

Abstract
It is widely accepted that energy intake restriction without essential nutrient deficiency delays the onset of aging and extends life span. The mechanism underlying this phenomenon is still unknown though a number of different, nonmutually exclusive explanations have been proposed. In each of these, different facets of physiology play the more significant role in the mechanism of aging retardation. Some examples include the altered lipid composition model, the immune response model and models describing changes in endocrine function. In this paper we propose the hypothesis that metabolic reprogramming is the key event in the mechanism of dietary restriction, and the physiological effects at the cellular, tissue and organismal level may be understood in terms of this initial event.
Dietary restriction (DR) is the most successful intervention tested to date in mammals which greatly extends maximum life span and keeps animals ‘younger longer’ [1-3]. Consequently, any hypothesis about the etiology of aging must reconcile the effects of DR on aging. With increased knowledge of the mechanism of DR, we stand to gain a considerable insight into the process of aging.
We propose that a change in the regulation of energy metabolism in response to DR is the primary step in the retardation of aging (fig. 1). First we describe the evidence in support of metabolic reprogramming, a switch to an altered metabolic state, by DR in mice. Next we consider evidence for metabolic shifts in other model organisms where life span is extended by DR or by genetic manipulation. Then we focus on changes in mitochondrial energy metabolism with age and DR in mammals. Next we will explore the effects of altered mitochondrial function in the context of reactive oxygen species (ROS) generation and oxidative stress. Finally we describe the metabolic and morphological changes in white adipose tissue that we believe are a result of altered mitochondrial function. We propose that the activation of adipose tissue through metabolic reprogramming is critical to the mechanism of DR and that it leads to the changes in the animal physiology that are described in the models indicated above.
Fig. 1.
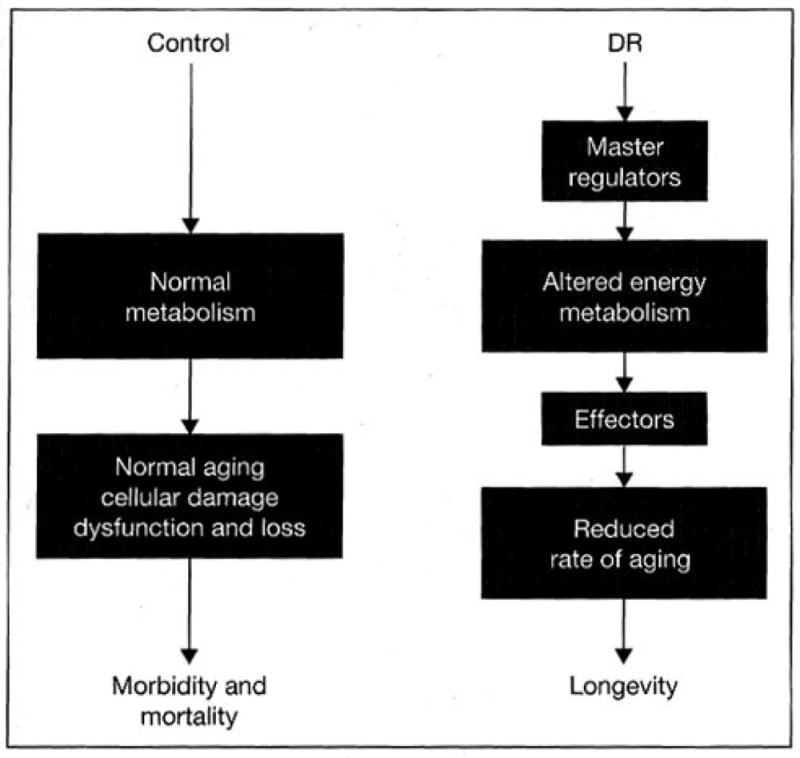
Master regulators respond to DR and induce shifts in metabolism in the restricted organism. These regulators may include the transcriptional coactivator PGC-1α and members of the nuclear receptor family PPAR-α and PPAR-γ. Effectors that respond to the altered metabolic state are involved in tissue-specific changes that ultimately lead to changes at the organismal level, delaying aging and promoting longevity.
Metabolic Reprogramming in Tissues from Dietary-Restricted Animals
The inverse linear relationship between calorie intake and life span in mice [4] suggests that genes central to energy metabolism may be critical in the underlying mechanism of DR in mammals. We have examined transcriptional changes with age and with DR in multiple tissues and find shifts in the expression of genes encoding proteins involved in energy metabolism to be a prominent feature of DR. We propose that these shifts directly contribute to the longevity of the animal. Our studies have primarily focused on postmitotic tissues because these are most vulnerable to the effects of age. Analysis of the transcriptional response to DR in these tissues is likely to reveal clues to the mechanism of aging retardation.
In skeletal muscle, a decrease in metabolic activity with age is suggested through a reduction in the expression of genes involved in energy metabolism [5]. This extends to genes associated with mitochondrial function as well as genes involved in glycolysis and glycogen synthesis, and large reductions in expression were also observed for genes involved in fatty acid biosynthesis. We observed a striking attenuation of these age-related changes in gene expression in skeletal muscle from DR animals compared to age-matched controls. In particular, we observed a transcriptional shift toward increased energy metabolism and increased biosynthesis. The expression of genes involved in glycolysis and gluconeogenesis was increased, as was the expression of transcripts associated with fatty acid metabolism. The increased expression of peroxisome proliferator-activated receptor γ (PPAR- γ) may contribute to the increased insulin sensitivity in skeletal muscle of the DR animals [6].
In the heart, lipid metabolism and fatty acid oxidation (FAO) are the major energy source in adults [7]. In old age, genes involved in lipid transport, lipolysis and FAO are downregulated and genes involved in carbohydrate metabolism are upregulated, resulting in an overall shift in metabolism [8]. These metabolic alterations, which are also observed in pathological heart conditions [9], are completely or partially prevented by DR [8], In addition, we observed a significant change with DR in the expression of key genes that are not affected with age. It is important to emphasize that this latter type of DR effect is distinct from the prevention of age-associated transcriptional changes. Genes that shift in expression with DR but do not change with age may provide clues to the mechanism of aging retardation by DR and may lead to the identification of primary regulators. In the heart, this group includes many nuclear genes encoding components of the electron transport system (ETS) that show a striking and coordinated upregulation with DR [unpubl. data].
In our earlier experiments we sought to characterize the tissue-specific transcriptional changes with age and examine the effect of DR. We identified two groups of genes that are regulated by DR: one group is regulated by age at the transcriptional level and DR either partially or completely compensates for the age-induced changes; the other group is regulated specifically by DR and does not show age-dependent changes in gene expression. Clearly the genes that are regulated specifically by DR and not affected with age are reasonable candidates in the mechanism of life span extension. The potential involvement of genes that are regulated by age in the mechanism of DR is less easily deduced. In analyzing these data it became clear that in order to dissect out the DR-specific transcriptional changes we would be better served looking at young to mid-age mice where the age-related changes in transcription would be less significant. Specifically, we examined the transcriptional changes with fasting, short-term DR (23 days) or long-term DR (9 months) in epididymal white adipose tissue from 10-month-old mice [10]. Here again we identified metabolic reprogramming as a prominent feature. White adipose tissue is remarkably refractory to both fasting and short-term DR but undergoes a dramatic transformation in response to long-term DR. This is in contrast to similar experiments in the liver where many of the DR-induced changes were observed with short-term DR [11].
In white adipose tissue, long-term DR increases the expression of genes involved in the glycolytic pathway, the lipolytic pathway, amino acid metabolism and mitochondrial energy metabolism in young mice [10] suggesting an activation of energy metabolism. Again, these shifts in gene expression are not compensatory in the delay of aging and may be involved in the mechanism of aging retardation. One of the more striking findings was the concerted increase in expression of 26 nuclear genes encoding mitochondrial ETS proteins. We also identified a dramatic decrease in the expression of genes encoding inflammatory molecules (56 genes); these alterations may play an important role in the protection against inflammation derived from white adipose tissue and in life span extension by DR [12].
The transcriptional shifts observed in each of these tissues are indicative of metabolic reprogramming which we believe is a key component of the mechanism of aging retardation by DR (fig. 1). The coordinated increase in expression of genes encoding components of mitochondrial ETS in both heart and adipose tissue is striking. Several aging studies in yeast, worms and flies support a role for metabolic regulation in longevity. We discuss the evidence below, with a particular focus on mitochondrial energy metabolism. Studies in transgenic and wild-type mice further support our hypothesis that shifts in energy metabolism can affect a broad spectrum of phenotypes and support our proposal that metabolic shifts are key elements in the mechanism of DR.
Evidence for Metabolic Reprogramming in Organisms with Extended Life Span
In Saccharomyces cerevisiae, life span extension by DR induces an active regulated response [13] and there is a shift in metabolism in the restricted organisms toward increased respiration [14], The increase in respiration is associated with a decrease in ROS production and this is thought to be indicative of increased mitochondrial uncoupling [15]. Although yeasts are facultative anaerobes, the influence of mitochondrial perturbations on life span under aerobic conditions indicates that manipulation of mitochondrial function directly influences longevity. The retrograde response pathway in yeast provides a mechanism for communication of changes in mitochondrial function to the nucleus [16]. This pathway has been linked to adaptive regulation of metabolism and the stress response [17] and its activation induces the expression of cytoplasmic, mitochondrial and peroxisomal metabolic genes [18] and life span extension [19]. These studies demonstrate that changes in mitochondrial function are transmitted to the nucleus and induce pathways that will provide a compensatory metabolic change. The fact that changes in mitochondrial efficiency can and do exert large-scale changes in gene expression and metabolic regulation supports the idea that a program as complex as life span extension by DR could conceivably be initiated in such a manner. Interestingly, mitochondrial signaling seems to converge with the nutrient-sensing TOR (target of rapamycin) pathway in yeast, where TOR inhibition activates the transcription factors involved in the retrograde response [20]. There is evidence that this cross talk may be conserved in mammalian systems where mitochondrial deficiency stabilizes the interaction between TOR and the inhibitory regulatory associated protein of mTOR (Raptor) protein [21]. These findings demonstrate that there is an open line of communication between mitochondrial efficiency and the nutrient-sensing TOR pathway, allowing for an integration of signaling pathways and a coordinated metabolic response. Inhibition of TOR signaling extends life span in yeast [22,23], worms [24] and flies [25]. Reduced TOR signaling in mice by knockout of the TOR effector S6K1 increases FAO and negatively regulates insulin signaling [26]. It will be interesting to see what role TOR signaling plays in life span extension by DR.
In Caenorhabditis elegans, a systematic RNA interference screen to identify gene alterations that affect life span has uncovered a complex relationship between mitochondrial function and longevity [27], and inhibition of mitochondrial function early in development extends life span in this organism [28], More recent studies in worms have clearly identified a role for metabolic regulators in longevity [29, 30], These studies involve RNA interference knockdown of specific gene products starting from the egg hatching stage. This is a situation where the animal must survive in the absence or depletion of the requisite pathway and while novel factors that influence life span have been discovered, no information about any metabolic compensatory mechanisms in response to this inhibition in the targeted animal has been gleaned. It is possible that the inhibition of mitochondrial oxidative phosphorylation at an early stage induces alternative energy metabolism pathways in these animals, resetting the longevity of the animal [31]. This may explain why inhibiting respiratory chain components in the adult animal does not affect life span [28].
DR does not appear to alter the metabolic rate in C. elegans [32] and genetic analysis indicates that life span extension by DR is independent of DAF-16, the forkhead transcription factor involved in the insulin/IGF pathway [33]. A number of components downstream of DAF-16 have been shown to influence metabolism and life span. These include the DAF-12 nuclear hormone receptor [34] and its coregulator DIN-1 [35], and the DAF-15 regulator of TOR (homologue of mammalian Raptor) [36]. These factors conceivably represent points of convergence of signaling by glucose/insulin, lipophilic factors and amino acid limitation-sensing factors. Such cross talk between pathways would permit regulation of the appropriate coordinated metabolic response to the nutritional status of the animal.
Several studies in Drosophila have also demonstrated that life span may be extended by genes involved in metabolic regulation. Flies with a mutation in INDY (‘I’m not dead yet’), a Krebs cycle intermediate transporter, are long-lived [37], These animals do not show a change in metabolic rate [38], reminiscent of the results of some DR studies [39], although the presumed changes in metabolism have yet to be characterized. Disruption of ecdysone steroid hormone signaling in Drosophila also extends life span [40]. In the absence of this hormone, the ecdysone receptor complex interacts with transcriptional repressors Rpd3 and Sin3 [41]. What is interesting about this, from our point of view, is that reduction of Rpd3 extends life span in flies [42] and reduction in Sin3 causes upregulation of genes involved in the oxidative metabolism of fatty acid to acetyl-CoA and genes involved in mitochondrial oxidative phosphorylation [43]. The similarity between the ecdysone receptor complex and the nuclear hormone complexes in mammalian systems has led to speculation that the mammalian counterparts may also participate in the regulation of aging [44].
As in worms, life span is extended in flies by reduction of insulin signaling [45, 46], Here again the mechanism of life span extension in insulin-signaling-deficient animals is not thought to be the same as that for DR, but the evidence suggests that there are common elements [47], perhaps downstream of the fork-head transcription factor dFOXO. The reduction of blood glucose and enhanced insulin sensitivity in DR mammals hints at the involvement of altered insulin signaling in the mechanism of aging retardation. We do not dispute this but suggest that the changes in insulin signaling and sensitivity are secondary to the metabolic shift in these animals. Studies on long-lived transgenic mouse models indicate that the effects of DR are not fully explained by reduced growth hormone/IGF-1 axis activity [48-51].
Overexpression dFOXO in the fat body of flies extends life span [52, 53], Forkhead transcription factors are downstream of the insulin signaling pathway and in worms regulate metabolism and the stress response [54, 55]. These findings point to the importance of the fat body in whole-body regulation of metabolism and longevity and indicate that secreted factors are involved in the mechanism of life span extension in these transgenic animals. Even though there is evidence to suggest that DR acts independently of FOXO transcription factors [33], factors downstream of FOXO appear to be common to both insulin/IGF longevity pathways and life span extension by DR, in particular members of the nuclear receptor family and possibly factors influencing the TOR nutrient signaling pathway. There is now mounting evidence that signals from white adipose tissue in mammals can influence whole-body metabolism and life span. These quantitative and qualitative changes in adipose tissue may be critically involved in the mechanism of aging retardation by DR (see below).
Taken together, the evidence presented here confirms that life span may be influenced by regulation of metabolism, that mitochondrial efficiency influences the metabolic state and that a communication network exists to coordinate changes in mitochondrial function with regulation of metabolism. These data support the concept that metabolic reprogramming could be an initial event in the mechanism of life span extension by DR and that many genetic manipulations that extend life span may also be viewed in this way.
Mitochondria in Aging and Dietary Restriction
Mitochondria are the key organelle in substrate utilization and energy production. DR directly affects mitochondrial function, increasing the expression of components of the ETS as well as genes involved in fatty acid transport and β-oxidation [8, 10] and there is a clear reduction in the production of ROS [56-61]. DR enhances mitochondrial oxidative capacity in liver and skeletal muscle in rats [62]. ROS are generated continuously as part of normal mitochondrial function [63]. One inbuilt mechanism to combat ROS accumulation is through uncoupling of the mitochondrial membrane potential by proton leak [64], However, studies with mitochondria isolated from liver and skeletal muscle from age-matched control and restricted rats demonstrate that ROS production in mitochondria is reduced even though there is no change in proton leak [65, 66], These studies touch on the role of mitochondrial uncoupling proteins UCP2 and UCP3 and raise questions as to how mitochondrial function is altered by DR. The role of ROS in aging and DR will be discussed in the following section.
In heart, skeletal muscle and white adipose tissue, expression of the mitochondrial uncoupling protein UCP3 is elevated by DR [8, 10, 66]. Studies in mice overexpressing UCP3 support a role for this protein in energy balance and lipid metabolism [67]. Although the physiological role of UCP3 is controversial [68-70], increased UCP3 expression augments FAO and decreases ROS production without uncoupling respiration [71 ] and enhances the capacity for fatty acid transport and FAO in skeletal muscle [72]. These data argue that UCP3 is not merely a mitochondrial uncoupling protein involved in the regulation of the proton leak. Elevated free fatty acids induce UCP3 expression consistent with UCP3 playing a role in the use of free fatty acid as a fuel [73]. In one model [68], UCP3 works with the carnitine palmitoyl transferases, CPT-1 and CPT-2, to cycle fatty acid anions through the mitochondria. The increase in CPT-1 expression in restricted tissues supports a role for UCP3 in fatty acid transport, providing increased capacity for fatty acid metabolism.
Studies in type 2 diabetes have implicated mitochondrial dysfunction in this disorder [74, 75] providing a link between mitochondrial function and whole-body endocrine signaling. Mitochondrial abnormalities lead to neuromuscular disorders known as mitochondrial myopathies and encephalomyopathies [76], as well as heart disease [77]. Mitochondrial function declines with age in humans [78]; however, the extent of the contribution of mitochondrial function to the onset of age-related pathologies like diabetes and heart disease is not yet clear. Tissue-specific disruption of the respiratory chain in mouse hearts causes a switch from fatty acid to glucose metabolism that precedes the inevitable heart failure in these animals [79], This demonstrates that changes in mitochondrial function are sufficient to implement large-scale metabolic changes in mice in vivo.
Recent studies have demonstrated that mice with a mitochondrial mutator phenotype develop several age-associated disorders providing strong support for a model in which mitochondrial function is a determinant of aging [80, 81]. Finally, analyses of individual mice have revealed a positive association between metabolic intensity and life span [82], One issue that complicates studies involving isolated mitochondria is that the experimental setup measures the maximum capacity of the isolated mitochondria in an environment that is experimentally determined, but cannot reveal the actual in vivo differences in mitochondrial function where the intracellular environment may not be equivalent in the organism as a whole. Nevertheless, together these data support a key role for mitochondrial energy metabolism in the control of life span.
Stress, Oxidative Stress and Longevity
Mice [83], rats [84] and monkeys [85] subjected to DR demonstrate decreased body temperatures indicative of altered energy balance. Reduction in oxidative stress is a feature of DR in rodents [57, 86] and may be a direct result of this metabolic reprogramming. DR attenuates the age-associated increase in rates of mitochondrial ROS generation in multiple tissues and reduces the accrual of oxidative damage [58-61, 87, 88], Mitochondrial function is preserved with age in DR animals, and the loss of mitochondrial membrane fluidity is delayed [89] compared to control animals. Reduction of mitochondrial H202 production and oxidative damage to mtDNA in rat gastrocnemius muscle with DR has been described [90] and we have reported that DR in monkeys lowers oxidative damage in skeletal muscle [91]. More recently, the role of ROS has broadened to encompass the control of normal cellular functions (e.g. transcriptional control, signal transduction) and cell death pathways [92], These data reveal a potential role of ROS in aging that is independent of damage induction. It has been proposed that DR, by lowering ROS, attenuates age-associated increases in the binding activities of redox-sensitive transcriptional factors (e.g. HIF-1, NF-κB, AP-1) [93], These factors may be important in the mechanism of aging retardation by DR where reduced ROS production prevents these signaling molecules from implementing the changes we see in the transcriptional profiles of aging animals.
In the course of our analysis of mouse microarray databases, a number of interesting candidates were identified including PPAR coactivator 1a (PGC-1α, and the redox-sensitive transcription factors HIF-1α and NF-κB. Surprisingly, RT-PCR analysis demonstrated that DR has little effect on genes from the sirtuin and forkhead transcription factor families, genes that have been associated with longevity in lower organisms [unpubl. data]. Instead, SIRT1 and FOXO3 are regulated post-transcriptionally by DR [94] [unpubl. data]. SIRT1 has subsequently been associated with DR in cell culture models [94] and activation of SIRT1 is thought to be a key feature in the mechanism of DR, although this has yet to be conclusively shown in mice. FOXO3 is a homologue of the worm longevity factor DAF-16, a component of the insulin signaling pathway. FOXO3 has been linked to cell survival and the stress response in mice and is associated with both SIRT1 and p53 [95, 96].
The regulation and activation of factors associated with the stress response has led us to ask if other elements associated with the stress response might also play a role in DR’s action. We performed a screen to identify kinases activated by DR in the mouse heart. We used tissue from 10-month-old animals to eliminate the influence of age-dependent changes. Interestingly, we identified a number of kinases that are regulated by DR both in terms of total protein levels and degree of modification. Among these are JNK and GSK3β, which are respectively regulated by DR in multiple tissues [unpubl. data].
JNK signaling enhances resistance to oxidative stress and extends life span in worms and flies [97, 98], In mice, JNK plays a role in insulin signaling and obesity [99, 100], and affects insulin resistance in the liver and insulin production in the pancreas [101]. Factors downstream of JNK include FOXO [97, 98], which is required for JNK-dependent life span extension in worms and flies, and PPAR-γ [102], Interestingly, activation of JNK under conditions of oxidative stress is initiated in the mitochondria [103] suggesting that there is direct communication between this organelle and effectors of the stress response. These findings prompt further investigation into a role for JNK in the mechanism of life span extension by DR. It is noted that JNK-dependent life span extension in worms requires DAF-16/FOXO [98] and that life span extension by DR does not [33]; however, constitutive activation of JNK is not a genetic mimic of DR. Regulation of JNK by DR potentially provides a link between stress resistance pathways and longevity, perhaps by influencing factors downstream of FOXO.
The mitogen-activated protein kinase p38 is responsive to numerous stimuli, including environmental stress and cytokine signaling [104]. Activation of p38 increases insulin sensitivity in skeletal muscle in a manner that is independent of contractile induced insulin sensitivity [105], p38 activates the transcriptional coactivator PGC-1α by phosphorylation, thereby regulating the induction of mitochondrial respiration in muscle [106, 107] where PGC-1α plays a role in fiber type switching [108]. The yeast homologue of GSK3β is involved in nutrient sensing and the stress response [109], In mammals, GSK3β is a negative regulator of JNK [110] and is involved in insulin sensitivity in skeletal muscle [111]. Interestingly both JNK and p38 are activated by ROS signaling from the mitochondria [112]. It is unclear how kinases usually associated with the stress response are activated by DR; however, longevity and stress resistance have been linked in most genetic studies performed to date.
The Role of Adipose Tissue in Aging Retardation by Dietary Restriction
Recent studies have highlighted the importance of white adipose tissue in overall metabolic regulation and data from our laboratory and others suggest that the changes in white adipose tissue observed in animals on DR are of particular significance. In mice, long-term DR induces morphological and transcriptional alterations. The mass of epididymal white adipose tissue is reduced by 75%, which appears to be due to a reduction in cell size [10]. DR suppresses the expression of over 50 genes in inflammation and promotes structural remodeling of the cytoskeleton, extracellular matrix and vasculature [12]. It is probable that reductions in systemic inflammatory tone caused by DR may underlie its ability to oppose a broad spectrum of age-associated diseases including cancers and cardiovascular disease. We contend that a key consequence of the metabolic reprogramming induced by DR is the alteration in adipose tissue physiology and metabolism.
Aging is associated with alterations in body fat distribution, obesity and insulin resistance [113, 114]. High levels of leptin are observed with obesity in humans and rodents [115, 116]. DR reduces plasma insulin and leptin levels [117, 118], and opposes the development of age-related insulin and leptin resistance [119, 120]. Transgenic mice lacking the insulin receptor in adipose tissue have reduced adiposity and display a modestly extended longevity compared to DR [121]. These data argue that disruption of IGF signaling in adipose tissue alone is sufficient to extend life span and mirrors the experiments in worms and flies where fat-body-specific knockdown or overexpression of components of the insulin signaling pathway affect life span [52, 53, 122].
The concept of adipose tissue as an endocrine organ has come into focus recently [123]. Elevated serum levels of adipose tissue secretory products have been associated with numerous pathologies including cardiovascular disease, insulin resistance and diabetes. Resistin and adiponectin are adipocyte secretory proteins that negatively and positively regulate insulin sensitivity, respectively [124-126]. Resistin expression was upregulated by DR [10] and adiponectin was not significantly altered. While the significance of the DR-induced changes is not yet clear, the fact that adipocyte-derived signaling molecules are directly affected by DR lends support to the idea that changes in adipose tissue by DR can be transmitted throughout the organism.
DR-induced transcriptional alterations in white adipose tissue included increased expression of genes involved in adipocyte differentiation. Both PPAR-γ and SIRT1 have previously been implicated in this process [127] but it is as yet unclear if either is playing a role in DR-induced changes observed in white adipose tissue. Histological examination of white adipose tissue from mice on DR confirmed the presence of multilocular adipocytes which may represent an intermediate phenotype between white and brown adipocytes [10]. The metabolic shifts observed are consistent with this, including the increased expression of the β3-adrenergic receptor and UCP3. In white adipose tissue, activation of the β-adrenergic receptors leads to mobilization of fat stores and regulates the release of several adipokines [128]. In brown fat, β-adrenergic receptor activation leads to increased expression of the thermogenic uncoupler UCP1 via p38 and the transcriptional coactivator PGC-1α [129]. Ordinarily, PGC-1α protein levels are barely detectable in white adipose tissue but are increased in white adipose tissue from DR animals [unpubl. data]. The increase in PGC-1α may be critical in the activation of adipose tissue by DR. Adenovirus-driven expression of PGC-1α increased the expression of ETS components and FAO enzymes in human adipocytes, and transcription profiling indicated a metabolic activation of the fat cells [130]. Adenovirus-induced hyperleptinemia reduces fat stores in normal rats and increased the capacity for fat oxidation [131]. In these animals, expression of PGC-1α was dramatically increased, as was the expression of gene targets of PGC-1α, and electron microscopy revealed changes in mitochondrial number and morphology. These data describe a striking similarity between the effect of DR and the effect of upregulation of PGC-1α on adipose tissue.
We believe that the changes observed in white adipose tissue are fundamental to the mechanism of life span extension by DR. Age-related changes in adiposity correlate with systemic oxidative stress in humans and mice, and in cultured adipocytes, elevation of fatty acids increased oxidative stress and caused dysregulated production of adipokines [132]. The pharmacological induction of β-oxidation is currently being explored as a treatment for obesity and diabetes [133]; both of these disorders are prevented by DR. We suggest that the DR-induced shift in metabolism in white adipose tissue provides an increased capacity for FAO and permits the mobilization of fat stores without increasing oxidative damage through altered mitochondrial function and the induction of UCP3. The activation of white adipose tissue in this manner influences whole-body physiology in a manner that promotes longevity: in our model we predict that changes in levels of adipokines and other adipose secretory factors systemically influence metabolism, endrocine and immune function and that quantitative and qualitative changes in serum lipids affect nuclear receptor signaling in multiple tissues and influence lipid composition throughout the organism.
PGC-1α is a Candidate Factor in the Mechanism of Aging Retardation by Dietary Restriction
We have presented evidence that DR induces metabolic shifts in multiple tissues and that the influence of metabolism on longevity is conserved across species. Based on these observations and because mitochondrial function has been linked to aging and life span extension by DR [134], we examined our heart microarray data set and looked for regulators of mitochondrial function as potential effectors of DR in mice. PGC-1α is a critical transcriptional coactivator of mitochondrial function that is responsive to changes in energy demands [135, 136]. It induces mitochondrial biogenesis and the expression of genes involved in multiple mitochondrial pathways. Overexpression of PGC-1α stimulates the mitochondrial antioxidant defense system in vascular endothelial cells [137], PGC-1α has been associated with glucose regulation, the insulin signaling pathway and has been implicated in diabetes [138, 139] and obesity [140], conditions that are prevented by DR.
Microarray analysis demonstrates that expression of PGC-1α is increased in hearts from DR mice and that there is a coordinated increase in expression of targets of PGC-1α activity [unpubl. data]. In both heart [8] and adipose tissue [10], we observe a clear trend of upregulation of nuclear genes encoding components of the ETS, many of which are targets of PGC-1α. Expression of PGC-1α is also elevated in epididymal white adipose tissue of young/mid-age DR animals [unpubl. data], indicating that induction of PGC-1α is part of a regulated metabolic response to DR.
Apart from its role in mitochondrial regulation, PGC-1α acts as a transcriptional coactivator of the PPAR nuclear receptor family. The PPARs have been linked to obesity and metabolic regulation and play a central role in the cross talk between glucose and lipid homeostasis [141]. Metabolic integration of FAO, carbohydrate metabolism, energy uncoupling and whole-body insulin sensitivity is attained through the coordinated activity of PPAR-α, PPAR-γ and PPAR-δ in adipose and liver tissues where PGC-1α levels are elevated by DR. Transcriptional analysis of the effect of DR in wild-type and PPAR-α knockout mice has revealed that 19% of the transcriptional changes in the liver are dependent on PPAR-α [ 142] stressing the importance of this nuclear receptor in the mechanism of DR.
Studies in PGC-1α null mice confirm the role of PGC-1α in adaptive energy metabolism [143, 144], In the liver, PGC-1α is associated with FOXO1, one of the mammalian DAF-16/dFOXO homologues, and is involved in hepatic insulin signaling [145]. FOXO1 is involved in PPAR-γ regulation in adipocytes, and there is a complex interplay between these factors in adipocyte differentiation [146], PPAR-γ is also regulated by mTOR, and PPAR-γ activity is dependent on amino acid sufficiency [147], Another factor that appears to provide a connection between PGC-1α, FOXO and PPARs is SIRT1. SIRT1 regulates PPAR-γ in adipocytes [148] and is involved in PGC-1α activation in the insulin signaling pathway in the liver [149, 150]. Control of gene expression in this manner, where transcriptional coactivators and repressor factors are the targets for numerous signaling pathways, provides a strategy that permits the functional integration of multiple distinct biological programs [151].
Conclusion
The central role of energy metabolism in longevity has been a unifying feature in our work and in aging research. Here we propose a model for the mechanism of DR where metabolic reprogramming, the coordinate induction of an altered metabolic state, is an early event in the mechanism of life span extension by DR. We predict that tissue-specific changes in energy metabolism occur through PGC-1α and the PPAR nuclear receptor family. These shifts in energy metabolism induce a move from fat storage to fat mobilization, influence stress pathway signaling and ROS production. Activation of adipose tissue is a critical event in the mechanism of life span extension and leads to altered adipokine and lipid signaling and reduced systemic inflammation. The influence of metabolic reprogramming on endocrine and immune function leads to a reduced rate of aging. It is clear from the data described here that this model is highly simplified. Many if not all of the pathways and factors described here have been shown to be interconnected and are influenced through multiple inputs. The key to our model is the initial event, which is the shift in how energy is generated and how fuel is utilized, and that this occurs through small changes in activity of metabolic regulators to influence the balance of fuel utilization without deregulating nutrient homeostasis in the animal as a whole. In thinking of the mechanism of DR in this way, it is possible to extrapolate and understand some transgenic models of longevity in the context of metabolic regulation and also to see where the effect of nonphysiological genetic manipulations on life span could be misleading.
References
- 1.Weindruch RH, Walford RL. The Retardation of Aging and Disease by Dietary Restriction. Springfield: Thomas; 1988. [Google Scholar]
- 2.Masoro EJ. Caloric restriction. Aging (Milano) 1998;10:173–174. [PubMed] [Google Scholar]
- 3.Masoro EJ. Caloric restriction: a key to understanding and modulating aging. In: Vijg J, editor. Research Profiles in Aging. Vol. 1 Amsterdam: Elsevier; 2002. [Google Scholar]
- 4.Weindruch R, Walford RL, Fligiel S, Guthrie D. The retardation of aging in mice by dietary restriction: longevity, cancer, immunity and lifetime energy intake. J Nutr. 1986;116:641–654. doi: 10.1093/jn/116.4.641. [DOI] [PubMed] [Google Scholar]
- 5.Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- 6.Kintscher U, Law RE. PPARgamma-mediated insulin sensitization: the importance of fat versus muscle. Am J Physiol Endocrinol Metab. 2005;288:E287–E291. doi: 10.1152/ajpendo.00440.2004. [DOI] [PubMed] [Google Scholar]
- 7.Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004;95:568–578. doi: 10.1161/01.RES.0000141774.29937.e3. [DOI] [PubMed] [Google Scholar]
- 8.Lee CK, Allison DB, Brand J, Weindruch R, Prolla TA. Transcriptional profiles associated with aging and middle age-onset caloric restriction in mouse hearts. Proc Natl Acad Sci USA. 2002;99:14988–14993. doi: 10.1073/pnas.232308999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finck BN, Lehman JJ, Barger PM, Kelly DP. Regulatory networks controlling mitochondrial energy production in the developing, hypertrophied, and diabetic heart. Cold Spring Harb Symp Quant Biol. 2002;67:371–382. doi: 10.1101/sqb.2002.67.371. [DOI] [PubMed] [Google Scholar]
- 10.Higami Y, Pugh TD, Page GP, Allison DB, Prolla TA, Weindruch R. Adipose tissue energy metabolism: altered gene expression profile of mice subjected to long-term caloric restriction. FASEB J. 2004;18:415–417. doi: 10.1096/fj.03-0678fje. [DOI] [PubMed] [Google Scholar]
- 11.Cao SX, Dhahbi JM, Mote PL, Spindler SR. Genomic profiling of short- and long-term caloric restriction effects in the liver of aging mice. Proc Natl Acad Sci USA. 2001;98:10630–10635. doi: 10.1073/pnas.191313598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higami Y, Barger JL, Page GP, Allison DB, Smith SR, Prolla TA, Weindruch R. Energy restriction lowers the expression of genes linked to inflammation, the cytoskeleton, the extracellular matrix, and angiogenesis in mouse adipose tissue. J Nutr. 2006;136:343–352. doi: 10.1093/jn/136.2.343. [DOI] [PubMed] [Google Scholar]
- 13.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- 15.Barros MH, Bandy B, Tahara EB, Kowaltowski AJ. Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae. J Biol Chem. 2004;279:49883–49888. doi: 10.1074/jbc.M408918200. [DOI] [PubMed] [Google Scholar]
- 16.Jazwinski SM. The retrograde response links metabolism with stress responses, chromatin-dependent gene activation, and genome stability in yeast aging. Gene. 2005;354:22–27. doi: 10.1016/j.gene.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 17.Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell. 2004;14:1–15. doi: 10.1016/s1097-2765(04)00179-0. [DOI] [PubMed] [Google Scholar]
- 18.Epstein CB, Waddle JA, Hale WT, Dave V, Thornton J, Macatee TL, Garner HR, Butow RA. Genome-wide responses to mitochondrial dysfunction. Mol Biol Cell. 2001;12:297–308. doi: 10.1091/mbc.12.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jazwinski SM. Metabolic control and gene dysrcgulation in yeast aging. Ann NY Acad Sci. 2000;908:21–30. doi: 10.1111/j.1749-6632.2000.tb06632.x. [DOI] [PubMed] [Google Scholar]
- 20.Komeili A, Wedaman KP, O’Shea EK, Powers T. Mechanism of metabolic control: target of rapamycin signaling links nitrogen quality to the activity of the Rtg1 and Rtg3 transcription factors. J Cell Biol. 2000;151:863–878. doi: 10.1083/jcb.151.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 22.Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- 23.Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–184. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- 25.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in DrosophilaM by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 27.Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screcn identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 28.Dillin A, Hsu AL, Arantes-Olivcira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- 29.Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, Lee SS. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005;19:1544–1555. doi: 10.1101/gad.1308205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen M, Hsu AL, Dillin A, Kenyon C. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screcn. PLos Genet. 2005;1:e17. doi: 10.1371/journal.pgen.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rea S, Johnson TE. A metabolic model for life span determination in Caenorhabditis elegans. Dev Cell. 2003;5:197–203. doi: 10.1016/s1534-5807(03)00242-9. [DOI] [PubMed] [Google Scholar]
- 32.Houthoofd K, Braeckman BP, Lenaerts I, Brys K, De Vreese A, Van Eygen S, Vanfleteren JR. No reduction of metabolic rate in food restricted Caenorhabditis elegans. Exp Gerontol. 2002;37:1359–1369. doi: 10.1016/s0531-5565(02)00172-9. [DOI] [PubMed] [Google Scholar]
- 33.Houthoofd K, Braeckman BP, Johnson TE, Vanfleteren JR. Life extension via dietary restriction is independent of the Ins/IGF-1 signalling pathway in Caenorhabditis elegans. Exp Gerontol. 2003;38:947–954. doi: 10.1016/s0531-5565(03)00161-x. [DOI] [PubMed] [Google Scholar]
- 34.Gerisch B, Weitzel C, Kober-Eisermann C, Rottiers V, Antebi A. A hormonal signaling pathway influencing C. elegans metabolism, reproductive development, and life span. Dev Cell. 2001;1:841–851. doi: 10.1016/s1534-5807(01)00085-5. [DOI] [PubMed] [Google Scholar]
- 35.Ludewig AH, Kober-Eisermann C, Weitzel C, Bethke A, Neubert K, Gerisch B, Hutter H, Antebi A. A novel nuclear receptor/coregulator complex controls C. elegans lipid metabolism, larval development, and aging. Genes Dev. 2004;18:2120–2133. doi: 10.1101/gad.312604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- 37.Rogina B, Reenan RA, Nilsen SP, Helfand SL. Extended life-span conferred by cotransporter gene mutations in Drosophila. Science. 2000;290:2137–2140. doi: 10.1126/science.290.5499.2137. [DOI] [PubMed] [Google Scholar]
- 38.Marden JH, Rogina B, Montooth KL, Helfand SL. Conditional tradeoffs between aging and organismal performance of Indy long-lived mutant flies. Proc Natl Acad Sci USA. 2003;100:3369–3373. doi: 10.1073/pnas.0634985100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramsey JJ, Harper ME, Weindruch R. Restriction of energy intake, energy expenditure, and aging. Free Radic Biol Med. 2000;29:946–968. doi: 10.1016/s0891-5849(00)00417-2. [DOI] [PubMed] [Google Scholar]
- 40.Simon AF, Shih C, Mack A, Benzer S. Steroid control of longevity in Drosophila melanogaster. Science. 2003;299:1407–1410. doi: 10.1126/science.1080539. [DOI] [PubMed] [Google Scholar]
- 41.Tsai CC, Kao HY, Yao TP, McKeown M, Evans RM. SMRTER, a Drosophila nuclear receptor coregulator, reveals that EcR-mcdiated repression is critical for development. Mol Cell. 1999;4:175–186. doi: 10.1016/s1097-2765(00)80365-2. [DOI] [PubMed] [Google Scholar]
- 42.Rogina B, Helfand SL, Frankel S. Longevity regulation by Drosophila Rpd3 deacetylase and caloric restriction. Science. 2002;298:1745. doi: 10.1126/science.1078986. [DOI] [PubMed] [Google Scholar]
- 43.Pile LA, Spellman PT, Katzenberger RJ, Wasserman DA. The SIN3 deacetylase complex represses genes encoding mitochondrial proteins: implications for the regulation of energy metabolism. J Biol Chem. 2003;278:37840–37848. doi: 10.1074/jbc.M305996200. [DOI] [PubMed] [Google Scholar]
- 44.Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003;299:1346–1351. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- 45.Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- 46.Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292:107–110. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- 47.Clancy DJ, Gems D, Hafen E, Leevers SJ, Partridge L. Dietary restriction in long-lived dwarf flies. Science. 2002;296:319. doi: 10.1126/science.1069366. [DOI] [PubMed] [Google Scholar]
- 48.Shimokawa I, Higami Y, Tsuchiya T, Otani H, Komatsu T, Chiba T, Yamaza H. Life span extension by reduction of the growth hormone-insulin-like growth factor-1 axis: relation to caloric restriction. Faseb J. 2003;17:1108–1109. doi: 10.1096/fj.02-0819fje. [DOI] [PubMed] [Google Scholar]
- 49.Bartke A, Wright JC, Mattison JA, Ingram DK, Miller RA, Roth GS. Extending the lifespan of long-lived micc. Nature. 2001;414:412. doi: 10.1038/35106646. [DOI] [PubMed] [Google Scholar]
- 50.Masternak MM, Al-Regaiey K, Bonkowski MS, Panici J, Sun L, Wang J, Przybylski GK, Bartke A. Divergent effects of caloric restriction on gene expression in normal and long-lived mice. J Gerontol A Biol Sci Med Sci. 2004;59:784–788. doi: 10.1093/gerona/59.8.b784. [DOI] [PubMed] [Google Scholar]
- 51.Masternak MM, Al-Regaiey KA, Del Rosario Lim MM, Jimenez-Ortega V, Panici JA, Bonkowski MS, Bartke A. Effects of caloric restriction on insulin pathway gene expression in the skeletal muscle and liver of normal and long-lived GHR-KO mice. Exp Gerontol. 2005;40:679–684. doi: 10.1016/j.exger.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 52.Hwangbo DS, Gersham B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 2004;429:562–566. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- 53.Giannakou ME, Goss M, Junger MA, Hafen E, Leevers SJ, Partridge L. Long-lived Drosophila with overexpressed dFOXO in adult fat body. Science. 2004;305:361. doi: 10.1126/science.1098219. [DOI] [PubMed] [Google Scholar]
- 54.Murphy CT, McCarroll SA, Bargmann Cl, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 55.Lee SS, Kennedy S, Tolonen AC, Ruvkun G. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 2003;300:644–647. doi: 10.1126/science.1083614. [DOI] [PubMed] [Google Scholar]
- 56.Masoro EJ, Shimokawa I, Yu BP. Retardation of the aging processes in rats by food restriction. Ann NY Acad Sci. 1991;621:337–352. doi: 10.1111/j.1749-6632.1991.tb16990.x. [DOI] [PubMed] [Google Scholar]
- 57.Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu BP, Chen JJ, Kang CM, Choe M, Maeng YS, Kristal BS. Mitochondrial aging and lipoperoxidative products. Ann NY Acad Sci. 1996;786:44–56. doi: 10.1111/j.1749-6632.1996.tb39050.x. [DOI] [PubMed] [Google Scholar]
- 59.Lass A, Sohal BH, Weindruch R, Forster MJ, Sohal RS. Caloric restriction prevents age-associated accrual of oxidative damage to mouse skeletal muscle mitochondria. Free Radic Biol Med. 1998;25:1089–1097. doi: 10.1016/s0891-5849(98)00144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Merry BJ. Calorie restriction and age-related oxidative stress. Ann NY Acad Sci. 2000;908:180–198. doi: 10.1111/j.1749-6632.2000.tb06646.x. [DOI] [PubMed] [Google Scholar]
- 61.Nagai M, Takahashi R, Goto S. Dietary restriction initiated late in life can reduce mitochondrial protein carbonyls in rat livers: Western blot studies. Biogerontology. 2000;1:321–328. doi: 10.1023/a:1026590819033. [DOI] [PubMed] [Google Scholar]
- 62.Barazzoni R, Zanetti M, Bosutti A, Biolo G, Vitali-Serdoz L, Stebel M, Guarnieri G. Moderate caloric restriction, but not physiological hyperleptinemia per se, enhances mitochondrial oxidative capacity in rat liver and skeletal muscle-tissue-specific impact on tissue triglyceride content and AKT activation. Endocrinology. 2005;146:2098–2106. doi: 10.1210/en.2004-1396. [DOI] [PubMed] [Google Scholar]
- 63.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 64.Brand MD. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp Gerontol. 2000;35:811–820. doi: 10.1016/s0531-5565(00)00135-2. [DOI] [PubMed] [Google Scholar]
- 65.Hagopian K, Harper ME, Ram JJ, Humble SJ, Weindruch R, Ramsey JJ. Long-term calorie restriction reduces proton leak and hydrogen peroxide production in liver mitochondria. Am J Physiol Endocrinol Metab. 2005;288:E674–E684. doi: 10.1152/ajpendo.00382.2004. [DOI] [PubMed] [Google Scholar]
- 66.Bevilacqua L, Ramsey JJ, Hagopian K, Weindruch R, Harper ME. Long-term caloric restriction increases UCP3 content but decreases proton leak and reactive oxygen species production in rat skeletal muscle mitochondria. Am J Physiol Endocrinol Metab. 2005;289:E429–E438. doi: 10.1152/ajpendo.00435.2004. [DOI] [PubMed] [Google Scholar]
- 67.Clapham JC, Arch JR, Chapman H, Haynes A, Lister C, Moore GB, Piercy V, Carter SA, Lehner I, Smith SA, Beeley LJ, Godden RJ, Herrity N, Skehel M, Changani KK, Hockings PD, Reid DG, Squires SM, Hatcher J, Trail B, Latcham J, Rastan S, Harper AJ, Cadenas S, Buckingham JA, Brand MD, Abuin A. Mice overexpressing human uncoupling protein-3 in skeletal muscle are hyperphagic and lean. Nature. 2000;406:415–418. doi: 10.1038/35019082. [DOI] [PubMed] [Google Scholar]
- 68.Himms-Hagen J, Harper ME. Physiological role of UCP3 may be export of fatty acids from mitochondria when fatty acid oxidation predominates: a hypothesis. Exp Biol Med (Maywood) 2001;226:78–84. doi: 10.1177/153537020122600204. [DOI] [PubMed] [Google Scholar]
- 69.Schrauwen P, Hesselink MK. The role of uncoupling protein 3 in fatty acid metabolism: protection against lipotoxicity? Proc Nutr Soc. 2004;63:287–292. doi: 10.1079/PNS2003336. [DOI] [PubMed] [Google Scholar]
- 70.Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005;2:85–93. doi: 10.1016/j.cmet.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 71.MacLellan JD, Gerrits MF, Gowing A, Smith PJ, Wheeler MB, Harper ME. Physiological increases in uncoupling protein 3 augment fatty acid oxidation and decrease reactive oxygen species production without uncoupling respiration in muscle cells. Diabetes. 2005;54:2343–2350. doi: 10.2337/diabetes.54.8.2343. [DOI] [PubMed] [Google Scholar]
- 72.Bezaire V, Spriet LL, Campbell S, Sabet N, Gerrits M, Bonen A, Harper ME. Constitutive UCP3 overexpression at physiological levels increases mouse skeletal muscle capacity for fatty acid transport and oxidation. Faseb J. 2005;19:977–979. doi: 10.1096/fj.04-2765fje. [DOI] [PubMed] [Google Scholar]
- 73.Weigle DS, Selfridge LE, Schwartz MW, Seeley RJ, Cummings DE, Havel PJ, Kuijper JL, Beltran del Rio H. Elevated free fatty acids induce uncoupling protein 3 expression in muscle: a potential explanation for the effect of fasting. Diabetes. 1998;47:298–302. doi: 10.2337/diab.47.2.298. [DOI] [PubMed] [Google Scholar]
- 74.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 76.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rosenberg P. Mitochondrial dysfunction and heart disease. Mitochondrion. 2004;4:621–628. doi: 10.1016/j.mito.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 78.Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci USA. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hansson A, Hance N, Dufour E, Rantanen A, Hultenby K, Clayton DA, Wibom R, Larsson NG. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc Natl Acad Sci USA. 2004;101:3136–3141. doi: 10.1073/pnas.0308710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 81.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 82.Speakman JR, Talbot DA, Selman C, Snart S, McLaren JS, Redman P, Krol E, Jackson DM, Johnson MS, Brand MD. Uncoupled and surviving: individual mice with high metabolism have greater mitochondrial uncoupling and live longer. Aging Cell. 2004;3:87–95. doi: 10.1111/j.1474-9728.2004.00097.x. [DOI] [PubMed] [Google Scholar]
- 83.Koizumi A, Tsukada M, Wada Y, Masuda H, Weindruch R. Mitotic activity in mice is suppressed by energy restriction-induced torpor. J Nutr. 1992;122:1446–1453. doi: 10.1093/jn/122.7.1446. [DOI] [PubMed] [Google Scholar]
- 84.Duffy PH, Feuers R, Nakamura KD, Leakey J, Hart RW. Effect of chronic caloric restriction on the synchronization of various physiological measures in old female Fischer 344 rats. Chronobiol Int. 1990;7:113–124. doi: 10.3109/07420529009056963. [DOI] [PubMed] [Google Scholar]
- 85.Lane MA, Baer DJ, Rumpler WV, Weindruch R, Ingram DK, Tilmont EM, Cutler RG, Roth GS. Calorie restriction lowers body temperature in rhesus monkeys, consistent with a postulated anti-aging mechanism in rodents. Proc Natl Acad Sci USA. 1996;93:4159–4164. doi: 10.1073/pnas.93.9.4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Merry BJ. Oxidative stress and mitochondrial function with aging – The effects of calorie restriction. Aging Cell. 2004;3:7–12. doi: 10.1046/j.1474-9728.2003.00074.x. [DOI] [PubMed] [Google Scholar]
- 87.Sohal RS, Agarwal S, Candas M, Forster MJ, Lai H. Effect of age and caloric restriction on DNA oxidative damage in different tissues of C57BL/6 mice. Mech Ageing Dev. 1994;76:215–224. doi: 10.1016/0047-6374(94)91595-4. [DOI] [PubMed] [Google Scholar]
- 88.Sohal RS, Ku HH, Agarwal S, Forster MJ, Lai H. Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech Ageing Dev. 1994;74:121–133. doi: 10.1016/0047-6374(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 89.Chen JJ, Yu BP. Alterations in mitochondrial membrane fluidity by lipid peroxidation products. Free Radic Biol Med. 1994;17:411–418. doi: 10.1016/0891-5849(94)90167-8. [DOI] [PubMed] [Google Scholar]
- 90.Drew B, Phaneuf S, Dirks A, Selman C, Gredilla R, Lezza A, Barja G, Leeuwenburgh C. Effects of aging and caloric restriction on mitochondrial energy production in gastrocnemius muscle and heart. Am J Physiol Regul Integr Comp Physiol. 2003;284:R474–R480. doi: 10.1152/ajpregu.00455.2002. [DOI] [PubMed] [Google Scholar]
- 91.Zainal TA, Oberley TD, Allison DB, Szweda LI, Weindruch R. Caloric restriction of rhesus monkeys lowers oxidative damage in skeletal muscle. Faseb J. 2000;14:1825–1836. doi: 10.1096/fj.99-0881com. [DOI] [PubMed] [Google Scholar]
- 92.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 93.Kim HJ, Jung KJ, Yu BP, Cho CG, Choi JS, Chung HY. Modulation of redox-sensitive transcription factors by calorie restriction during aging. Mech Ageing Dev. 2002;123:1589–1595. doi: 10.1016/s0047-6374(02)00094-5. [DOI] [PubMed] [Google Scholar]
- 94.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 95.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 96.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacctylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 97.Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115–125. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 98.Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis RJ, Tissenbaum HA. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc Natl Acad Sci USA. 2005;102:4494–4499. doi: 10.1073/pnas.0500749102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 100.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 101.Kaneto H, Nakatani Y, Kawamori D, Miyatsuka T, Matsuoka TA, Matsuhisa M, Yamasaki Y. Role of oxidative stress, endoplasmic reticulum stress, and c-Jun N-terminal kinase in pancreatic beta-cell dysfunction and insulin resistance. Int J Biochem Cell Biol. 2005;37:1595–1608. doi: 10.1016/j.biocel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 102.Park KS, Lee RD, Kang SK, Han SY, Park KL, Yang KH, Song YS, Park HJ, Lee YM, Yun YP, Oh KW, Kim DJ, Yun YW, Hwang SJ, Lee SE, Hong JT. Neuronal differentiation of embryonic mid-brain cells by upregulation of peroxisome proliferator-activated receptor-gamma via the JNK-dependent pathway. Exp Cell Res. 2004;297:424–433. doi: 10.1016/j.yexcr.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 103.Dougherty CJ, Kubasiak LA, Frazier DP, Li H, Xiong WC, Bishopric NH, Webster KA. Mitochondrial signals initiate the activation of c-Jun N-terminal kinase (JNK) by hypoxia-reoxygenation. Faseb J. 2004;18:1060–1070. doi: 10.1096/fj.04-1505com. [DOI] [PubMed] [Google Scholar]
- 104.Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969–1978. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Geiger PC, Wright DC, Han DH, Holloszy JO. Activation of p38 MAP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol Metab. 2005;288:E782–E788. doi: 10.1152/ajpendo.00477.2004. [DOI] [PubMed] [Google Scholar]
- 106.Barger PM, Browning AC, Garner AN, Kelly DP. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: a potential role in the cardiac metabolic stress response. J Biol Chem. 2001;276:44495–44501. doi: 10.1074/jbc.M105945200. [DOI] [PubMed] [Google Scholar]
- 107.Fan M, Rhee J, St-Pierre J, Handschin C, Puigserver P, Lin J, Jaeger S, Erdjument-Bromage H, Tempst P, Spiegelman BM. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: modulation by p38 MAPK. Genes Dev. 2004;18:278–289. doi: 10.1101/gad.1152204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 109.Hirata Y, Andoh T, Asahara T, Kikuchi A. Yeast glycogen synthase kinase-3 activates Msn2p-dependent transcription of stress responsive genes. Mol Biol Cell. 2003;14:302–312. doi: 10.1091/mbc.E02-05-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu S, Yu S, Hasegawa Y, Lapushin R, Xu HJ, Woodgett JR, Mills GB, Fang X. Glycogen synthase kinase 3beta is a negative regulator of growth factor-induced activation of the c-Jun N-terminal kinase. J Biol Chem. 2004;279:51075–51081. doi: 10.1074/jbc.M408607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dokken BB, Sloniger JA, Henriksen EJ. Acute selective glycogen synthase kinase-3 inhibition enhances insulin signaling in prediabetic insulin-resistant rat skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E1188–E1194. doi: 10.1152/ajpendo.00547.2004. [DOI] [PubMed] [Google Scholar]
- 112.Horbinski C, Chu CT. Kinase signaling cascades in the mitochondrion: a matter of life or death. Free Radic Biol Med. 2005;38:2–11. doi: 10.1016/j.freeradbiomed.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 113.Enzi G, Gasparo M, Biondetti PR, Fiore D, Semisa M, Zurlo F. Subcutaneous and visceral fat distribution according to sex, age, and overweight, evaluated by computed tomography. Am J Clin Nutr. 1986;44:739–746. doi: 10.1093/ajcn/44.6.739. [DOI] [PubMed] [Google Scholar]
- 114.Fraze E, Chiou YA, Chen YD, Reaven GM. Age-related changes in postprandial plasma glucose, insulin, and free fatty acid concentrations in nondiabetic individuals. J Am Geriatr Soc. 1987;35:224–228. doi: 10.1111/j.1532-5415.1987.tb02313.x. [DOI] [PubMed] [Google Scholar]
- 115.Ma XH, Muzumdar R, Yang XM, Gabriely I, Berger R, Barzilai N. Aging is associated with resistance to effects of leptin on fat distribution and insulin action. J Gerontol A Biol Sci Med Sci. 2002;57:B225–B231. doi: 10.1093/gerona/57.6.b225. [DOI] [PubMed] [Google Scholar]
- 116.Wang ZW, Pan WT, Lee Y, Kakuma T, Zhou YT, Unger RH. The role of leptin resistance in the lipid abnormalities of aging. Faseb J. 2001;15:108–114. doi: 10.1096/fj.00-0310com. [DOI] [PubMed] [Google Scholar]
- 117.Masoro EJ. Caloric restriction and aging: an update. Exp Gerontol. 2000;35:299–305. doi: 10.1016/s0531-5565(00)00084-x. [DOI] [PubMed] [Google Scholar]
- 118.Shimokawa I, Higami Y. A role for leptin in the antiaging action of dietary restriction: a hypothesis. Aging (Milano) 1999;11:380–382. doi: 10.1007/BF03339816. [DOI] [PubMed] [Google Scholar]
- 119.Barzilai N, Banerjee S, Hawkins M, Chen W, Rossetti L. Caloric restriction reverses hepatic insulin resistance in aging rats by decreasing visceral fat. J Clin Invest. 1998;101:1353–1361. doi: 10.1172/JCI485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fernandez-Galaz C, Fernandez-Agullo T, Perez C, Peralta S, Arribas C, Andres A, Carrascosa JM, Ros M. Long-term food restriction prevents ageing-associated central leptin resistance in Wistar rats. Diabetologia. 2002;45:997–1003. doi: 10.1007/s00125-002-0851-4. [DOI] [PubMed] [Google Scholar]
- 121.Barger JL, Walford RL, Weindruch R. The retardation of aging by caloric restriction: its significance in the transgenic era. Exp Gerontol. 2003;38:1343–1351. doi: 10.1016/j.exger.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 122.Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003;115:489–502. doi: 10.1016/s0092-8674(03)00889-4. [DOI] [PubMed] [Google Scholar]
- 123.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- 124.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 125.Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab. 2002;13:84–89. doi: 10.1016/s1043-2760(01)00524-0. [DOI] [PubMed] [Google Scholar]
- 126.Mora S, Pessin JE. An adipocentric view of signaling and intracellular trafficking. Diabetes Metab Res Rev. 2002;18:345–356. doi: 10.1002/dmrr.321. [DOI] [PubMed] [Google Scholar]
- 127.Picard F, Guarente L. Molecular links between aging and adipose tissue. Int J Obes (Lond) 2005;29(suppl 1):S36–S39. doi: 10.1038/sj.ijo.0802912. [DOI] [PubMed] [Google Scholar]
- 128.Collins S, Cao W, Robidoux J. Learning new tricks from old dogs: beta-adrenergic receptors teach new lessons on firing up adipose tissue metabolism. Mol Endocrinol. 2004;18:2123–2131. doi: 10.1210/me.2004-0193. [DOI] [PubMed] [Google Scholar]
- 129.Cao W, Daniel KW, Robidoux J, Puigserver P, Medvedev AV, Bai X, Floering LM, Spiegelman BM, Collins S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol Cell Biol. 2004;24:3057–3067. doi: 10.1128/MCB.24.7.3057-3067.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tiraby C, Tavernier G, Lefort C, Larrouy D, Bouillaud F, Ricquier D, Langin D. Acquirement of brown fat cell features by human white adipocytes. J Biol Chem. 2003;278:33370–33376. doi: 10.1074/jbc.M305235200. [DOI] [PubMed] [Google Scholar]
- 131.Orci L, Cook WS, Ravazzola M, Wang MY, Park BH, Montesano R, Unger RH. Rapid transformation of white adipocytes into fat-oxidizing machines. Proc Natl Acad Sci USA. 2004;101:2058–2063. doi: 10.1073/pnas.0308258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bogacka I, Ukropcova B, McNeil M, Gimble JM, Smith SR. Structural and functional consequences of mitochondrial biogenesis in human adipocytes in vitro. J Clin Endocrinol Metab. 2005;90:6650–6656. doi: 10.1210/jc.2005-1024. [DOI] [PubMed] [Google Scholar]
- 134.Merry BJ. Molecular mechanisms linking calorie restriction and longevity. Int J Biochem Cell Biol. 2002;34:1340–1354. doi: 10.1016/s1357-2725(02)00038-9. [DOI] [PubMed] [Google Scholar]
- 135.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor alpha (PPARalpha) signaling in the gene regulatory control of energy metabolism in the normal and diseased heart. J Mol Cell Cardiol. 2002;34:1249–1257. doi: 10.1006/jmcc.2002.2061. [DOI] [PubMed] [Google Scholar]
- 136.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 137.Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005;66:562–573. doi: 10.1016/j.cardiores.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 138.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 139.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Semple RK, Crowley VC, Sewter CP, Laudes M, Christodoulides C, Considine RV, Vidal-Puig A, O’Rahilly S. Expression of the thermogenic nuclear hormone receptor coactivator PGC-1alpha is reduced in the adipose tissue of morbidly obese subjects. Int J Obes Relat Metab Disord. 2004;28:176–179. doi: 10.1038/sj.ijo.0802482. [DOI] [PubMed] [Google Scholar]
- 141.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 142.Corton JC, Apte U, Anderson SP, Limaye P, Yoon L, Latendresse J, Dunn C, Everitt JI, Voss KA, Swanson C, Kimbrough C, Wong JS, Gill SS, Chandraratna RA, Kwak MK, Kensler TW, Stulnig TM, Steffensen KR, Gustafsson JA, Mehendale HM. Mimetics of caloric restriction include agonists of lipid-activated nuclcar receptors. J Biol Chem. 2004;279:46204–46212. doi: 10.1074/jbc.M406739200. [DOI] [PubMed] [Google Scholar]
- 143.Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 144.Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLos Biol. 2005;3:e 101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, Spiegelman BM. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 146.Dowell P, Otto TC, Adi S, Lane MD. Convergence of peroxisome proliferator-activated receptor gamma and Foxol signaling pathways. J Biol Chem. 2003;278:45485–45491. doi: 10.1074/jbc.M309069200. [DOI] [PubMed] [Google Scholar]
- 147.Kim JE, Chen J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes. 2004;53:2748–2756. doi: 10.2337/diabetes.53.11.2748. [DOI] [PubMed] [Google Scholar]
- 148.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 150.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1alpha. J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 151.Spiegelman BM, Heinrich R. Biological control through regulated transcriptional coactivators. Cell. 2004;119:157–167. doi: 10.1016/j.cell.2004.09.037. [DOI] [PubMed] [Google Scholar]