

Abstract
The dermal extracellular matrix (ECM) comprises the bulk of skin and confers strength and resiliency. In young skin, fibroblasts produce and adhere to the dermal ECM, which is composed primarily of type I collagen fibrils. Adherence allows fibroblasts to spread and exert mechanical force on the surrounding ECM. In this state, fibroblasts display a “youthful” phenotype characterized by maintenance of the composition and structural organization of the dermal ECM. During aging, fibroblast-ECM interactions become disrupted due to fragmentation of collagen fibrils. This disruption causes loss of fibroblast spreading and mechanical force, which inextricably lead to an “aged” phenotype; fibroblasts synthesize less ECM proteins and more matrix-degrading metalloproteinases. This imbalance of ECM homeostasis further drives collagen fibril fragmentation in a self-perpetuating cycle. This article summarizes age-related changes in the dermal ECM and the mechanisms by which these changes alter the interplay between fibroblasts and their extracellular matrix microenvironment that drive the aging process in human skin.
Keywords: Aging, Collagen, CCN1, MMP-1, Skin
Introduction
Skin, like all organs in the human body, undergoes sequential and often cumulative alterations with the passage of time. Uniquely, the “aged” phenotype of skin may be accelerated by environmental factors, most notably, chronic exposure to ultraviolet (UV) irradiation from the sun and, therefore, is not a singular consequence that inevitably ensues during chronological aging (Rittié and Fisher 2015). Clinically, the aged phenotype of skin may be described as wrinkled, sagging, and generally less elastic and resilient than its youthful counterpart, although variations within this phenotype exist between natural, chronological aging and sun-induced premature aging, or photoaging. Chronologically aged skin, for example, is characteristically thin, dry, and finely wrinkled, while photoaged skin is typically leathery and lax with coarse wrinkles and uneven pigmentation (Sjerobabski-Masnec and Situm 2010). “Broken”-appearing blood vessels (telangiectasia) and brown spots (lentigines) subsequent to repeated exposure to UV irradiation may also be present (Sjerobabski-Masnec and Situm 2010). Unsurprisingly, the origin of such cosmetic alterations lies at the molecular level. Histological examination of the extracellular matrix (ECM) environment of aged skin reveals aberrant changes from that of young skin. Dermal thinning is observed, and elastin fibers, which are thin and single-stranded in young skin, become progressively beaded and lacking in the terminal fibers that extend into the epidermis (Montagna and Carlisle 1979). Similarly, type I collagen, the most abundant structural protein in skin, undergoes organizational and structural changes during the aging process that reduce its overall strength. Namely, collagen fibrils in aged skin display high levels of degradation and fragmentation and are replenished by dermal fibroblasts at diminishing rates (Mitchell 1967; Lovell et al. 1987; Rittié and Fisher 2002). These changes in collagen fibers with respect to aging can be visualized in Fig. 1. Furthermore, hydrophilic ECM biomolecules including the non-sulfated glycoseaminoglycan (GAG), hyaluronic acid, may be present in lower quantities, while the proteoglycans (PGs), decorin and versican, show a reduction in the molecular size of their polysaccharide chains (Carrino et al. 2011; Lee et al. 2016).
Fig. 1.

Fragmentation of collagen fibrils within dermis of aged or photoaged skin causes collapse of fibroblasts. a Transmission electron micrograph of fibroblast (colored pink for clarity) within dermis of sun-protected skin from young adult. Note extended cytoplasm (X) close proximity to abundant collagen fibrils (arrows) (nucleus (N), original magnification ×2000). b Transmission electron micrograph of fibroblast (colored pink for clarity) within dermis of photodamaged skin. Note collapse of cytoplasm inward toward nucleus (N) and lack of adjacent collagen fibrils (asterisks) (original magnification ×2000). c Scanning electron micrograph of collagen fibrils in young adult skin. Note densely packed long fibrils, without apparent fragmentation (original magnification ×10,000). d Scanning electron micrograph of collagen fibrils in photodamaged human skin. Note large gaps and numerous fragmented fibrils (original magnification ×8000). Inset shows higher magnification of fragmented ends of fibrils (arrows) (original magnification ×12,500). Reprinted with permission from Fisher et al. Arch Dermatol. 2008;144(5):666–672. 10.1001/archderm.144.5.666
Although the mechanical integrity imparted to the ECM by elastin, collagen, and hydrophilic biomolecules may seem immediately apparent, their role in mediating biological processes is acknowledged but not fully understood. Specifically, within the ECM, collagen is known to provide tensile strength and resistance to plastic deformation; elastin affords extensibility and reversible recoil such that tissues may tolerate repetitive mechanical insults (Gosline et al. 2002). PGs and GAGs sequester water molecules that dampen mechanical forces, thereby cushioning tissues under compression (Culav et al. 1999). As these forces are perceived by the ECM, they are communicated to cells via transmembrane surface adhesion receptors, or integrins, that connect with the intracellular actin cytoskeleton (Wang and Ingber 1994), (Burridge and Chrzanowska-Wodnicka 1996), and, in doing so, allow the ECM to elegantly direct a variety of cellular processes including differentiation, proliferation, adhesion, migration, and apoptosis (Ridley et al. 2003; Kim et al. 2011). However, as the skin ages and the ECM begins to manifest the aforementioned deleterious effects, communication lines between the matrix and the fibroblast population responsible for maintaining tissue homoeostasis are impaired. Additionally, pro-inflammatory mediators bound to the intact ECM are released, protected stem cell niches are compromised, and an age-associated cellular phenotype is induced, all of which feed into a viscous cycle of further matrix destruction and fibroblast apoptosis.
This article will discuss the molecular pathways leading to collagen degradation secondary to UV irradiation and chronological aging of skin. We will further highlight the consequences that these changes to the ECM microenvironment have on fibroblast and stem cell populations housed within the dermis. Lastly, we will comment on the systemic implications of prolonged exposure to the assortment of growth factors, proteases, cytokines, and chemokines that ensue with cellular aging of dermal fibroblasts.
Dermal extracellular matrix structural protein changes with age
The dermis of human skin is composed of two distinct regions, the upper, loosely constructed papillary dermis, and the lower, densely compacted reticular dermis. Although the border between the two regions is indiscriminant, their elastin and collagen compositions and organizations make them histologically distinct. As illustrated in Fig. 2, mature collagen molecules are composed of three intertwining α chains, each characterized by the distinctive amino acid triplet repeat Gly–X–Y, where Gly is glycine and X and Y may be any residue, but are most frequently proline and hydroxyproline, respectively, and flanked by two non-collagenous segments, or telopeptides (Exposito et al. 2010). While the spacing of Gly at every third repeat position is responsible for establishing the signature triple helical structure of collagen, the amino acid variability at the X and Y positions, in addition to the frequency and length of non-collagenous domains, are responsible for determining the specific type of collagen (28 of which have been identified in vertebrates to date) (Heino 2007). Type III collagen, for example, is a homotrimer of α1(III) chains supercoiled around one another in a relatively low diameter fibril and constitutes 20% of the total collagen concentration in human skin, most of which is localized the mesh-like papillary dermis (Meigel et al. 1977). The remaining 80% of dermal collagen is type I, a heterotrimer molecule composed of two α1(I) chains and one α2(I) chain, which adopt an antiparallel fibril arrangement in the reticular dermis to produce dense fibers (~90 nm in diameter) displaying longitudinal periodic banding with a frequency of approximately 67 nm (Braun-Falco and Rupec 1964), (Fang et al. 2012).
Fig. 2.

Schematic diagram of a collagen fibril illustrating the distinct triple helical structure of the α-chains. Representative portions of three individual α-chains, each with the triple amino acid repeat Gly-X-Y, are shown in the magnified section. Each chain is comprised of a glycine (black), proline (position ‘X,’ red), and hydroxyproline (position ‘Y,’ blue) residue. Hydrogen bonds (dashed black lines) between the N–H of glycine on each chain and the carbonyl of proline on an adjacent chain establish the triple helix
Post-translational modifications of type I collagen include enzyme-mediated intracellular conversion of varying fractions of the lysine (Lys) residues within the α1(I) and α2(I) chains to hydroxylysine (Hyl) via lysyl hydroxylase and extracellular conversion of Lys and Hyl into aldehydes (Lysald or Hylald) via lysyl oxidase (LOX) (Tanzer 1973). These enzyme-mediated modifications are illustrated in Fig. 3. Following secretion of procollagen and its conversion to mature collagen by proteolytic removal of the N– and C–terminal propeptides, the LOX-directed conversion of Lys/Hyl into Lysald/Hylald enables spontaneous, non-reducible inter- and intra-peptide crosslinking that stabilizes dermal collagen and confers resistance to proteolytic cleavage (Herchenhan et al. 2015). LOX is similarly vital in generating crosslinks that prevent excessive elasticity and facilitate matrix deposition within elastin fibers (Liu et al. 2004). Primary elastin sequences (i.e., tropoelastin, the protein form secreted by fibroblasts) contains the requisite Lys residues (K) for crosslink formation within KA– (AAKAAKA) or KP– (PGAGVKPGKGP) type domains that display an alternating pattern with hydrophobic domains rich in glycine, proline, valine, and alanine (Brownaugsburger et al. 1995). The spontaneous reaction between Lys/Hyl and an aldehyde-modified variant, Lysald/Hylald, into an irreversible crosslink domain, lysinonorleucine, is shown in Fig. 4.
Fig. 3.

Enzyme-mediated modifications of collagen molecules. Intracellular processing includes the conversion of lysine into hydroxylysine via lysyl hydroxylase (red), while extracellular lysly oxidase (orange) is required to convert the amine moeity of lysine (or hydroxylysine) into an aldehyde functional group
Fig. 4.

The mechanism for lysinonorleucine crosslink formation between collagen/elastin fibrils. The coupling of protonated Lys (or Hyl) with Lysald (or Hylald) occurs spontaneously following aldehyde-generation via LOX. Reduction of the reversible Schiff base intermediate generates an irreversible lysinonorleucine crosslink junction
The overarching theme in dermal aging is disruption of the balance between degradation and formation of the ECM support structures, collagen and elastin, leading to accumulation of crosslink fragments, loss of dermal elasticity, and impaired dermal fibroblast function. Under normal conditions, elastin displays a very low rate of turnover; the protein structures laid down during fetal development must sustain up to thousands of millions of cycles of stretch and recoil over a lifetime, making them an easy target to accumulate age-related damage (Rosenbloom et al. 1993; Pasquali-Ronchetti and Baccarani-Contri 1997). Similarly, the turnover of collagen is relatively infrequent; on average, the estimated half-life of mature, crosslinked collagen is 15 years in human skin (Verzijl et al. 2000). However, the wound response triggers rapid collagen degradation by matrix metalloproteases (MMPs) to allow migration of immune cells into the dermal compartment (Pilcher et al. 1997; Nissinen and Kahari 2015). This degradation of collagen during the early phase of the wound response is followed by induction of new collagen synthesis by fibroblasts to restore lost collagen in the dermal ECM.
Oxidative stress promotes dermal aging
In contrast to the wound healing response, exposure to solar UV irradiation indirectly initiates keratinocyte and fibroblast synthesis of MMPs without subsequent upregulation of new collagen production, thereby resulting in net collagen loss and ECM fragmentation (Evans et al. 2004; Quan et al. 2013a). Induction of MMPs by UV irradiation is mediated by photochemical reactions that convert absorption of UV energy by cellular macromolecules into reactive oxygen species (ROS), such as superoxide anion (O2−) and hydrogen peroxide (H2O2). Similarly, in the absence of UV irradiation, fibroblasts and keratinocytes continuously produce ROS from molecular oxygen during aerobic respiration and metabolism (Harman 1981; Harman 1992; Poljsak and Milisav 2012). With the passage of time, small amounts of ROS exceed anti-oxidant defenses and drive production of collagen-degrading MMPs.
Regardless of the source, ROS accumulation has deleterious effects on the dermal ECM. Indeed, the oxidants inactivate protein tyrosine phosphatases (PTPs), allowing net phosphorylation of receptor tyrosine kinases (RTKs) and activation of downstream signaling pathways, including 3 families of mitogen activated protein kinases (MAPKs), namely, ERK (extracellular signal-regulated kinase), p38, and JNK (c-Jun N– terminal kinase) (Rittié and Fisher 2002). These MAPKs are essential for the expression of c-Fos and c-Jun, which associate to form transcription factor activator protein 1 (AP-1). AP-1 plays a critical role in transcriptional up-regulation of MMP1, MMP3, and MMP9, (Kim et al. 2010) and overexpression of these MMPs leads to increased fragmentation of the dermal ECM. Specifically, MMP-1 initiates cleavage of type I and type III collagen fibrils at a specific location within their central triple helix, and, once cleaved, the fibrils are further degraded by MMP-3 and MMP-9 (Fisher et al. 1996; Brennan et al. 2003). Oxidant exposure also induces CCN1 (cysteine-rich protein 61) in dermal fibroblasts through AP-1 dependent activation of the CCN1 gene promoter (Qin et al. 2014). CCN1 has been shown to up-regulate MMP-1 via interaction with αVβ3 integrin, and thereby contributes to aberrant dermal collagen homeostasis in human skin (Qin et al. 2013). These ROS-directed signaling pathways are illustraded in Fig. 5.
Fig. 5.
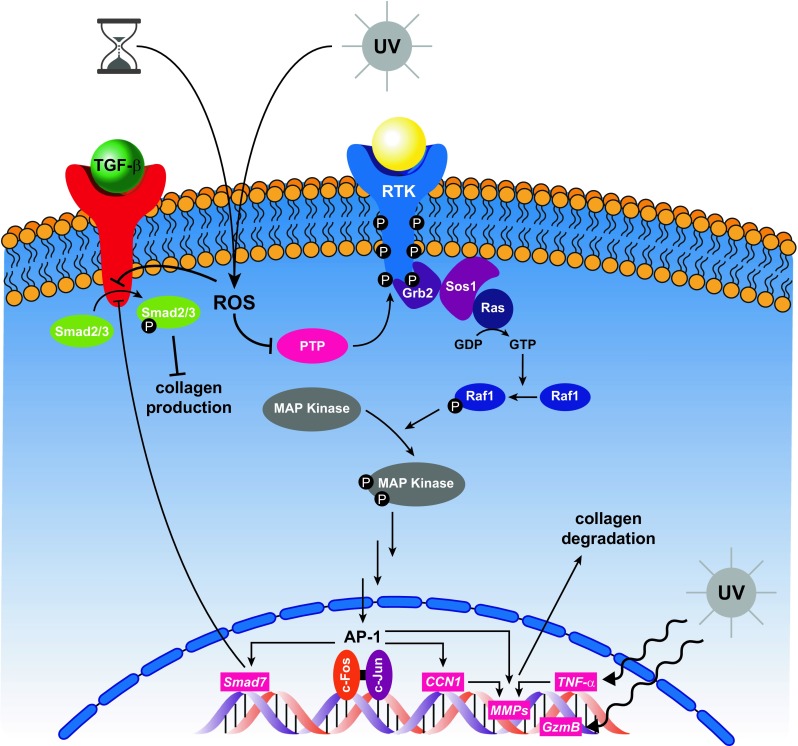
Schematic illustration of the consequences of chronological- and photo-aging on intracellular pathways that regulate collagen homeostasis. The overall impact of time and UV irradiation on dermal fibroblasts is upregulation of MMPs, resulting in collagen degradation and concomitant downregulation of collagen synthesis. ROS (reactive oxygen species), PTP (protein tyrosine phosphatases), RTK (receptor tyrosine kinase), Grb2 (growth factor receptor-bound protein 2), Sos1 (son of sevenless homolog 1), MAPK (mitogen activated protein kinase), AP-1 (activated protein 1), CCN1 (cysteine-rich protein 61), MMP (matrix metalloprotease), TNF- α (tumor necrosis factor alpha), GzmB (granzyme B), TGF-β (transforming growth factor beta)
In addition to activating MMPs via the MAPK pathway, UV irradiation induces transcription of tumor necrosis factor (TNF)-α, a pleiotropic cytokine that upregulates cellular production of MMP-1 and MMP-3 and has been shown to increase collagen degradation in human skin (Bashir et al. 2009; Ågren et al. 2015). Moreover, formation of AP-1 by the MAPK pathway induces Smad7, which inhibits phosphorylation of Smad2/3 by transforming growth factor (TGF)-β type 1 receptor, thereby blocking TGF-β signaling and inhibiting collagen production by dermal fibroblasts (Quan et al. 2005). Lastly, chronic exposure to UV irradiation has been linked with upregulation of granzyme B (GzmB), a serine protease that cleaves decorin. Since decorin binds collagen in the same region as the MMP-1 cleavage site, its presence affords collagen protection against protease degradation (Geng et al. 2006), as illustrated in GzmB knockout mice, which display significantly reduced wrinkle formation following long-term exposure to UVA irradiation (Parkinson et al. 2015). When GzmB is overexpressed, less decorin is available to shield collagen from protease cleavage, giving MMP-1 open access to fragment the crosslinked collagen fibrils.
Dermal cellular changes with age
The collective impact of ECM degradation in aged skin is a shift in fibroblast morphology from flattened, spread, and making contact with numerous intact collagen fibers to collapsed with little cytoplasm and lacking direct association with the surrounding fragmented collagen fibrils (Fisher et al. 2002; Varani et al. 2006). Indeed, network connectivity and cell surface area are reduced by 80% and 75%, respectively, in fibroblasts located within age-associated dermal microenvironments (AADMs) (Varani et al. 2006). The nature of collagen crosslink formation also undergoes age-associated remodeling as the proportion of LOX-derived crosslinks decreases relative to the number of glycation-mediated crosslinks in a process whereby sugar, most notably glucose, is inserted between collagen molecules in lieu of direct coupling of amino acid side chains on adjacent proteins (Szauter et al. 2005; Gautieri et al. 2017). Formation of these intermolecular collagen–sugar–collagen crosslinks is harmful on two accords. On a macromolecular scale, they increase tissue stiffness, and on the molecular scale, they reduce the capacity of collagen for binding hyaluronic acid and GAGs such as decorin (Reihsner et al. 2000; Ahmed et al. 2017).
Secondary to these deleterious effects in network structure, and thus cell-ECM connectivity, fibroblast within the AADM undergo alterations in their secretory protein profile that establish a positive feedback loop that promotes further ECM degradation. Specifically, in vivo analysis has shown that fibroblast within an AADM express significantly higher levels of endogenous oxidants, proteases, and CCNs and lower levels of pro-collagen and hyaluronic acid than fibroblasts in young skin (Longas et al. 1986; Griffiths et al. 1993; Talwar et al. 1995; Fisher et al. 2009; Quan et al. 2011a). Interestingly, the “aged” phenotype can be derived in young fibroblasts (i.e., those harvested from young skin and undergoing ≤10 passages) through serial passages of the cells to the point of replicative senescence or by culturing the cells in a three-dimensional network composed of fragmented collagen (Longas et al. 1986; Griffiths et al. 1993; Talwar et al. 1995; Fisher et al. 2009; Quan et al. 2011a; Quan et al. 2012). In each scenario, MMP-1 levels were significantly elevated, and in the case of replicative senescence, knockdown of CCN1 partially restored collagen homeostasis (Quan et al. 2012).
CCN proteins in human skin aging
CCN1 is an ECM-associated matricellular protein and one of the six members of the CCN family, a group of functionally distinct proteins that share a similar predicted modular secondary structure (Bork 1993; Chen and Lau 2009). Connective tissue growth factor (CCN2), nephroblastoma overexpressed (CCN3), Wnt-induced secreted protein-1 (CCN4), Wnt-induced secreted protein-2 (CCN5), and Wnt-induced secreted protein-3 (CCN6) constitute the remaining five members (Lau and Lam 1999; Leask and Abraham 2003; Perbal 2004). Importantly, CCN proteins have been shown to regulate a diverse range of cellular functions from migration, proliferation, differentiation, and apoptosis to angio- and chondrogenesis, synthesis of ECM proteins, cell-matrix interactions, and cell adhesion (Leask and Abraham 2006). CCN1, like all members of the CCN family, is composed of an N–terminal secretion signal peptide followed by 4 conserved structural domains: the insulin-like growth factor binding protein (IGFBP) domain, the von Willebrand factor type C (VWC) domain, the thrombospondin type 1 (TSP1) domain, and the C–terminal (CT) domain that contains a cysteine knot (Bork 1993).
Evaluation of the molecular mechanisms surrounding CCN1 induction of MMP-1 expression in dermal fibroblasts has indicated that the N–terminal domains (IGFBP, VWC, and TSP1) interact with αVβ3 integrins in a concerted manner; removal of any one of these modular domains renders the protein incapable of upregulating MMP-1, but the CT domain is functionally dispensable (Qin et al. 2013). Additionally, ECM–CCN1 interaction may be a further requirement for functional activity, since signal peptide-deleted, non-secreted CCN1 did not affect MMP-1 expression (Qin et al. 2013). As mentioned previously, fibroblasts within an AADM, as well as those passaged to replicative senescence display elevated levels of CCN1, a finding that is correlated with significant reduction in type I collagen production, elevated levels of MMP-1, and increased collagen lattice fragmentation (Quan et al. 2012). Further investigation into the role of CCN1 in collagen dysregulation has revealed that CCN1 down regulates the TGF-β type II receptor and induces the AP-1 transcription factor, resulting in impaired fibroblast responsiveness to TGF-β signaling and upregulation of MMP-1, respectively (Quan et al. 2006). Since TGF-β signaling is the major driver for production of collagen and other ECM proteins by dermal fibroblasts, its inhibition constitutes a multipronged assault on ECM homeostasis (Quan et al. 2006). Moreover, CCN2, a key downstream effector of the TGF-β pathway is significantly reduced in aged skin (Quan et al. 2010). ROS have also been shown to downregulate CCN2, as well as CCN4 and CCN 5, but their functional roles in aged dermis have yet to be determined (Qin et al. 2014).
The dermal matrisome
The information discussed above points to the novel perspective that alterations to the dermal ECM composition and organization are a major driving force for human skin aging. Recently, the term “matrisome” has been coined to describe the group of heterogeneous ECM components and ECM-associated proteins (Naba et al. 2012; Hynes and Naba 2012). Specifically, the matrisome is composed of two large groups, the core matrisome and the matrisome-associated proteins (Naba et al. 2016). The core matrisome may further be divided into 3 subcategories, collagens (the most abundant proteins), proteoglycans, and ECM glycoproteins, which includes the CCN family of proteins. Matrisome-associated proteins are composed of ECM-bound secreted factors such as TGF-β and cytokines, in addition to the ECM regulators, MMPs and AADMs. Therefore, the biology of human skin aging can be viewed largely as a disorder of the dermal matrisome. Our current knowledge of the impact of aging on the human skin matrisome is very limited; comprehensive understanding of age-related changes in human skin matrisomal proteins, and mechanisms that bring about these alterations, will likely yield important, novel insights into the molecular basis of skin aging and age-related skin diseases.
Outlook: Dermal ECM deterioration and decline of health during aging
An emerging body of evidence suggests that the “aged” phenotype of dermal fibroblasts is a consequence not of their cellular age but of lost connections with the ECM stemming from years of gradual breakdown by MMPs, which is driven by oxidative metabolism and accelerated by oxidative assaults stemming from solar UV irradiation. Central to this hypothesis is the observed ability of fibroblasts harvested from individuals ≥80 years of age to retain their capacity for functional activation. Specifically, injection of space-filling, crosslinked hyaluronic acid into aged skin markedly improves fibroblast spreading with concomitant upregulation of the TGF-β/CCN2 pathway leading to increased collagen production, which evolves into dense bundles of mature collagen with characteristic D-spacing (Quan et al. 2013b). Furthermore, topical treatment with retinol, a metabolite of Vitamin A, increases collagen production and decreases MMP-1 expression in human aged skin (Griffiths et al. 1993; Varani et al. 2000; Quan et al. 2011b).
The phenotypic, cellular, and biochemical alterations linked to fragmentation and disorganization of the dermal ECM may have repercussions well beyond those of cosmetic alterations to the skin. For example, age-associated degradation of dermal crosslinked collagen and elastin, manifests in the skin as fragility and reduced capacity for wound healing (Vandekerkhof et al. 1994). Stability of the ECM modulates the availability of many bioactive molecules bound within. As crosslinked collagen molecules are fragmented, larger quantities of pro-inflammatory mediators may be released. In addition, loss of contact with the dermal ECM due to its fragmentation induces production of pro-inflammatory mediators and MMPs by fibroblasts. This self-perpetuating cycle produces increasingly larger quantities of bioactive molecules that, upon ECM degradation, have the ability to enter circulation for transport throughout the body (Quan et al. 2006; Qin et al. 2014). Given that the surface area of skin is the largest of any organ in humans, potential serum transport of these bioactive molecules raises the intriguing possibility that deleterious age-related alterations in the dermal ECM may contribute to systemic aging. Indeed, a recent report provides evidence in support of a role of age-related decline of skin barrier function in systemic aging in mice (Hu et al. 2017).
Future research should strive to determine the systemic pathological consequences of dermal aging and explore the possibility that preventative or therapeutic interventions that target dermal structural integrity may not only improve skin appearance but also general health during aging.
Acknowledgements
This work was supported by funding from the National Institute of Health (grant R01-AG051849 and RO1-AG054835 to GJF and TQ and T32-AM07197 to MAC – PI: JT Elder).
Abbreviations
- UV
ultraviolet
- ECM
extracellular matrix
- GAG
glycoseaminoglycan
- PG
prostaglandin
- Lys
lysine
- Hyl
hydroxylysine
- LOX
lysyl oxidase
- MMP
matrix metalloprotease
- ROS
reactive oxygen species
- PTP
protein tyrosine phosphatase
- RTK
receptor tyrosine kinase
- MAPK
mitogen activated protein kinase
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun N-terminal kinase
- AP-1
activated protein 1
- CCN1
cysteine-rich protein 61
- TNF- α
tumor necrosis factor alpha
- TGF- β
transforming growth factor beta
- GzmB
granzyme B
- AADM
age-associated dermal microenvironment
- IGFBP
insulin-like growth factor binding protein
- VWC
von Willebrand factor type C
- TSP1
thrombospondin type 1
References
- Ågren MS, Schnabel R, Christensen LH, Mirastschijski U. Tumor necrosis factor-α-accelerated degradation of type I collagen in human skin is associated with elevated matrix metalloproteinase (MMP)-1 and MMP-3 ex vivo. Eur J Cell Biol. 2015;94:12–21. doi: 10.1016/j.ejcb.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed T, Nash A, Clark KEN, Ghibaudo M, de Leeuw NH, Potter A, Stratton R, Birch HL, Casse RE, Bozec L. Combining nano-physical and computational investigations to understand the nature of “aging” in dermal collagen. Int J Nanomedicine. 2017;12:3303–3314. doi: 10.2147/IJN.S121400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir MM, Sharma MR, Werth VP. TNF-α production in the skin. Arch Dermatol Res. 2009;301:87–91. doi: 10.1007/s00403-008-0893-7. [DOI] [PubMed] [Google Scholar]
- Bork P. The modular architecture of a new family of growth-regulators related to connective-tissue growth-factor. FEBS Lett. 1993;327:125–130. doi: 10.1016/0014-5793(93)80155-N. [DOI] [PubMed] [Google Scholar]
- Braun-Falco O, Rupec M. Some observations on dermal collagen fibrils in ultra-thin sections. J Invest Dermatol. 1964;42:15–19. doi: 10.1038/jid.1964.6. [DOI] [PubMed] [Google Scholar]
- Brennan M, Bhatti H, Nerusu KC, Bhagavathula N, Kang SW, Fisher GJ, Varani J, Voorhees JJ. Matrix metalloproteinase-1 is the major collagenolytic enzyme responsible for collagen damage in UV-irradiated human skin. Photochem Photobiol. 2003;78:43–48. doi: 10.1562/0031-8655(2003)078<0043:MMITMC>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Brownaugsburger P, Tisdale C, Broekelmann T, Sloan C, Mecham RP. Identification of an elastin cross-linking domain that joins 3 peptide chains - possible role in nucleated assembly. J Biol Chem. 1995;270:17778–17783. doi: 10.1074/jbc.270.30.17778. [DOI] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Carrino DA, Calabro A, Darr AB, Dours-Zimmermann MT, Sandy JD, Zimmermann DR, Sorrell JM, Hascall VC, Caplan AI. Age-related differences in human skin proteoglycans. Glycobiology. 2011;21:257–268. doi: 10.1093/glycob/cwq162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol. 2009;41:771–783. doi: 10.1016/j.biocel.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culav EM, Clark CH, Merrilees MJ. Connective tissues: matrix composition and its relevance to physical therapy. Phys Ther. 1999;79:308–319. [PubMed] [Google Scholar]
- Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Exposito JY, Valcourt U, Cluzel C, Lethias C. The fibrillar collagen family. Int J Mol Sci. 2010;11:407–426. doi: 10.3390/ijms11020407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M, Goldstein EL, Turner AS, Les CM, Orr BG, Fisher GJ, Welch KB, Rothman ED, Holl MMB. Type I collagen D-spacing in fibril bundles of dermis, tendon, and bone: bridging between nano- and micro-level tissue hierarchy. ACS Nano. 2012;6:9503–9514. doi: 10.1021/nn302483x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher GJ, Datta SC, Talwar HS, Wang ZQ, Varani J, Kang S, Voorhees JJ. Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature. 1996;379:335–339. doi: 10.1038/379335a0. [DOI] [PubMed] [Google Scholar]
- Fisher GJ, Kang SW, Varani J, Bata-Csorgo Z, Wan YS, Datta S, Voorhees JJ. Mechanisms of photoaging and chronological skin aging. Arch Dermatol. 2002;138:1462–1470. doi: 10.1001/archderm.138.11.1462. [DOI] [PubMed] [Google Scholar]
- Fisher GJ, Varani J, Voorhees JJ (2008). Looking older: fibroblast collapse and therapeutic implications. Arch Dermatol 144:666–672 [DOI] [PMC free article] [PubMed]
- Fisher GJ, Quan T, Purohit T, Shao Y, Cho MK, He T, Varani J, Kang S, Voorhees JJ. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am J Pathol. 2009;174:101–114. doi: 10.2353/ajpath.2009.080599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautieri A, Passini FS, Silvan U, Guizar-Sicairos M, Carimati G, Volpi P, Moretti M, Schoenhuber H, Redaelli A, Berli M, Snedeker JG. Advanced glycation end-products: mechanics of aged collagen from molecule to tissue. Matrix Biol. 2017;59:95–108. doi: 10.1016/j.matbio.2016.09.001. [DOI] [PubMed] [Google Scholar]
- Geng Y, McQuillan D, Roughley PJ. Slrp interaction can protect collagen fibrils from cleavage by collagenases. Matrix Biol. 2006;25:484–491. doi: 10.1016/j.matbio.2006.08.259. [DOI] [PubMed] [Google Scholar]
- Gosline J, Lillie M, Carrington E, Guerette P, Ortlepp C, Savage K. Elastic proteins: biological roles and mechanical properties. Philos Trans R Soc Lond Ser B Biol Sci. 2002;357:121–132. doi: 10.1098/rstb.2001.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths CEM, Russman AN, Majmudar G, Singer RS, Hamilton TA, Voorhees JJ. Restoration of collagen formation in photodamaged human skin by tretinoin (retinoic acid) N Engl J Med. 1993;329:530–535. doi: 10.1056/NEJM199308193290803. [DOI] [PubMed] [Google Scholar]
- Harman D. The aging process. Proc Natl Acad Sci U S A. 1981;78:7124–7128. doi: 10.1073/pnas.78.11.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Free-radical theory of aging. Mutat Res. 1992;275:257–266. doi: 10.1016/0921-8734(92)90030-S. [DOI] [PubMed] [Google Scholar]
- Heino J. The collagen family members as cell adhesion proteins. BioEssays. 2007;29:1001–1010. doi: 10.1002/bies.20636. [DOI] [PubMed] [Google Scholar]
- Herchenhan A, Uhlenbrock F, Eliasson P, Weis M, Eyre D, Kadler KE, Magnusson SP, Kjaer M. Lysyl oxidase activity is required for ordered collagen fibrillogenesis by tendon cells. J Biol Chem. 2015;290:16440–16450. doi: 10.1074/jbc.M115.641670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu LZ, Mauro TM, Dang EL, Man G, Zhang J, Lee DL, Wang G, Feingold KR, Elias PM, Man MQ. Epidermal dysfunction leads to an age-associated increase in levels of serum inflammatory cytokines. J Invest Dermatol. 2017;137:1277–1285. doi: 10.1016/j.jid.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO, Naba A. Overview of the matrisome – an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol. 2012;4:a004903. doi: 10.1101/cshperspect.a004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Ryu H-C, Kim J-H. Low-dose UVB irradiation stimulates matrix metalloproteinase-1 expression via a BLT2-linked pathway in HaCaT cells. Exp Mol Med. 2010;42:833–841. doi: 10.3858/emm.2010.42.12.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Turnbull J, Guimond S. Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol. 2011;209:139–151. doi: 10.1530/JOE-10-0377. [DOI] [PubMed] [Google Scholar]
- Lau LF, Lam SCT. The CCN family of angiogenic regulators: the integrin connection. Exp Cell Res. 1999;248:44–57. doi: 10.1006/excr.1999.4456. [DOI] [PubMed] [Google Scholar]
- Leask A, Abraham DJ. The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem Cell Biol. 2003;81:355–363. doi: 10.1139/o03-069. [DOI] [PubMed] [Google Scholar]
- Leask A, Abraham DJ. All in the ccn family: essential matricellular signaling modulators emerge from the bunker. J Cell Sci. 2006;119:4803–4810. doi: 10.1242/jcs.03270. [DOI] [PubMed] [Google Scholar]
- Lee DH, Oh J-H, Chung JH. Glycosaminoglycan and proteoglycan in skin aging. J Dermatol Sci. 2016;83:174–181. doi: 10.1016/j.jdermsci.2016.05.016. [DOI] [PubMed] [Google Scholar]
- Liu XQ, Zhao Y, Gao JG, Pawlyk B, Starcher B, Spencer JA, Yanagisawa H, Zuo J, Li TS. Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nature Genet. 2004;36:178–182. doi: 10.1038/ng1297. [DOI] [PubMed] [Google Scholar]
- Longas MO, Russell CS, He X-Y. Chemical alterations of hyaluronic acid and dermatan sulfate detected in aging human skin by infrared spectroscopy. Biochim Biophys Acta. 1986;884:265–269. doi: 10.1016/0304-4165(86)90172-8. [DOI] [PubMed] [Google Scholar]
- Lovell CR, Smolenski KA, Duance VC, Light ND, Young S, Dyson M. Type I and III collagen content and fibre distribution in normal human skin during ageing. Br J Dermatol. 1987;117:419–428. doi: 10.1111/j.1365-2133.1987.tb04921.x. [DOI] [PubMed] [Google Scholar]
- Meigel WN, Gay S, Weber L. Dermal architecture and collagen type distribution. Arch Dermatol Res. 1977;259:1–10. doi: 10.1007/BF00562732. [DOI] [PubMed] [Google Scholar]
- Mitchell RE. Chronic solar dermatosis: a light and electron microscopic study of the dermis. J Invest Dermatol. 1967;48:203–220. doi: 10.1038/jid.1967.33. [DOI] [PubMed] [Google Scholar]
- Montagna W, Carlisle K. Structural changes in aging human skin. J Invest Dermatol. 1979;73:47–53. doi: 10.1111/1523-1747.ep12532761. [DOI] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. 2012;11:M111.014647. doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO. The extracellular matrix: tools and insights for the "omics" era. Matrix Biol. 2016;49:10–24. doi: 10.1016/j.matbio.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissinen LM, Kahari VM. Collagen turnover in wound repair-a macrophage connection. J Invest Dermatol. 2015;135:2350–2352. doi: 10.1038/jid.2015.246. [DOI] [PubMed] [Google Scholar]
- Parkinson LG, Toro A, Zhao HY, Brown K, Tebbutt SJ, Granville DJ. Granzyme B mediates both direct and indirect cleavage of extracellular matrix in skin after chronic low-dose ultraviolet light irradiation. Aging Cell. 2015;14:67–77. doi: 10.1111/acel.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquali-Ronchetti I, Baccarani-Contri M. Elastic fiber during development and aging. Microsc Res Tech. 1997;38:428–435. doi: 10.1002/(SICI)1097-0029(19970815)38:4<428::AID-JEMT10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Perbal B. CCN proteins: multifunctional signalling regulators. Lancet. 2004;363:62–64. doi: 10.1016/S0140-6736(03)15172-0. [DOI] [PubMed] [Google Scholar]
- Pilcher BK, Dumin JA, Sudbeck BD, Krane SM, Welgus HG, Parks WC. The activity of collagenase-1 is required for keratinocyte migration on a type I collagen matrix. J Cell Biol. 1997;137:1445–1457. doi: 10.1083/jcb.137.6.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poljsak B, Milisav I. The neglected significance of "antioxidative stress". Oxidative Med Cell Longev. 2012;Article ID 480895:12. doi: 10.1155/2012/480895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin ZP, Fisher GJ, Quan TH. Cysteine-rich protein 61 (CCN1) domain-specific stimulation of matrix metalloproteinase-1 expression through alpha V beta 3 integrin in human skin fibroblasts. J Biol Chem. 2013;288:12386–12394. doi: 10.1074/jbc.M112.424358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin ZP, Robichaud P, He TY, Fisher GJ, Voorhees JJ, Quan TH. Oxidant exposure induces cysteine-rich protein 61 (CCN1) via c-jun/ap-1 to reduce collagen expression in human dermal fibroblasts. PLoS One. 2014;9:e115402. doi: 10.1371/journal.pone.0115402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan TH, He TY, Voorhees JJ, Fisher GJ. Ultraviolet irradiation induces Smad7 via induction of transcription factor AP-1 in human skin fibroblasts. J Biol Chem. 2005;280:8079–8085. doi: 10.1074/jbc.M409647200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan TH, He TY, Shao Y, Lin L, Kang SW, Voorhees JJ, Fisher GJ. Elevated cysteine-rich 61 mediates aberrant collagen homeostasis in chronologically aged and photoaged human skin. Am J Pathol. 2006;169:482–490. doi: 10.2353/ajpath.2006.060128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan TH, Shao Y, He TY, Voorhees JJ, Fisher GJ. Reduced expression of connective tissue growth factor (CTGF/CCN2) mediates collagen loss in chronologically aged human skin. J Invest Dermatol. 2010;130:415–424. doi: 10.1038/jid.2009.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan TH, Qin ZP, Robichaud P, Voorhees JJ, Fisher GJ. CCN1 contributes to skin connective tissue aging by inducing age-associated secretory phenotype in human skin dermal fibroblasts. J Cell Commun Signal. 2011;5:201–207. doi: 10.1007/s12079-011-0144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan TH, Qin ZP, Shao Y, Xu YR, Voorhees JJ, Fisher GJ. Retinoids suppress cysteine-rich protein 61 (CCN1), a negative regulator of collagen homeostasis, in skin equivalent cultures and aged human skin in vivo. Exp Dermatol. 2011;20:572–576. doi: 10.1111/j.1600-0625.2011.01278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan T, Qin Z, Voorhees JJ, Fisher GJ. Cysteine-rich protein 61 (CCN1) mediates replicative senescence-associated aberrant collagen homeostasis in human skin fibroblasts. J Cell Biochem. 2012;113:3011–3018. doi: 10.1002/jcb.24179. [DOI] [PubMed] [Google Scholar]
- Quan T, Little E, Quan H, Qin Z, Voorhees JJ, Fisher GJ. Elevated matrix metalloproteinases and collagen fragmentation in photodamaged human skin: impact of altered extracellular matrix microenvironment on dermal fibroblast function. J Invest Dermatol. 2013;133:1362–1366. doi: 10.1038/jid.2012.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan T, Wang F, Shao Y, Rittié L, Xia W, Orringer JS, Voorhees JJ, Fisher GJ. Enhancing structural support of the dermal microenvironment activates fibroblasts, endothelial cells, and keratinocytes in aged human skin in vivo. J Invest Dermatol. 2013;133:658–667. doi: 10.1038/jid.2012.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reihsner R, Melling M, Pfeiler W, Menzel E-J. Alterations of biochemical and two-dimensional biomechanical properties of human skin in diabetes mellitus as compared to effects of in vitro non-enzymatic glycation. Clin Biomech. 2000;15:379–386. doi: 10.1016/S0268-0033(99)00085-6. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Rittié L, Fisher GJ. UV-light-induced signal cascades and skin aging. Ageing Res Rev. 2002;1:705–720. doi: 10.1016/S1568-1637(02)00024-7. [DOI] [PubMed] [Google Scholar]
- Rittié L, Fisher GJ. Natural and sun-induced aging of human skin. Cold Spring Harb Perspect Med. 2015;5:a015370. doi: 10.1101/cshperspect.a015370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom J, Abrams WR, Mecham R. Extracellular matrix 4: the elastic fiber. FASEB J. 1993;7:1208–1218. doi: 10.1096/fasebj.7.13.8405806. [DOI] [PubMed] [Google Scholar]
- Sjerobabski-Masnec I, Situm M. Skin aging. Acta Clin Croat. 2010;49:515–518. [PubMed] [Google Scholar]
- Szauter KM, Cao TY, Boyd CD, Csiszar K. Lysyl oxidase in development, aging and pathologies of the skin. Pathol Biol. 2005;53:448–456. doi: 10.1016/j.patbio.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Talwar HS, Griffiths CEM, Fisher GJ, Hamilton TA, Voorhees JJ. Reduced type I and type III procollagens in photodamaged adult human skin. J Invest Dermatol. 1995;105:285–290. doi: 10.1111/1523-1747.ep12318471. [DOI] [PubMed] [Google Scholar]
- Tanzer ML. Cross-linking of collagen. Science. 1973;180:561–566. doi: 10.1126/science.180.4086.561. [DOI] [PubMed] [Google Scholar]
- Vandekerkhof PCM, Vanbergen B, Spruijt K, Kuiper JP. Age-related-changes in wound-healing. Clin Exp Dermatol. 1994;19:369–374. doi: 10.1111/j.1365-2230.1994.tb02684.x. [DOI] [PubMed] [Google Scholar]
- Varani J, Warner RL, Gharaee-Kermani M, Phan SH, Kang SW, Chung JH, Wang ZQ, Datta SC, Fisher GJ, Voorhees JJ. Vitamin a antagonizes decreased cell growth and elevated collagen-degrading matrix metalloproteinases and stimulates collagen accumulation in naturally aged human skin. J Invest Dermatol. 2000;114:480–486. doi: 10.1046/j.1523-1747.2000.00902.x. [DOI] [PubMed] [Google Scholar]
- Varani J, Dame MK, Rittié L, Fligiel SEG, Kang S, Fisher GJ, Voorhees JJ. Decreased collagen production in chronologically aged skin - roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. 2006;168:1861–1868. doi: 10.2353/ajpath.2006.051302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzijl N, DeGroot J, Thorpe SR, Bank RA. Shaw JN, Lyons TJ, Bijlsma JWJ, Lafeber F, Baynes JW, TeKoppele JM. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275:39027–39031. doi: 10.1074/jbc.M006700200. [DOI] [PubMed] [Google Scholar]
- Wang N, Ingber DE. Control of cytoskeletal mechanics by extracellular matrix, cell shape, and mechanical tension. Biophys J. 1994;66:2181–2189. doi: 10.1016/S0006-3495(94)81014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]