

Abstract
Bone metastases occur in ~70% of metastatic breast cancer patients often leading to skeletal injuries. Current treatments are mainly palliative and underscore the unmet clinical need for improved therapies. In this study, we provide preclinical evidence for an antimetastatic therapy based on targeting integrin β3 (β3) which is selectively induced on breast cancer cells in bone by the local bone microenvironment. In a preclinical model of breast cancer, β3 was strongly expressed on bone metastatic cancer cells but not primary mammary tumors or visceral metastases. In tumor tissue from breast cancer patients, β3 was significantly elevated on bone metastases relative to primary tumors from the same patient (n=42). Mechanistic investigations revealed that TGF-β signaling through SMAD2/SMAD3 was necessary for breast cancer induction of β3 within the bone. Using a micelle-based nanoparticle therapy that recognizes integrin αvβ3 (αvβ3-MPs of ~12.5nm), we demonstrated specific localization to breast cancer bone metastases in mice. Using this system for targeted delivery of the chemotherapeutic docetaxel, we showed that bone tumor burden could be reduced significantly with less bone destruction and less hepatotoxicity compared to equimolar doses of free docetaxel. Furthermore, mice treated with αvβ3-MP-docetaxel exhibited a significant decrease in bone-residing tumor cell proliferation compared to free docetaxel. Taken together, our results offer preclinical proof of concept for a method to enhance delivery of chemotherapeutics to breast cancer cells within the bone by exploiting their selective expression of integrin αvβ3 at that metastatic site.
Keywords: Breast Cancer, Bone Metastases, Integrins, Nanoparticles, Drug Therapy
Introduction
Bone metastases occur in approximately 70% of metastatic breast cancer patients (1), often leading to the development of significant skeletal complications, such as pathological fracture, spinal cord compression, or bone pain (2). Current treatments – radiation, surgery, chemotherapy, and antiresorptive drugs – can improve quality of life but are rarely curative, with limited effect on overall survival (3-5). Treating breast cancer bone metastases has proven difficult due to frequent dissemination of metastases throughout the skeleton, a lack of tumor-specific targets expressed across breast cancer subtypes, and the chemo-protective nature of the bone microenvironment (2,6,7).
One approach to overcome these problems has been to enhance drug delivery to bone, most commonly by using bone matrix-targeted hydroxyapatite-avid bisphosphonates (8-10). Bisphosphonates are standard of care in patients with bone metastases or osteoporosis for their ability to inhibit osteoclast function and formation (1,2,10). While osteoclast inhibition can attenuate tumor-associated bone destruction with secondary effects on bone tumor growth, it does not improve overall survival in metastatic breast cancer patients (11,12). In line with this, bisphosphonate-targeted chemotherapy delivery can reduce mouse models of breast cancer-associated bone loss more effectively than free-drug chemotherapy (13,14). However, bisphosphonate-targeted drug delivery indiscriminately accumulates on all skeletal bone matrix and primarily targets osteoclasts and neighboring marrow cells near the bone matrix (8,15). To improve drug uptake by bone metastases, we sought to target tumor cells in the bone microenvironment more directly.
One targeting candidate of interest was the integrin αvβ3. Integrins are heterodimeric cell surface receptors, composed of an α and β subunit from a large family of subunits, that bind to components of the extracellular matrix or to other cells (16). Integrin are expressed in a cell-specific and context-dependent manner, and are critical to all aspects of cancer progression and metastasis through effects on migration, invasion, and cell survival (16-18). Specifically, integrin αvβ3 is composed of the tightly regulated integrin subunit β3 and the more widely expressed αv subunit. Expression of αvβ3 on most cells in the body is typically low, but is elevated on several cancer types (17), as well as a variety of cell types that are important within the tumor microenvironment, such as osteoclasts (11,12), neo-angiogenic endothelium (19), and tumor-promoting macrophages (20). The expression of αvβ3 on these cells led to the investigation of αvβ3 as a therapeutic target for cancer treatment (21). In several clinical trials, pharmacological inhibition with αvβ3 antagonists as a single-agent cancer therapy did not demonstrate a significant clinical effect (22,23). We, and others, have demonstrated that the inefficiency of αvβ3 inhibitors in advanced cancer may be due to the inherent complexities of integrin signaling and off-target effects of αvβ3 antagonism (20,24).
Rather than aiming to antagonize αvβ3 function, we sought to utilize αvβ3 as a molecular target for enhancing drug delivery to cells of interest within the bone. Integrin β3 and αvβ3 expression has been observed on human breast cancer bone metastases (25-27), and ectopic overexpression of tumoral β3 on breast cancer has been demonstrated to enhance tumor establishment in bone (26,28). However, for the purpose of targeting αvβ3, physiological expression on breast cancer during tumor growth and metastasis has not been defined. Here, we report that the bone microenvironment preferentially induces integrin β3 on breast cancer metastases, as compared to the primary tumor or visceral metastases, and we identify TGF-β to be responsible for this induction. Utilizing this information, we evaluated nanoparticle-mediated drug delivery targeted against integrin αvβ3. We recently developed phospholipid/polysorbate-80 micelle nanoparticles (MPs, ~12.5 nm) for their small size and unique mechanism of “contact-facilitated drug delivery” (29); in the present study, we demonstrate that integrin αvβ3-targeted micelle nanoparticles (αvβ3-MPs) carrying the chemotherapeutic docetaxel reduce bone metastases and tumor-associated bone destruction more effectively than free-docetaxel. Collectively, we demonstrate that αvβ3 is a tumor target on breast cancer bone metastases and provide support for safer, more effective therapies against this often incurable disease by targeting integrin αvβ3.
Materials and Methods
Animals
Animal studies were performed according to the guidelines established by the Washington University Institutional Animal Care and Use Committee (IACUC). PyMT-Bo1 tumor cells were implanted into female C57BL/6 mice, or 4T1 tumor cells were implanted into female BALB/c mice. All mice were obtained from The Jackson Laboratory, and injected at 6 to 7 weeks of age. All rodents were housed according to the guidelines of the Division of Comparative Medicine, Washington University School of Medicine. In collaboration with Dr. Theresa Guise, histological bone sections from female athymic nude mice injected with MDA-MB-231 human breast cancer cells were obtained from an experiment previously described (30).
Cell lines
The murine C57BL/6 PyMT-Bo1 luminal B breast cancer cell line (stably expressing GFP and firefly luciferase genes) was originally isolated from a transgenic MMTV-PyMT breast tumor, as previously validated and described (20). The murine BALB/c 4T1 triple-negative breast cancer cell line was purchased from ATCC, as previously described (31). Human MDA-MB-231 breast cancer cells (HTB-26) were purchased from ATCC (30). All cells were maintained at sub-confluence in DMEM with 10% FBS and 0.5% penicillin–streptomycin, in a humidified chamber at standard culture conditions. Low-passage stocks were used and regularly tested for Mycoplasma and maintenance of growth characteristics.
Murine cancer models
To establish orthotopic mammary fat pad (MFP) tumors, 0.1×106 tumor cells in 50μL PBS were injected into MFP tissue of 7-week-old female mice. To establish experimental secondary metastases, 0.05×106 tumor cells in 50uL PBS were intracardiac injected into the left ventricular chamber of 6-week-old female, with one exception; in collaboration with Dr. Theresa Guise, human MDA-MB-231 tumor cells were intracardiac injected (0.1×106 tumor cells in 100μL PBS) into 4-week-old female athymic nude mice, as previously described (30).
Synthesis of αvβ3-targeted micelle nanoparticles
Phospholipid/polysorbate 80 micelle nanoparticles (MPs) were prepared as a microfluidized suspension of 20% (v/v), combining polysorbate tween 80 (Sigma Aldrich, Inc.) with a 2.0% (w/v) of a surfactant comixture, and 1.7% (w/v) glycerin in pH 6.5 carbonate buffer, as previously described (29). Optionally, the surfactant comixture included 2.28mol% of docetaxel-prodrug (DTX-PD), and/or 0.15mol% of αvβ3-targeted quinolone nonpeptide coupled to phosphatidylethanolamine-PEG2000, with the remaining mol% lecithin. Docetaxel was modified into an Sn2 lipase-labile phosphatidylcholine DTX-PD as previously described (32). For fluorescent labeling, rhodamine-conjugated to phosphatidylethanolamine (0.1 mol%) was incorporated into the lipid surfactant. The surfactant components for each formulation were combined with the polysorbate, buffer, and glycerin mixtures and were homogenized at 20,000 psi for 4 minutes at 4C with a microfluidics (M110s; Microfluidics, Inc). The nanoparticles were preserved under inert gas in sterile sealed vials until use. Dynamic light scattering showed a nominal particle size of 12.5±0.8 nm, with polydispersities 0.290±0.03, and an average electrophoretic zeta potential of -3.82±1.23 mV.
The nanoparticle-targeting quinolone nonpeptide specific for integrin αvβ3 was originally developed by Bristol-Myers Squibb Medical Imaging (US patent 6,511,648 and related patents) and coupled to phosphatidylethanolamine-PEG2000, as previously described (29). The quinolone nonpeptide was initially characterized as the 111In-DOTA conjugate RP478 and cyan 5.5 homologue TA145 (33). This αvβ3-targeting ligand is selective for cells expressing αvβ3 (IC50 = 12 nM), as compared to IC50>10 μM for ανβ5, α5β1, or αIIββ3 (34). Furthermore, the affinity of the ανβ3-targeting ligand increases 15-fold for activated integrin αvβ3 receptor in the presence of Mn2+ (33). In vitro, pretreating human endothelial with the human-specific integrin αvβ3 antibody (LM609) competitively inhibited binding of this αvβ3-targeting ligand (33). This αvβ3-targeted ligand has been applied to the surface of large, vascular-constrained perfluorocarbon nanoparticles (~250nm), which displayed a specific affinity for αvβ3-expressing angiogenic blood vessels and αvβ3-expressing melanoma cells, in contrast to nanoparticle controls (32,35,36). The in vivo specificity of this αvβ3-targeting ligand has been further demonstrated through competitive pretreatment with unlabeled αvβ3-targeting ligand, which inhibited αvβ3-targeted nanoparticle binding (35).
Bioluminescence imaging (BLI)
In vivo and ex vivo BLI was performed on IVIS50 (PerkinElmer, Waltham, MA) as previously described (20). Total photon flux (photons/sec) was measured from fixed regions of interest (ROIs) over entire mouse or manually around ex vivo organs using Living Image 2.6, as indicated. Mice with outstanding chest BLI intensity indicative of a failed intracardiac injection or with ineffective D-luciferin administration were excluded from all analyses. Investigators were blinded to treatment groups during all BLI analyses.
Rhodamine-labeled MP colocalization with breast cancer bone metastases
Tumor-bearing mice day 8 post-intracardiac injected with PyMT-Bo1 cells, or age-matched tumor-free mice, were treated with a single dose of rhodamine-labeled MPs (either non-targeted or αvβ3-targeted) at a nanoparticle dose of 2nmol/g mouse weight. Investigators were blinded to treatment groups during intravenous MP and αvβ3-MP injections. MPs were allowed to circulate for 3 hours in vivo, before sacrifice and tissue collection. Unbound MPs were cleared from circulation via cardiac perfusion with 30mL of PBS.
For fluorescent analysis of nanoparticle colocalization within bone, fresh-frozen long bones in optimum cutting temperature embedding medium (Tissue-Tek) were sectioned 5μm thick onto cryofilm tape (Section Lab Co., Ltd, Hiroshima, Japan) at the histology core of the Washington University Musculoskeletal Research Center. Cryofilm tape sections were fixed in -20°C acetone for 5 minutes and air-dried for 10 minutes. Slides were briefly rehydrated with PBS, and then mounted with ProLong Gold with DAPI (Invitrogen).
Fluorescent imaging and analysis
Fluorescent images were captured on a Photometrics CoolSNAP MYO camera connected to a Nikon Eclipse Ti-E microscope, acquired with the 4x Plan Fluor PhL DL objective through a TxRED HYQ filter cube and DAPI filter cube. The TxRED fluorescent channel was equally set using fluorescent look-up-table (LUT) to the same minimum and maximum values, gamma=1. Fluorescent colocalization analysis was performed using NIS-Elements AR (Nikon Canada, Inc., Richmond, BC, Canada). Bone metastatic region of interest (ROI) was drawn at the tumor/marrow boundary, as defined by DAPI visualization of nuclei density. Using the object count function in NIS-Elements AR, fluorescent nanoparticle colocalization was calculated via the ratio of rhodamine-positive pixel area by the total tumor ROI pixel area [(number of pixels of positively labeled objects within the fixed tumor area) / (total number of pixels within the fixed tumor area) × 100].
Treatment of murine breast cancer metastases
Using the experimental metastasis model, PyMT-Bo1 metastases were established within female C57BL/6 mice. BLI analysis confirmed the establishment of bone metastases on day 3 post-intracardiac injection, and metastatic-bearing mice were randomly sorted and treatment started. Docetaxel (LC Laboratories, Woburn, MA) was prepared at 10 mg/mL in a tween80/ethanol/saline (20:13:67, v/v/v) solution for drug solubility, and diluted to 0.5 mg/mL DTX or 1.0 mg/mL in saline for intravenous administration. For nanoparticle treatment, mice received either a cumulative dose of 5.55 mg/kg docetaxel, an equimolar dose of nanoparticle-encapsulated docetaxel-prodrug (2.2 μmol/kg), or an equal amount of saline or cargo-free αvβ3-MP, fractionally administered intravenously every three days. A priori comparisons of interest were between saline-treated mice as compared to DTX or αvβ3-MP/DTX-PD, or between saline-treated mice compared to the nanoparticle control treatments.
In collaboration with Dr. Theresa Guise, histological bone sections were obtained from female athymic nude mice bearing MDA-MB-231 bone metastases, treated daily with the TGF-β Receptor I kinase inhibitor, 60 mg/kg/d SD-208 (Epichem Pty Ltd, Murdoch University, Australia) or vehicle (1% methylcellulose), from an experiment previously published (30).
Serum chemistry and hematologic analysis
Blood was collected by submandibular vein puncture into Microtainer EDTA tubes (BD Biosciences) for hematologic analysis (Hemavet 950 FS; Drew Scientific, Inc., Dallas, TX) or Microtainer serum separator tubes (BD Biosciences) for serum chemistry analysis on a Liasys 330 AMS Diagnostic liquid chemistry analyzer (Weston, FL). Investigators were blinded to treatment groups during analysis.
MTT assay
The MTT assay was performed as described previously (31). Signal intensity normalized to 0% for the viability of cells at time of drug addition, and 100% for the viability of vehicle control treated cells at 72 hr.
Radiography
Osteolytic lesions were imaged by X-Ray imaging system (Faxitron, Buffalo Grove, IL), and lesion area within the tibiofemoral joint was quantified using ImageJ (NIH, Bethesda, Maryland).
Immunohistological staining
Freshly removed tissue was fixed in 10% neutral buffered formalin for 24 hours. Bone was decalcified in 14% EDTA for 10 days. Tissue was paraffin embedded and sectioned 5μm thick at the histology core of the Washington University Musculoskeletal Research Center. Standard tartrate-resistant acidic phosphatase (TRAP) staining or hematoxylin and eosin (H&E) staining was performed by the musculoskeletal histology core of the Washington University Musculoskeletal Research Center.
For immunohistochemistry, all slides were stained in parallel, using identical staining conditions. Paraffin tissue slides were prepared by immersing slides in xylene and rehydrating tissue in 100% ethanol, 95% ethanol, 70% ethanol, 50% ethanol, and deionized water washing steps. Slides were immersed in EDTA antigen retrieval buffer (1mM EDTA, 0.05% Tween 20, PH 8.0) at 50 °C overnight. Slides were treated with dual endogenous enzyme block (Dako), TBS/0.1% Tween-20 (TBST) wash buffer, and 10 minutes of serum-free protein block (Dako). Slides were stained with the following primary antibodies: anti-integrin β3 antibody (D7X3P, 1:200, Cell Signaling), anti-integrin αv (ab179475, 1:500, Abcam), isotype control rabbit IgG (ab27472, Abcam) antibody, or biotinylated anti-PCNA antibody (PC10, 1:100, eBioscience). Following primary antibody incubation, slides were extensively washed in TBST. Either Anti-Rabbit EnVision+ System-HRP (Dako) or Vectastain Elite ABC HRP kit (Vector Laboratories, Burlingame, California) was used as the secondary antibody, followed by Liquid DAB+ (3,3’-Diaminobenzidine) Substrate system (Dako), according to the manufacturer’s protocol. Nuclear hematoxylin counterstain was applied, followed by dehydration through 70% ethanol, 95% ethanol, 100% ethanol, and xylene. Slides were mounted with Cytoseal XYL (Thermo Scientific).
Histological imaging and analysis
Histological slides were imaged on either an Olympus NanoZoomer 2.0-HT System or on a Zeiss AxioScan.Z1. In each experiment, post-image analysis was limited to changes in brightness or contrast, gamma = 1, which were applied equally to all images. Sections stained with integrin β3 or proliferating cell nuclear antigen (PCNA) were quantified using Visiopharm software (Hørsholm, Denmark), which allows for recognition and quantification of DAB-stained tissue areas. A supervised bayesian pixel classifier was used to classify an image based on three distinct categories: DAB staining, hematoxylin staining (nuclei), and unstained tissue and other background structures. Integrin β3 expression from each sample was calculated within 5 to 10 random high-powered fields (hpf) of 100-200 cells within the tumors. Values expressed as the percentage of integrin β3 expression (area of total DAB-positive staining) divided by the tumor area in the hpf. PCNA proliferation index was quantified by calculating the number of PCNA-positive cells divided by the total number of cells within the entire bone metastatic region. ImageJ software (NIH, Bethesda, Maryland) was used to measure tumor area, and to quantitate osteoclasts (OCs) (defined as a TRAP positive, multinucleated cell on a bone surface) per millimeter of bone surface (mmB.S.) at the tumor/bone interface (N.OC/mmB.S.). Histological analysis of bone metastatic tumor burden (tumor area / total bone marrow area) was calculated by measuring the tumor area within the metaphysis of the tibiofemoral joint, divided by the total marrow area of the metaphysis.
Flow cytometry and fluorescence activated cell sorting (FACS)
In vitro tumor cells were lifted with 1x Versene (Invitrogen). Ex vivo tumor cells were collected from the MFP or the bone, and prepared into single-cell suspensions for flow cytometry analysis as previously described (20). Ex vivo cells were stained with PE-conjugated anti-mouse integrin β3 (1:200, clone: 2C9.G2, BD Pharmingen), fixed and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s protocol, and then with AlexaFluor488-conjugated anti-human/mouse cytokeratin 18 (1:100, clone LDK18, eBioscience). Data acquisition was performed on the LSRFortessa (BD Biosciences) and FlowJo software version 10.1 (Tree Star) was used for analysis, and fluorescent compensation using UltraComp eBeads (eBioscience) according to the manufacturer’s protocol. All flow cytometry data presented as median fluorescent intensity (MFI). Ex vivo flow cytometry analysis of bone samples with insufficient number of tumor cells (<500 events) were excluded.
For fluorescence activated cell sorting (FACS), in vitro tumor cells were lifted with 1x Versene (Invitrogen) and stained for surface expression of integrin β3 as described above. Tumor cells were sorted into two populations using a BD FACSAria-II cell sorter (BD Biosciences): integrin β3 negative (β3-) cells (based on the fluorescent intensity of unstained cells) and integrin β3-expressing (β3+) cells. In addition, control cells sorted without β3 discrimination were also collected (β3-all). After sorting, each population was counted for live/dead cells, and 0.05×106 live tumor cells in 50uL PBS were intracardiac injected into the 6-week-old female mice.
Pharmacological inhibition of signaling pathways
Tumor cells were pretreated for 1 hour with pharmacological inhibitors: cells were pretreated for 1 hour with the following pharmacological inhibitors: TGFβ-receptor I kinase inhibitor, specific for the site necessary for SMAD2/SMAD3 phosphorylation (SMAD2/3i, SB431542, 20 μM, Sigma-Aldrich); p38 MAP kinase (MAPK) inhibitor (p38i, SB203580, 20 μM, Cell Signaling); MEK1/2 (MAPK/ERK Kinase) inhibitor (MEK1/2i, U0126, 20 μM, Cell Signaling); c-Jun N-terminal kinase (JNK) inhibitor (JNKi, SP600125, 50 μM, Sigma-Aldrich). After 1 hour of pretreatment, cells were treated with 2 ng/mL of murine TGF-β1 (R&D systems) or vehicle treatment. Cells were all cultured in 0.1% DMSO.
Western blot analysis
Whole cell lysates from tumor cells were collected in RIPA buffer (Cell signaling) in the presence of Halt phosphatase inhibitor cocktail (Thermo Scientific) at 4°C. Protein samples were separated on 10% Mini-PROTEAN TGX polyacrylamide gels (Bio-Rad) and transferred onto an Immobilon-P polyvinylidene difluoride membrane (EMD Millipore). Membranes were incubated with phosphorylated-SMAD2/phosphorylated-SMAD3 (pSMAD2/pSMAD3, D27F4), total SMAD2/SMAD3 (D7G7), integrin β3 (D7X3P), or β-actin (13E5), followed by horseradish peroxidase-conjugated anti-rabbit secondary antibody (all from Cell Signaling). All antibodies were diluted and used according to the manufacturer’s protocol. Bands were developed by enhanced chemiluminescence.
Real-time quantitative PCR (qPCR) analysis
Total RNA from cells was isolated with the RNeasy Mini Plus kit (Qiagen). Complementary DNA was made using the SuperScript II first-strand synthesis system for qPCR (Invitrogen). qPCR was performed using SYBR Advantage mix (Bio-Rad) as previously described (20), with mouse-specific primers for mRNA genes of interest: Itgb1 forward: 5’-GCAGGTGTCGTGTTTGTGAATGCT-3’, Itgb1 reverse: 5’-ACAAGTTGGCCCTTGAAACTTGGG-3’, Itgb3 forward: 5’-TTCAATGCCACCTGCCTCAACAAC-3’, Itgb3 reverse: 5’-ACGCACCTTGGCCTCGATACTAAA-3’, Itgb5 forward: 5’-TGTTCAGCTACACAGAACTGCCCA-3’, Itgb5 reverse: 5’-TTTGGAACTTGGCAAACTCTCGGC-3’, Itgb6 forward: 5’-TCAAACCTCCTTCACTCCTGCCAT-3’, Itgb6 reverse: 5’-TGAATCTGCTGGTTGCTCCAGACT-3’, Itgb8 forward: 5’-CAACTGCATCCAGGAGCTGA-3’, Itgb8 reverse: 5’-AACCAAGACGGAAGTCACGG-3’, Itga2 forward: 5’-AGCCCGTGATCTTTCCTAAAC-3’, Itga2 reverse: 5’-GCAGCCACAGAGTAACCTAAA-3’, Itga2b forward: 5’-ACATTGAGGGCTTTGAGAGGCT-3’, Itga2b reverse: 5’-TTGCCACAGGCAACATCACG-3’, Itga3 forward: 5’-GCGGAAGGACTGGGATTTAT-3’, Itga3 reverse: 5’-GATGATGTCCGTGGGATGTAG-3’, Itgav forward: 5’- AGAAGACGTTGGGCCTATTG-3’, Itgav reverse: 5’-TGTAAGGCCACTGGAGATTTAG-3’, Gapdh forward: 5’-AGGTCGGTGTGAACGGATTTG-3’, Gapdh reverse: 5’-TGTAGACCATGTAGTTGAGGTCA-3’. Target gene expression was normalized against the housekeeping gene GAPDH (Gapdh), and data were analyzed using the ΔΔCt method.
Panel of cytokines and growth factors
Tumor cells were cultured for 24 hours with the following murine factors: 2 ng/mL TGF-β1 (R&D Systems), 2 ng/mL TGF-β2 (R&D Systems), 50 ng/mL Sonic Hedgehog (Shh) (PeproTech, #315-22), 50 ng/mL WNT-3A (PeproTech, #315-20), 100 ng/mL Insulin-Like Growth Factor 1 (IGF-1) (PeproTech, #250-19), 50 ng/mL Epidermal growth factor (EGF) (PeproTech, #315-09), 50 ng/mL Fibroblast Growth Factor 2 (FGF2 or bFGF) (PeproTech, #450-33), 100 ng/mL Osteopontin (OPN) (Leinco, #O121), 10 ng/mL Interleukin 4 (IL-4) (R&D systems), 200 ng/mL Stromal Cell-derived Factor 1α (SDF-1α or CXCL12) (Biolegend), 10 ng/mL Interleukin 6 (IL-6) (R&D systems).
Patient breast cancer and matching bone metastatic biopsies
Matching primary breast and bone metastatic biopsies were both taken at the time of metastatic diagnosis, from patients without detectable bone metastases at diagnosis but detectable bone metastases at least 6 months after diagnosis. Data was obtained in accordance with the guidelines established by the Washington University Institutional Review Board (IRB #201102394) and WAIVER of Elements of Consent per 45 CFR 46.116 (d). All patient information was de-identified prior to sharing with investigators. All of the human research activities and all activities of the IRBs designated in the Washington University (WU) Federal Wide Assurance (FWA), regardless of sponsorship, are guided by the ethical principles in “The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects Research of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research.”
Tissue samples all displayed detectable tumor cells, as previously determined by examination of cellular morphology by a board-certified pathologist. Immunohistochemistry for integrin β3 was performed as described in the previous section. Tumor-associated β3 expression was semi-quantitatively scored in a blinded manner, in which the scorer did not have access to disease classification or clinical annotation, using the histoscore (H-score) system: H-score = Σ (i × %), where ‘i’ is the staining intensity (0 to 3 scale), and ‘%’ is the percentage of tumor cells stained at each intensity, ranging 0% to 100%. We observed an expression pattern of tumoral β3 consistent with a previous literature description of β3 expression in human breast cancer: primarily localized along the cell plasma membrane at sites of tumor-tumor and tumor-stroma contact (37). In addition to the β3 expression on bone metastases, we also observed β3 staining on some host cells within the primary and bone tumor microenvironments, including tumor-infiltrating immune cells (β3 expression colocalized with CD68-stained cells, predominately expressed on monocytes and macrophages), tumor-associated blood vessels, and osteoclasts, consistent with previous observations (11,12,19,20).
Breast cancer subtyping was based on ER,PR,HER2 status, where luminal A was ER+,PR±,HER2- (n=18), luminal B was ER+,PR±,HER2+ (n=7), HER2-enriched was ER-,PR-,HER2+ (n=2), and triple-negative was ER-,PR-,HER2- (n=7). Eight breast cancer carcinomas had incomplete genetic subtyping and were excluded from molecular subtype analysis.
Statistical analysis
All data shown as mean with error bars representing SEM. All sample sizes reported in the study are the minimum number of samples. For animal studies, sample sizes were estimated according to our previous experience. Statistical differences were analyzed using either a two-tailed t-test, ANOVA with Tukey’s test for a posteriori (post-hoc) multiple comparisons, or a two-tailed unpaired t-test with Bonferroni correction for a priori comparisons between a control group and experimental treatment groups of interest. Assumptions for ANOVA and t-test (independent samples, approximately normal distributions) for samples n>5 were sufficiently met, or used if a random sample of n≤5 were selected from an approximately normally distributed population. Non-normally distributed data was analyzed using a two-tailed Mann–Whitney U-test or a two-tailed Wilcoxon signed-rank test for matched-pairs. All tests were considered significant at P<0.05, or in case of k comparisons, P<0.05/k. Data analyses were complete using Prism 6 (GraphPad Software).
Results
Breast cancer cells overexpress integrin β3 within the bone microenvironment
To determine the potential of targeting αvβ3 at different metastatic sites, we evaluated breast cancer expression of integrin αv or β3 at the primary site as compared to secondary metastases. Using the murine breast cancer cell line PyMT-Bo1 that models the luminal B subtype, primary MFP tumors were established by orthotopic injection, or metastases within the bone and visceral organs were established by intracardiac injection, into separate cohorts of mice. Tumors were removed 12 days post-injection and stained for integrin β3 or integrin αv expression using immunohistochemistry. Bone metastases expressed elevated levels of β3, as compared to MFP tumors or visceral metastases within the lung or kidney (Fig. 1A and Supplemental Fig. S1A). Unlike the selective upregulation of integrin β3 on bone metastases, we observed expression of integrin αv across all primary and metastatic tumors (Supplemental Fig. S1B). To determine whether the observed β3 expression was tumor-cell specific, we used flow cytometry to quantify cell surface β3 expression. Using PyMT-Bo1 cells or murine 4T1 triple-negative breast cancer cells, MFP tumors or bone metastases were established by orthotopic injection or intracardiac injection, respectively. Isolated tumor cells were identified based on cytokeratin 18 (CK18+) expression (Supplemental Fig. S1C). In both cell lines, tumor cells from bone metastases expressed significantly elevated β3 levels, as compared to MFP tumor cells (Fig. 1B,C).
Figure 1.

Integrin β3 is preferential induced on breast cancer cells within the bone microenvironment. (A) Immunohistochemistry for β3 on PyMT-Bo1 mammary tumors or metastases, established by orthotopic or intracardiac injection into separate cohorts of mice, respectively. Tumors were removed 12 days post-injection. Representative images (left), quantification of DAB-stained tumors (right), n=4 per group. Scale=50 μm. One-way ANOVA with Tukey’s post-hoc test, **** P<0.0001. (B,C) Mammary tumors or bone metastases were established as previously described. Isolated tumor cells were identified based on cytokeratin 18 expression (CK18+) and evaluated by flow cytometry for surface β3 expression. Representative samples (left). (B) PyMT-Bo1 cells, 13 days post-injection, n=3 mammary, n=4 bone. (C) 4T1 cells, 15 days post-injection, n=5 mammary, n=4 bone. Two-tailed unpaired t-test, *** P<0.001, ** P<0.01. Data presented as mean±SEM.
To assess the translational implications of this observation, we examined β3 expression on biopsies from 42 breast cancer patients, comparing patient-matched primary tumors to bone metastases. Immunohistochemistry for β3 was semi-quantitatively scored based on the extent and intensity of tumor-associated β3 expression. Across nearly all patients, human bone metastases expressed significantly higher levels of tumor-associated β3, as compared to the matching primary tumor (Fig. 2A and Supplemental Fig. S1D). Furthermore, this elevated expression of bone metastatic β3 was observed across all breast cancer patient subtypes: luminal A, luminal B, HER2-enriched, and triple-negative (Fig. 2B).
Figure 2.

Immunohistochemistry for β3 on patient-matched primary breast cancer and bone metastatic biopsies. (A) Semi-quantitatively analysis of the extent and intensity of tumor-associated β3 expression, using the histoscore (H-score) system, n=42 matched-pairs, two-tailed Wilcoxon signed-rank test, **** P<0.0001. (B) Subdivision of Fig. 2A by molecular subtype (see Materials and Methods for details). Representative images of patient-matched primary tumors and bone metastases (right). Scale=50 μm. Two-tailed paired t-test, **** P<0.0001, *** P<0.001, ** P<0.01, * P<0.05. Data presented as mean±SEM.
Tumoral β3 is induced in the bone microenvironment and mediated by TGF-β signaling
We next asked whether bone metastases express elevated β3 due to preferential seeding and colonization of a β3hi tumor cell subpopulation in the bone. Using fluorescence-activated cell sorting (FACS), PyMT-Bo1 cells were sorted into three different subpopulations based on surface β3 expression: β3-negative cells (β3-), β3-expressing cells (β3+), and control cells sorted without β3 discrimination (β3-all). These PyMT-Bo1 subpopulations were collected and injected intracardially into separate groups of mice, which all developed bone metastases (Fig. 3A). Flow cytometry revealed that regardless of initial β3 status, each FACS subpopulation developed into bone metastases that strongly expressed tumoral β3 (Fig. 3B). This result demonstrates that elevated expression of bone metastatic β3 is not due to preferential seeding of β3hi tumor cells to the bone, and suggests that tumoral β3 is induced in the bone microenvironment.
Figure 3.

Bone microenvironment and TGF-β stimulation induce tumoral β3. (A) In vitro PyMT-Bo1 cells were FACS sorted into three groups based on basal β3 expression: β3-, β3+, and control β3-all. Immediately after collection, cells were intracardiac injected into separate groups of mice. (B) 13 days post-injection, isolated bone metastatic PyMT-Bo1 cells were identified by CK18+ and evaluated by flow cytometry for surface β3 expression. Representative samples (left), n=4 β3-, n=4 β3-all, n=5 β3+ (right). One-way ANOVA with Tukey’s post-hoc test, ns=(not significant). (C,D) qPCR analysis of β3 (Itgb3) mRNA expression by PyMT-Bo1 or 4T1 cells cultured in vitro, following 24 hours stimulation with the listed factors (see Materials and Methods for details). One of two biological replicates, each in technical duplicate. One-way ANOVA with Tukey’s post-hoc test, **** P<0.0001. Data presented as mean±SEM.
To ascertain pathways that might be responsible for this induction, we evaluated a panel of cytokines and growth factors present in the bone microenvironment for their effect on β3 expression. Of the tested factors, only members of the TGF-β family upregulated integrin β3 expression, observed in both PyMT-Bo1 and 4T1 cells (Fig. 3C,D). To assess the specificity of TGF-β-mediated upregulation of integrin β3 as compared to other related integrin subunits, we evaluated how TGF-β alters expression of integrin αv, as well as a subfamily of β-integrin subunits that can also heterodimerize with integrin αv (integrin subunits β1, β5, β6 and β8). In addition, we evaluated integrin αIIb (glycoprotein-IIb), which is largely restricted to platelets and megakaryocyte-lineage cells but can also heterodimerize with integrin β3 (16), and integrin α2, which facilitates adhesion to type I collagen and is upregulated on prostate cancer lines in response to TGF-β (38).
Collectively, we evaluated gene expression of integrin subunits β1, β3, β5, β6, β8, αv, αIIb, and α2 on PyMT-Bo1 and 4T1 cells, following 24 hours of in vitro TGF-β stimulation. Integrin β3 was the most upregulated subunit in both PyMT-Bo1 and 4T1 cells (Supplemental Figure S2A,B). In PyMT-Bo1 cells, integrin α2 was the next most upregulated subunit, though 5-fold less than the level of β3 induction (Supplemental Figure S2A). In 4T1 cells, β5 and α2 were the next most upregulated subunits, though 4-fold less than the level of β3 induction (Supplemental Figure S2A). TGF-β-mediated changes in αv, αIIb, and the other β-integrin subunits were either unchanged, reduced, or displayed a less-than 2-fold upregulation (Supplemental Figure S2A,B). These results provide support for TGF-β as a potent inducer of integrin β3 as compared to these other integrin subunits.
TGF-β signals through TGF-β Receptor 1 (TGFβR1), which canonical phosphorylates the transcription factors SMAD2/SMAD3. In addition, TGFβR1 can activate ‘non-canonical’ signaling pathways, most commonly p38 MAP kinase (p38), MAPK/ERK Kinase-1&-2 (MEK1/2), and c-Jun N-terminal kinase (JNK) (39). To evaluate how TGF-β induces β3, tumor cells were treated with TGF-β in combination with pharmacological inhibitors for p38, MEK1/2, JNK, or TGFβRI kinase activity at the site specific for SMAD2/SMAD3 phosphorylation (see Fig. 4 for details). In both PyMT-Bo1 and 4T1, only pharmacological inhibition of SMAD2/SMAD3 phosphorylation completely ablated β3 upregulation. This was observed by flow cytometry for surface β3 expression (Fig. 4A,B) and qPCR analysis for β3 mRNA levels (Supplemental Fig. S3A). Western blot analysis confirmed selective inhibition of SMAD2/SMAD3 phosphorylation (Fig. 4C) and suppression of total β3 protein (Fig. 4D) with TGFβRI kinase inhibition. To determine whether TGF-β signaling was responsible for tumoral β3 induction in vivo, mice bearing human MDA-MB-231 triple-negative breast cancer bone metastases were treated daily with a pharmacological TGFβRI kinase inhibitor. TGFβRI kinase inhibition significantly suppressed β3 expression on MDA-MB-231 breast cancer bone metastases (Fig. 4E), demonstrating the role of TGF-β signaling in β3 induction on breast cancer cells within the bone microenvironment. TGFβRI kinase inhibition resulted in an approximately 1.6-fold reduction in MDA-MB-231 bone metastatic tumor burden (Supplemental Fig. S3B), consistent with a previous report (30).
Figure 4.

TGF-β induces tumoral β3 through TGFβRI-mediated phosphorylation of SMAD2/SMAD3. (A,B) In vitro tumor cells treated with either 2 ng/mL TGF-β1 or DMSO control, in the presence of a pharmacological inhibitor: SMAD2/3i (20μM SB431542), MEK1/2i (20 μM U0126), p38i (20μM SB203580), or JNKi (50μM SP600125). After 48 hours, flow cytometry for surface β3 expression was evaluated on (A) PyMT-Bo1 cells, or (B) 4T1 cells. Representative experiment (left), n=3 biological replicates (right). One-way ANOVA with Tukey’s post-hoc test, with denoted significance in relation to DMSO control, **** P<0.0001, *** P<0.001, * P<0.05. (C,D) Western blot analysis of in vitro PyMT-Bo1 cells treated as previously described, for (C) 3 hours, or (D) 24 hours. (E) Mice intracardiac injected with MDA-MB-231 cells were treated daily with a TGFβRI kinase inhibitor of SMAD2/3 phosphorylation (SD-208, 60 mg/kg/d) or vehicle control (1% methylcellulose) for 28 days. Immunohistochemistry for β3 with representative images (left) and quantification of DAB-stained bone metastases, n=4 (right). Scale=100 μm. Two-tailed unpaired t-test, *** P<0.001. Data presented as mean±SEM.
Integrin αvβ3-targeted micelle nanoparticles colocalize with breast cancer bone metastases
To evaluate whether bone metastatic induction of β3 could be therapeutically exploited, we sought to examine the potential of nanotherapy targeting integrin αvβ3. We used single-lipid phospholipid/polysorbate-80 micelle nanoparticles (MPs, ~12.5 nm), coated with a well-characterized quinolone nonpeptide targeting ligand with enhanced specificity for activated αvβ3 (Supplemental Fig. S4A). The specificity of this targeting ligand for integrin αvβ3 has been demonstrated through a variety of assays both in vitro and in vivo (32-36) (see Materials and Methods for details). To test the effectiveness of integrin αvβ3 as a target for enhancing drug delivery to bone metastases, rhodamine-labeled αvβ3-targeted MPs (αvβ3-MPs) or non-targeted MPs were administered intravenously into mice bearing PyMT-Bo1 bone metastases. Immunofluorescent analysis of bone metastases revealed strong colocalization of αvβ3-MPs with bone metastases, while non-targeted MPs displayed only minor colocalization (Fig. 5A). Furthermore, accumulation of either αvβ3-MPs or MPs within tumor-free bone marrow was negligible (Supplemental Fig. S4B). Together, these results demonstrate the targeting potential and specificity of αvβ3-MPs for bone metastases.
Figure 5.

Micelle nanoparticles targeting integrin αvβ3 colocalize with breast cancer bone metastases and are designed to carry the chemotherapeutic docetaxel. (A) Mice bearing PyMT-Bo1 bone metastases were intravenously injected with rhodamine-labeled (red) non-targeted MPs or αvβ3-MPs. After 3 hours, bones were removed and fresh-frozen sections were immunofluorescently imaged. DAPI nuclear counterstain (blue). Representative images (left), quantification of fluorescent colocalization with the bone metastases, n=3 per group (right). Scale=200 μm. Two-tailed unpaired t-test, *** P<0.001. (B) Schematic of αvβ3-MP-mediated “contact-facilitated drug delivery.” Upon hemifusion, phospholipid-conjugated docetaxel-prodrug (DTX-PD) transfers to the target cell’s plasma membrane, where bioactive DTX is enzymatically liberated by phospholipases and released directly into the cytoplasm. (C) In vitro PyMT-Bo1 cell viability via MTT assay after 72 hours of DTX or DTX-PD treatment. n=2 biological replicates, each in technical triplicate. (D) Treatment schematic of mice intracardiac injected with PyMT-Bo1 cells. Representative ex vivo BLI of tumor-bearing organs on day 12. (E) Following the schematic in Fig. 5D, mice were treated with increasing concentrations of docetaxel, with the cumulative dose as indicated. Day 12 ex vivo PyMT-Bo1 metastatic tumor burden by BLI analysis, n=6. Bone, Kidney, Lung: two-tailed unpaired t-test. Liver: two-tailed Mann–Whitney U-test. All with Bonferroni correction for a priori multiple comparisons between control 0 mg/kg and each DTX treatment group, ** P<(0.01/4), * P<(0.05/4), ns=(not significant). Data presented as mean±SEM.
Next, we sought to evaluate the therapeutic efficacy of αvβ3-MP-mediated drug delivery. Nanotherapeutics often suffer from a premature loss of drug payloads during circulation and diminished intracellular drug bioavailability due to endosomal entrapment (40). To overcome these problems, a lipase-labile phospholipid-prodrug concept was employed, in which drug cargo is coupled to the Sn2 acyl-chain of phosphatidylcholine. This phospholipid-prodrug can be stably incorporated into the MP membrane during self-assembly and provides drug retention during circulation (35). Upon αvβ3-MP binding to activated αvβ3 on target cells, a hemifusion complex forms between MPs and the plasma membrane, enabling endocytosis-independent “contact-facilitated drug delivery” (see Fig. 5B for details) (36,41).
The chemotherapeutic agent selected for nanoparticle-delivery was docetaxel (DTX), a potent microtubule inhibitor employed as a first-line agent against breast cancer (42). We modified DTX as an Sn2 lipase-labile phospholipid-prodrug (DTX-PD) to enable αvβ3-MP delivery (Fig. 5B) (32). In vitro, PyMT-Bo1 cells were markedly sensitive to both DTX and DTX-PD, with a bioequivalent IC50 of 5.5 nM (Fig. 5C). Unfortunately, chemotherapeutics such as DTX rarely eradicate bone metastases in human patients (3,4) and can induce off-target toxicity such as hair loss, neuropathy, pancytopenia, and liver toxicity (42). To test the efficacy of DTX against PyMT-Bo1 cells in vivo, mice were intracardiac injected with PyMT-Bo1 cells. BLI analysis confirmed equivalent establishment of bone metastases on day 3 post-intracardiac injection, and mice bearing PyMT-Bo1 metastases received intravenous DTX treatments beginning on day 4 post-intracardiac injection (see Fig. 5D). Animals experienced dose-dependent DTX-induced hematological toxicity (Supplemental Fig. S5A), yet metastatic tumor burden in the liver, kidney, or bone was not attenuated by DTX treatment, and lung metastatic tumor burden was only significantly reduced at the highest DTX dose of 20 mg/kg/week (Fig. 5E). Thus, while DTX was potently suppressive against PyMT-Bo1 in vitro, DTX displayed limited efficacy against PyMT-Bo1 metastases in vivo.
αvβ3-MP-mediated drug delivery of docetaxel attenuates bone metastases
To evaluate the therapeutic potential of αvβ3-targeted delivery of DTX-PD against PyMT-Bo1 bone metastases, mice were treated with DTX, or an equimolar amount of DTX-PD encapsulated by αvβ3-MP (αvβ3-MP/DTX-PD). In parallel, two control nanoparticle treatments were evaluated to assess the in vivo specificity of αvβ3-MP targeting, and the specificity of docetaxel-mediated tumor-suppressive effects: non-targeted MP/DTX-PD and cargo-free αvβ3-MP without DTX-PD cargo, respectively.
As before, BLI analysis on day 3 post-intracardiac injection confirmed PyMT-Bo1 bone metastatic establishment and treatment began on day 4, with a cumulative dose of 5.55 mg/kg DTX or equimolar dose of MP-encapsulated DTX-PD (see Fig. 5D). On day 12, ex vivo BLI analysis of bone metastases revealed that αvβ3-MP/DTX-PD significantly attenuated bone metastatic tumor burden, while no significant attenuation was observed with DTX treatment (Fig. 6A,B). Consistent with this result, histological analysis of bone tumor burden (Fig. 6C) and x-ray analysis of tumor-associated bone loss (Fig. 6D) revealed a significant reduction with αvβ3-MP/DTX-PD treatment as compared to saline, and no significant attenuation with DTX. BLI analysis of visceral metastases within the liver, lungs, kidneys, and brain revealed that neither αvβ3-MP/DTX-PD nor DTX resulted in significant attenuation (Supplemental Fig. S5B), supporting our previous observations concerning bone-specific β3 expression and resistance of PyMT-Bo1 metastases to DTX.
Figure 6.

Integrin αvβ3-targeted nanotherapy attenuates breast cancer bone metastases. Following the schematic described in Fig. 5D, mice were treated with either a cumulative dose of 5.55 mg/kg docetaxel (DTX), an equimolar dose of docetaxel-prodrug encapsulated by αvβ3-MP (αvβ3-MP/DTX-PD), or saline. Analyses completed on samples collected on day 12 post-injection. (A) Representative in vivo BLI of mice bearing PyMT-Bo1 bone metastases, matching day 3 and day 12 post-injected mice from each group (B) Ex vivo PyMT-Bo1 bone metastatic tumor burden by BLI analysis, n=9. (C) Histological analysis of tumor burden within the tibiofemoral joint, n=4. (D) X-ray analysis of osteolytic bone destruction, n=9. (B-D) Scale=1 mm, two-tailed unpaired t-test with Bonferroni correction for a priori multiple comparisons between saline and each experimental treatment, * P<(0.05/2). (E) Serum chemistry analysis of BUN, AST, and ALT, n=5. Gray range illustrates the normal reference range (mean±2*SD) for female C57BL/6 mice bearing PyMT-Bo1 metastases. (F) TRAP staining for quantification of osteoclast number per millimeter of bone surface (N.OC/mmB.S.) at the tumor (T), bone (B) interface, n=4. (G) Quantification of PCNA-positive cells within the bone metastatic region, n=3. (F-G) Scale=50 μm, one-way ANOVA with Tukey’s post-hoc test, ** P<0.01, * P<0.05, ns=(not significant). All images are representative and data presented as mean±SEM.
Hepatotoxicity was tested by serum liver function tests for aspartate transaminase (AST) and alanine transaminase (ALT). DTX treatment resulted in elevated levels of AST and ALT outside of the normal ranges, while αvβ3-MP/DTX-PD treatment displayed no evidence of hepatotoxicity (Fig. 6E). Both renal function, assessed by blood urea nitrogen (BUN), and whole blood counts were within the normal range among mice treated with saline, DTX, or αvβ3-MP/DTX-PD (Fig. 6E and Supplemental Fig. S5C).
Neither non-targeted MP/DTX-PD nor cargo-free αvβ3-MP treatment significantly altered bone metastatic tumor burden or bone destruction, demonstrating the necessity of αvβ3-MP targeting and the specificity of docetaxel in mediating the tumor-suppressive effects of this nanotherapy (Supplemental Fig. S6A-D). Control αvβ3-MP treated mice displayed elevated levels of AST, but ALT, renal, and hematological toxicities were within the normal range for αvβ3-MP and non-targeted MP/DTX-PD treatments (Supplemental Fig. S6E,F).
To evaluate the potential mechanism of αvβ3-MP/DTX-PD anti-bone metastatic activity, we quantified osteoclast number at the tumor-bone interface, which was unchanged between saline, DTX, and αvβ3-MP/DTX-PD treatments (Fig. 6F), demonstrating that αvβ3-MP/DTX-PD attenuates tumor-induced bone loss independent of inhibition of osteoclast formation or osteoclast killing. To assess whether tumor cells responded to the effects of the chemotherapy, we measured tumor proliferation by immunohistochemistry for proliferating cell nuclear antigen (PCNA). Bone metastatic tumor cells treated with αvβ3-MP/DTX-PD displayed a significantly lower proliferation index than saline or DTX (Fig. 6G). Collectively, this work provides support for safer, more effective drug delivery against breast cancer bone metastases through exploitation of αvβ3 expression.
Discussion
Breast cancer bone metastases often irreversibly damage the skeleton, severely impacting quality of life and overall survival (2). Treating bone metastases has proven difficult, in part due to the bone microenvironment’s status as a chemo-protective niche (7), and coupled with inefficient delivery of drug to tumor cells in bone (43). While drug or nanoparticle conjugation to hydroxyapatite-avid molecules like bisphosphonates have provided positive results against mouse models of bone metastases (13,14), they lack direct specificity for tumor cells (8,15). In this study, we provide new evidence for integrin β3 as a molecular target on breast cancer bone metastases in mice and humans, and demonstrate the potential for improving therapeutic efficacy against this metastatic site via αvβ3-targeted approaches.
We first investigated differences in the physiological expression of integrin β3 on breast cancer tumors growing in different organs, in search of a tumor target on cells within the bone microenvironment. We demonstrated that orthotopic injection of murine breast cancer cell lines into the MFP establishes primary tumors with weak β3 expression. Intracardiac injection establishes metastases within the lung and kidney with similarly weak β3 expression, but establishes bone metastases with significantly stronger β3 expression, observed by both immunohistochemistry and flow cytometry of tumoral surface expression. In contrast, integrin αv was strongly expressed on tumors at all evaluated sites. We employed the widely-published intracardiac injection model to establish experimental bone and visceral metastases in these experiments. While this model does not recapitulate all steps of the metastatic cascade, circulating tumor cells still need to extravasate from the vessels to establish within the bone and other organs.
We also evaluated β3 expression on 42 human bone metastatic tissue samples, as compared to matching primary breast cancer biopsies from the same patient. Across nearly all patients, tumor-associated β3 expression was significantly higher on bone metastases. Furthermore, we show for the first time that β3 is highly expressed on breast cancer bone metastases of all subtypes, including those with the worst overall survival that have proven difficult to target (44), supporting the translational potential of targeting β3 in bone metastatic breast cancer patients.
Based on previous studies demonstrating that ectopic overexpression of β3 enhances tumor cell establishment in bone (26,28), we considered the possibility that breast cancer bone metastases display elevated β3 due to preferential bone colonization of a tumor cell subpopulation with high basal β3 expression. We established bone metastases from FACS-sorted tumor cell subpopulations, which were initially positive or negative for basal β3 expression. Here, we demonstrated that initial β3 expression is dispensable for establishing breast cancer bone metastases with elevated β3 expression. This finding does not contradict the role of tumoral αvβ3 in enhancing bone colonization (26,28), as there are numerous ligands for αvβ3 within the bone microenvironment (17,18); rather, it indicates that tumoral β3 can be induced during the establishment of bone metastases, suggesting that β3 expression might be regulated by the bone microenvironment itself.
The bone is a natural reservoir for TGF-β, which is stored in the matrix as an inactive latent complex that must be liberated for bioactivation (45). Bioactive TGF-β plays a critical role in both the homeostatic and pathologic bone microenvironment, regulating osteolytic destruction and pathologic muscle weakness (30,46). TGF-β has been shown to induce β3 expression on normal and transformed mammary epithelial cells (47-49), but the physiological regulation of tumoral β3 in vivo has not been explored. Here, we demonstrated that TGF-β strongly induces tumoral β3, and analysis of other integrin subunits of interest revealed that TGF-β induces integrin β3 more strongly than any other. Furthermore, we determined that TGF-β induces β3 through TGFβRI phosphorylation of SMAD2/SMAD3, evaluated at the transcriptional, total protein, and cell surface expression level. And for the first time, we demonstrate that TGF-β signaling is required for integrin β3 induction by breast cancer cells within the bone.
We were surprised by the lack of tumoral β3 upregulation in the MFP tumor or visceral metastases, where TGF-β can be present. This observation suggests that other microenvironments either lack a sufficient amount of bioactive TGF-β necessary for tumoral β3 induction, or contain inhibitors of TGF-β or SMAD2/SMAD3 signaling that prevent TGF-β-mediated induction of tumoral β3. Studies are in progress to define the molecular mechanism underlying the low expression of tumoral β3 outside of the bone.
Selective elevation of β3 on bone metastases prompted us to explore αvβ3-targeted nanotherapy as a means to overcome the barriers limiting effective treatment. We selected phospholipid/polysorbate-80 micelle nanoparticles (MPs) for their small size (~12.5 nm) and unique mechanism of drug delivery (see Fig. 5B for details), which overcomes two problems that often hinder effective nanotherapy: poor intracellular drug bioavailability and premature loss of drug during circulation (40). Micelle nanoparticles deliver phospholipid-prodrug cargo directly to the target cell’s plasma membrane, where bioactive free-drug is enzymatically liberated directly into the cytoplasm by phospholipase activity (36,41). Furthermore, phospholipid-prodrug is stably incorporated into the MP-single lipid layer, thereby minimizing passive drug loss during circulation (35). Administering rhodamine-labeled MPs or αvβ3-MPs into mice bearing breast cancer bone metastases resulted in stronger αvβ3-MP colocalization with bone metastases, compared to a 6.5-fold reduction in colocalization with non-targeted MPs. To examine the importance of MP’s small size in bone metastatic colocalization, we also evaluated the colocalization potential of significantly larger αvβ3-targeted perfluorocarbon nanoparticles (~250nm) (35). We observed that fluorescently-labeled αvβ3-targeted perfluorocarbon nanoparticles were much less effective at localizing to the center of bone metastases (data not shown). These observations prompted us to proceed with αvβ3-MP-mediated drug delivery, and we selected the chemotherapeutic docetaxel (DTX) as cargo for lipase-labile phospholipid-prodrug modification (DTX-PD).
While PyMT-Bo1 breast cancer cells are equally sensitive to both DTX and DTX-PD in vitro, PyMT-Bo1 metastases were strikingly resistant in vivo, with bone metastatic tumor burden essentially unaffected by DTX treatment even at the highest, most toxic dose tested (20 mg/kg/week). These observations highlight the difficulty of achieving an inhibitory dose of chemotherapy at metastatic sites in vivo. To test the efficacy of MP-encapsulated DTX-PD, mice bearing PyMT-Bo1 metastases were treated with a suboptimal DTX dose (5.55mg/kg/week), 3.6-fold lower than the previously tested 20 mg/kg/week DTX dose which did not significantly attenuate bone metastases. At this suboptimal dose, we observed a significant attenuation of bone metastatic tumor burden and tumor-associated bone loss with αvβ3-MP/DTX-PD treatment, as compared to no significant attenuation by equimolar DTX treatment. Furthermore, neither cargo-free αvβ3-MP treatment nor non-targeted MP/DTX-PD treatment significantly altered bone metastatic tumor burden or osteolytic bone destruction. The observation that micelle coating with the αvβ3-targeting ligand was necessary for bone metastatic colocalization, coupled with fact that αvβ3-MP/DTX-PD failed to attenuate metastases outside of β3-expressing bone metastases, provides additional support for the in vivo specificity of the αvβ3-targeting ligand for integrin αvβ3, in line with previous reports (32-36).
To examine the potential mechanism of αvβ3-MP-mediated activity, we quantified osteoclast number at the tumor/bone interface, which is reflective of osteoclast formation and serves as a useful indicator of bone resorption. Previous studies have found that osteoclast formation is markedly sensitive to inhibition by DTX, even at nanomolar concentrations (50). Osteoclast number at the tumor/bone interface was similar between αvβ3-MP/DTX-PD and saline treatment groups, demonstrating that αvβ3-MP/DTX-PD treatment attenuates bone metastases and tumor-associated bone loss independent of inhibiting osteoclast formation or direct osteoclast killing. It is possible that there remains an effect of αvβ3-MP/DTX-PD treatment on osteoclast function that was not revealed by histomorphometric analyses.
We evaluated tumor cell proliferation through expression quantification of proliferating cell nuclear antigen (PCNA), a DNA polymerase processivity factor required for DNA synthesis during replication. DTX inhibits tumor proliferation through effects on microtubule stabilization and disruption of mitotic spindle assembly (42). We found that αvβ3-MP/DTX-PD treatment significantly decreased PCNA expression on bone-residing tumor cells, as compared to saline and DTX treatment. Taken together with the osteoclast histomorphometry result, these observations suggest that αvβ3-MP/DTX-PD attenuated bone metastases due to enhanced therapeutic efficacy against bone-residing tumor cells.
We observed that DTX administration resulted in increased hepatotoxicity, while hematological values and liver function tests remained within normal limits following αvβ3-MP/DTX-PD treatment. This work thus provides support for safer, more effective drug delivery against bone metastases through exploitation of αvβ3 expression on breast cancer cells within bone. Going forward, αvβ3-targeted drug cargo could be modified to fit specific treatment needs.
We recognize the limitation of αvβ3-targeted nanotherapy against breast cancer metastases outside of the bone. Nevertheless, bone metastases occur in approximately 70% of metastatic breast cancer patients and represent the only metastatic site in approximately 30% of patients, suggesting that a substantial number of patients could benefit from this approach (1,44). Collectively, we provide support for integrin αvβ3-targeted drug delivery as a bone-specific therapeutic strategy to address the unmet clinical need for effective treatments against breast cancer bone metastases.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
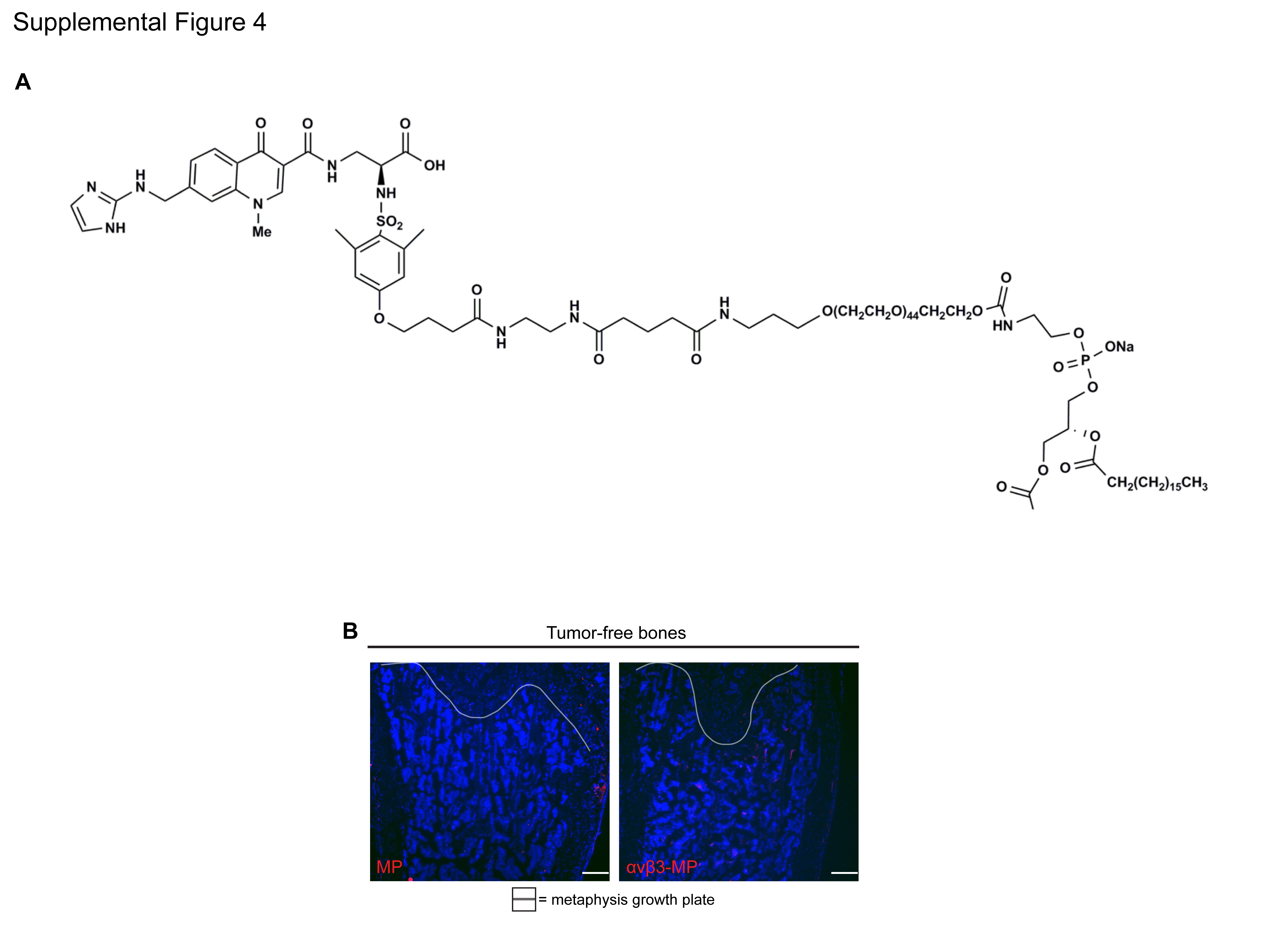{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Sheila Stewart, Daniel Link, Roberta Faccio, Fanxin Long, David DeNardo, and Vivek Arora, Jingyu Xiang, Takayuki Kobayashi, and Bhavna Murali for their valuable suggestions. We thank Lynne Marsala, Julie Prior, Crystal Idleburg, and Samantha Coleman for their expert technical assistance.
Financial support: This research was supported in whole or part by grants from the CA154737 (K.N. Weilbaecher / D. Pan / G.M. Lanza), CA100730 (K.N. Weilbaecher), CA097250 (K.N. Weilbaecher), HL122471 (G.M. Lanza), HL112518 (G.M. Lanza), HL113392 (G.M. Lanza), HHSN26820140042C (G.M. Lanza), P30CA091842 (G.A. Colditz), CA143057 (T.A. Guise), CA69158 (T.A. Guise), and training grants 5T32GM007067-39 (M.H. Ross), T32AR060719 (M.H. Ross), 5T32CA113275-07 (A.K. Esser), and GM07200 (G.C. Fox). Additional funding support provided by grants from the St. Louis Men’s Group Against Cancer and the Siteman Cancer Center. Imaging and analysis of human breast cancer breast and bone biopsies was performed on the Zeiss Axio Scan.Z1 through the use of Washington University Center for Cellular Imaging (WUCCI), supported by Washington University School of Medicine, the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CDI-CORE-2015-505), the National Institute for Neurological Disorders and Stroke (NS086741), and by the Foundation for Barnes Jewish Hospital. Technical support was provided by the Washington University Musculoskeletal Research Center (P30AR057235), the Hope Center Alafi Neuroimaging Lab (Shared Instrumentation Grant S10 RR027552), the Molecular Imaging Center at Washington University (P50 CA094056), and the St. Louis Breast Tissue Registry (funded by The Department of Surgery at Washington University School of Medicine, St. Louis, MO).
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest.
References
- 1.Buijs JT, van der Pluijm G. Osteotropic cancers: from primary tumor to bone. Cancer Lett. 2009;273:177–93. doi: 10.1016/j.canlet.2008.05.044. [DOI] [PubMed] [Google Scholar]
- 2.Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12:6243s–9s. doi: 10.1158/1078-0432.CCR-06-0931. [DOI] [PubMed] [Google Scholar]
- 3.Niikura N, Liu J, Hayashi N, Palla SL, Tokuda Y, Hortobagyi GN, et al. Treatment outcome and prognostic factors for patients with bone-only metastases of breast cancer: a single-institution retrospective analysis. Oncologist. 2011;16:155–64. doi: 10.1634/theoncologist.2010-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahn SG, Lee HM, Cho SH, Lee SA, Hwang SH, Jeong J, et al. Prognostic factors for patients with bone-only metastasis in breast cancer. Yonsei Med J. 2013;54:1168–77. doi: 10.3349/ymj.2013.54.5.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suva LJ, Washam C, Nicholas RW, Griffin RJ. Bone metastasis: mechanisms and therapeutic opportunities. Nat Rev Endocrinol. 2011;7:208–18. doi: 10.1038/nrendo.2010.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Croucher PI, McDonald MM, Martin TJ. Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer. 2016;16:373–86. doi: 10.1038/nrc.2016.44. [DOI] [PubMed] [Google Scholar]
- 7.Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14:2519–26. doi: 10.1158/1078-0432.CCR-07-2223. [DOI] [PubMed] [Google Scholar]
- 8.Dang L, Liu J, Li F, Wang L, Li D, Guo B, et al. Targeted Delivery Systems for Molecular Therapy in Skeletal Disorders. Int J Mol Sci. 2016;17:428. doi: 10.3390/ijms17030428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tautzenberger A, Kovtun A, Ignatius A. Nanoparticles and their potential for application in bone. Int J Nanomedicine. 2012;7:4545–57. doi: 10.2147/IJN.S34127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cole LE, Vargo-Gogola T, Roeder RK. Targeted delivery to bone and mineral deposits using bisphosphonate ligands. Adv Drug Deliv Rev. 2016;99:12–27. doi: 10.1016/j.addr.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Bakewell SJ, Nestor P, Prasad S, Tomasson MH, Dowland N, Mehrotra M, et al. Platelet and osteoclast beta3 integrins are critical for bone metastasis. Proc Natl Acad Sci U S A. 2003;100:14205–10. doi: 10.1073/pnas.2234372100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan EA, Schneider JG, Baroni TE, Uluckan O, Heller E, Hurchla MA, et al. Dissection of platelet and myeloid cell defects by conditional targeting of the beta3-integrin subunit. FASEB J. 2010;24:1117–27. doi: 10.1096/fj.09-138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller K, Clementi C, Polyak D, Eldar-Boock A, Benayoun L, Barshack I, et al. Poly(ethylene glycol)-paclitaxel-alendronate self-assembled micelles for the targeted treatment of breast cancer bone metastases. Biomaterials. 2013;34:3795–806. doi: 10.1016/j.biomaterials.2013.01.052. [DOI] [PubMed] [Google Scholar]
- 14.Ye WL, Zhao YP, Li HQ, Na R, Li F, Mei QB, et al. Doxorubicin-poly (ethylene glycol)-alendronate self-assembled micelles for targeted therapy of bone metastatic cancer. Sci Rep. 2015;5:14614. doi: 10.1038/srep14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carbone EJ, Rajpura K, Allen BN, Cheng E, Ulery BD, Lo KW. Osteotropic nanoscale drug delivery systems based on small molecule bone-targeting moieties. Nanomedicine. 2016;13:37–47. doi: 10.1016/j.nano.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 16.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 17.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider JG, Amend SR, Weilbaecher KN. Integrins and bone metastasis: integrating tumor cell and stromal cell interactions. Bone. 2011;48:54–65. doi: 10.1016/j.bone.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–71. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 20.Su X, Esser AK, Amend SR, Xiang J, Xu Y, Ross MH, et al. Antagonizing Integrin beta3 Increases Immunosuppression in Cancer. Cancer Res. 2016;76:3484–95. doi: 10.1158/0008-5472.CAN-15-2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16:201–18. doi: 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim KB, Prieto V, Joseph RW, Diwan AH, Gallick GE, Papadopoulos NE, et al. A randomized phase II study of cilengitide (EMD 121974) in patients with metastatic melanoma. Melanoma Res. 2012;22:294–301. doi: 10.1097/CMR.0b013e32835312e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alva A, Slovin S, Daignault S, Carducci M, Dipaola R, Pienta K, et al. Phase II study of cilengitide (EMD 121974, NSC 707544) in patients with non-metastatic castration resistant prostate cancer, NCI-6735. A study by the DOD/PCF prostate cancer clinical trials consortium. Invest New Drugs. 2012;30:749–57. doi: 10.1007/s10637-010-9573-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ley K, Rivera-Nieves J, Sandborn WJ, Shattil S. Integrin-based therapeutics: biological basis, clinical use and new drugs. Nat Rev Drug Discov. 2016;15:173–83. doi: 10.1038/nrd.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liapis H, Flath A, Kitazawa S. Integrin alpha V beta 3 expression by bone-residing breast cancer metastases. Diagn Mol Pathol. 1996;5:127–35. doi: 10.1097/00019606-199606000-00008. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Y, Bachelier R, Treilleux I, Pujuguet P, Peyruchaud O, Baron R, et al. Tumor alphavbeta3 integrin is a therapeutic target for breast cancer bone metastases. Cancer Res. 2007;67:5821–30. doi: 10.1158/0008-5472.CAN-06-4499. [DOI] [PubMed] [Google Scholar]
- 27.Page JM, Merkel AR, Ruppender NS, Guo R, Dadwal UC, Cannonier SA, et al. Matrix rigidity regulates the transition of tumor cells to a bone-destructive phenotype through integrin beta3 and TGF-beta receptor type II. Biomaterials. 2015;64:33–44. doi: 10.1016/j.biomaterials.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sloan EK, Pouliot N, Stanley KL, Chia J, Moseley JM, Hards DK, et al. Tumor-specific expression of alphavbeta3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006;8:R20. doi: 10.1186/bcr1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soodgupta D, Pan D, Cui G, Senpan A, Yang X, Lu L, et al. Small Molecule MYC Inhibitor Conjugated to Integrin-Targeted Nanoparticles Extends Survival in a Mouse Model of Disseminated Multiple Myeloma. Mol Cancer Ther. 2015;14:1286–94. doi: 10.1158/1535-7163.MCT-14-0774-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, et al. Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat Med. 2015;21:1262–71. doi: 10.1038/nm.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiang J, Hurchla MA, Fontana F, Su X, Amend SR, Esser AK, et al. CXCR4 Protein Epitope Mimetic Antagonist POL5551 Disrupts Metastasis and Enhances Chemotherapy Effect in Triple-Negative Breast Cancer. Mol Cancer Ther. 2015;14:2473–85. doi: 10.1158/1535-7163.MCT-15-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan D, Schmieder AH, Wang K, Yang X, Senpan A, Cui G, et al. Anti-angiogenesis therapy in the Vx2 rabbit cancer model with a lipase-cleavable Sn 2 taxane phospholipid prodrug using alpha(v)beta(3)-targeted theranostic nanoparticles. Theranostics. 2014;4:565–78. doi: 10.7150/thno.7581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sadeghi MM, Krassilnikova S, Zhang J, Gharaei AA, Fassaei HR, Esmailzadeh L, et al. Detection of injury-induced vascular remodeling by targeting activated alphavbeta3 integrin in vivo. Circulation. 2004;110:84–90. doi: 10.1161/01.CIR.0000133319.84326.70. [DOI] [PubMed] [Google Scholar]
- 34.Harris TD, Kalogeropoulos S, Nguyen T, Liu S, Bartis J, Ellars C, et al. Design, synthesis, and evaluation of radiolabeled integrin alpha v beta 3 receptor antagonists for tumor imaging and radiotherapy. Cancer Biother Radiopharm. 2003;18:627–41. doi: 10.1089/108497803322287727. [DOI] [PubMed] [Google Scholar]
- 35.Pan D, Pham CT, Weilbaecher KN, Tomasson MH, Wickline SA, Lanza GM. Contact-facilitated drug delivery with Sn2 lipase labile prodrugs optimize targeted lipid nanoparticle drug delivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8:85–106. doi: 10.1002/wnan.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soman NR, Lanza GM, Heuser JM, Schlesinger PH, Wickline SA. Synthesis and characterization of stable fluorocarbon nanostructures as drug delivery vehicles for cytolytic peptides. Nano Lett. 2008;8:1131–6. doi: 10.1021/nl073290r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zutter MM, Mazoujian G, Santoro SA. Decreased expression of integrin adhesive protein receptors in adenocarcinoma of the breast. Am J Pathol. 1990;137:863–70. [PMC free article] [PubMed] [Google Scholar]
- 38.Kostenuik PJ, Singh G, Orr FW. Transforming growth factor beta upregulates the integrin-mediated adhesion of human prostatic carcinoma cells to type I collagen. Clin Exp Metastasis. 1997;15:41–52. doi: 10.1023/a:1018484323210. [DOI] [PubMed] [Google Scholar]
- 39.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moros M, Mitchell SG, Grazu V, de la Fuente JM. The fate of nanocarriers as nanomedicines in vivo: important considerations and biological barriers to overcome. Curr Med Chem. 2013;20:2759–78. doi: 10.2174/0929867311320220003. [DOI] [PubMed] [Google Scholar]
- 41.Kaneda MM, Sasaki Y, Lanza GM, Milbrandt J, Wickline SA. Mechanisms of nucleotide trafficking during siRNA delivery to endothelial cells using perfluorocarbon nanoemulsions. Biomaterials. 2010;31:3079–86. doi: 10.1016/j.biomaterials.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones SE, Erban J, Overmoyer B, Budd GT, Hutchins L, Lower E, et al. Randomized phase III study of docetaxel compared with paclitaxel in metastatic breast cancer. J Clin Oncol. 2005;23:5542–51. doi: 10.1200/JCO.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 43.Soundrapandian C, Sa B, Datta S. Organic-inorganic composites for bone drug delivery. AAPS PharmSciTech. 2009;10:1158–71. doi: 10.1208/s12249-009-9308-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diessner J, Wischnewsky M, Stuber T, Stein R, Krockenberger M, Hausler S, et al. Evaluation of clinical parameters influencing the development of bone metastasis in breast cancer. BMC Cancer. 2016;16:307. doi: 10.1186/s12885-016-2345-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horner A, Kemp P, Summers C, Bord S, Bishop N, Kelsall A, et al. Expression and distribution of transforming growth factor-β isoforms and their signaling receptors in growing human bone. Bone. 1998;23:95–102. doi: 10.1016/s8756-3282(98)00080-5. [DOI] [PubMed] [Google Scholar]
- 46.Chiechi A, Waning DL, Stayrook KR, Buijs JT, Guise TA, Mohammad KS. Role of TGF-beta in breast cancer bone metastases. Adv Biosci Biotechnol. 2013;4:15–30. doi: 10.4236/abb.2013.410A4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parvani JG, Galliher-Beckley AJ, Schiemann BJ, Schiemann WP. Targeted inactivation of beta1 integrin induces beta3 integrin switching, which drives breast cancer metastasis by TGF-beta. Mol Biol Cell. 2013;24:3449–59. doi: 10.1091/mbc.E12-10-0776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galliher AJ, Schiemann WP. Beta3 integrin and Src facilitate transforming growth factor-beta mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006;8:R42. doi: 10.1186/bcr1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Desgrosellier JS, Lesperance J, Seguin L, Gozo M, Kato S, Franovic A, et al. Integrin alphavbeta3 drives slug activation and stemness in the pregnant and neoplastic mammary gland. Dev Cell. 2014;30:295–308. doi: 10.1016/j.devcel.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takahashi M, Mizoguchi T, Uehara S, Nakamichi Y, Yang S, Naramoto H, et al. Docetaxel inhibits bone resorption through suppression of osteoclast formation and function in different manners. J Bone Miner Metab. 2009;27:24–35. doi: 10.1007/s00774-008-0013-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.