

In multiple sclerosis, increasing abnormality of the magnetization transfer ratio (MTR) is seen with increasing proximity to the lateral ventricles. Brown et al. identify an abnormal periventricular MTR gradient in individuals with a recent clinically isolated syndrome. Its presence is independent of lesions, and predicts early conversion to multiple sclerosis.
Keywords: multiple sclerosis, magnetization transfer ratio, normal-appearing white matter
Abstract
In established multiple sclerosis, tissue abnormality—as assessed using magnetization transfer ratio—increases close to the lateral ventricles. We aimed to determine whether or not (i) these changes are present from the earliest clinical stages of multiple sclerosis; (ii) they occur independent of white matter lesions; and (iii) they are associated with subsequent conversion to clinically definite multiple sclerosis and disability. Seventy-one subjects had MRI scanning a median of 4.6 months after a clinically isolated optic neuritis (49 females, mean age 33.5 years) and were followed up clinically 2 and 5 years later. Thirty-seven healthy controls (25 females, mean age 34.4 years) were also scanned. In normal-appearing white matter, magnetization transfer ratio gradients were measured 1–5 mm and 6–10 mm from the lateral ventricles. In control subjects, magnetization transfer ratio was highest adjacent to the ventricles and decreased with distance from them; in optic neuritis, normal-appearing white matter magnetization transfer ratio was lowest adjacent to the ventricles, increased over the first 5 mm, and then paralleled control values. The magnetization transfer ratio gradient over 1–5 mm differed significantly between the optic neuritis and control groups [+0.059 percentage units/mm (pu/mm) versus −0.033 pu/mm, P = 0.010], and was significantly steeper in those developing clinically definite multiple sclerosis within 2 years compared to those who did not (0.132 pu/mm versus 0.016 pu/mm, P = 0.020). In multivariate binary logistic regression the magnetization transfer ratio gradient was independently associated with the development of clinically definite multiple sclerosis within 2 years (magnetization transfer ratio gradient odds ratio 61.708, P = 0.023; presence of T2 lesions odds ratio 8.500, P = 0.071). At 5 years, lesional measures overtook magnetization transfer ratio gradients as significant predictors of conversion to multiple sclerosis. The magnetization transfer ratio gradient was not significantly affected by the presence of brain lesions [T2 lesions (P = 0.918), periventricular T2 lesions (P = 0.580) or gadolinium-enhancing T1 lesions (P = 0.724)]. The magnetization transfer ratio gradient also correlated with Expanded Disability Status Scale score 5 years later (Spearman r = 0.313, P = 0.027). An abnormal periventricular magnetization transfer ratio gradient occurs early in multiple sclerosis, is clinically relevant, and may arise from one or more mechanisms that are at least partly independent of lesion formation.
Introduction
In patients with established multiple sclerosis, it has recently been shown that magnetization transfer ratio (MTR) is increasingly abnormal in normal-appearing white matter (NAWM) and white matter lesions close to the lateral ventricles (Liu et al., 2015). Several mechanisms may be responsible for this periventricular MTR gradient, and it is unclear whether the underlying process differs from those leading to white matter lesion formation. It is also not known if such a periventricular predilection for non-lesional white matter pathology occurs early in multiple sclerosis or is associated with subsequent disease activity. An early, clinically-relevant change that is independent of lesions might represent an early treatment target.
About 80% of patients with multiple sclerosis initially present with a clinically isolated syndrome (CIS) such as optic neuritis. About two-thirds of patients with a CIS have asymptomatic brain white matter lesions visible on MRI, and these are associated with a substantially greater risk of subsequently developing clinically definite multiple sclerosis (CDMS; 80% compared with 20% for those who do not have such lesions; Fisniku et al., 2008). However, white matter lesions are not the only abnormality found early in patients with multiple sclerosis; grey matter lesions are also seen (Calabrese et al., 2007; Nielsen et al., 2013), and NAWM and grey matter are both abnormal when assessed using quantitative MRI measures (Fernando et al., 2005; Gallo et al., 2005; Rocca et al., 2008; Henry et al., 2009).
By looking for an abnormal periventricular MTR gradient in patients who have recently had a CIS, we would be able to establish if it is seen from the earliest clinical stages of multiple sclerosis and, given that patients usually have very few white matter lesions immediately after a CIS, determine if it is closely associated with white matter lesions. Using previously acquired MTR data from a longitudinal study of patients with a recent CIS, we aimed to (i) look for gradients in NAWM abnormalities around the lateral ventricles and, if present; (ii) to determine whether or not they were associated with white matter lesions; or (iii) were associated with the subsequent risk of developing multiple sclerosis and disability.
Materials and methods
Participants
From a prospectively recruited CIS cohort (Fernando et al., 2005) we included data from 81 patients who had presented with a clinically isolated optic neuritis, who had no previous history of neurological symptoms, were aged between 16 and 50 years at symptom onset, had 3D fast spoiled gradient echo (FSPGR) as part of their baseline MRI assessments and MTR 3 months later. Participants were recruited from Moorfields Eye Hospital (London) by a single experienced neuro-ophthalmologist. They were evaluated clinically and with MRI a median of 1.4 (range 0.1–3.5) months after optic neuritis onset (‘baseline’) and underwent a further scan (including MTR) on the same scanner 3 months later (median 4.6; mean 4.8; range 2.0–7.5 months after optic neuritis onset); patients were followed up clinically 2 and 5 years later (Brownlee et al., 2015) and had Expanded Disability Status Scale (EDSS) and paced serial addition test (PASAT, 3-s intervals; Cutter et al., 1999) scores assessed at 5 years. This work therefore describes cross-sectional MTR data (collected 4.6 months after symptom onset) with clinical data at 4.6 months plus 2 and 5 years after symptom onset. We also studied a group of 39 healthy control subjects with no known neurological disorder or clinical follow-up. Demographic and clinical data are reported in Table 1.
Table 1.
Participant characteristics
| Healthy controls | Patients with optic neuritis | |
|---|---|---|
| n | 37 | 71 |
| Mean age, years ± SD (range) | 34.4 ± 4.8 (27–47) | 33.5 ± 6.7 (20–48) |
| female : male | 25 : 12 | 49 : 22 |
| Median baseline EDSS (range) | - | 1 (0–3) |
| Number with abnormal T2 scan (excluding symptomatic lesion) | - | 55 |
| Number with periventricular lesions | - | 46 |
| Number with Gd-enhancing lesions | - | 22 |
| Mean T2 lesion volume (ml) ± SD | - | 1.47 ± 3.32 |
| Mean brain parenchymal fraction ± SD | 0.84 ± 0.04 | 0.85 ± 0.03 |
| Number attaining CDMS at time of MTR scan | - | 8 |
| Number attaining McDonald multiple sclerosis at time of MTR scan | - | 33 |
| Number with clinical follow-up at 2 and 5 years | - | 58 |
| Number converting to CDMS within 2 years of optic neuritis (of 58 with clinical follow-up) | - | 18 |
| Number converting to McDonald multiple sclerosis within 2 years of optic neuritis (of 58 with clinical follow-up) | - | 40 |
| Number converting to CDMS within 5 years of optic neuritis (of 58 with clinical follow-up) | - | 31 |
| Number converting to McDonald multiple sclerosis within 5 years of optic neuritis (of 58 with clinical follow-up) | - | 45 |
| Median (range) EDSS score at 5-year follow-up (assessed in 50/58) | - | 1 (0-8.5) |
| Median (range) PASAT score at 5-year follow-up (assessed in 31/58) | - | 46.5 (17-59) |
In the optic neuritis group, MTR scans were undertaken 3 months after first enrolment in the study, and so lesion measures from scans obtained at 3 months are shown. Volumetric brain scans were acquired at baseline but not at 3 months. PASAT = Paced Serial Addition Test, 3-s intervals.
All participants gave written informed consent and the study was approved by our local institutional ethics committees.
Clinical assessments
Multiple sclerosis was diagnosed on the basis of further relapses (CDMS; Poser et al., 1983) and using the 2010 McDonald criteria (Polman et al., 2011).
MRI
All magnetic resonance studies were performed on a 1.5 T GE Signa Echospeed scanner (General Electric Medical Systems). The following sequences were acquired: (i) an FSPGR scan of the whole brain (124 × 1.5 mm slices; matrix, 256 × 160, interpolated to a final in-plane resolution of 1.1 mm, repetition time = 10.9 ms, echo time = 4.2 ms, inversion time = 450 ms) for volumetric measures; (ii) dual echo proton-density/T2 scans of the whole brain (46 × 3 mm contiguous axial-oblique slices parallel to the anterior/posterior commissural line, matrix 256 × 256, field of view 24 × 18 cm, repetition time = 3200 ms, echo time = 15/90 ms), for the evaluation of white matter lesions; (iii) T1-weighted pre- and post-gadolinium (0.1 mmol/kg body weight) spin echo sequences of the brain (46 × 3 mm contiguous axial-oblique slices parallel to the anterior/posterior commissural line, matrix 256 × 256, field of view 24 × 18 cm, repetition time = 600 ms, echo time = 17 ms) to evaluate the presence of pathologically enhancing lesions; and (iv) MTR data using a dual-echo, spin-echo sequence of the whole brain (28 × 5 mm contiguous), axial-oblique slices parallel to the anterior/posterior commissural line with an interleaved sequence described by Barker et al. (1996) [repetition time = 1720 ms, echo time = 30/80 ms, number of excitations 0.75, matrix 256 × 128, field of view 24 × 24 cm, magnetization transfer-weighted by the application of a pre-saturation pulse (Hamming apodized 3 lobe sinc pulse, duration 64 ms, flip angle 1430°, and a peak amplitude of 14.6 μT giving a normal bandwidth of 62.5 Hz, applied 1 kHz from the water resonance)]. MTR maps were calculated on a voxel-by-voxel basis using the short echo data because of its higher signal-to-noise compared to the longer echo data. Proton density and T2-weighted images are included in this sequence, intrinsically registered to the magnetization transfer data, and were used to facilitate registrations to native magnetization transfer space as described below. The FSPGR scan was acquired at baseline (when magnetization transfer imaging was not performed); all other scans and lesion counts used for this analysis were acquired 3 months later (when the FSPGR was not repeated; Fernando et al., 2005).
Image analysis
Magnetization transfer ratio map calculation
For each participant, MTR maps were calculated using the following equation: MTR (in percentage units, pu) = [(MTRoff − MTRon) / MToff.) × 100]. The interleaved nature of the MTR sequence used meant that co-registration of MTon and MToff data was not required. MTR values were extracted from each NAWM and lesion ring using FSLstats (http://www.fmrib.ox.ac.uk/fsl/).
Tissue segmentation
T1-hypointensities were outlined using a semi-automated edge-finding tool (JIM v6.0, Xinapse systems, Aldwincle, UK) on the 3D FSPGR images and these masks were used to perform lesion filling on the 3D FSPGR scans (Chard et al., 2010; Prados et al., 2016). After lesion filling the FSPGR scans were segmented into grey matter, white matter and CSF probability maps using the new segmentation pipeline in Statistical Parametric Mapping 12 (SPM12; Wellcome Trust Centre for Neuroimaging, London, UK; www.fil.ion.ucl.ac.uk/). Brain parenchymal fraction was calculated using SPM12-derived tissue segmentations for use as a covariate in the statistical models (Chard et al., 2010).
Generation of normal-appearing white matter masks
White matter lesions were identified on the dual echo proton-density/T2-weighted scans, and outlined using JIM v6.0 (Xinapse systems, Aldwincle, UK) by one investigator (W.J.B.). Using NiftyReg (Modat et al., 2010, 2014) the T2-weighted image was linearly co-registered with the T2-weighted image embedded in the magnetization transfer sequence, thus aligning the white matter lesion masks with the MTR maps. NiftyReg implements a symmetric and inverse-consistent registration ensuring that the results are unbiased towards the directionality of the registration process because the forward and backward transformations are optimized concurrently in an inverse-consistent manner. The symmetric full affine approach (Modat et al., 2014) with 12 degrees of freedom is based on the asymmetric block-matching approach (Ourselin et al., 2001).
The proton density and T2 images embedded in the magnetization transfer sequence were used to generate a pseudo-T1 image, by subtracting the proton density scans from the T2 images (Hickman et al., 2002). For each participant, the 3D FSPGR scan was then linearly co-registered to the pseudo-T1 using NiftyReg (Modat et al., 2014) and the transformation applied to the grey matter, white matter and CSF tissue segmentation maps. In MTR space the grey matter, white matter and CSF maps were binarized using a probabilistic threshold of >90% (Samson et al., 2014). In MTR space, white matter lesion masks were dilated by two voxels (to account for perilesional MTR abnormalities; Vrenken et al., 2006), and the dilated lesion masks were subtracted from the thresholded white matter tissue probability maps to produce NAWM masks.
Segmentation of normal-appearing white matter into concentric periventricular rings
Given the voxel size of magnetization transfer images (1 × 1 × 5 mm), to reduce the potential for partial volume effects between CSF and white matter all analyses were limited to two axial slices orthogonal to the wall of the lateral ventricles, i.e. a slice immediately above the insula, and the slice immediately below this. To consistently identify these slices in all participants, a position marker was placed in the lateral ventricles just superior to the insula on a MNI152 brain template (Grabner et al., 2006). The MNI152 brain template was registered to each participant’s native MTR space with a non-linear transformation as previously described (Muhlert et al., 2013) using NiftyReg (Modat et al., 2010). The position of the marker was checked and corrected as needed by one investigator (J.W.L.B.) and then the two axial slices were selected relative to this template marker.
To identify the lateral ventricles we used a previously described approach (Liu et al., 2015). Briefly, a mask of the lateral ventricles was created in MNI152 space using the Wake Forest University School of Medicine PickAtlas toolbox (Maldjian et al., 2003) and then registered to each participant’s MTR native space using the previously computed transformation. The lateral ventricle mask was then intersected with each participant's CSF mask on the two previously selected axial slices. These masks were checked and manually edited by one investigator (J.W.L.B.) to ensure anatomical accuracy, and then sequentially dilated in the axial plane by 1 voxel (1 mm) using DilM (part of the FSL software package (http://www.fmrib.ox.ac.uk/fsl/) producing concentric rings around the lateral ventricles (Fig. 1). The first two rings were discarded to limit partial volume effects from CSF as each of these contained CSF in over half of participants (first ring 117/120 participants; second ring 63/120). As per previous work (Liu et al., 2015), white matter lesional MTR and the percentage of lesioned white matter were computed for the next 10 rings. Residual CSF was identified in the first of these 10 rings in 10 patients with optic neuritis and two controls and so these participants were excluded, leaving 71 patients with optic neuritis and 37 healthy controls.
Figure 1.

MTR image processing. (A) NAWM from two 5 mm axial slices were extracted at (B) and immediately below (C) the level of the insula. Rings of concentric MTR values from the NAWM around the ventricles were then extracted (D and E). The first two rings were excluded to minimize partial volume effects.
We also recalculated the mean NAWM and grey matter MTR (as reported in Fernando et al., 2005) for use as covariates in the statistical models.
Statistical analyses
Statistical analyses were performed using SPSS [IBM SPSS version 22 for Windows (SPSS, Inc., Chicago, IL, USA)]. Demographic data are presented as mean (standard deviation, SD) and MTR data as mean (standard error, SE), while EDSS values are presented as median (range). In each participant, mean MTR values were calculated in the 1 mm rings in both axial slices, and then averaged across both slices, weighted by the number of voxels in each ring. After discarding the first two rings (see above), we calculated periventricular MTR gradients over the next 5 mm closest to the ventricles (i.e. NAWM MTR in ring five − NAWM MTR in ring one) and deep MTR gradients over the subsequent 5 mm (i.e. NAWM MTR in ring 10 − NAWM MTR in ring six), dividing both by the number of intervals to give the MTR change in percentage units (pu) per mm. This division was consistent with previous work (Liu et al., 2015), and coincided with the point at which the MTR gradients in the optic neuritis and control groups converged (Fig. 2A).
Figure 2.


Periventricular NAWM MTR in healthy control and optic neuritis groups. (A) NAWM MTR of rings in healthy controls (HC, blue) and patients with optic neuritis 4.6 months after symptom onset (ON, red). Mean ± 2 SE. MTR is expressed as percentage units (pu). Ring 1 is closest to the ventricular surface. (B) NAWM MTR in healthy controls (HC, blue) and patients with optic neuritis 4.6 months after symptom onset (ON), divided into those with (yellow) and without (green) asymptomatic lesions on their baseline scan. Mean ± 2 SE is shown. MTR is expressed as percentage units (pu). Ring 1 is closest to the ventricular surface. (C) NAWM MTR in patients with optic neuritis 4.6 months after symptom onset (ON) plus asymptomatic lesions, divided into those with (n = 46, yellow circles) and without (n = 9, green circles) periventricular lesions. Mean ± 2 SE. MTR is expressed as percentage units (pu). Ring 1 is closest to the ventricular surface. (D) NAWM MTR in healthy controls (HC, blue) and patients with optic neuritis 4.6 months after symptom onset (ON), divided into those who did convert to CDMS within 2 years (yellow) and those that did not (green). Mean ± 2 SE is shown. MTR is expressed as percentage units (pu). Ring 1 is closest to the ventricular surface.
In the optic neuritis cohort, 58/71 patients were followed-up 2 and 5 years after their baseline MRI scan. These patients were subdivided into those who developed CDMS or McDonald multiple sclerosis within 2 years of their optic neuritis. We also examined those developing CDMS and McDonald multiple sclerosis within 5 years of their optic neuritis. To assess associations with gadolinium-enhancing lesions, the optic neuritis cohort was subdivided into those with or without enhancing lesions. To explore associations with white matter lesions, the optic neuritis cohort was also subdivided into those who did or did not have asymptomatic T2 white matter lesions and, in those with lesions, those who did or did not have periventricular lesions. Independent sample t-tests and ANOVA tests were used to compare clinical and MRI measures between groups. For the MTR gradient measures, the same comparisons were also performed using general linear models firstly correcting for brain parenchymal fraction (to control for the possible effects of atrophy on periventricular MTR gradients), then correcting for mean NAWM and grey matter MTR (to ensure diffuse MTR changes were not driving MTR gradients). Spearman correlation was used to explore the relationship between MTR gradients and both EDSS and PASAT scores. Factors predicting conversion to CDMS status were examined with multivariate binary logistic regression. Results were considered statistically significant at the P < 0.05 level.
Results
There were no significant demographic differences between the clinically isolated optic neuritis group and controls (Table 1). At the 3-month scan, 55/71 (77.5%) of the optic neuritis group had white matter lesions, 46/71 (64.8%) had periventricular lesions and 22/71 (31.0%) had gadolinium-enhancing lesions. Thirteen patients were lost to follow-up. After 2 years, 18/58 (31%) had experienced a further clinical relapse and so developed CDMS while 40/58 (69%) had McDonald multiple sclerosis. Five years after optic neuritis, these figures had risen to 31/58 (53.5%) and 45/58 (77.6%), respectively.
Periventricular magnetization transfer ratio gradients in optic neuritis compared with controls
Table 2 and Fig. 2A show the mean and SE NAWM MTR per ring in the optic neuritis and control groups. The mean MTR of each NAWM ring was lower in the optic neuritis group than in healthy controls. In controls, mean MTR was highest in the rings nearest the ventricles and declined with distance from them. In contrast, in the optic neuritis group, mean MTR was lowest in the ring nearest the ventricles, increased over the first 5 mm and then paralleled control values.
Table 2.
Mean ring MTR in healthy controls and patients with optic neuritis
| Ring | Mean ± SE MTR in pu | Mean ± SE MTR in pu | |||
|---|---|---|---|---|---|
| Healthy controls | All optic neuritis | Optic neuritis subgroups | |||
| Convert to CDMS <2 y | Convert to CDMS 2–5 y | Did not convert to CDMS | |||
| (n = 37) | (n = 71) | (n = 18) | (n = 13) | (n = 27) | |
| 1 | 38.364 ± 0.138 | 37.670 ± 0.102 | 37.371 ± 0.183 | 37.831 ± 0.180 | 37.805 ± 0.203 |
| P = 0.000 | P = 0.000 | P = 0.062 | P = 0.013 | ||
| 2 | 38.334 ± 0.113 | 37.836 ± 0.085 | 37.639 ± 0.166 | 37.773 ± 0.205 | 37.951 ± 0.156 |
| P = 0.001 | P = 0.001 | P = 0.020 | P = 0.043 | ||
| 3 | 38.254 ± 0.095 | 37.912 ± 0.080 | 37.862 ± 0.176 | 37.946 ± 0.209 | 37.903 ± 0.133 |
| P = 0.007 | P = ns | P = ns | P = ns | ||
| 4 | 38.293 ± 0.080 | 37.959 ± 0.075 | 37.944 ± 0.158 | 37.942 ± 0.190 | 37.918 ± 0.132 |
| P = 0.003 | P = ns | P = ns | P = ns | ||
| 5 | 38.232 ± 0.080 | 37.936 ± 0.076 | 37.901 ± 0.163 | 37.916 ± 0.204 | 37.859 ± 0.126 |
| P = 0.009 | P = ns | P = ns | P = ns | ||
| 6 | 38.109 ± 0.087 | 37.827 ± 0.077 | 37.822 ± 0.162 | 37.807 ± 0.209 | 37.765 ± 0.129 |
| P = 0.017 | P = ns | P = ns | P = ns | ||
| 7 | 38.006 ± 0.083 | 37.751 ± 0.074 | 37.751 ± 0.166 | 37.772 ± 0.194 | 37.706 ± 0.124 |
| P = 0.025 | P = ns | P = ns | P = ns | ||
| 8 | 38.024 ± 0.084 | 37.774 ± 0.073 | 37.754 ± 0.174 | 37.805 ± 0.192 | 37.740 ± 0.115 |
| P = 0.027 | P = ns | P = ns | P = ns | ||
| 9 | 38.113 ± 0.084 | 37.864 ± 0.075 | 37.805 ± 0.187 | 37.942 ± 0.192 | 37.822 ± 0.117 |
| P = 0.030 | P = ns | P = ns | P = ns | ||
| 10 | 38.187 ± 0.085 | 37.952 ± 0.078 | 37.850 ± 0.191 | 38.048 ± 0.198 | 37.945 ± 0.123 |
| P = 0.044 | P = ns | P = ns | P = ns | ||
Independent sample t-tests were used to compare the healthy control and whole optic neuritis groups; one-way ANOVA to compare the optic neuritis subgroups. pu = percentage units; ns = not significant. Bold P-values < 0.05.
Over the first 5 mm from the ventricles, we found significantly different MTR gradients between the optic neuritis and control groups (optic neuritis: mean +0.059 pu/mm ± 0.028; controls mean −0.033 pu/mm ± 0.028, t = −2.62, P = 0.010). This difference remained significant after adjustment for brain parenchymal fraction (P = 0.009). Conversely, the MTR gradient over the next five rings showed no significant difference between the groups (optic neuritis: +0.003 pu/mm ± 0.008, controls −0.009 pu/mm ± 0.012, P = 0.541).
Associations of periventricular magnetization transfer ratio gradients with lesions
In the optic neuritis group, over the first 5 mm, periventricular MTR gradients did not differ significantly between those with or without asymptomatic white matter lesions seen on a T2-weighted scan from the same scanning session (+0.058 pu/mm ± 0.024 versus +0.063 pu/mm ± 0.043, respectively, P = 0.918). The periventricular MTR gradient in the optic neuritis group who did not have brain lesions showed a non-significant trend to be higher than healthy controls (+0.063 pu/mm ± 0.043 versus −0.033 pu/mm ± 0.028, respectively, P = 0.064) (Fig. 2B). The presence of periventricular lesions seen on a T2-weighted scan had no significant effect on the periventricular gradient [0.068 pu/mm ± 0.028 (with periventricular lesions) versus 0.043 pu/mm ± 0.030 (without periventricular lesions), P = 0.580, Fig. 2C]. The mean gradient in those with and without gadolinium-enhancing lesions similarly did not differ significantly (0.067 pu/mm ± 0.040 versus 0.055 pu/mm ± 0.025, respectively, P = 0.794).
Associations of periventricular magnetization transfer ratio gradients with conversion to multiple sclerosis and disability
Table 3 and Fig. 2D show the gradients over the first and second 5 mm of NAWM extending from the ventricles in the optic neuritis groups who did or did not develop CDMS within 2 years.
Table 3.
Periventricular (1–5 mm) and deep (6–10 mm) MTR gradients in NAWM of patients with optic neuritis and healthy controls
| Clinical classification at 2 years | n | MTR gradient | |
|---|---|---|---|
| Mean ± SE (pu/mm) | |||
| Periventricular (1 to 5 mm) | Deep (6 to 10 mm) | ||
| Healthy controls | 37 | −0.033 ± 0.028 | −0.009 ± 0.012 |
| Optic neuritis | |||
| Did not convert to CDMS | 40 | 0.016 ± 0.029 | 0.020 ± 0.010 |
| Converted to CDMS | 18 | 0.132 ± 0.041* | −0.010 ± 0.019 |
| Did not convert to McDonald multiple sclerosis | 18 | 0.015 ± 0.048 | 0.021 ± 0.015 |
| Converted to McDonald multiple sclerosis | 40 | 0.069 ± 0.028** | 0.006 ± 0.011 |
Optic neuritis group divided according to conversion to CDMS status within 2 years. *Converters versus healthy controls P = 0.001, converters versus non-converters P = 0.020, non-converters versus healthy controls P = 0.221. Optic neuritis group also divided according to conversion to McDonald multiple sclerosis within 2 years. **Converters versus healthy controls P = 0.014; otherwise, no significant differences in gradients detected.
The periventricular MTR gradient in the group who developed CDMS within 2 years differed from healthy controls and the optic neuritis group who did not convert to CDMS within 2 years (Table 3; overall effect P = 0.006; converters versus healthy controls P = 0.001, converters versus non-converters P = 0.020, non-converters versus healthy controls P = 0.221). The periventricular MTR gradient also differed between those who converted to CDMS within 5 years and healthy controls, while the difference between converters and non-converters did not reach significance; overall effect P = 0.026; converters versus healthy controls P = 0.007, converters versus non-converters P = 0.123, non-converters versus healthy controls P = 0.304. The differences between those who developed multiple sclerosis and healthy controls remained significant when McDonald MRI criteria were used instead of CDMS (Table 3). The group effect on the periventricular MTR gradient did not materially change after including brain parenchymal fraction in the statistical models. The group effect on the periventricular MTR gradient also remained significant after adjustment for mean NAWM MTR (P = 0.014) and mean grey matter MTR (P = 0.022). When both NAWM MTR and grey matter MTR were added, the model remained significant (P = 0.021) as did differences in MTR gradients between converters and non-converters (P = 0.037) and between converters and healthy controls (P = 0.006).
We also examined whether or not periventricular MTR gradients over 1–5 mm had an effect on conversion to CDMS independent of the presence of lesions. A multivariate binary logistic regression showed that the MTR gradient over 1-5 mm was independently associated with conversion to CDMS at two years [MTR gradient: odds ratio (OR) 61.708 P = 0.023; presence of T2 lesions OR 8.500 P = 0.071]. A similar pattern was seen when the MTR gradient was compared to periventricular T2 lesions (MTR gradient: OR 44.100 P = 0.029; presence of periventricular T2 lesions OR 5.84 P = 0.040). However, the MTR gradient did not predict conversion to CDMS over 5 years.
There was no significant difference in 6–10 mm MTR gradients between optic neuritis groups who did or did not develop CDMS at 2 or 5 years (P = 0.124 and P = 0.231, respectively).
Unlike the periventricular MTR gradient, the mean MTR of band 1 or 5 in the optic neuritis group converting to CDMS within 2 years was not significantly higher than in those not converting to CDMS (Supplementary material).
Associations of magnetization transfer ratio gradients with disability
A correlation was found between the MTR gradient over the first 5 mm and EDSS status 5 years later (Spearman r = 0.313, P = 0.027; EDSS measured in 50/58 patients at this time point). No significant correlation was found between inner MTR gradient and PASAT score at 5 years (P = 0.815), although this was only undertaken in 31/58 patients at this time point.
No significant correlations were found between EDSS 5 years later and mean MTR in band 1 (P = 0.165) or band 5 (P = 0.540), NAWM MTR (P = 0.506) or grey matter MTR (P = 0.109).
Lesion measures
The mean MTR of lesional voxels, like the NAWM MTR, was lowest at the ventricular margin and increased with distance from it (Fig. 3). Lesion volumes are shown in Fig. 4.
Figure 3.
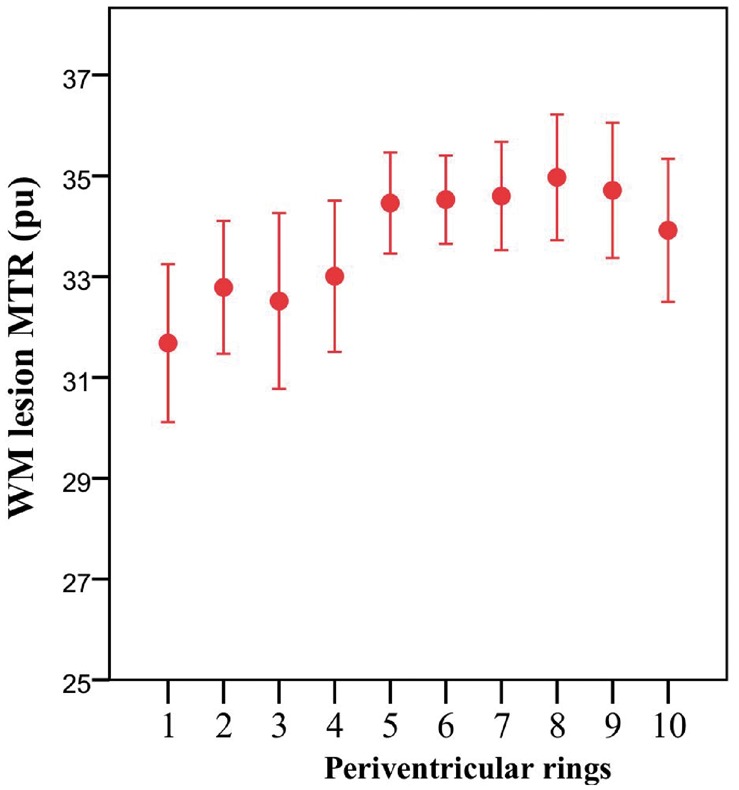
Periventricular white matter lesional MTR. Mean MTR of white matter (WM) lesional voxels in patients with optic neuritis 4.6 months after symptom onset (ON). Mean ± 2 SE. MTR is expressed as percentage units (pu). Ring 1 is closest to the ventricular surface.
Figure 4.
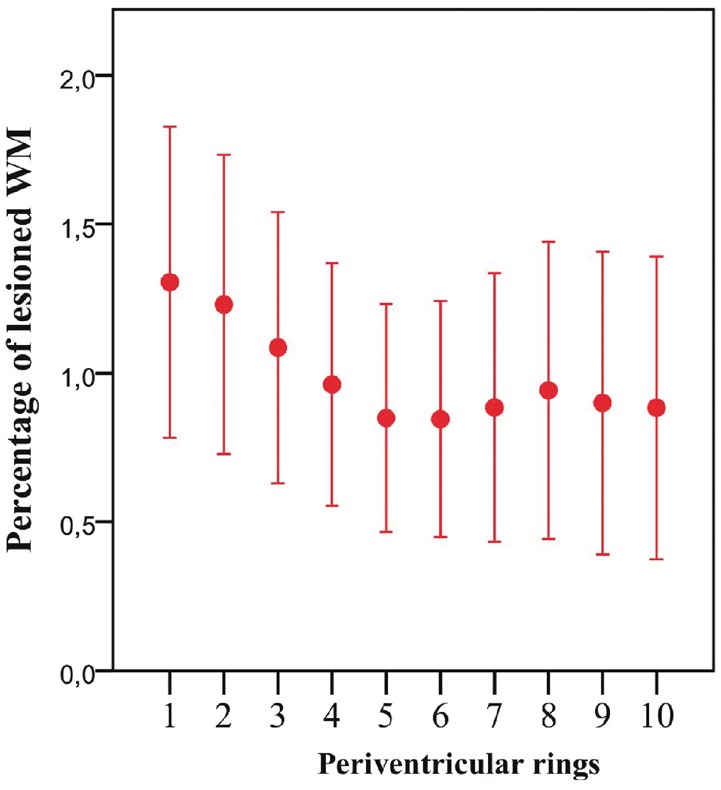
Percentage periventricular lesional white matter. Percentage of lesional white matter (WM) per ring in patients with optic neuritis 4.6 months after symptom onset (ON). Mean ± 2 SE. Ring 1 is closest to the ventricular surface.
Discussion
In a previous study in patients with established relapsing-remitting and secondary progressive multiple sclerosis, we found that multiple sclerosis effects on NAWM MTR increased close to the lateral ventricles (Liu et al., 2015). However, we could not determine how early in the clinical course of multiple sclerosis this abnormal periventricular MTR gradient occurred, whether or not it was due to a mechanism independent of white matter lesion formation, or if it was associated with subsequent disease activity. In the present study we found that an abnormal periventricular MTR gradient is present within 5 months of a clinically isolated optic neuritis, is not dependent on the presence of white matter lesions and is associated with the subsequent risk of developing multiple sclerosis and disability. Both the periventricular NAWM MTR gradient and white matter lesions independently predicted conversion to CDMS over 2 years. This raises the possibility that lesions and NAWM periventricular abnormalities in early multiple sclerosis may arise from different but nevertheless clinically relevant pathophysiological processes.
In the optic neuritis group we looked for significant differences in the periventricular MTR gradients between those with and without asymptomatic white matter lesions, those with and without periventricular white matter lesions, and those with and without gadolinium-enhancing lesions, and found none. Consistent with previous work (Liu et al., 2015) MTR in lesions also showed periventricular gradients similar to those in NAWM. Collectively, this strongly suggests that abnormal gradients in NAWM MTR are not dependent on the presence of white matter lesions. In keeping with previous results (Barkhof et al., 1997) periventricular lesions predicted conversion to CDMS over 2 years, and in a multivariate logistic regression the MTR gradient over 1–5 mm also independently predicted conversion to CDMS over 2 years. The identification of an early abnormal periventricular MTR gradient that is not directly related to lesion formation, but linked with clinical outcomes suggests that there is a pathological process distinct to that underlying lesion formation.
The pathological basis of the abnormal periventricular MTR gradient is uncertain. MTR correlates with myelin and axonal density (Schmierer et al., 2007) and may also be reduced by tissue oedema and inflammation (Dousset et al., 1992; Gareau et al., 2000). To the best of our knowledge, there have been no histopathological studies looking for a gradient of pathology in extra-lesional periventricular white matter. Several mechanisms could underlie the periventricular MTR changes seen, perhaps in combination, but without knowing the underlying pathological substrate we can only speculate which are responsible.
Considering first factors external to the brain, CSF-mediated and ependymal processes may both influence multiple sclerosis pathology, particularly in tissues close to the surface. CSF from patients with a CIS or multiple sclerosis has been found to cause neuronal death in vitro (Alcazar et al., 2000; Vidaurre et al., 2014), and when sampled at the time of relapse in relapsing-remitting multiple sclerosis induces oligodendrocyte apoptosis (Menard et al., 1998). In addition, leucocytes can enter the CSF space through the choroid plexus, and in patients with multiple sclerosis the concentration of the tight junction protein claudin-3—which helps maintain the blood-CSF barrier integrity—is lower when compared with healthy controls (Kooij et al., 2014). ‘Granular ependymitis’ has been reported at post-mortem in some patients with multiple sclerosis; however, while it is associated with 1 in 10 periventricular lesions it is also evident distant from lesions, and is seen in non-inflammatory CNS conditions (Adams et al., 1987).
Within the brain, there are several mechanisms through which a periventricular pathological gradient might arise. Axons and oligodendrocytes are susceptible to hypoxia, and the periventricular venous watershed renders this region intrinsically susceptible to hypoperfusion and hypoxia (Andeweg, 1996); periventricular hypoperfusion is significantly greater in multiple sclerosis than controls and is evident even after a CIS (Dewar et al., 2003; Varga et al., 2009; Beggs, 2013). White matter lesions nearly always form around veins, and the high venular density around the lateral ventricles likely accounts for white matter lesions’ periventricular predilection (Brownell and Hughes, 1962; Adams et al., 1987; Narayanan et al., 1997; Evangelou et al., 2000). However, we think that lesions themselves are unlikely to directly explain the white matter periventricular MTR gradient, as we have excluded lesions plus a two-voxel layer NAWM perilesional cuff, and extending this to a four-voxel layer cuff in our previous work did not materially alter the results (Liu et al., 2015). A tract-mediated effect of lesions may also contribute to gradients: adjacent to the lateral ventricles run multiple white matter tracts, e.g. those in the corpus callosum, and so it may be expected the remote effects of axonal transection in lesions (Trapp et al., 1998) will be more apparent in regions with high compared with low densities of parallel tracts. Again, we think that it is unlikely that this explains our present results as the periventricular MTR gradient did not significantly differ between those who did and those who did not have additional asymptomatic brain lesions, and the mean lesion load was low (∼1.5 ml).
Elucidating the responsible mechanism(s) may have significant implications for early multiple sclerosis treatment. For example, in established multiple sclerosis, neuropathological work has shown a cortical gradient in neuronal loss (Magliozzi et al., 2010) where an MTR gradient has also been identified (Samson et al., 2014). If a gradient in axonal loss were confirmed around the ventricles, one might infer that the in vivo periventricular MTR findings indicate a neurodegenerative process that should be targeted from the earliest clinical stages of multiple sclerosis.
There are a few study limitations worth noting. First, we used previously acquired MTR data with lower resolution (1 × 1 × 5 mm) than that used in our recent study examining periventricular gradients in established multiple sclerosis (1 × 1 × 1 mm; Liu et al., 2015) and given this, partial volume effects were a greater concern. To minimize these, we restricted our analyses to two axial slices perpendicular to the lateral ventricular wall, so avoiding oblique CSF–white matter boundaries within periventricular voxels, and through plane smoothing of periventricular MTR gradients (recalling that these were seen over the first 5 mm around the ventricles, and the axial slices were 5 mm thick). Despite this, the first two periventricular rings still contained CSF in the majority of participants, and so these rings were also excluded from subsequent analyses. Noting that the periventricular MTR gradient seen in previous work was steepest close to the ventricles (Liu et al., 2015), it is likely that excluding these rings will have significantly reduced our sensitivity to gradients. In 10/81 participants with optic neuritis and 2/39 healthy controls, there was still CSF in the third ring so these participants were also excluded from the analyses, further reducing the power of the study to detect periventricular gradients. While we were very careful to avoid partial volume effects between CSF and white matter, partial volume between periventricular rings will tend to smooth MTR gradients, and so reduce sensitivity to disease effects. Similarly, restricting our analyses to two axial slices, while limiting the risk of MTR gradients being smoothed through plane, substantially reduced the volume of each ring studied when compared to our previous study using 1 × 1 × 1 mm MTR data; this too is likely to have reduced sensitivity to disease effects. To allow for brain atrophy, which could exacerbate this further, we included brain parenchymal fraction as a covariate when looking for differences in MTR gradients between clinical subgroups, and found no material difference in the results. To provide context with established MTR metrics and confirm that the difference in MTR gradient between converters and non-converters was not simply driven by diffuse tissue differences, we added NAWM and grey matter MTR as covariates and the MTR gradient remained a significant factor.
By 5 years, ∼55% of the optic neuritis group developed CDMS, while at the time of their MTR scan only ∼10% had done so (Table 1). The pattern of group differences in periventricular MTR gradient were still apparent, albeit with less statistical significance, following exclusion of those converting before the MTR scan (Supplementary material). As such, we think it is unlikely that the apparent predictive power of the periventricular MTR gradient for conversion to CDMS within 5 years is simply due to such a gradient being present in those who had already developed CDMS by the time of their MTR scan. Thirteen of the 71 patients with optic neuritis studied at baseline did not have clinical follow-up. There were no significant differences in any baseline demographic features between those who were or were not followed up (Supplementary Table 1), therefore we believe this is unlikely to have biased our results in favour of detecting differences in periventricular MTR gradients between groups, but it may have reduced our sensitivity to them.
While we have assessed cross-sectional differences in MTR measures between the healthy control and optic neuritis groups, it would be of considerable interest to see how these differences evolve with time, and whether or not these changes relate more closely to clinical outcomes. Some longitudinal MTR data are available for the present cohorts (healthy controls n = 18, optic neuritis n = 44), but the resolution of the 2D MTR scans (1 × 1 × 5 mm) means that it is not possible to accurately align the same axial slices over time. When we looked for changes in periventricular MTR gradients over time without registering scans, we found no significant change over 5 years.
It is well recognized that a substantial number of patients with multiple sclerosis may have significant cognitive deficits (Langdon, 2011). However, only about half (31/58) of those followed-up at 5 years underwent cognitive testing, of whom only 12 had developed CDMS. Given this, it is perhaps unsurprising that we did not find a correlation with periventricular MTR gradients in this cohort, and this would perhaps be better examined in a group of patients with progressive multiple sclerosis who are more likely to have cognitive impairment (Langdon, 2011).
The parent cohort’s principal presenting CIS was optic neuritis, and for consistency we excluded three non-optic neuritis participants who would otherwise have been eligible for our study. In some cohorts, optic neuritis appears to carry a lower risk of conversion to CDMS when compared with other presentations (Tintore et al., 2015) and it would be of interest to see if periventricular gradients differ dependent on the type of CIS.
While inner periventricular MTR gradients independently contributed to the prediction of conversion to CDMS and McDonald multiple sclerosis, there was substantial overlap in the range of values between groups (Supplementary material) and no clear threshold was found beyond which conversion to multiple sclerosis by 5 years was inevitable. As such measurement of periventricular MTR gradients using the methods employed in this study is unlikely to be useful in clinical practice. However, this does not negate the clinical relevance of the pathological processes underlying abnormal MTR gradients, which—independent of those leading to white matter lesion formation—may be a potential target for treatments.
In conclusion, our findings show that an abnormal periventricular MTR gradient occurs soon after a CIS and is associated with subsequent conversion to multiple sclerosis and disability. The abnormal periventricular MTR gradient was not significantly affected by the presence of white matter lesions, and therefore seems likely to arise from an at least partly independent mechanism. Histopathological studies are warranted to elucidate the nature of these MTR gradients.
Supplementary Material
Acknowledgements
The authors thank the patients who took part in the study, the Multiple Sclerosis Society of Great Britain and Northern Ireland and the National Institute for Health Research University College London Hospitals Biomedical Research Centre for financial support.
Funding
The Queen Square MS centre is supported by the MS Society of Great Britain and Northern Ireland. J.W.L.B is funded through a Next Generation Fellowship funded by the Grant Charity of the Freemason’s. M.P. is supported by the non-profit Karol Wojtila Association (Lavagna, Italy). F.P. is funded by the National Institute for Health Research University College London Hospitals Biomedical Research Centre (NIHR BRC UCLH/UCL High Impact Initiative-BW.mn.BRC10269). S.O. receives funding from the EPSRC (EP/H046410/1, EP/J020990/1, EP/K005278), the MRC (MR/J01107X/1), the NIHR Biomedical Research Unit (Dementia) at UCL and the NIHR BRC UCLH/UCL (BW.mn.BRC10269). W.J.B. is funded by the Neurological Foundation of New Zealand and the United Kingdom Multiple Sclerosis Society. D.C. receives research support from the MS Society of Great Britain and Northern Ireland and the National Institute for Health Research (NIHR) University College London Hospitals (UCLH) Biomedical Research Centre.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- CDMS
clinically definite multiple sclerosis
- CIS
clinically isolated syndrome
- EDSS
Expanded Disability Status Scale
- FSPGR
fast spoiled gradient echo
- MTR
magnetization transfer ratio
- NAWM
normal-appearing white matter
References
- Adams CW, Abdulla YH, Torres EM, Poston RN. Periventricular lesions in multiple sclerosis: their perivenous origin and relationship to granular ependymitis. Neuropathol Appl Neurobiol 1987; 13: 141–52. [DOI] [PubMed] [Google Scholar]
- Alcazar A, Regidor I, Masjuan J, Salinas M, Alvarez-Cermeno JC. Axonal damage induced by cerebrospinal fluid from patients with relapsing-remitting multiple sclerosis. J Neuroimmunol 2000; 104: 58–67. [DOI] [PubMed] [Google Scholar]
- Andeweg J. The anatomy of collateral venous flow from the brain and its value in aetiological interpretation of intracranial pathology. Neuroradiology 1996; 38: 621–8. [DOI] [PubMed] [Google Scholar]
- Barker GJ, Tofts PS, Gass A. An interleaved sequence for accurate and reproducible clinical measurement of magnetization transfer ratio. Magn Reson Imaging 1996; 14: 403–11. [DOI] [PubMed] [Google Scholar]
- Barkhof F, Filippi M, Miller DH, Scheltens P, Campi A, Polman CH, et al. Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain 1997; 120 (Pt 11): 2059–69. [DOI] [PubMed] [Google Scholar]
- Beggs CB. Venous hemodynamics in neurological disorders: an analytical review with hydrodynamic analysis. BMC Med 2013; 11: 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownell B, Hughes JT. The distribution of plaques in the cerebrum in multiple sclerosis. J Neurol Neurosurg Psychiatry 1962; 25: 315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee WJ, Swanton JK, Altmann DR, Ciccarelli O, Miller DH. Earlier and more frequent diagnosis of multiple sclerosis using the McDonald criteria. J Neurol Neurosurg Psychiatry 2015; 86: 584–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese M, De Stefano N, Atzori M, Bernardi V, Mattisi I, Barachino L, et al. Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis. Arch Neurol 2007; 64: 1416–22. [DOI] [PubMed] [Google Scholar]
- Chard DT, Jackson JS, Miller DH, Wheeler-Kingshott CA. Reducing the impact of white matter lesions on automated measures of brain gray and white matter volumes. J Magn Reson Imaging 2010; 32: 223–8. [DOI] [PubMed] [Google Scholar]
- Cutter GR, Baier ML, Rudick RA, Cookfair DL, Fischer JS, Petkau J, et al. Development of a multiple sclerosis functional composite as a clinical trial outcome measure. Brain 1999; 122 (Pt 5): 871–82. [DOI] [PubMed] [Google Scholar]
- Dewar D, Underhill SM, Goldberg MP. Oligodendrocytes and ischemic brain injury. J Cereb Blood Flow Metab 2003; 23: 263–74. [DOI] [PubMed] [Google Scholar]
- Dousset V, Grossman RI, Ramer KN, Schnall MD, Young LH, Gonzalez-Scarano F, et al. Experimental allergic encephalomyelitis and multiple sclerosis: lesion characterization with magnetization transfer imaging. Radiology 1992; 182: 483–91. [DOI] [PubMed] [Google Scholar]
- Evangelou N, Esiri MM, Smith S, Palace J, Matthews PM. Quantitative pathological evidence for axonal loss in normal appearing white matter in multiple sclerosis. Ann Neurol 2000; 47: 391–5. [PubMed] [Google Scholar]
- Fernando KT, Tozer DJ, Miszkiel KA, Gordon RM, Swanton JK, Dalton CM, et al. Magnetization transfer histograms in clinically isolated syndromes suggestive of multiple sclerosis. Brain 2005; 128 (Pt 12): 2911–25. [DOI] [PubMed] [Google Scholar]
- Fisniku LK, Brex PA, Altmann DR, Miszkiel KA, Benton CE, Lanyon R, et al. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain 2008; 131 (Pt 3): 808–17. [DOI] [PubMed] [Google Scholar]
- Gallo A, Rovaris M, Riva R, Ghezzi A, Benedetti B, Martinelli V, et al. Diffusion-tensor magnetic resonance imaging detects normal-appearing white matter damage unrelated to short-term disease activity in patients at the earliest clinical stage of multiple sclerosis. Arch Neurol 2005; 62: 803–8. [DOI] [PubMed] [Google Scholar]
- Gareau PJ, Rutt BK, Karlik SJ, Mitchell JR. Magnetization transfer and multicomponent T2 relaxation measurements with histopathologic correlation in an experimental model of MS. J Magn Reson Imaging 2000; 11: 586–95. [DOI] [PubMed] [Google Scholar]
- Grabner G, Janke AL, Budge MM, Smith D, Pruessner J, Collins DL. Symmetric atlasing and model based segmentation: an application to the hippocampus in older adults. Med Image Comput Comput Assisted Interv 2006; 9 (Pt 2): 58–66. [DOI] [PubMed] [Google Scholar]
- Henry RG, Shieh M, Amirbekian B, Chung S, Okuda DT, Pelletier D. Connecting white matter injury and thalamic atrophy in clinically isolated syndromes. J Neurol Sci 2009; 282: 61–6. [DOI] [PubMed] [Google Scholar]
- Hickman SI, Barker GJ, Molyneux PD, Miller DH. Technical note: the comparison of hypointense lesions from ‘pseudo-T1' and T1-weighted images in secondary progressive multiple sclerosis. Mult Scler 2002; 8: 433–5. [DOI] [PubMed] [Google Scholar]
- Kooij G, Kopplin K, Blasig R, Stuiver M, Koning N, Goverse G, et al. Disturbed function of the blood-cerebrospinal fluid barrier aggravates neuro-inflammation. Acta Neuropathol 2014; 128: 267–77. [DOI] [PubMed] [Google Scholar]
- Langdon DW. Cognition in multiple sclerosis. Curr Opin Neurol 2011; 24: 244–9. [DOI] [PubMed] [Google Scholar]
- Liu Z, Pardini M, Yaldizli O, Sethi V, Muhlert N, Wheeler-Kingshott CA, et al. Magnetization transfer ratio measures in normal-appearing white matter show periventricular gradient abnormalities in multiple sclerosis. Brain 2015; 138 (Pt 5): 1239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 2010; 68: 477–93. [DOI] [PubMed] [Google Scholar]
- Maldjian JA, Laurienti PJ, Kraft RA, Burdette JH. An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage 2003; 19: 1233–9. [DOI] [PubMed] [Google Scholar]
- Menard A, Pierig R, Pelletier J, Bensa P, Belliveau J, Mandrand B, et al. Detection of a gliotoxic activity in the cerebrospinal fluid from multiple sclerosis patients. Neurosci Lett 1998; 245: 49–52. [DOI] [PubMed] [Google Scholar]
- Modat M, Cash DM, Daga P, Winston GP, Duncan JS, Ourselin S. Global image registration using a symmetric block-matching approach. J Med Imaging 2014; 1: 024003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modat M, Ridgway GR, Taylor ZA, Lehmann M, Barnes J, Hawkes DJ, et al. Fast free-form deformation using graphics processing units. Comput Methods Programs Biomed 2010; 98: 278–84. [DOI] [PubMed] [Google Scholar]
- Muhlert N, Sethi V, Schneider T, Daga P, Cipolotti L, Haroon HA, et al. Diffusion MRI-based cortical complexity alterations associated with executive function in multiple sclerosis. J Magn Reson Imaging 2013; 38: 54–63. [DOI] [PubMed] [Google Scholar]
- Narayanan S, Fu L, Pioro E, De Stefano N, Collins DL, Francis GS, et al. Imaging of axonal damage in multiple sclerosis: spatial distribution of magnetic resonance imaging lesions. Ann Neurol 1997; 41: 385–91. [DOI] [PubMed] [Google Scholar]
- Nielsen AS, Kinkel RP, Madigan N, Tinelli E, Benner T, Mainero C. Contribution of cortical lesion subtypes at 7T MRI to physical and cognitive performance in MS. Neurology 2013; 81: 641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ourselin S, Roche A, Subsol G, Pennec X, Ayache N. Reconstructing a 3D structure from serial histological sections. Image Vis Comput 2001; 19: 25–31. [Google Scholar]
- Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011; 69: 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poser CM, Paty DW, Scheinberg L, McDonald WI, Davis FA, Ebers GC, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol 1983; 13: 227–31. [DOI] [PubMed] [Google Scholar]
- Prados F, Cardoso MJ, Kanber B, Ciccarelli O, Kapoor R, Gandini Wheeler-Kingshott CA, et al. A multi-time-point modality-agnostic patch-based method for lesion filling in multiple sclerosis. Neuroimage 2016; 139: 376–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca MA, Agosta F, Sormani MP, Fernando K, Tintore M, Korteweg T, et al. A three-year, multi-parametric MRI study in patients at presentation with CIS. J Neurol 2008; 255: 683–91. [DOI] [PubMed] [Google Scholar]
- Samson RS, Cardoso MJ, Muhlert N, Sethi V, Wheeler-Kingshott CA, Ron M, et al. Investigation of outer cortical magnetisation transfer ratio abnormalities in multiple sclerosis clinical subgroups. Mul Scler 2014; 20: 1322–30. [DOI] [PubMed] [Google Scholar]
- Schmierer K, Tozer DJ, Scaravilli F, Altmann DR, Barker GJ, Tofts PS, et al. Quantitative magnetization transfer imaging in postmortem multiple sclerosis brain. J Magn Reson Imaging 2007; 26: 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintore M, Rovira A, Rio J, Otero-Romero S, Arrambide G, Tur C, et al. Defining high, medium and low impact prognostic factors for developing multiple sclerosis. Brain 2015; 138 (Pt 7): 1863–74. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998; 338: 278–85. [DOI] [PubMed] [Google Scholar]
- Varga AW, Johnson G, Babb JS, Herbert J, Grossman RI, Inglese M. White matter hemodynamic abnormalities precede sub-cortical gray matter changes in multiple sclerosis. J Neurol Sci 2009; 282: 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidaurre OG, Haines JD, Katz Sand I, Adula KP, Huynh JL, McGraw CA, et al. Cerebrospinal fluid ceramides from patients with multiple sclerosis impair neuronal bioenergetics. Brain 2014; 137 (Pt 8): 2271–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrenken H, Geurts JJ, Knol DL, Polman CH, Castelijns JA, Pouwels PJ, et al. Normal-appearing white matter changes vary with distance to lesions in multiple sclerosis. AJNR Am J Neuroradiol 2006; 27: 2005–11. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.