase Sortase A from Staphylococcus aureus specifically recognizes the consensus sequence LPXTG (Sortag) that can be placed at the C terminus of a protein of interest.82 A nucleophilic attack of the thiol at C148 on the enzyme cleaves the amide bond between glycine and threonine of the Sortag, leading to a thioacyl intermediate. A second nucleophilic attack by an incoming glycine peptide carrying a payload of choice results in the site‐specific functionalization of proteins through a native amide bond.82, 83 Moreover, Sortase A has been applied for the N‐terminal modification of proteins and shows substantial promiscuity with respect to nucleophilic substrates.84 Witte and co‐workers used Sortase A to generate nanobody dimers through C‐to‐C fusion, as well as bispecific nanobodies against GFP and mouse class II MHC products in yields of up to 90 %.85
In addition to that, Sortase A has been used to site‐specifically attach fluorophores and radiotracers for single photon emission computed tomography (SPECT) and positron emission tomography (PET) imaging in vivo (Figure 10 a).86 In a recent study, Sortase A labelling was combined with an engineered unpaired cysteine to achieve double functionalization of nanobodies and site‐specifically fluorescently labelled nanobody dimers (Figure 10 b).87 A drawback of using Sortase A to functionalize nanobodies is the reversibility of the amide‐bond formation and the resulting need for a high substrate excess to drive the reaction towards completion.
Figure 10.
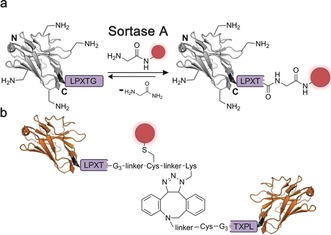
Sortase A functionalization of nanobodies. a) The transpeptidase Sortase A catalyzes the reversible formation of an amide bond between threonine of the Sortag LPXTG (purple) and a glycine‐functionalized probe. b) Sortase A functionalization have been combined with maleimide chemistry and strain‐promoted azide–alkyne cycloaddition (SPAAC) to generate fluorescently labelled nanobody dimers binding to class II major histocompatibility complex (MHC) and/or CD11b proteins. The nanobody is depicted in gray or orange, the introduced functionality in red. The crystal structure of a GFP‐binding nanobody is used in (a) and (b) [PDB ID: 3G9A].17
The lipoic acid ligase (LpIA) is an enzyme that recognizes a 13 amino acid peptide tag, the lipoic acid acceptor peptide (LAP, GFEIDKVWYDKADA), and ligates lipoic acid to the side chain of a lysine residue (Figure 11 a).89 By introducing mutations in the lipoic acid binding pocket of the enzyme, mutant enzyme variants could be generated that accept unnatural substrates instead of lipoic acid. The enzyme has since been used in numerous applications to attach biotin, fluorophores, or other labels to the peptide tag. In this case, a previously generated mutant of the enzyme was used that can attach a bioorthogonal aryl‐aldehyde handle to the acceptor peptide.90 Gray et al. applied this to the modification of an anti‐HER2 nanobody with a dinitrophenyl moiety.91 By incubating HER2‐positive cancer cells with the nanobody and an anti‐dinitrophenyl IgG antibody, the authors could trigger an immune response leading to antibody‐dependent cytotoxicity (Figure 11 b). The functionalized nanobody serves as an immunotherapeutic that mediates between the cancer antigen and the immune system.
Figure 11.
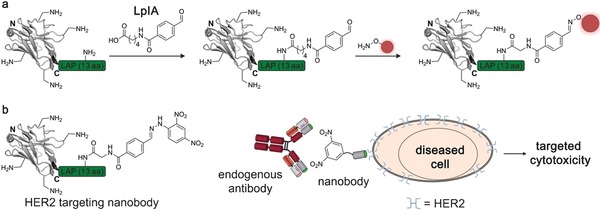
Functionalization of nanobodies by using the lipoic acid ligase (LpIA). a) LpIA recognizes the 13 amino acid LAP tag (green) and ligates an aldehyde‐containing lipoic acid derivative to the side chain of a lysine residue. A subsequent oxime‐forming reaction enables site‐specific functionalization of the nanobody. b) A nanobody‐based activation immunotherapeutic is shown. An anti‐HER2 nanobody was site‐specifically functionalized with a dinitrophenyl (DNP) moiety using LpIA. The DNP acts as an endogenous antibody‐recruiting domain, facilitating a targeted immune response upon HER2 binding of the nanobody. The nanobody is depicted in gray, the introduced functionality in red. The crystal structure of a GFP‐binding nanobody is used in (a) and (b) [PDB ID: 3G9A].17
Recently, our research groups have added to the toolbox of site‐specific chemoenzymatic protein functionalization92 with Tub‐tag labeling. This is versatile method that allows the C‐terminal attachment of small unnatural tyrosine derivatives using the recombinant enzyme tubulin tyrosine ligase (TTL) (Figure 12 a). The derivatives carry unique chemical entities like azides, aldehydes, and iodides, thus making Tub‐tag compatible with several well‐established bioorthogonal reactions. In detail, the TTL recognizes a 14 amino acid recognition sequence (Tub‐tag, VDSVEGEGEEEGEE) fused to the C terminus of a protein of interest and covalently attaches the tyrosine derivative of choice in an ATP‐dependent reaction. Moreover, based on the enlarged catalytic cavity formed by the enzyme during the catalytic cycle (Figure 12 b), we were able to significantly broaden the substrate scope of Tub‐tag labeling.93 This allowed us to advance the versatile two‐step method to a fast one‐step labelling strategy that makes use of fluorescent coumarin and biotin derivatives as TTL substrates (Figure 12 c, d). Since the C terminus of nanobodies is most distant from their antigen‐binding region, we envisioned Tub‐tag labelling as a well‐suited technology for nanobody functionalization. Therefore, we recombinantly expressed a number of Tub‐tagged nanobodies and generated nanobody‐based immunoprecipitation tools and super‐resolution probes with minimal linkage errors.92a, 93
Figure 12.
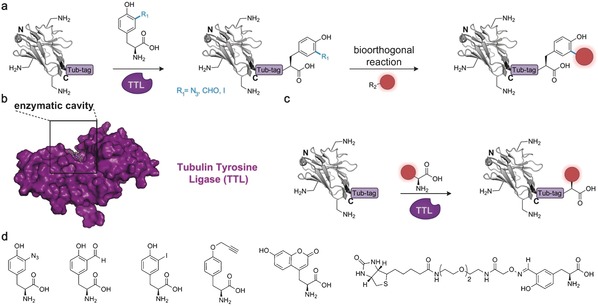
Tub‐tag labelling for the functionalization of nanobodies and recombinant antigen‐binding proteins. a) Versatile two‐step labelling of nanobodies. TTL‐mediated incorporation of tyrosine derivatives containing chemical reporters enables successive conjugation to a functional probe (red) using a bioorthogonal reaction. b) The TTL (PDB ID: 4IHJ)88 forms an extended cavity during the catalytic cycle. This cavity allows lead to a broad substrate tolerance. c) Fluorescent or biotinylated substrates of TTL enable the efficient one‐step labelling of biomolecules. d) Substrates of TTL include ortho‐and para‐functionalized tyrosine derivatives, fluorescent coumarin amino acids. and large biotinylated tyrosine derivatives. The nanobody is depicted in gray, the introduced functionality in red, and TTL in purple. The crystal structure of a GFP‐binding nanobody is used in (a) and (c) [PDB ID: 3G9A].17
Amber Suppression
Another prominent technology for the site‐specific labelling of proteins is amber suppression.94 Here, an unnatural amino acid carrying a unique chemical entity (a bioorthogonal group) is incorporated into a random site within the sequence of the target protein using an engineered expression machinery. In a second step, a payload of choice is site‐specifically attached to the bioorthogonal group. One of the major advantages of amber suppression is the high variety of unnatural amino acids that can be incorporated using the system.95 Recently, the unnatural amino acid AmAzZLys was incorporated into a nanobody against the epidermal growth factor receptor (EGFR).96 AmAzZLys is a lysine derivative that contains a benzylic amine and azide, which allowed subsequent double functionalization of the nanobody with a fluorophore and a 5 kDa PEG chain (Figure 13 a). Moreover, the benzylic azide was used to perform photoinduced crosslinking to EGFR upon antigen binding (Figure 13 b). However, amber suppression is technically demanding and results in a significantly lower expression yield compared to the wild‐type nanobody, thus limiting the usage of amber suppression for nanobody functionalization.96
Figure 13.
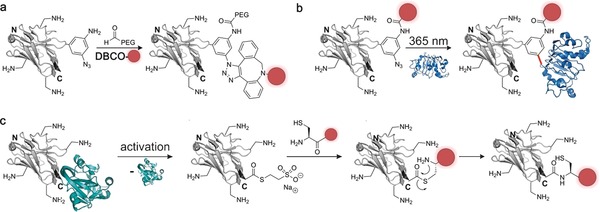
Methods that have been used for the site‐specific labelling of nanobodies (gray). a) Amber suppression was used to install the amino acid AmAzZLys into a nanobody. The benzylic amine of the unnatural amino acid was reacted with an aldehyde‐containing PEG chain and the benzylic azide with a dibenzocyclooctyl fluorophore, resulting in a doubly functionalized nanobody. b) AmAzZLys was incorporated into a nanobody targeting the epidermal growth factor receptor (EGFR, PDB ID: 3P0Y99b). The amino group was used for fluorescent labelling, while the azide was activated by UV irradiation, and photo‐cross‐linking between the nanobody and EGFR was performed. c) Expressed protein ligation (EPL) shown for the C‐terminal functionalization of nanobodies. The respective nanobody is expressed as an intein fusion (intein shown in turquoise, PDB ID: 4GIG99a). Activation with a reducing agent like DTT or mercaptoethanol results in the formation of a highly reactive thioester. Transthioesterification initiated by nucleophilic attack of a cysteine‐containing probe, followed by a S‐to‐N acyl transfer results in the formation of a stable amide bond and the site‐specific functionalization of the nanobody. The nanobody is depicted in gray, the introduced functionality in red, EGFR in blue, and intein in turquoise. The crystal structure of a GFP‐binding nanobody is used in (a), (b), and (c) [PDB ID: 3G9A].17
Expressed Protein Ligation and Protein trans‐Splicing
Expressed protein ligation (EPL) is a technology that is based on the naturally occurring splicing of proteins.97 A protein of interest is expressed as a fusion with a mutated intein that generates a highly reactive C‐terminal thioester upon activation with a reducing agent like 2‐mercaptoethanol or dithiothreitol. In a following ligation reaction to a peptide carrying an N‐terminal cysteine, a new peptide bond is generated through S‐to‐N acyl transfer.97 Most of the time, the cysteine peptide is synthesized by solid‐phase peptide synthesis (SPPS), which enables the straightforward incorporation of functional probes and payloads. Therefore, EPL has broad application for the site‐specific functionalization of proteins and nanobodies (Figure 13 c).74b An alkyne‐modified cysteine was recently incorporated to a vascular cell adhesion molecule 1 (VCAM‐1)‐binding nanobody by EPL and further modified with biotin by a subsequent copper‐catalyzed click reaction.98
Optimizing the expression and ligation protocol allowed the generation of decent amounts of nanobody with high functionalization yields of up to 100 %. In another study, EPL was used for the double functionalization of a vascular‐tumor‐targeting nanobody with polymersomes for drug delivery and biotin.100 While EPL facilitates the C‐terminal functionalization of proteins, Bachmann et al. recently made use of the GOS‐TerL intein and a protein trans‐splicing reaction to functionalize a GFP‐binding nanobody with a synthetic fluorophore at its N terminus.101 However, when choosing EPL or intein‐fusion strategies, refolding from inclusion bodies is often required to achieve functional proteins, which significantly increases experimental effort.102
In recent years, huge efforts have been made to broaden the toolbox of conjugation methods for the functionalization of nanobodies and other antigen‐binding proteins. While all of the methods discussed in this review have their pros and cons, the methodological versatility enables the interested scientist to pick one of the validated techniques according to the requirements of the individual application. Nevertheless, to fully maintain protein function, the site‐specific C‐terminal functionalization of nanobodies has proven beneficious compared to unselective labeling strategies. Owing to reduced engineering effort and high conjugation yields, the labeling of unpaired cysteine residues and chemoenzymatic functionalization strategies are particularly suitable for achieving this goal.
In addition to the functionalization of nanobodies and other binders, their subsequent cellular delivery is of high interest to the scientific community. Recent achievements are discussed in the next section.
6. Cellular Delivery of Small Antigen‐Binding Proteins
The direct cellular delivery of nanobodies and antigen‐binding proteins is a long‐standing goal in cell biology and medicine, since it would offer a non‐integrative way to analyze and manipulate cellular processes, protein–protein interactions, and protein function.103 Therefore, intense research has been invested within recent decades to develop general methods to achieve this goal.11c, 104 In principle, intracellular functional antigen‐binding proteins can be obtained by transfecting cells, for example with the use of intrabodies that are optimized for intracellular expression.11b However, the functional cytosolic expression of binders remains challenging and their functionalization with small molecules like affinity tags, fluorophores, and drugs is not possible when using such techniques.11b,11c, 104a, 105
In this sense, the delivery of functional antigen‐binding proteins into living cells would vastly expand the methodological repertoire of antigen‐binding proteins for intracellular use.107 Supercharged proteins are a class of engineered proteins that are able to penetrate mammalian cells.108 In 2016, Bruce et al. made use of this concept and successfully engineered nanobodies against GFP, HER2, and β‐lactamase in order to make them cell‐permeable.109 To accomplish this, they “resurfaced” the nanobodies, that is, mutated several amino acid residues on the surface of the proteins to basic residues, resulting in net positive charges of +14 and +15 for the nanobodies overall. These polycationic nanobodies could then penetrate cells (incubated with 250–500 nm nanobody) and showed localization in the cytosol and not in endosomes (Figure 14 a). While they demonstrated that the engineering does not impact the nanobody structure or the ability of the GFP‐binding nanobody to bind its antigen, it is difficult to estimate what effect the high net charge could have on localization and antigen binding in a generalistic manner. Besides making the proteins directly cell‐permeable, delivery of proteins can also be achieved using biophysical methods like microinjection and electroporation110 and by using one of the increasingly important carrier‐based delivery systems.105a In principle, the carrier‐based delivery of cargo to cells follows two main pathways; endocytosis‐dependent uptake (Figure 14 b) and the transduction across the cell membrane (Figure 14 c).
Figure 14.
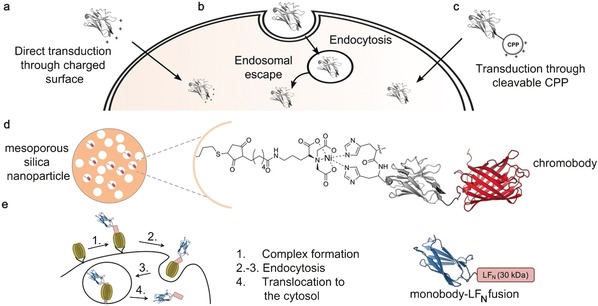
Depiction of delivery mechanisms that have been used for the cellular uptake of nanobodies. Nanobodies have been delivered into cells through the use of a) charged surface mediated transduction, b) endocytosis, or c) cyclic cell‐penetrating peptides (CPPs). The nanobody is depicted in gray, the introduced functionality in red. d) Thiol‐containing mesoporous silica nanoparticles (MSNs) were conjugated to a maleimide‐functionalized nitrilotriacetic acid (NTA) linker. Activation with a metal ion (shown for Ni) facilitated binding of a His6‐chromobody. The MSN–chromobody complex showed endosomal entrapment upon cellular incubation, thus necessitating endosomal escape triggers like the peptide INF7 to enable cytosolic distribution of the chromobody. e) A cell delivery system based on the anthrax lethal toxin. Recombinant antigen‐binding proteins (shown for a monobody) are expressed as a fusion with the 30 kDa N‐terminal domain of the toxin enzyme lethal factor (LFN). The protective‐antigen (PA)‐based pore‐forming transporter (PA oligomers shown in green) is bound to a host‐cell receptor. The binder‐LFN forms a complex with the transporter (step 1) and endocytosis is initiated (step 2 and 3). Due to the acidic environment in the endosome, the PA oligomers form a transmembrane pore, unfolds the binder‐LFN fusion protein and initiates its translocation to the cytosol (step 4). The nanobody is depicted in gray, RFP (PDB ID: 1GGX106) in red, and the monobody in blue (PDB ID: 3RZW18). The crystal structure of a GFP‐binding nanobody is used in (a), (b), (c), and (d) [PDB ID: 3G9A].17
Endocytosis is the predominant pathway described for the delivery of biomolecules.111 Here, extracellular macromolecules pass through the plasma membrane via encapsulation in vesicles and become trapped in endosomes, which can lead to lysosomal degradation. To be available inside the cell, the biomolecule then needs to escape from the endosome. This can be achieved through lipid or osmotic pressure mediated destabilization of the membrane or translocation of the cargo through transmembrane pores.111, 112 In 2016, Chiu and co‐workers developed large‐pore mesoporous silica nanoparticles (MSNs) that are functionalized with nitrilotriacetic acid (NTA) groups at the internal surface (Figure 14 d).113 Activation of the complex with various metal ions enabled the covalent attachment of a His6‐tagged GFP‐chromobody. Incubation of living cells with these complexes at a concentration of 25 nm resulted in endocytic uptake and endosomal entrapment of the conjugate. Although a small amount of chromobody was able to escape the endosomes (1–2 %), probably due to a proton sponge effect generated by the His6‐tag, the use of endosomal‐escape triggers like fusogenic peptide INF7, acidity, DMSO, or chloroquine was necessary to obtain a decent amount ( 17 %) of chromobody within the cytosol. Importantly, intracellular co‐localization of the chromobody with its antigen confirmed full functionality of the antigen‐binding protein, and the MSN‐NTA complex allows combination with any His‐tagged protein. One year later, Röder et al. combined fluorescently labelled nanobodies (via NHS‐esters) with a number of different nanoparticle‐forming oligoaminoamides equipped with succinoyl tetraethylene pentamine units that trigger endosomal release.114 Incubation of the encapsulated nanobodies with HeLa cells at 3.6 μm concentrations resulted in either receptor‐specific or non‐specific endocytic uptake. Through this strategy, they were able to achieve high co‐localization with the antigen and intracellular availability of the nanobody of up to 60 %. Nevertheless, endosomal escape remained a bottleneck, as indicated by a significant amount of nanoparticles entrapped in cellular vesicles.
In the past few years, a receptor‐dependent delivery system based on the anthrax lethal toxin has been developed and applied for the delivery of a number of biomolecules.115 In nature, protective antigen (PA) binds to anthrax receptors on human cells and oligomerizes to form heptamers or octamers. Once lethal factor (LF) binding occurs, the whole complex is endocytosed. Subsequently, the acidic milieu of the endosomes initiates a rearrangement of PA, leading to the formation of a PA pore in the membrane that allows translocation of the LF to the cytosol. Liao et al. made use of this system and chemoenzymatically attach LF to an affibody, a DARPin, and a monobody by using Sortase A (Figure 14 e).116 In this way, they were able to achieve delivery of these recombinant binders to the cytosol with maintained antigen‐binding properties (cells were incubated with pm–nm concentrations of recombinant binder and 20 nm PA). Nevertheless, translocation through the PA pore requires protein unfolding and subsequent refolding, thus limiting this approach to antigen‐binding proteins that are readily folded within the reductive environment of the cytosol. In this sense, most of the aforementioned examples for the cellular delivery of nanobodies and antigen‐binding proteins require endosomal escape mechanisms that constitute a major bottleneck for addressing intracellular antigens in living cells.
The use of cell‐penetrating peptides (CPPs) might circumvent this limitation since they have been shown to enable the direct cellular uptake of functional full‐length proteins.117 Therefore, the generation of cell‐penetrating antigen‐binding proteins by CPP fusion is the next logical step to fulfill the goal of immediate bioavailability and antigen binding. Herce and Schumacher et al. recently made use of this concept and ligated linear and cyclic HIV‐derived TAT and deca‐arginine (R10) peptides to the C terminus of two different GFP‐binding nanobodies by EPL (Figure 15 a).118 Subsequent studies revealed that the conjugates produced transduction rates of up to 95 % in cells from different cell lines when incubated with low μm concentrations (10 μm). Moreover, the uptake initiated by R10 peptides is up to three times increased compared to that initiated by TAT, and cyclization of the peptides further increases uptake efficiency. Based on these findings, cyclic R10 peptides are most suitable for the generation of cell‐permeable nanobodies. cR10 conjugates were then applied for the co‐transport of GFP and GFP fusion proteins with a size of up to 83 kDa, including the therapeutically relevant Mecp2 protein.119 Moreover, the cell‐permeable nanobodies have been used to visualize protein–protein interactions in living cells by slightly adopting the previously published three‐hybrid assay.48 These experiments show that cell‐permeable nanobodies are powerful tools for cell biology and the delivery of recombinant and therapeutically relevant proteins. Since arginine‐rich CPPs localize to the nucleolus and bind to negatively charged RNA, addressing targets within the cytosol requires the rapid cleavage of the CPP after cellular uptake. Therefore, Herce and Schumacher et al. site‐specifically conjugated a fluorophore via EPL to the C terminus of the nanobody and used the cysteine created at the EPL junction for linkage of the CPP via a disulfide (Figure 15 b). Uptake studies and fluorescence microscopy revealed efficient internalization of the Cy5‐labelled nanobody without nucleolar enrichment, thus indicating reductive CPP cleavage within cytosol (Figure 15 c). This conjugate was used for the visualization of antigens in living cells, and constitutes a very promising tool for intracellular immunostaining and immunomanipulation.
Figure 15.
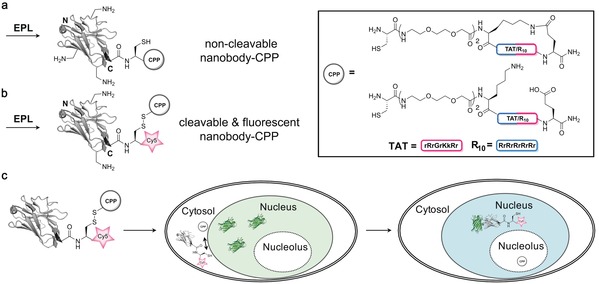
Charge‐induced membrane transduction of nanobodies initiated by cell penetrating peptides (CPPs). a–b) Expressed protein ligation (EPL) of nanobodies was used to site‐specifically conjugate CPPs through a) a stable amide bond to give non‐cleavable nanobody–CPP conjugates or b) a cleavable disulfide that gets reduced within the reductive cytosolic environment (cleavable and fluorescent nanobody–CPP). c) Incubation with different cell lines revealed efficient uptake in up to 95 % of the cells when incubated with low μm concentrations. Upon incubation of the Cy5‐labelled cleavable nanobody with cells, the conjugate crosses the cellular membrane into the cytosol, CPP cleavage is initiated, and the fluorescently labelled nanobody binds its nuclear antigen GFP. The nanobody is depicted in gray, GFP in green, and Cy5 in red. The crystal structure of a GFP‐binding nanobody is used in (a), (b), and (c) [PDB ID: 3G9A].17
7. Conclusion
Ever since their discovery, nanobodies have emerged as powerful antigen binders that, together with conventional antibodies and other classes of antigen‐binding proteins, form a versatile toolbox for biochemistry, cell biology, and beyond. Nanobodies are characterized by their small size, increased solubility and stability compared to other antigen‐binding proteins, and enlarged CDR loops that open the door towards previously inaccessible antigens. The fusion of nanobodies to fluorescent proteins initiated the generation of highly sophisticated, functional binders that have been further advanced through conjugation to organic fluorophores, tracers, and drugs, as well as the immobilization of nanobodies to solid supports. While such conjugates were originally synthesized using NHS chemistry, resulting in heterogeneous mixtures, strategies for the site‐specific functionalization of nanobodies have been developed, leading to products with improved biophysical properties. New approaches for the cellular delivery of functional antigen‐binding proteins allows their use within the cellular environment, which constitutes a major step in live‐cell immunolabelling and antigen manipulation. Taken together, the versatility of antigen‐binding proteins, methods for their functionalization, and strategies for their cellular delivery forms a powerful basis for the generation of next‐generation diagnostics and therapeutic tools with striking properties.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Jonas Helma completed his graduate and doctoral studies at the Ludwig‐Maximilians‐University Munich in the group of Heinrich Leonhardt, focusing on recombinant nanobodies and their use in biochemistry and cell biology. Since 2017, he has been co‐leader of an EXIST‐funded research group at the LMU Munich, together with Dominik Schumacher, developing techniques for site‐specific antibody conjugates in research and therapy.

Biographical Information
Anselm F. L. Schneider completed his undergraduate studies and master's degree in Biochemistry at the Freie Universität Berlin, travelling to the University of Oxford for his master's thesis with a scholarship from the Deutscher Akademischer Austauschdienst. Since 2016, he has been working towards a PhD in the laboratory of Christian Hackenberger, supported by a PhD fellowship from the Fonds der chemischen Industrie.

Biographical Information
Heinrich Leonhardt studied Biochemistry at the Freie Universität Berlin and finished his doctoral studies at the Max‐Planck‐Institute for Molecular Genetics in 1989 in the group of Thomas A. Trautner. After postdoctoral training at the Harvard Medical School in Boston, he started his own research group at the Franz‐Volhard‐Klinik and the Max‐Delbrück‐Centre for Molecular Medicine in Berlin in 1995. Since 2002, he has held a Professorship at the Faculty of Biology of the Ludwig‐Maximilians‐University Munich. The main topic of his research group is the role and regulation of DNA modifications in mice and man in development and disease.

Biographical Information
Dominik Schumacher studied chemistry and business chemistry at the University of Düsseldorf. In 2013, he joined Christian Hackenberger's group for his Ph.D. studies, which were funded by the Kekule scholarship of the Fonds der Chemischen Industrie. Since 2017, he has been co‐leader of an EXIST‐funded research group at the LMU Munich together with Jonas Helma. His work focusses on the development of site‐specific protein functionalization, in particular antigen‐binding proteins with defined activities.

Biographical Information
Christian P. R. Hackenberger completed his graduate studies at the universities of Freiburg and UW Madison and his doctoral studies in 2003 at the RWTH Aachen in the group of Carsten Bolm. After a postdoctoral position at MIT, he started his own group at the Freie Universität Berlin in 2005. In 2012, he was appointed Leibniz‐Humboldt Professor for Chemical Biology at the Leibniz‐Research Institute for Molecular Pharmacology and the Humboldt Universität zu Berlin. His group works on the development new chemoselective and biorthogonal reactions and novel approaches to functional protein synthesis and delivery.

Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SPP1623) to C.P.R.H. (HA 4468/9‐1) and H.L. (LE 721/13‐2), the Einstein Foundation Berlin (Leibniz‐Humboldt Professorship) and the Boehringer‐Ingelheim Foundation (Plus 3 award) to C.P.R.H., the Fonds der Chemischen Industrie (FCI) to C.P.R.H. and to D.S. (Kekulé fellowship) and A.F.L.S. (Chemiefonds fellowship), and the Nanosystems Initiative Munich (NIM) to H.L.
D. Schumacher, J. Helma, A. F. L. Schneider, H. Leonhardt, C. P. R. Hackenberger, Angew. Chem. Int. Ed. 2018, 57, 2314.
References
- 1. Vidarsson G., Dekkers G., Rispens T., Front. Immunol. 2014, 5, 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Coombs R. R., Mourant A. E., Race R. R., Br. J. Exp. Pathol. 1945, 26, 255–266; [PMC free article] [PubMed] [Google Scholar]
- 2b. Yalow R. S., Berson S. A., J. Clin. Invest. 1960, 39, 1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Köhler G., Milstein C., Nature 1975, 256, 495–497; [DOI] [PubMed] [Google Scholar]
- 3b. Jones P. T., Dear P. H., Foote J., Neuberger M. S., Winter G., Nature 1986, 321, 522–525; [DOI] [PubMed] [Google Scholar]
- 3c. Morrison S. L., Johnson M. J., Herzenberg L. A., Oi V. T., Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Schumacher D., Hackenberger C. P., Curr. Opin. Chem. Biol. 2014, 22, 62–69; [DOI] [PubMed] [Google Scholar]
- 4b. Schumacher D., Hackenberger C. P., Leonhardt H., Helma J., J. Clin. Immunol. 2016, 36, 100–107; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Agarwal P., Bertozzi C. R., Bioconjugate Chem. 2015, 26, 176–192; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Junutula J. R., Raab H., Clark S., Bhakta S., Leipold D. D., Weir S., Chen Y., Simpson M., Tsai S. P., Dennis M. S., Lu Y., Meng Y. G., Ng C., Yang J., Lee C. C., Duenas E., Gorrell J., Katta V., Kim A., McDorman K., Flagella K., Venook R., Ross S., Spencer S. D., Wong W. L., Lowman H. B., Vandlen R., Sliwkowski M. X., Scheller R. H., Polakis P., Mallet W., Nat. Biotechnol. 2008, 26, 925–932; [DOI] [PubMed] [Google Scholar]
- 4e. Mantaj J., Jackson P. J. M., Rahman K. M., Thurston D. E., Angew. Chem. Int. Ed. 2017, 56, 462–488; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 474–502. [Google Scholar]
- 5. Patterson D. M., Nazarova L. A., Prescher J. A., ACS Chem. Biol. 2014, 9, 592–605. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Engvall E., Perlmann P., Immunochemistry 1971, 8, 871–874; [DOI] [PubMed] [Google Scholar]
- 6b. Van Weemen B. K., Schuurs A. H., FEBS Lett. 1971, 15, 232–236. [DOI] [PubMed] [Google Scholar]
- 7. Helma J., Cardoso M. C., Muyldermans S., Leonhardt H., J. Cell Biol. 2015, 209, 633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. H. W. Schroeder, Jr. , Cavacini L., J. Allergy Clin. Immunol. 2010, 125, S41–S52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Williams W. V., Weiner D. B., Biologically active peptides: Design, synthesis and utilization, Technomic Publishing Company, 1993. Lancaster, PA; [Google Scholar]
- 9b. Riechmann L., Muyldermans S., J. Immunol. Methods 1999, 231, 25–38. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Holliger P., Hudson P. J., Nat. Biotechnol. 2005, 23, 1126–1136; [DOI] [PubMed] [Google Scholar]
- 10b. Hudson P. J., Souriau C., Nat. Med. 2003, 9, 129–134. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Beck A., Wurch T., Bailly C., Corvaia N., Nat. Rev. Immunol. 2010, 10, 345–352; [DOI] [PubMed] [Google Scholar]
- 11b. Marschall A. L., Dubel S., Boldicke T., MAbs 2015, 7, 1010–1035; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Marschall A. L., Frenzel A., Schirrmann T., Schungel M., Dubel S., MAbs 2011, 3, 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frenzel A., Hust M., Schirrmann T., Front. Immunol. 2013, 4, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Hamers-Casterman C., Atarhouch T., Muyldermans S., Robinson G., Hamers C., Songa E. B., Bendahman N., Hamers R., Nature 1993, 363, 446–448; [DOI] [PubMed] [Google Scholar]
- 13b. Könning D., Zielonka S., Grzeschik J., Empting M., Valldorf B., Krah S., Schröter C., Sellmann C., Hock B., Kolmar H., Curr. Opin. Struct. Biol. 2016, 45, 10–16. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Massa S., Xavier C., De Vos J., Caveliers V., Lahoutte T., Muyldermans S., Devoogdt N., Bioconjugate Chem. 2014, 25, 979–988; [DOI] [PubMed] [Google Scholar]
- 14b. Pleiner T., Bates M., Trakhanov S., Lee C.-T., Schliep J. E. E., Chug H., Böhning M., Stark H., Urlaub H., Görlich D., eLIFE 2015, 4, e11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ward E. S., Gussow D., Griffiths A. D., Jones P. T., Winter G., Nature 1989, 341, 544–546. [DOI] [PubMed] [Google Scholar]
- 16. Muyldermans S., Annu. Rev. Biochem. 2013, 82, 775–797. [DOI] [PubMed] [Google Scholar]
- 17. Kirchhofer A., Helma J., Schmidthals K., Frauer C., Cui S., Karcher A., Pellis M., Muyldermans S., Casas-Delucchi C. S., Cardoso M. C., Leonhardt H., Hopfner K. P., Rothbauer U., Nat. Rev. Mol. Cell Biol. 2010, 17, 133–138. [DOI] [PubMed] [Google Scholar]
- 18. Koide A., Wojcik J., Gilbreth R. N., Hoey R. J., Koide S., J. Mol. Biol. 2012, 415, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eigenbrot C., Ultsch M., Dubnovitsky A., Abrahmsen L., Hard T., Proc. Natl. Acad. Sci. USA 2010, 107, 15039–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rauth S., Hinz D., Borger M., Uhrig M., Mayhaus M., Riemenschneider M., Skerra A., Biochem. J. 2016, 473, 1563–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Flajnik M. F., Kasahara M., Nat. Rev. Genet. 2010, 11, 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. De Genst E., Silence K., Decanniere K., Conrath K., Loris R., Kinne J., Muyldermans S., Wyns L., Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fuh G., Wu P., Liang W. C., Ultsch M., Lee C. V., Moffat B., Wiesmann C., J. Biol. Chem. 2006, 281, 6625–6631. [DOI] [PubMed] [Google Scholar]
- 24. Revets H., De Baetselier P., Muyldermans S., Expert Opin. Biol. Ther. 2005, 5, 111–124. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Nguyen V. K., Hamers R., Wyns L., Muyldermans S., EMBO J. 2000, 19, 921–930; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Stijlemans B., Conrath K., Cortez-Retamozo V., Van Xong H., Wyns L., Senter P., Revets H., De Baetselier P., Muyldermans S., Magez S., J. Biol. Chem. 2004, 279, 1256–1261; [DOI] [PubMed] [Google Scholar]
- 25c. Spinelli S., Desmyter A., Verrips C. T., de Haard H. J., Moineau S., Cambillau C., Nat. Struct. Mol. Biol. 2006, 13, 85–89. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Chothia C., Novotny J., Bruccoleri R., Karplus M., J. Mol. Biol. 1985, 186, 651–663; [DOI] [PubMed] [Google Scholar]
- 26b. Harmsen M. M., Ruuls R. C., Nijman I. J., Niewold T. A., Frenken L. G., de Geus B., Mol. Immunol. 2000, 37, 579–590; [DOI] [PubMed] [Google Scholar]
- 26c. Maass D. R., Sepulveda J., Pernthaner A., Shoemaker C. B., J. Immunol. Methods 2007, 324, 13–25; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26d. Muyldermans S., Atarhouch T., Saldanha J., Barbosa J. A., Hamers R., Protein Eng. 1994, 7, 1129–1135; [DOI] [PubMed] [Google Scholar]
- 26e. Vu K. B., Ghahroudi M. A., Wyns L., Muyldermans S., Mol. Immunol. 1997, 34, 1121–1131. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Rothbauer U., Zolghadr K., Tillib S., Nowak D., Schermelleh L., Gahl A., Backmann N., Conrath K., Muyldermans S., Cardoso M. C., Leonhardt H., Nat. Methods 2006, 3, 887–889; [DOI] [PubMed] [Google Scholar]
- 27b.C. Hemmer, S. Djennane, L. Ackerer, K. Hleibieh, A. Marmonier, S. Gersch, S. Garcia, E. Vigne, V. Komar, M. Perrin, C. Gertz, L. Belval, F. Berthold, B. Monsion, C. Schmitt-Keichinger, O. Lemaire, B. Lorber, C. Gutierrez, S. Muyldermans, G. Demangeat, C. Ritzenthaler, Plant. Biotechnol. J 2017, DOI: https://doi.org/10.1111/pbi.12819;
- 27c. Conrath K., Vincke C., Stijlemans B., Schymkowitz J., Decanniere K., Wyns L., Muyldermans S., Loris R., J. Mol. Biol. 2005, 350, 112–125. [DOI] [PubMed] [Google Scholar]
- 28. Cortez-Retamozo V., Backmann N., Senter P. D., Wernery U., De Baetselier P., Muyldermans S., Revets H., Cancer Res. 2004, 64, 2853–2857. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Rouet R., Dudgeon K., Christie M., Langley D., Christ D., J. Biol. Chem. 2015, 290, 11905–11917; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29b. Ma X., Barthelemy P. A., Rouge L., Wiesmann C., Sidhu S. S., J. Mol. Biol. 2013, 425, 2247–2259. [DOI] [PubMed] [Google Scholar]
- 30. Vincke C., Loris R., Saerens D., Martinez-Rodriguez S., Muyldermans S., Conrath K., J. Biol. Chem. 2009, 284, 3273–3284. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Van Audenhove I., Gettemans J., EBioMedicine 2016, 8, 40–48; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31b. Coppieters K., Dreier T., Silence K., de Haard H., Lauwereys M., Casteels P., Beirnaert E., Jonckheere H., de Wiele C. Van, Staelens L., Hostens J., Revets H., Remaut E., Elewaut D., Rottiers P., Arthritis Rheum. 2006, 54, 1856–1866; [DOI] [PubMed] [Google Scholar]
- 31c. Verheesen P., de Kluijver A., van Koningsbruggen S., de Brij M., de Haard H. J., van Ommen G. J., van der Maarel S. M., Verrips C. T., Hum. Mol. Genet. 2006, 15, 105–111. [DOI] [PubMed] [Google Scholar]
- 32. Dreier B., Plückthun A., Methods Mol. Biol. 2012, 805, 261–286. [DOI] [PubMed] [Google Scholar]
- 33. Koide S., Koide A., Lipovsek D., Methods Enzymol. 2012, 503, 135–156. [DOI] [PubMed] [Google Scholar]
- 34. Gebauer M., Skerra A., Methods Enzymol. 2012, 503, 157–188. [DOI] [PubMed] [Google Scholar]
- 35.
- 35a. Al-Khodor S., Price C. T., Kalia A., Kwaik Y. A., Trends Microbiol. 2010, 18, 132–139; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35b. Li J., Mahajan A., Tsai M. D., Biochemistry 2006, 45, 15168–15178; [DOI] [PubMed] [Google Scholar]
- 35c. Plückthun A., Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [DOI] [PubMed] [Google Scholar]
- 36. Gross G. G., Junge J. A., Mora R. J., Kwon H. B., Olson C. A., Takahashi T. T., Liman E. R., Ellis-Davies G. C., McGee A. W., Sabatini B. L., Roberts R. W., Arnold D. B., Neuron 2013, 78, 971–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Löfblom J., Feldwisch J., Tolmachev V., Carlsson J., Stahl S., Frejd F. Y., FEBS Lett. 2010, 584, 2670–2680. [DOI] [PubMed] [Google Scholar]
- 38. Silverman G. J., Goodyear C. S., Siegel D. L., Transfusion 2005, 45, 274–280. [DOI] [PubMed] [Google Scholar]
- 39.
- 39a. Nord K., Gunneriusson E., Ringdahl J., Stahl S., Uhlen M., Nygren P. A., Nat. Biotechnol. 1997, 15, 772–777; [DOI] [PubMed] [Google Scholar]
- 39b. Lofdahl P. A., Nord O., Janzon L., Nygren P. A., N. Biotechnol. 2009, 26, 251–259. [DOI] [PubMed] [Google Scholar]
- 40. Flower D. R., Biochem. J. 1996, 318, 1–14.8761444 [Google Scholar]
- 41. Dmitriev O. Y., Lutsenko S., Muyldermans S., J. Biol. Chem. 2016, 291, 3767–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Monegal A., Ami D., Martinelli C., Huang H., Aliprandi M., Capasso P., Francavilla C., Ossolengo G., de Marco A., Protein Eng. Des. Sel. 2009, 22, 273–280. [DOI] [PubMed] [Google Scholar]
- 43. Desmyter A., Spinelli S., Roussel A., Cambillau C., Curr. Opin. Struct. Biol. 2015, 32, 1–8. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. Dantas-Barbosa C., Brigido M. D. M., Maranhao A. Q., Int. J. Mol. Sci. 2012, 13, 5420–5440; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44b. Pardon E., Laeremans T., Triest S., Rasmussen S. G., Wohlkonig A., Ruf A., Muyldermans S., Hol W. G., Kobilka B. K., Steyaert J., Nat. Protoc. 2014, 9, 674–693; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44c. Baker M., Nat. Methods 2011, 8, 623–626. [DOI] [PubMed] [Google Scholar]
- 45.
- 45a. de Marco A., Methods Mol. Biol. 2014, 1129, 125–135; [DOI] [PubMed] [Google Scholar]
- 45b. Thomassen Y. E., Verkleij A. J., Boonstra J., Verrips C. T., J. Biotechnol. 2005, 118, 270–277; [DOI] [PubMed] [Google Scholar]
- 45c. Rahbarizadeh F., Rasaee M. J., Forouzandeh M., Allameh A. A., Mol. Immunol. 2006, 43, 426–435. [DOI] [PubMed] [Google Scholar]
- 46.
- 46a. Ghahroudi M. A., Desmyter A., Wyns L., Hamers R., Muyldermans S., FEBS Lett. 1997, 414, 521–526; [DOI] [PubMed] [Google Scholar]
- 46b. Barthelemy P. A., Raab H., Appleton B. A., Bond C. J., Wu P., Wiesmann C., Sidhu S. S., J. Biol. Chem. 2008, 283, 3639–3654; [DOI] [PubMed] [Google Scholar]
- 46c. Rothbauer U., Zolghadr K., Muyldermans S., Schepers A., Cardoso M. C., Leonhardt H., Mol. Cell. Proteomics 2008, 7, 282–289. [DOI] [PubMed] [Google Scholar]
- 47. van der Linden R. H., Frenken L. G., de Geus B., Harmsen M. M., Ruuls R. C., Stok W., de Ron L., Wilson S., Davis P., Verrips C. T., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1999, 1431, 37–46. [DOI] [PubMed] [Google Scholar]
- 48. Herce H. D., Deng W., Helma J., Leonhardt H., Cardoso M. C., Nat. Commun. 2013, 4, 2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Caussinus E., Kanca O., Affolter M., Nat. Struct. Mol. Biol. 2011, 19, 117–121. [DOI] [PubMed] [Google Scholar]
- 50. De Clercq S., Zwaenepoel O., Martens E., Vandekerckhove J., Guillabert A., Gettemans J., Cell Mol. Life Sci. 2013, 70, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ward A. B., Szewczyk P., Grimard V., Lee C. W., Martinez L., Doshi R., Caya A., Villaluz M., Pardon E., Cregger C., Swartz D. J., Falson P. G., Urbatsch I. L., Govaerts C., Steyaert J., Chang G., Proc. Natl. Acad. Sci. USA 2013, 110, 13386–13391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Truttmann M. C., Wu Q., Stiegeler S., Duarte J. N., Ingram J., Ploegh H. L., J. Biol. Chem. 2015, 290, 9087–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.
- 53a. Loris R., Marianovsky I., Lah J., Laeremans T., Engelberg-Kulka H., Glaser G., Muyldermans S., Wyns L., J. Biol. Chem. 2003, 278, 28252–28257; [DOI] [PubMed] [Google Scholar]
- 53b. Lam A. Y., Pardon E., Korotkov K. V., Hol W. G., Steyaert J., J. Struct. Biol. 2009, 166, 8–15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53c. Domanska K., Vanderhaegen S., Srinivasan V., Pardon E., Dupeux F., Marquez J. A., Giorgetti S., Stoppini M., Wyns L., Bellotti V., Steyaert J., Proc. Natl. Acad. Sci. USA 2011, 108, 1314–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. De Genst E., Chan P. H., Pardon E., Hsu S. T., Kumita J. R., Christodoulou J., Menzer L., Chirgadze D. Y., Robinson C. V., Muyldermans S., Matagne A., Wyns L., Dobson C. M., Dumoulin M., J. Phys. Chem. B 2013, 117, 13245–13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.
- 55a. Li G., Zhu M., Ma L., Yan J., Lu X., Shen Y., Wan Y., ACS Appl. Mater. Interfaces 2016, 8, 13830–13839; [DOI] [PubMed] [Google Scholar]
- 55b. Li H., Sun Y., Elseviers J., Muyldermans S., Liu S., Wan Y., Analyst 2014, 139, 3718–3721. [DOI] [PubMed] [Google Scholar]
- 56.
- 56a. Chudakov D. M., Matz M. V., Lukyanov S., Lukyanov K. A., Physiol. Rev. 2010, 90, 1103–1163; [DOI] [PubMed] [Google Scholar]
- 56b. Kremers G. J., Gilbert S. G., Cranfill P. J., Davidson M. W., Piston D. W., J. Cell Sci. 2011, 124, 157–160; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56c. Schneider A. F., Hackenberger C. P., Curr. Opin. Biotechnol. 2017, 48, 61–68. [DOI] [PubMed] [Google Scholar]
- 57.
- 57a. Skube S. B., Chaverri J. M., Goodson H. V., Cytoskeleton 2010, 67, 1–12; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57b. Jensen E. C., Anat. Rec. 2012, 295, 2031–2036. [DOI] [PubMed] [Google Scholar]
- 58. Harris L. J., Larson S. B., Hasel K. W., McPherson A., Biochemistry 1997, 36, 1581–1597. [DOI] [PubMed] [Google Scholar]
- 59.
- 59a. Csanády L., Chan K. W., Seto-Young D., Kopsco D. C., Nairn A. C., Gadsby D. C., J. Gen. Physiol. 2000, 116, 477–500; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59b. Han W., Rhee J. S., Maximov A., Lin W., Hammer R. E., Rosenmund C., Sudhof T. C., J. Biol. Chem. 2005, 280, 5089–5100; [DOI] [PubMed] [Google Scholar]
- 59c. Hosein R. E., Williams S. A., Haye K., Gavin R. H., J. Eukaryotic Microbiol. 2003, 50, 403–408; [DOI] [PubMed] [Google Scholar]
- 59d. Rappoport J. Z., Simon S. M., Traffic 2008, 9, 1250–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Burgess A., Lorca T., Castro A., PLoS One 2012, 7, e45726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Traenkle B., Emele F., Anton R., Poetz O., Haeussler R. S., Maier J., Kaiser P. D., Scholz A. M., Nueske S., Buchfellner A., Romer T., Rothbauer U., Mol. Cell. Proteomics 2015, 14, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Helma J., Schmidthals K., Lux V., Nuske S., Scholz A. M., Krausslich H. G., Rothbauer U., Leonhardt H., PLoS One 2012, 7, e50026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zolghadr K., Gregor J., Leonhardt H., Rothbauer U., Methods Mol. Biol. 2012, 911, 569–575. [DOI] [PubMed] [Google Scholar]
- 64. Guizetti J., Schermelleh L., Mantler J., Maar S., Poser I., Leonhardt H., Muller-Reichert T., Gerlich D. W., Science 2011, 331, 1616–1620. [DOI] [PubMed] [Google Scholar]
- 65. Ries J., Kaplan C., Platonova E., Eghlidi H., Ewers H., Nat. Methods 2012, 9, 582–584. [DOI] [PubMed] [Google Scholar]
- 66. Leduc C., Si S., Gautier J., Soto-Ribeiro M., Wehrle-Haller B., Gautreau A., Giannone G., Cognet L., Lounis B., Nano Lett. 2013, 13, 1489–1494. [DOI] [PubMed] [Google Scholar]
- 67. Klooster R., Maassen B. T., Stam J. C., Hermans P. W., Haaft M. R. T., Detmers F. J., de Haard H. J., Post J. A., Verrips C. T., J. Immunol. Methods 2007, 324, 1–12. [DOI] [PubMed] [Google Scholar]
- 68.
- 68a. Frauer C., Leonhardt H., Nucleic Acids Res. 2009, 37, e22; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68b. Trinkle-Mulcahy L., Boulon S., Lam Y. W., Urcia R., Boisvert F. M., Vandermoere F., Morrice N. A., Swift S., Rothbauer U., Leonhardt H., Lamond A., J. Cell Biol. 2008, 183, 223–239; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68c. Pichler G., Leonhardt H., Rothbauer U., Methods Mol. Biol. 2012, 911, 475–483; [DOI] [PubMed] [Google Scholar]
- 68d. Pollithy A., Romer T., Lang C., Muller F. D., Helma J., Leonhardt H., Rothbauer U., Schuler D., Appl. Environ. Microbiol. 2011, 77, 6165–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nevoltris D., Lombard B., Dupuis E., Mathis G., Chames P., Baty D., ACS Nano 2015, 9, 1388–1399. [DOI] [PubMed] [Google Scholar]
- 70. Ma L., Sun Y., Kang X., Wan Y., Biosens. Bioelectron. 2014, 61, 165–171. [DOI] [PubMed] [Google Scholar]
- 71. Massa S., Xavier C., Muyldermans S., Devoogdt N., Expert Opin. Drug Delivery 2016, 13, 1149–1163. [DOI] [PubMed] [Google Scholar]
- 72. Platonova E., Winterflood C. M., Junemann A., Albrecht D., Faix J., Ewers H., Methods 2015, 88, 89–97. [DOI] [PubMed] [Google Scholar]
- 73. Doronina S. O., Toki B. E., Torgov M. Y., Mendelsohn B. A., Cerveny C. G., Chace D. F., DeBlanc R. L., Gearing R. P., Bovee T. D., Siegall C. B., Francisco J. A., Wahl A. F., Meyer D. L., Senter P. D., Nat. Biotechnol. 2003, 21, 778–784. [DOI] [PubMed] [Google Scholar]
- 74.
- 74a. Kim Y., Ho S. O., Gassman N. R., Korlann Y., Landorf E. V., Collart F. R., Weiss S., Bioconjugate Chem. 2008, 19, 786–791; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74b. Hackenberger C. P., Schwarzer D., Angew. Chem. Int. Ed. 2008, 47, 10030–10074; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 10182–10228; [Google Scholar]
- 74c. Gunnoo S. B., Madder A., ChemBioChem 2016, 17, 529–553. [DOI] [PubMed] [Google Scholar]
- 75.
- 75a. Chatalic K. L., Veldhoven-Zweistra J., Bolkestein M., Hoeben S., Koning G. A., Boerman O. C., de Jong M., van Weerden W. M., J. Nucl. Med. 2015, 56, 1094–1099; [DOI] [PubMed] [Google Scholar]
- 75b. Fridy P. C., Thompson M. K., Ketaren N. E., Rout M. P., Anal. Biochem. 2015, 477, 92–94; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75c. Sadeqzadeh E., Rahbarizadeh F., Ahmadvand D., Rasaee M. J., Parhamifar L., Moghimi S. M., J. Controlled Release 2011, 156, 85–91; [DOI] [PubMed] [Google Scholar]
- 75d. van Brussel A. S., Adams A., Oliveira S., Dorresteijn B., El Khattabi M., Vermeulen J. F., van der Wall E., Mali W. P., Derksen P. W., van Diest P. J., van Bergen En Henegouwen P. M., Mol. Imaging Biol. 2016, 18, 535–544; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75e. Vugmeyster Y., Entrican C. A., Joyce A. P., Lawrence-Henderson R. F., Leary B. A., Mahoney C. S., Patel H. K., Raso S. W., Olland S. H., Hegen M., Xu X., Bioconjugate Chem. 2012, 23, 1452–1462. [DOI] [PubMed] [Google Scholar]
- 76.
- 76a. Lotze J., Reinhardt U., Seitz O., Beck-Sickinger A. G., Mol. BioSyst. 2016, 12, 1713–2012; [DOI] [PubMed] [Google Scholar]
- 76b. Drake P. M., Rabuka D., Curr. Opin. Chem. Biol. 2015, 28, 174–180; [DOI] [PubMed] [Google Scholar]
- 76c. Schneider A. F. L., Hackenberger C. P. R., Curr. Opin. Biotechnol. 2017, 48, 61–68; [DOI] [PubMed] [Google Scholar]
- 76d. Chen X., Wu Y. W., Org. Biomol. Chem. 2016, 14, 5417–5439; [DOI] [PubMed] [Google Scholar]
- 76e. Falck G., Müller K. M., Antibodies 2018, 7, 2073–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.
- 77a. Li H., Yan J., Ou W., Liu H., Liu S., Wan Y., Biosens. Bioelectron. 2015, 64, 111–118; [DOI] [PubMed] [Google Scholar]
- 77b. Zhu M., Gong X., Hu Y., Ou W., Wan Y., J. Transl. Med. 2014, 12, 352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Griffin M., Casadio R., Bergamini C. M., Biochem. J. 2002, 368, 377–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Strop P., Liu S. H., Dorywalska M., Delaria K., Dushin R. G., Tran T. T., Ho W. H., Farias S., Casas M. G., Abdiche Y., Zhou D., Chandrasekaran R., Samain C., Loo C., Rossi A., Rickert M., Krimm S., Wong T., Chin S. M., Yu J., Dilley J., Chaparro-Riggers J., Filzen G. F., O'Donnell C. J., Wang F., Myers J. S., Pons J., Shelton D. L., Rajpal A., Chem. Biol. 2013, 20, 161–167. [DOI] [PubMed] [Google Scholar]
- 80. Dennler P., Bailey L. K., Spycher P. R., Schibli R., Fischer E., ChemBioChem 2015, 16, 861–867. [DOI] [PubMed] [Google Scholar]
- 81.
- 81a. Dennler P., Chiotellis A., Fischer E., Bregeon D., Belmant C., Gauthier L., Lhospice F., Romagne F., Schibli R., Bioconjugate Chem. 2014, 25, 569–578; [DOI] [PubMed] [Google Scholar]
- 81b. Siegmund V., Schmelz S., Dickgiesser S., Beck J., Ebenig A., Fittler H., Frauendorf H., Piater B., Betz U. A., Avrutina O., Scrima A., Fuchsbauer H. L., Kolmar H., Angew. Chem. Int. Ed. 2015, 54, 13420–13424; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13618–13623. [Google Scholar]
- 82. Mao H., Hart S. A., Schink A., Pollok B. A., J. Am. Chem. Soc. 2004, 126, 2670–2671. [DOI] [PubMed] [Google Scholar]
- 83. Popp M. W., Antos J. M., Grotenbreg G. M., Spooner E., Ploegh H. L., Nat. Chem. Biol. 2007, 3, 707–708. [DOI] [PubMed] [Google Scholar]
- 84. Theile C. S., Witte M. D., Blom A. E., Kundrat L., Ploegh H. L., Guimaraes C. P., Nat. Protoc. 2013, 8, 1800–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Witte M. D., Cragnolini J. J., Dougan S. K., Yoder N. C., Popp M. W., Ploegh H. L., Proc. Natl. Acad. Sci. USA 2012, 109, 11993–11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.
- 86a. Massa S., Vikani N., Betti C., Ballet S., Vanderhaegen S., Steyaert J., Descamps B., Vanhove C., Bunschoten A., van Leeuwen F. W., Hernot S., Caveliers V., Lahoutte T., Muyldermans S., Xavier C., Devoogdt N., Contrast Media Mol. Imaging 2016, 11, 328–339; [DOI] [PubMed] [Google Scholar]
- 86b. Rashidian M., Keliher E., Dougan M., Juras P. K., Cavallari M., Wojtkiewicz G. R., Jacobsen J., Edens J. G., Tas J. M., Victora G., Weissleder R., Ploegh H., ACS Cent. Sci. 2015, 1, 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rashidian M., Wang L., Edens J. G., Jacobsen J. T., Hossain I., Wang Q., Victora G. D., Vasdev N., Ploegh H., Liang S. H., Angew. Chem. Int. Ed. 2016, 55, 528–533; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 538–543. [Google Scholar]
- 88. Prota A. E., Magiera M. M., Kuijpers M., Bargsten K., Frey D., Wieser M., Jaussi R., Hoogenraad C. C., Kammerer R. A., Janke C., Steinmetz M. O., J. Cell. Biochem. J. Cell. Biol. 2013, 200, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Puthenveetil S., Liu D. S., White K. A., Thompson S., Ting A. Y., J. Am. Chem. Soc. 2009, 131, 16430–16438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cohen J. D., Zou P., Ting A. Y., ChemBioChem 2012, 13, 888–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gray M. A., Tao R. N., DePorter S. M., Spiegel D. A., McNaughton B. R., ChemBioChem 2016, 17, 155–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.
- 92a. Schumacher D., Helma J., Mann F. A., Pichler G., Natale F., Krause E., Cardoso M. C., Hackenberger C. P., Leonhardt H., Angew. Chem. Int. Ed. 2015, 54, 13787–13791; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13992–13996; [Google Scholar]
- 92b. Leonhardt H., Hackenberger C. P., Helma J., Schumacher D., (Ludwig-Maximilians-Universitaet Muenchen, Forschungsverbund Berlin E.V.), WO2016066749 A1, 2016.
- 93. Schumacher D., Lemke O., Helma J., Gerszonowicz L., Waller V., Stoschek T., Durkin M. D., Budisa N., Leonhardt H., Keller B. G., Hackenberger C. P. R., Chem. Sci. 2017, 8, 3471–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.
- 94a. Wang L., Brock A., Herberich B., Schultz P. G., Science 2001, 292, 498–500; [DOI] [PubMed] [Google Scholar]
- 94b. Liu W., Brock A., Chen S., Chen S., Schultz P. G., Nat. Methods 2007, 4, 239–244; [DOI] [PubMed] [Google Scholar]
- 94c. Agostini F., Voller J. S., Koksch B., Acevedo-Rocha C. G., Kubyshkin V., Budisa N., Angew. Chem. Int Ed. 2017, 56, 9680–9703; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9810-9835; [Google Scholar]
- 94d. Brabham R., Fascione M. A., ChemBioChem 2017, 18, 1973–1983. [DOI] [PubMed] [Google Scholar]
- 95. Liu C. C., Schultz P. G., Annu. Rev. Biochem. 2010, 79, 413–444. [DOI] [PubMed] [Google Scholar]
- 96. Yamaguchi A., Matsuda T., Ohtake K., Yanagisawa T., Yokoyama S., Fujiwara Y., Watanabe T., Hohsaka T., Sakamoto K., Bioconjugate Chem. 2016, 27, 198–206. [DOI] [PubMed] [Google Scholar]
- 97. Muir T. W., Sondhi D., Cole P. A., Proc. Natl. Acad. Sci. USA 1998, 95, 6705–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ta D. T., Redeker E. S., Billen B., Reekmans G., Sikulu J., Noben J. P., Guedens W., Adriaensens P., Protein Eng. Des. Sel. 2015, 28, 351–363. [DOI] [PubMed] [Google Scholar]
- 99.
- 99a. Dearden A. K., Callahan B., Roey P. V., Li Z., Kumar U., Belfort M., Nayak S. K., Protein Sci. 2013, 22, 557–563; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99b. Schaefer G., Haber L., Crocker L. M., Shia S., Shao L., Dowbenko D., Totpal K., Wong A., Lee C. V., Stawicki S., Clark R., Fields C., Phillips G. D. L., Prell R. A., Danilenko D. M., Franke Y., Stephan J. P., Hwang J., Wu Y., Bostrom J., Sliwkowski M. X., Fuh G., Eigenbrot C., Cancer Cell 2011, 20, 472–486. [DOI] [PubMed] [Google Scholar]
- 100. Debets M. F., Leenders W. P., Verrijp K., Zonjee M., Meeuwissen S. A., Otte-Holler I., van Hest J. C., Macromol. Biosci. 2013, 13, 938–945. [DOI] [PubMed] [Google Scholar]
- 101. Bachmann A. L., Mootz H. D., J. Pept. Sci. 2017, 23, 624–630. [DOI] [PubMed] [Google Scholar]
- 102. Muralidharan V., Muir T. W., Nat. Methods 2006, 3, 429–438. [DOI] [PubMed] [Google Scholar]
- 103. Fu A., Tang R., Hardie J., Farkas M. E., Rotello V. M., Bioconjugate Chem. 2014, 25, 1602–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.
- 104a. Freund G., Sibler A. P., Desplancq D., Oulad-Abdelghani M., Vigneron M., Gannon J., Van Regenmortel M. H., Weiss E., MAbs 2013, 5, 518–522; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104b. Kimura H., Hayashi-Takanaka Y., Stasevich T. J., Sato Y., Histochem. Cell Biol. 2015, 144, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.
- 105a. Biocca S., Ruberti F., Tafani M., Pierandrei-Amaldi P., Cattaneo A., Biotechnology 1995, 13, 1110–1115; [DOI] [PubMed] [Google Scholar]
- 105b. Desplancq D., Freund G., Conic S., Sibler A. P., Didier P., Stoessel A., Oulad-Abdelghani M., Vigneron M., Wagner J., Mely Y., Chatton B., Tora L., Weiss E., Exp. Cell Res. 2016, 342, 145–158; [DOI] [PubMed] [Google Scholar]
- 105c. Sato Y., Mukai M., Ueda J., Muraki M., Stasevich T. J., Horikoshi N., Kujirai T., Kita H., Kimura T., Hira S., Okada Y., Hayashi-Takanaka Y., Obuse C., Kurumizaka H., Kawahara A., Yamagata K., Nozaki N., Kimura H., Sci. Rep. 2013, 3, 2436; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105d. Worn A., Pluckthun A., J. Mol. Biol. 2001, 305, 989–1010. [DOI] [PubMed] [Google Scholar]
- 106. Wall M. A., Socolich M., Ranganathan R., Nat. Struct. Biol. 2000, 7, 1133–1138. [DOI] [PubMed] [Google Scholar]
- 107.
- 107a. Akishiba M., Takeuchi T., Kawaguchi Y., Sakamoto K., Yu H.-H., Nakase I., Takatani-Nakase T., Madani F., Gräslund A., Futaki S., Nat. Chem. 2017, 9, 751–761; [DOI] [PubMed] [Google Scholar]
- 107b. Fuchs S. M., Raines R. T., ACS Chem. Biol. 2007, 2, 167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.
- 108a. Cronican J. J., Thompson D. B., Beier K. T., McNaughton B. R., Cepko C. L., Liu D. R., ACS Chem. Biol. 2010, 5, 747–752; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108b. McNaughton B. R., Cronican J. J., Thompson D. B., Liu D. R., Proc. Natl. Acad. Sci. USA 2009, 106, 6111–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bruce V. J., Lopez-Islas M., McNaughton B. R., Protein Sci. 2016, 25, 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.
- 110a.Y. Komarova, J. Peloquin, G. Borisy, CSH Protoc 2007, DOI: https://doi.org/10.1101/pdb.prot4657; [DOI] [PubMed]
- 110b. Kang S., Kim K. H., Kim Y. C., Sci.Rep. 2015, 5, 15835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Khalil I. A., Kogure K., Akita H., Harashima H., Pharmacol. Rev. 2006, 58, 32–45. [DOI] [PubMed] [Google Scholar]
- 112.
- 112a. D′Astolfo D. S., Pagliero R. J., Pras A., Karthaus W. R., Clevers H., Prasad V., Lebbink R. J., Rehmann H., Geijsen N., Cell 2015, 161, 674–690; [DOI] [PubMed] [Google Scholar]
- 112b. Krantz B. A., Trivedi A. D., Cunningham K., Christensen K. A., Collier R. J., J. Mol. Biol. 2004, 344, 739–756. [DOI] [PubMed] [Google Scholar]
- 113. Chiu H. Y., Deng W., Engelke H., Helma J., Leonhardt H., Bein T., Sci. Rep. 2016, 6, 25019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Röder R., Helma J., Preiss T., Rädler J. O., Leonhardt H., Wagner E., Pharm. Res. 2017, 34, 161–174. [DOI] [PubMed] [Google Scholar]
- 115. Rabideau A. E., Pentelute B. L., ACS Chem. Biol. 2016, 11, 1490–1501. [DOI] [PubMed] [Google Scholar]
- 116. Liao X., Rabideau A. E., Pentelute B. L., ChemBioChem 2014, 15, 2458–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.
- 117a. Nischan N., Herce H. D., Natale F., Bohlke N., Budisa N., Cardoso M. C., Hackenberger C. P., Angew. Chem. Int. Ed. 2015, 54, 1950–1953; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1972–1976; [Google Scholar]
- 117b.A recent review on CPPs: Brock R., Bioconjug. Chem. 2014, 25, 863—868. [DOI] [PubMed] [Google Scholar]
- 118. Herce H. D., Schumacher D., Schneider A. F. L., Ludwig A. K., Mann F. A., Fillies M., Kasper M. A., Reinke S., Krause E., Leonhardt H., Cardoso M. C., Hackenberger C. P. R., Nat. Chem. 2017, 9, 762–771. [DOI] [PubMed] [Google Scholar]
- 119.
- 119a. Amir R. E., Van den Veyver I. B., Wan M., Tran C. Q., Francke U., Zoghbi H. Y., Nat. Genet. 1999, 23, 185–188; [DOI] [PubMed] [Google Scholar]
- 119b. Rett A., Krankenschwester 1966, 19, 121–122. [PubMed] [Google Scholar]
- 120. Pruszynski M., Koumarianou E., Vaidyanathan G., Revets H., Devoogdt N., Lahoutte T., Zalutsky M. R., Nucl. Med. Biol. 2013, 40, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]