

Abstract
Lung immaturity is the major cause of morbidity and mortality in premature infants, especially those born <28 weeks of gestation. These infants are at high risk of developing respiratory distress syndrome (RDS), a lung disease caused by insufficient surfactant production and immaturity of saccular/alveolar type II epithelial cells in the lung. RDS treatment includes oxygen and respiratory support that improve survival but also increase the risk for bronchopulmonary dysplasia (BPD), a chronic lung disease characterized by arrested alveolarization, airway hyperreactivity, and pulmonary hypertension. The mechanisms regulating normal alveolar development and how injury disrupts normal development to cause BPD are not well understood. We examined the role of the matricellular protein CCN5 (Cysteine-rich protein 61/Connective tissue growth factor/Nephroblastoma-overexpressed protein) in the development of BPD. Cultured non-proliferating alveolar type II cells expressed low levels of CCN5 protein, and displayed higher levels during proliferation. siRNA targeting of CCN5 reduced alveolar type II cell proliferation and migration in cell culture. In a mouse model of hyperoxia-induced BPD, CCN5 protein was increased only in proliferating alveolar type I cells. Alveolar epithelial cells co-expressing markers of type II cells and type I cells also appeared. The results suggest that hyperoxic injury in immature lungs induces proliferation of type I cells and trans-differentiation of type II cells into type I cells. We propose that the mechanism of the injury response in BPD includes CCN5 expression. Study of CCN5 in neonatal alveolar injury will further our understanding of BPD pathophysiology while providing a mechanistic foundation for therapeutic approaches.
Keywords: CCN5, RDS, BPD, Alveolar type I, Alveolar type II, Epithelial cells, Proliferation, Differentiation
Introduction
Lung immaturity is the major cause of morbidity and mortality in premature infants, especially those born <28 weeks gestation. Proper lung development from 24 to 28 weeks requires coordinated distal airway epithelial cell proliferation and differentiation. Infants born at this age are at high risk for development of respiratory distress syndrome (RDS), a lung disease caused by insufficient surfactant production and immaturity of the saccular/alveolar type II epithelial cells in the lung. RDS treatment includes oxygen and respiratory support, with exogenous surfactant replacement (Verder 2007). Respiratory support with oxygen and mechanical ventilation improves preterm infant survival but also increases their risk of developing bronchopulmonary dysplasia (BPD), a chronic lung disease characterized by arrested alveolarization, alveolar simplification, airway hyperreactivity, and pulmonary hypertension (Northway Jr. et al. 1967; D’Alessandro et al. 2017; Jobe and Steinhorn 2017; Keller et al. 2017). The mechanisms regulating normal alveolar development and BPD are not well understood.
The gas-exchanging structures of the lung are the saccules and alveoli. Saccular formation begins in the respiratory bronchioles of the lung in parallel with development of the alveolar capillary bed in infants born at 24–28 weeks of gestation, followed by the beginning of alveolar development (Adamson and Bowden 1975). Clinical interventions, such as high oxygen therapy in the first week of life, can adversely impact the saccular and alveolarization process, creating the clinical syndrome of BPD (Northway Jr. et al. 1967; D’Alessandro et al. 2017; Jobe and Steinhorn 2017; Keller et al. 2017). The major manifestations of hyperoxic lung injury in neonates include fewer and larger alveoli (alveolar simplification) and reduced development of the sub-alveolar microvascular bed. Airway hyperreactivity and pulmonary hypertension are common long-term problems. Several animal models of BPD, including mice, rats, and baboons, have been developed by exposing the saccular stage lung to hyperoxia. In these models hyperoxia exposure produced complete arrest of saccular formation and alveolar septation with fewer and larger alveoli. Even though the normal signals for the progression of saccular development and the subsequent septation required to form alveoli are unknown, it is well-recognized that hyperoxia disrupts this process.
In response to hyperoxic injury, alveolar type II epithelial cells in adult lungs can proliferate and differentiate to type I cells (Yee et al. 2014; Jansing et al. 2017). Since both type I and type II cells play crucial roles in respiratory function, it is important that the appropriate number of both cell types exist during normal lung development and also the healing from injurious exposures. Many factors regulate the trans-differentiation process between type I and II cells. Type I cells have been described as terminally differentiated cells derived from the progenitor type II cells. However, some studies have reported that under certain conditions, including following injury, type I cells have the ability to proliferate and trans-differentiate to type II cells (Mercurio and Rhodin 1976). The lack of information about the mechanisms and molecules responsible for response to hyperoxic injury and the dynamics of type I and II cell trans- differentiation in the developing lung remains a major impediment to developing effective therapies for pre-term infants.
We are interested in defining the role of the matricellular protein CCN5 during normal lung alveolar epithelial cell development and response to neonatal hyperoxic injury. CCN5 is a member of the Cysteine-rich 61/Connective tissue growth factor /Nephroblastoma-overexpressed (CCN) family of genes (Mason et al. 2004; Delmolino and Castellot Jr. 1997). The six members of this family are matricellular and nuclear proteins that have important functions in numerous cell and physiologic processes, including embryonic development, cell motility, proliferation, angiogenesis, and differentiation (Vaidya et al. 2017).
Several studies have provided functional evidence that CCN5 inhibits smooth muscle cell (SMC) proliferation and motility while promoting a differentiated SMC phenotype (Lake et al. 2003; Lake and Castellot Jr. 2003; Mason et al. 2004). In non-mesenchymal cells, the Banerjee laboratory has shown that CCN5 acts as a negative regulator of plasticity by inhibiting the epithelial-mesenchymal transition (EMT) in breast and pancreatic cancer cells (Banerjee and Banerjee 2012; Das et al. 2017).
Though the mechanism of action and biological activity profile of CCN5 in smooth muscle cells and cancer cells has received considerable attention, there are still several important gaps in our understanding of CCN5’s role in the pathobiology of many other tissues and diseases. In particular, no studies have examined CCN5 regulation and function in the developing lung during the alveolarization and in response to injury. In this study we found that CCN5 expression and proliferation are coordinately up-regulated during normal alveolarization and in response to hyperoxic injury in a murine model of oxygen-induced BPD.
Material and methods
Media
Dulbecco’s Modified Eagle’s low glucose medium and protease inhibitor cocktail, Triton-X, hydroxyl proline and bovine serum albumin were purchased from Sigma-Aldrich (St. Louis MO). Fetal Bovine Serum (Lot # ANB 18202A) was from BD Biosciences, L- glutamine, Pen/Strep, Invitrolon PVDF filter paper and NuPage 4–12% Bis-Tris pre-cast gels (1.0 mm X 12 wells) were obtained from Novex/Invitrogen. A siRNA cocktail of three siRNA sequences targeting CCN5, Silencer Negative Control scrambled siRNA and Silencer GAPDH siRNA were purchased from Ambion.
Reagents
Tris-Glycine-SDS 10X running buffer, transfer buffer (10X Tris-buffered saline washing buffer), TBS-Tween-20 (10X), Tris-EDTA buffer, and SDS sample buffer were from Boston Bio Products (Ashland, MA). Methanol 100% was from Fisher Scientific, ethanol 100% from Decon. Histology grade xylene and glass slip covers were from VWR. Conical tubes (15 and 50 ml) and 6 well culture plates were from BD Falcon; 100 mm petri dishes were from Fisher. Kodak film (X-OMAT Blue XB) and Restore Plus Western Blot Stripping Buffer were from Thermo Scientific. Mountain Plus DAPI was from Vector. Anti-CCN5 rabbit polyclonal antibody (cat#ab38317), anti-surfactant protein c (SPC) pro-protein (rabbit polyclonal antibody, ab90716), monoclonal antibody to beta Actin (HRP conjugated, cat# 20272), anti-T1α rabbit polyclonal antibody (cat#ab109059) and anti-Ki67 mouse monoclonal antibody (cat#ab6526) were all from Abcam (Cambridge, MA). Peroxidase-Conjugated Affinipure Goat Anti-Rabbit IgG (H+L), (111-035-144), secondary antibodies Alexa Fluor 488 (donkey anti mouse, cat# 715-547-003), Alexa Fluor 647 (donkey anti RABBIT, cat# 711-607-003), CY2 (donkey anti rabbit, cat# 711-225-152), CY3 (donkey anti mouse, cat# 715-165-151) and CY2 (goat anti mouse, cat# 115-225-003) were purchased from Jackson ImmunoResearch (West Grove PA). Monoject insulin syringes were from Kendall (Mansfield MA). Paraformaldehyde was from Fisher Scientific. Phosphate buffered saline was purchased from Gibco (Grand Island NY). OCT was obtained from Tissue Tek. Dispase was obtained from Sigma (St. Louis MO). Three pre-designed CCN5 siRNA sequences that targeted three different regions of CCN5 mRNA were purchased from ThermoFisher scientific (see below for additional information).
Cell cultures
The murine lung epithelial cell line MLE12 was purchased from the American Type Culture Collection (ATCC, VA). BCA Protein Assay Kit and RIPA buffer were obtained from Pierce/Thermo Scientific (Logan, UT). This cell line is derived from adult lung alveolar epithelial tumors created by expression of the SV40 large T antigen targeted to alveolar type II cells by the surfactant protein C promoter, and is commonly used as a model for Type II cells. MLE-12 cells exhibit characteristics of lung type II cells, including the expression of surfactant proteins B and C, formation of microvilli and multivesicular bodies, and expression of surfactant phospholipid. MLE-12 cells were plated at a density of 300 × 105 in 6 - well plates and cultured as follows: Two sets of cells were cultured in parallel. One set was incubated in DMEM plus 10% FBS, and allowed to proliferate exponentially. The other set was allowed to proliferate exponentially for 2 days and then media changed to serum free media for 24 h. Both sets were harvested at that time for Western Blot analysis.
Transfection with CCN5 siRNA
MLE-12 cells were transfected with siRNA using the transfection reagent Dharmafect 2. To knock down CCN5, three pre-designed CCN5 siRNA sequences that targeted three different regions of CCN5 mRNA were pooled and used [(gtcagggccacggagcttaggagaccttgg),(tgcttctggtatctgcagccccgctggggt), and (aactacagcagggctaggaccctcttgggc)]. The protocol for transfection of MLE-12 cells was adapted from the manufacturer’s guideline; all steps were done using RNAse- free pipette tips and RNase spray for decontaminating the work area. The protocol has two transfection steps. The first transfection was done when the cells were 30–50% confluent. One Eppendorf tube (tube 1) containing pooled CCN5 siRNA (5 μl siRNA in 95 μl serum-free DMEM/well) and a separate Eppendorf tube (tube 2) containing only the transfection reagent Dharmafect 2 (1 μl in 99 μl serum free DMEM/well) were prepared and both tubes incubated for 5 min at room temperature. The contents of tube 1 and tube 2 were mixed via trituration, incubated for 20 min at room temperature, and 800 μl of antibiotic free media was added to the mixture. This transfection media was added to the plated cells which were then incubated in 37 °C for 48 h. A second transfection step was done 48 h after the first transfection in the same manner, and the cells cultured for another 24 h resulting in a total siRNA exposure time of 72 h. At the time of the second transfection, the media were changed to serum free media. Scrambled siRNA was used as the control condition in each experiment. After 72 h of total exposure time, the cells were harvested for Western Blot analysis.
Western blot analysis
Cells were washed thrice with PBS, then 500 μl of RIPA buffer plus protease inhibitor mixture (1:50) was added to each well twice. Using a cell scraper, cells were scraped off the plates (on ice) and then aspirated into Eppendorf tubes, incubated on ice for 60 min with vortexing every 10 min. The tubes were spun at 12,000 g in a microfuge for 15 min at 4 °C and the supernatant transferred to new Eppendorf tubes and stored in −20 °C. The protein concentration in each sample was determined in duplicate using the BCA protein assay (Tang et al. 2007). For Western Blots the protein samples were heated at 70 °C for 10 min in sample reducing buffer to denature the protein. Proteins (20 μg per sample) were separated by electrophoresis using NuPage 4–12% Bis-Tris gels. Subsequently the proteins were transferred to a PVDF membrane. The membrane was blocked with 5% dry milk in TBST for 2 h at room temperature or at 4 °C overnight. The membrane was probed with primary and secondary antibodies, and then stripped and reprobed for beta actin. The primary antibodies were incubated overnight at 4 °C and the secondary antibodies were incubated for 2 h at room temperature. Blots were developed using Western Lightning Plus ECL and detected with Kodak film X-OMAT Blue XB. Densitometry was done for all blots and beta-actin was used as an internal control for protein loading.
Proliferation assay
Cell proliferation assays were performed as described in our previous work (Zhang et al. 1998). Briefly, 3 × 105 MLE12 cells were plated into 16-mm multi-well dishes. Cells were previously transfected with CCN5 siRNA or SCR siRNA using DharmaFECT transfection reagent as described above. Cells were growth-arrested in G0 using serum-free media, after which cultures were released from G0 by exposure to 10% FBS/DMEM. Cells were allowed to grow for the indicated time and then processed and counted in duplicate using a Coulter Counter (Fullerton, CA) after trypsinization.
Scratch wound assay
Transfected MLE12 cells were plated at confluence onto glass chamber slides. The following day a uniform straight scratch was made in the monolayer using a 200 μl yellow plastic pipette tip (Fisher Scientific, Pittsburgh, PA). Monolayers were washed gently, marked, and photographed using Nikon optics on an inverted microscope. After incubation for 12 h or 24 h at 37 °C, the cells were fixed in 1% paraformaldehyde and mounted. Nuclei were visualized using Hoechst 33258 labeling and then photographed with a fluorescence microscope.
Hyperoxia exposure
We used neonatal wild-type (C57/Bl6) mice for all studies. Mice were purchased from Charles River Laboratory. Animals were housed and cared for by the Division of Laboratory Animal Medicine at New England Medical Center. This facility conforms strictly to the current National Institutes of Health guidelines for animal care. They provide veterinary support and evaluation as needed. The animal use protocol was approved by the Institutional Animal Care and Use Committee.
The hyperoxia exposure was performed as previously published (Flecknoe et al. 2000). Briefly, five-day-old (P5) mice were exposed to room air or 90% O2 until P13 as follows. At day 1 age, the pups were assigned to new litters consisting of six pups for each mother. From days P5 – P13 of life (corresponding to the major period of murine alveolarization), one litter was exposed to 90% oxygen, using a Plexiglass chamber and one litter was exposed to room air. The exposure chamber was opened briefly twice a day to replenish food and water, clean the chambers, and rotate the mothers between the hyperoxic and normoxic environment to prevent maternal oxygen toxicity. Oxygen concentration was monitored continuously. On day P13 pups were used for studies as described below.
Lung fixation and extraction
Pups were sacrificed at day P13 and the trachea and lungs exposed by thoracotomy. Ethicon black nylon sutures were used to ligate the trachea and left main bronchus. The right main stem bronchus was intubated and 4% paraformaldehyde was instilled to inflation fix the right lung at 20 cm pressure. After fixation of the right lung both lungs were removed, the left lung was kept for western blot analysis, and right lung used for paraffin embedding, sectioning and immunofluorescent labeling.
Immunofluorescence
Sections (5 μm) were de-paraffinized and re-hydrated through xylene, and serial dilutions of ethanol and PBS. Antigen retrieval was done by incubating the slides in 10 mM Tris-EDTA for 15–20 min at 92.8 °C, followed by slow cooling to room temperature before transfer into PBS. Slides were washed twice for 5 min in TBS plus 0.025 Triton X-100, then sections were blocked for 2 h at room temperature with 10% normal serum with 1% BSA in TBS. Primary antibodies were added at different dilutions in TBS with 1% BSA onto each section, and incubated overnight at 4 °C. Slides were washed with TBST 0.025% Triton with gentle agitation three times for 5 min each. For fluorescent detection a fluorophore-conjugated secondary antibody diluted to the concentration recommended by the manufacturer in TBS with 1% BSA was added to the slide and incubated for 1 h in the dark at room temperature. Sections were then washed three times for 5 min and mounted with a coverslip with mounting medium containing DAPI. Pictures were taken using a Zeiss Axio fluorescence microscope and analyzed by AxioVersion software.
Flow cytometry
After lung extractions using the above mentioned protocol, we prepared a single cell suspension in Cell Staining Buffer using a protocol adapted from Biolegend for flow cytometry products (Menon et al. 2014). Cells were re-suspended in staining buffer up to ~15 ml and centrifuged at 350 × g for 5 min, discarding the supernatant. To reduce nonspecific immunofluorescence, cells were pre-incubated with 5–10 μg/ml purified anti-mouse CD16/CD32 antibody specific for Fcγ R III/II (BioLegend Cat. #101302, clone 93) on ice for 10 min. Following this blocking step, we added appropriate conjugated fluorescent primary antibodies (anti TTF1 1:500 and anti T1α 1:200), at predetermined optimum concentrations and incubated them on ice for 15–20 min in the dark. The cells were then washed twice with at 2–3 ml of Cell Staining Buffer and centrifuged at 350 × g for 5 min at 4 °C. The cell pellet was placed on ice and transported to the Tufts Flow Cytometry core facility within 5 min and analyzed immediately.
Results
CCN5 expression is significantly decreased in growth arrested MLE12 cells compared to exponentially growing cells
Studies in SMCs indicate that CCN5 plays an important role in maintaining the quiescence of SMCs (Lake et al. 2003). To determine if this function is also present in lung alveolar epithelial type II cells, we used Western blot analysis to analyze CCN5 expression in growth arrested compared to exponentially growing MLE12 cells. Unexpectedly, CCN5 was significantly decreased to 20% ± 2.2 (mean ± SEM, n = 5, p = 0.009) in growth arrested MLE12 cells compared to exponentially growing cells. Thus the relationship of CCN5 expression with cell proliferation in this lung type II epithelial cell line is opposite to the pattern seen in smooth muscle cells (Fig. 1a). Because CCN5 may act as a paracrine regulator of cells, we also measured CCN5 in conditioned media of growth arrested and exponentially growing MLE12 cells. Again, CCN5 was significantly decreased in conditioned media from growth arrested MLE1 cells (21.7% ± 6.19, mean ± SEM, n = 5, p = 0.03) compared to conditioned media from exponentially growing cells (Fig. 1b). These data show that growth-arrested pulmonary alveolar epithelial type II cells produce and secrete less CCN5 than exponentially growing cells, suggesting a different function for CCN5 in epithelial cells as opposed to smooth muscle cells and other epithelial cells as discussed previously.
Fig. 1.

a CCN5 expression in exponentially growing (Expo) and growth arrested (G0) MLE12 cells: Western blot analysis and densitometry of MLE12 lysate shows that CCN5 was significantly decreased in growth arrested compared to exponentially growing cells (20% ± 2.2 Mean ± SEM; N = 5. ** = P < 0.05). b CCN5 expression in conditioned media from exponentially growing (Expo) and growth arrested (G0) MLE12 cells: Western blot analysis and densitometry of MLE12 conditioned media shows that CCN5 was significantly decreased in conditioned media from growth arrested cells compared to that from exponentially growing cells (21.7 ± 6.19, Mean ± SEM; N = 5. * = P < 0.05). Statistical significance was evaluated using a paired two-tailed t-test
RNAi knock down of CCN5 in MLE12 inhibits cell proliferation and motility
We previously reported that overexpression of CCN5 protein levels in VSMC using a viral vector reduced proliferation and motility and that reducing CCN5 expression via siRNAs reversed the anti-proliferative function (Lake et al. 2003), suggesting that CCN5 plays an important inhibitory role in SMC proliferation. To more thoroughly explore the function of CCN5 in alveolar type II cells we employed RNA interference (RNAi) to knock down endogenous CCN5 expression in MLE12 cells. Western Blot analysis of cell lysates documented a significant decrease of CCN5 protein levels in CCN5 siRNA treated cells (64% ± 7.2 of scrambled siRNA-treated cells, means ± SEM, n = 3, P = 0.02) (Fig. 2a). We then assessed the effect of CCN5 knockdown on the proliferation and motility of MEL12 cells. We found that both proliferation and motility are decreased in CCN5 siRNA treated cells compared to SCR siRNA treated cells (Fig. 2b and c). These data suggest that, in contrast to its function in SMC, CCN5 may stimulate proliferation and motility in alveolar epithelial type II cells.
Fig. 2.
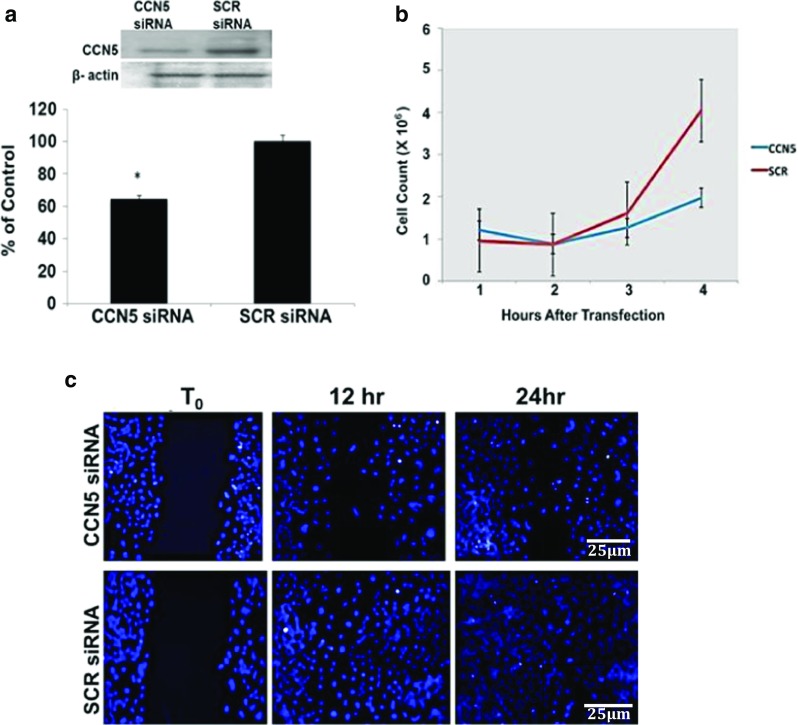
a RNAi knock down of CCN5 protein levels in exponentially growing MLE12 cells. Western blot analysis and densitometry shows the reduction of CCN5 in CCN5 siRNA treated cells compared to SCR siRNA treated cells (64% ± 7.2, Mean ± SEM; N = 3. * = p < 0.05). Statistical significance was evaluated using a paired two-tailed t-test. b Knockdown of CCN5 using siRNA inhibits proliferation in MLE12 cells. Cell counts of CCN5 siRNA-transfected MLE12 cells were performed at 12, 24, 72 and 96 h. CCN5 knockdown cells showed decreased proliferation compared to SCR-treated cells (data from 3 experiments). c Knockdown of CCN5 with siRNA inhibits motility of exponentially growing MLE12 cells. Initial pictures of the scratch wound were taken for reference. The monolayers were then incubated for 12 and 24 h at 37 °C, fixed, labeled with DAPI to visualize cell nuclei and identify the extent of cell movement into the wound. Cells treated with CCN5 siRNA showed decreased motility and decreased ability to cover the scratch area compared to SCR siRNA-treated cells. Scale bars shown apply to all panels
The effect of hyperoxia on CCN5 expression in early post-natal lungs
Having identified unique relationships of CCN5 expression to cell proliferation and migration in an adult alveolar type II cell line, we next investigated the role of CCN5 in the alveolarization phase of normal mouse lung development and in response to the hyperoxic injury that impedes alveolarization. We examined CCN5 expression in whole lungs of P13 mice pups exposed to either room air or hyperoxic conditions (90% oxygen) from P5 through P13. We have previously shown that this mouse model exhibits alveolar simplification, arrested alveolarization, and microvascular simplification typical of human BPD (Chetty et al. 2015). CCN5 protein levels were significantly decreased to 58.2% ± 18.9 (mean ± SEM, n = 6, p = 0.04) in hyperoxic lungs compared to room air lungs (Fig. 3).
Fig. 3.

CCN5 expression, measured by Western blot analysis, is decreased in oxygen-exposed neonatal mouse lungs compared to room air exposed lungs (58.2% ± 18.9, Mean + SEM; N = 6. * = P < 0.05). Statistical significance was evaluated using a paired two-tailed t-test
Alveolar epithelial cell-specific expression of CCN5 in P13 (postnatal day 13) room air and oxygen exposed lungs
To further characterize the expression of CCN5 in mouse lungs undergoing alveolarization under room air and hyperoxic conditions, we double-labeled paraffin embedded sections from room air and oxygen exposed lungs with fluorescent antibodies targeting SPC (a specific marker of alveolar epithelial type II cells) and CCN5. In room air lungs CCN5 expression was absent in SPC-positive cells (Fig. 4). The SPC-positive cells demonstrated the characteristic alveolar type II cell appearance (round epithelial cells with abundant SPC signal surrounding the nucleus in a doughnut shape; Fig. 4). In hyperoxia-exposed lungs, immunofluorescence for SPC showed two populations of SPC-positive cells. One type was positive for both SPC and CCN5 but instead of the characteristic shape of type II cells these cells were crescent or low oblong in shape, suggesting they were in progressive stages of flattening and thinning. The other type of SPC-positive cells continued to show the characteristic appearance of type II cells and continued to be negative for CCN5. This population appeared greatly reduced in number compared to room air lungs (Fig. 4). The appearance of two morphologically distinct cell types expressing SPC, with one cell type also positive for CCN5, suggests that oxygen injury of lungs induced alveolar epithelial type II cell differentiation towards type I cells through an intermediate cell type which is both SPC and CCN5 positive but with a shape approaching that of type I cells.
Fig. 4.

Immunofluorescence labeling of paraffin-embedded sections from 90% oxygen-exposed and room air neonatal mouse lungs. In room air CCN5 was not expressed by alveolar epithelial type II cells. In oxygen exposed lungs CCN5 was expressed in some SPC positive cells but these cells do not have the characteristic appearance of type II cells (arrows). Red (SPC), Green (CCN5). Scale bars shown apply to all panels
During oxygen-mediated lung injury, alveolar type II cells differentiate into type I cells via an intermediate proliferative cell type that expresses CCN5
In response to hyperoxic injury, adult type II cells have the ability to proliferate and differentiate to type 1 cells (Yee et al. 2014). Several investigators have observed alveolar epithelial cells that displayed characteristics of both type I and type II cells in culture plates (Flecknoe et al. 2000). This “intermediate” AEC type is thought to represent cells at an intermediate stage of trans-differentiation from type II and type I alveolar epithelial cells. In this study, we investigated the possibility that hyperoxic lung injury stimulates type II cells to differentiate into type I cells via an intermediate cell type and examined the possible involvement of CCN5 in the trans-differentiation process. Double labeling with the alveolar epithelial type II cell marker SPC, and the alveolar epithelial type I cell marker T1α showed no expression overlap in room temperature air lungs, while oxygen-exposed lungs showed saccular/alveolar epithelial cells that were positive for both proteins, suggesting an intermediate cell type that is positive for both SPC and T1α (Fig. 5). Cells positive for both SP-C and T1α exhibited the “intermediate” crescent or low oblong shape described above, but not the doughnut shape characteristic of type II cells.
Fig. 5.

Immunofluorescence labeling of sections from 90% oxygen-exposed and room air lungs. In room air there was no overlap between SPC and T1α, and SPC-positive cells had the characteristic appearance of type II cells. In oxygen-exposed lungs there was some overlap between SPC and T1α. Cells positive for SPC alone continued to have the characteristic type II cell appearance, while dual positive cells were flattened and elongated without the doughnut shape pattern of SPC expression (arrows). This indicates an intermediate cell type that expresses both markers. Left panel Green (SPC), Red (T1 α). Right panel Red (SPC), Green (T1 α). Scale bars shown apply to all panels
To check the proliferation activity of the alveolar epithelial cells during the trans-differentiation process in vivo, we double-labeled sections from oxygen exposed lungs using fluorescent-tagged antibodies against SPC and the proliferation marker Ki67. In hyperoxic sections some SPC positive cells also labeled positive for Ki67 (Fig. 6). Again, these double positive cells did not have the characteristic shape of type II cells (see Fig. 4). Double-labeling for Ki67 and CCN5 in hyperoxic lung sections show that all CCN5 positive cells also labeled with Ki67, indicating that they are actively proliferating (Fig. 6).
Fig. 6.

Immunofluorescence labeling sections from 90% oxygen-exposed. Left panel: In oxygen-exposed lungs some SPC positive cells, which do not show the characteristic appearance of type II cells exhibit proliferative activity. Green (SPC), Red (Ki67). Right panel: In oxygen exposed lungs all CCN5 positive cells show a proliferation activity. Green (CCN5), Red (Ki67). Scale bars shown apply to all panels
Fluorescent automated cell sorting (FACS) reveals three populations of alveolar epithelial cells after hyperoxia exposure
To further support the hypothesis of a type I-type II intermediate cell type, we used flow cytometry analysis to sort cells from extracted lungs. The flow cytometry data showed 3 groups of cells; one cell group was positive for the type II cell marker TTF1 and negative for the type I cell marker T1α, a second cell group was positive for the type I cell marker T1α but negative for the type II cell marker TTF1, and the third group was positive for both markers (Fig. 7a, b, c). To check CCN5 expression in each group of sorted cells we used Western blot analysis of cell lysates from each group. Western blot data showed that in room air samples, CCN5 was expressed in T1α positive cells but not in TTF1 positive cells, on the other hand, 90% oxygen exposed samples showed that CCN5 was expressed in T1α positive cells and double positive cells but not in TTF1 only positive cells (Fig. 8a). We also checked Ki67 expression using Western blot analysis. Our data showed that in room air samples Ki67 was expressed in T1α positive cells, but not by TTF1 positive cells. In oxygen exposed samples, Ki67 was strongly expressed in T1α positive and double positive cells, and very weakly expressed in TTF1 positive cells (Fig. 8b).
Fig. 7.

a Flow cytometry analysis showing negative controls (unstained cells) and positive controls (cells stained with only TTF1 or T1α). b Flow cytometry analysis of room air lungs: TTF1 positive cells (Type II cells), and T1α positive cells (Type I cells). c Flow cytometry analysis of 90% oxygen lungs: TTF1 positive cells (Type II cells), T1α positive cells (Type I cells) and double positive cells (intermediate cell type). Representative of 3 experiments
Fig. 8.

a CCN5 expression, measured by Western blot analysis. In room air, CCN5 is expressed in T1α positive cells but not in TTF1 positive cells. In 90% oxygen, CCN5 is expressed in T1α positive cells and in double positive cells but not in TTF1 positive cells. b Ki67 expression, measured by Western blot analysis. In room air, Ki67 is expressed in T1α positive cells but not in TTF1 positive cells. In oxygen, Ki67 is weakly expressed in TTF1 positive cells and strongly expressed in T1α positive cells, and double positive cells. Representative of 3 experiments
Discussion
When lung development is disturbed by injury, the development of normal lung architecture becomes disrupted, resulting in altered alveolar development. Depending on the severity of the architectural malformation, there may be serious consequences in terms of respiratory function, as well as long-term consequences for later life (Vrijlandt et al. 2005; Doyle et al. 2006; Baraldi and Filippone 2007). The alveolar structure of the lungs is composed of type II and type I epithelial cells. Type II cells, in addition to their critical role as producers of pulmonary surfactant, serve as progenitors of type I cells and transdifferentiate into type I cells to repair the alveolar epithelium of adult lungs when they are injured (Adamson and Bowden 1975; Yee et al. 2014). Primary cultures of type II cells prepared from adult lungs also rapidly undergo spontaneous trans-differentiation into type I-like cells (Flecknoe et al. 2000). Studies of alveolar type II cell fate frequently use lung injury models to induce epithelial damage. Such injury models initially reduce the population of alveolar type I cells, with the consequence of alveolar type II cell trans differentiation and proliferation to re-establish the type I cell population and a functional air–blood interface (Evans et al. 1973). In a study of the incorporation of [3 H]-thymidine into proliferating cells of NO2-challenged rat lungs, Evans and coworkers reported that 1 h after a radiolabeled pulse the population of radiolabeled alveolar epithelial cells (representing 35% of total lung parenchymal cells) was composed of 88% alveolar epithelial type II cells, less than 1% type I cells, and 12% of cells that could not be unambiguously assigned to one type or the other (Evans et al. 1973).
Previous work on CCN5 in mesenchyme-derived SMC shows that CCN5 acts as a cell cycle regulator that inhibits smooth muscle cell (SMC) proliferation and promotes SMC differentiation. Delmolino et al. originally identified CCN5 as heparin-induced gene in vascular smooth muscle cells (VSMC) (Delmolino and Castellot Jr. 1997). Shortly thereafter, another group showed that CCN5 is lost after cell transformation (Zhang et al. 1998). Since the original discovery of CCN5, several studies have examined its expression in different tissues and its potential function in tumors, cell localization, gene regulation, and function (Mason et al. 2004). Work from our laboratories previously used a rat carotid artery balloon injury model to demonstrate that CCN5 is highly expressed in quiescent (non-proliferating) VSMC in the uninjured rat aorta and 14 days after injury, but not in actively proliferating cells 2 days post-injury (Lake and Castellot Jr. 2003). These data suggest that CCN5 plays an important role in maintaining the quiescence of VSMC.
Recent investigations from the Banerjee laboratory suggested that CCN5 located both intracellularly (in the nucleus and cytoplasm) and extracellularly, is a negative regulator of plasticity. It prevents the EMT process in breast cancer cells and also pancreatic cancer cells. Some oncogenic signals, for example, miR-10b upregulation and activation of TGF-β- signaling, can accumulate during CCN5 upregulation in cancer cells. Loss of CCN5 activity may advance breast cancer growth, thus ectopic expression or exogenous provision of CCN5 protein may provide a novel therapeutic approach in breast cancer (Banerjee and Banerjee 2012; Das et al. 2017). Our work is consistent with the differential effect of CCN5 on epithelial cells vs. mesenchyme-derived cells.
In this study, we shed new light on fate of lung alveolar epithelial cells in the alveolarization stage which are exposed to hyperoxic injury, and the possible activity of the matricellular protein CCN5 in the injury response/repair process. We began by establishing the relationship of CCN5 expression with type II cell proliferation, using siRNA to silence CCN5 expression in the adult type II cell line MLE12 cells. MLE12 cells treated with siRNA targeting CCN5 showed a reduction in both proliferation and motility, effects that are the opposite of the reported function of CCN5 in SMCs. During alveolarization type II cells must undergo proliferation and motility, both to populate newly developing alveoli with type II cells and to move differentiating cells into position as type I cells. The fact that these two cell functions were affected in this manner with in vitro CCN5 knockdown also suggested the potential importance of CCN5 in alveolar epithelial cells during alveolar repair in response to lung injury.
We then studied the expression of CCN5 in the alveolar epithelium in vivo using a mouse model of BPD in which mouse pups are exposed to hyperoxia during the alveolarization phase. We found that CCN5 was significantly decreased in oxygen-exposed lungs compared to room air lungs. We used immunofluorescence microscopy to focus on the relationship of CCN5 expression with alveolar epithelial type II cell trans differentiation in response to hyperoxic injury, using alveolar type II and I cell markers (SPC, T1α) to follow the fate of type II cells and the concomitant expression of CCN5. We found that in room air CCN5 was not expressed in alveolar epithelial type II cells in lungs nearing the completion of alveolarization (P13). However, in oxygen-exposed lungs we identified a population of cells expressing both SPC and CCN5. Further, these cells did not show the characteristic morphology of type II cells, suggesting that these cells represent an intermediate population undergoing the trans-differentiation from type II to type I alveolar epithelial cells.
To support the intermediate cell type hypothesis, we used alveolar epithelial type II and I cell markers to document the trans-differentiation process. Oxygen-exposed lungs showed a population of cells expressing both the type II marker SPC and the type I marker T1α. These cells lacked the typical type II epithelial morphology. These findings are consistent with a cell type that is intermediate between type II and type I cells, further evidence of a trans-differentiation process in response to oxygen injury. Examination of proliferative activity by co labeling with Ki67 showed that these intermediate cells have proliferation activity and are CCN5 positive.
Alveolar epithelial cell populations were further documented using FACS analysis. Although TTF1 expression is also expressed in some distal bronchial epithelial cells, replaced SP-C labeling with TTF-1 labeling for this evaluation because TTF-1 gave a stronger signal that was more readily detected by FACS. This may have introduced some non-alveolar cells into our cell population, but we believe this to be minor as alveolar type II cells vastly outnumber other airway epithelial cells. We identified in lungs from room air treated mice two groups of cells: TTF1 positive cells and T1α cells. In oxygen exposed lungs, there were three groups of cells: TTF1 positive cells, T1α positive cells and double positive cells. Western blot analysis of cell lysates from each group showed that in room air, CCN5 was expressed in T1α positive cells but not in TTF1 positive cells. In oxygen samples, CCN5 was expressed in T1α positive cells and double positive cells. We also checked Ki67 expression. In the lung from the room air treatment group, Ki67 was expressed only in T1α positive cells, but in the lungs from the oxygen treatment group, Ki67 was expressed in T1α positive cells and double positive cells; however TTF1 positive cells showed a weak Ki67 expression. The fact that no Ki67 was seen in SPC- positive cells on immunofluorescence, but a weak signal for Ki67 was seen in western blot of TTF1 positive cells from oxygen lungs may reflect the increased sensitivity of western blot over immunofluorescence or it may be due to the presence of a small number of proliferating airway epithelial cells in the sample that were TTF1-positive. Ki67 expression is normally a representation of cells traversing the cell cycle in mitosis; however low levels of Ki67 have been identified bound to ribosomal RNA in non-proliferating cells (Bullwinkel et al. 2006). Thus our findings may reflect active proliferation by type I cells and transitional cells, while type II cells are mitotically quiescent as alveolarization disrupted by hyperoxia concludes, in agreement with the recent study by Yee et al. (2014).
In conclusion we find that CCN5 is expressed in proliferating alveolar type II cells from neonatal lungs, rather than quiescent cells, in distinct contrast to its expression pattern and function in smooth muscle cells and epithelial cell cancers of the breast and pancreas. CCN5 positively regulates type II cell motility, an important developmental event during alveolarization. CCN5 is expressed in vivo in type I alveolar epithelial cells and proliferating type II cells, but not non-proliferating type II cells at the conclusion of normal alveolarization. These CCN5-positive cells are also Ki67 positive, indicating proliferation of alveolar epithelial cells as alveolarization concludes. Oxygen injury to lungs during the alveolarization process causes alveolar epithelial type II cell trans-differentiation into type I cells, presumably as part of the repair process in injury. Further, we identified that this trans-differentiation proceeds through an intermediate cell type, which expresses CCN5 and shows proliferation activity. Our results emphasize the possible role of CCN5 in control of alveolar type I cell proliferation with alveolarization and in type II cell trans-differentiation in the repair response to injury occurring during alveolarization. The data introduce novel insights into alveolar epithelial cell maintenance with alveolarization and response to neonatal lung oxygen injury. This may have important translational impact as investigators seek to modulate the repair process. This study may provide significant clues for developing target approaches to benefit infants developing BPD.
Abbreviations
- BPD
bronchopulmonary dysplasia
- P
postnatal day
- SCR
scrambled siRNA
- siRNA
small interfering RNA
- SMCs
smooth muscle cells
- SPC
surfactant protein C
- TTF-1
thyroid transcription factor-1
- utSMC
uterine smooth muscle cell
- VSMC
vascular smooth muscle cells
Footnotes
Heber C. Nielsen and John J. Castellot, Jr. are Co-Senior Authors.
References
- Adamson IY, Bowden DH. Derivation of type 1 epithelium from type 2 cells in the developing rat lung. Lab Investig. 1975;32:736–745. [PubMed] [Google Scholar]
- Banerjee SK, Banerjee S. CCN5/WISP-2: A micromanager of breast cancer progression. J Cell Commun Signal. 2012;6:63–71. doi: 10.1007/s12079-012-0158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med. 2007;357:1946–1955. doi: 10.1056/NEJMra067279. [DOI] [PubMed] [Google Scholar]
- Bullwinkel J, Baron-Luhr B, Ludemann A, Wohlenberg C, Gerdes J, Scholzen T. Ki-67 protein is associated with ribosomal RNA transcription in quiescent and proliferating cells. J Cell Physiol. 2006;206:624–635. doi: 10.1002/jcp.20494. [DOI] [PubMed] [Google Scholar]
- Chetty A, Bennett M, Dang L, Nakamura D, Cao GJ, Mujahid S, Nielsen HC. Pigment epithelium-derived factor mediates impaired lung vascular development in neonatal hyperoxia. Am J Respir Cell Mol Biol. 2015;52:295–303. doi: 10.1165/rcmb.2013-0229OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro A, Nozik-Grayck E, Stenmark KR. Identification of Infants at Risk for Chronic Lung Disease at Birth. Potential for a Personalized Approach to Disease Prevention. Am J Respir Crit Care Med. 2017;196:951–952. doi: 10.1164/rccm.201706-1065ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Dhar K, Maity G, Sarkar S, Ghosh A, Haque I, Banerjee SK. Deficiency of CCN5/WISP-2-Driven Program in breast cancer Promotes Cancer Epithelial cells to mesenchymal stem cells and Breast Cancer growth. Sci Rep. 2017;7:1220. doi: 10.1038/s41598-017-00916-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmolino LM, Castellot JJ., Jr Heparin suppresses sgk, an early response gene in proliferating vascular smooth muscle cells. J Cell Physiol. 1997;173:371–379. doi: 10.1002/(SICI)1097-4652(199712)173:3<371::AID-JCP9>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Doyle LW, Faber B, Callanan C, Freezer N, Ford GW, Davis NM. Bronchopulmonary dysplasia in very low birth weight subjects and lung function in late adolescence. Pediatrics. 2006;118:108–113. doi: 10.1542/peds.2005-2522. [DOI] [PubMed] [Google Scholar]
- Evans MJ, Cabral LJ, Stephens RJ, Freeman G. Renewal of alveolar epithelium in the rat following exposure to NO2. Am J Pathol. 1973;70:175–198. [PMC free article] [PubMed] [Google Scholar]
- Flecknoe S, Harding R, Maritz G, Hooper SB. Increased lung expansion alters the proportions of type I and type II alveolar epithelial cells in fetal sheep. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1180–L1185. doi: 10.1152/ajplung.2000.278.6.L1180. [DOI] [PubMed] [Google Scholar]
- Jansing NL, McClendon J, Henson PM, Tuder RM, Hyde DM, Zemans RL. Unbiased Quantitation of Alveolar Type II to Alveolar Type I Cell Transdifferentiation during Repair after Lung Injury in Mice. Am J Respir Cell Mol Biol. 2017;57:519–526. doi: 10.1165/rcmb.2017-0037MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobe AH, Steinhorn R. Can We Define Bronchopulmonary Dysplasia? J Pediatr. 2017;188:19–23. doi: 10.1016/j.jpeds.2017.06.064. [DOI] [PubMed] [Google Scholar]
- Keller RL, Feng R, DeMauro SB, Ferkol T, Hardie W, Rogers EE, Moore PE. Bronchopulmonary Dysplasia and Perinatal Characteristics Predict 1-Year Respiratory Outcomes in Newborns Born at Extremely Low Gestational Age: A Prospective Cohort Study. J Pediatr. 2017;187:89–97.e83. doi: 10.1016/j.jpeds.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake AC, Bialik A, Walsh K, Castellot JJ., Jr CCN5 is a growth arrest-specific gene that regulates smooth muscle cell proliferation and motility. Am J Pathol. 2003;162:219–231. doi: 10.1016/S0002-9440(10)63813-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake AC, Castellot JJ., Jr CCN5 modulates the antiproliferative effect of heparin and regulates cell motility in vascular smooth muscle cells. Cell Commun Signal. 2003;1:5. doi: 10.1186/1478-811X-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason HR, Lake AC, Wubben JE, Nowak RA, Castellot JJ., Jr The growth arrest-specific gene CCN5 is deficient in human leiomyomas and inhibits the proliferation and motility of cultured human uterine smooth muscle cells. Mol Hum Reprod. 2004;10:181–187. doi: 10.1093/molehr/gah028. [DOI] [PubMed] [Google Scholar]
- Menon V, Thomas R, Ghale AR, Reinhard C, Pruszak J (2014) Flow cytometry protocols for surface and intracellular antigen analyses of neural cell types. J Vis Exp [DOI] [PMC free article] [PubMed]
- Mercurio AR, Rhodin JA. An electron microscopic study on the type I pneumocyte in the cat: differentiation. Am J Anat. 1976;146:255–271. doi: 10.1002/aja.1001460304. [DOI] [PubMed] [Google Scholar]
- Northway WH, Jr, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N Engl J Med. 1967;276:357–368. doi: 10.1056/NEJM196702162760701. [DOI] [PubMed] [Google Scholar]
- Tang S, Zhao J, Storhoff JJ, Norris PJ, Little RF, Yarchoan R, Hewlett IK. Nanoparticle-Based biobarcode amplification assay (BCA) for sensitive and early detection of human immunodeficiency type 1 capsid (p24) antigen. J Acquir Immune Defic Syndr. 2007;46:231–237. doi: 10.1097/QAI.0b013e31814a554b. [DOI] [PubMed] [Google Scholar]
- Vaidya R, Zambrano R, Hummler JK, Luo S, Duncan MR, Young K, Wu S. Recombinant CCN1 prevents hyperoxia-induced lung injury in neonatal rats. Pediatr Res. 2017;82:863–871. doi: 10.1038/pr.2017.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verder H. Nasal CPAP has become an indispensable part of the primary treatment of newborns with respiratory distress syndrome. Acta Paediatr. 2007;96:482–484. doi: 10.1111/j.1651-2227.2007.00263.x. [DOI] [PubMed] [Google Scholar]
- Vrijlandt EJ, Gerritsen J, Boezen HM, Duiverman EJ. Gender differences in respiratory symptoms in 19-year-old adults born preterm. Respir Res. 2005;6:117. doi: 10.1186/1465-9921-6-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee M, Buczynski BW, O'Reilly MA. Neonatal hyperoxia stimulates the expansion of alveolar epithelial type II cells. Am J Respir Cell Mol Biol. 2014;50:757–766. doi: 10.1165/rcmb.2013-0207OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Averboukh L, Zhu W, Zhang H, Jo H, Dempsey PJ, Liang P. Identification of rCop-1, a new member of the CCN protein family, as a negative regulator for cell transformation. Mol Cell Biol. 1998;18:6131–6141. doi: 10.1128/MCB.18.10.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]