

ABSTRACT
Fibroblast growth factor 23 (FGF23) production is regulated by both calciotropic hormones and inflammation. Consistent with this, elevated FGF23 levels are associated with inflammatory markers as well as parathyroid hormone (PTH) in various disease states, including chronic kidney disease (CKD). However, the molecular mechanisms underpinning Fgf23 transcription in response to these regulators are largely unknown. We therefore utilized chromatin immunoprecipitation followed by DNA sequencing (ChIP‐seq) data from an osteocyte cell line to identify potential regulatory regions of the Fgf23 gene. Based on ChIP‐seq analysis of enhancer‐associated histone modifications, including H3K4 methylation and H3K9 acetylation, we discovered several potential enhancers for Fgf23, one of which was located 16kb upstream of the gene's transcriptional start site. Deletion of this putative enhancer from the mouse genome using CRISPR‐Cas9 technology led to lower bone, thymus, and spleen expression of Fgf23 mRNA without altering circulating levels of the intact hormone, although as previously reported, only bone displayed significant basal expression. Nevertheless, lack of the −16kb enhancer blunted FGF23 upregulation in a tissue‐specific manner by the acute inflammatory inducers lipopolysaccharide (LPS), interleukin‐1‐beta (IL‐1β), and tumor necrosis factor‐alpha (TNFα) in bone, non‐osseous tissues, and in circulation. Lack of the −16kb enhancer also inhibited PTH‐induced bone Fgf23 mRNA. Moreover, the absence of this Fgf23 enhancer in an oxalate diet‐induced murine CKD model prevented the early onset induction of osseous, renal, and thymic Fgf23 mRNA levels and led to a significant blunting of elevated circulating intact FGF23 levels. These results suggest that −16kb enhancer mediates the induction of Fgf23 by inflammation and PTH and facilitates the increase in FGF23 expression in a murine model of CKD. As exemplified herein, these Fgf23 enhancer‐deleted mice will provide a unique model in which to study the role of FGF23 expression in inflammatory diseases. © 2017 The Authors. JBMR Plus is published by Wiley Periodicals, Inc. on behalf of the American Society for Bone and Mineral Research.
Keywords: PTH/VIT D/FGF23, CYTOKINES, OSTEOCYTES, TRANSCRIPTIONAL REGULATION, DISORDERS OF CALCIUM/PHOSPHATE METABOLISM, GENETIC ANIMAL MODELS
Introduction
Fibroblast growth factor 23 (FGF23) is a bone‐derived hormone that regulates phosphate homeostasis and vitamin D metabolism. FGF23 protein is synthesized as a full‐length, biologically active 32kDa glycoprotein that is biologically inactivated through cleavage by furin and furin‐like convertases.1, 2, 3, 4 Under physiological conditions, increased serum phosphate levels function to upregulate FGF23, which acts in turn at the kidney through both fibroblast growth factor receptors (FGFRs) and the co‐receptor alpha Klotho (αKlotho) to decrease levels and activity of the sodium phosphate cotransporters NPT2a and NPT2c.5 This action inhibits renal phosphate reabsorption6 and alters the metabolism of 1,25‐dihydroxyvitamin D3 (1,25(OH)2D3).2
Defects in the production or cleavage of FGF23 lead to altered phosphate and vitamin D levels in various diseases such as familial tumoral calcinosis (TC)7 or autosomal‐dominant hypophosphatemic rickets (ADHR).3 Elevated FGF23 levels have also been detected in various other diseases in which the role or the mechanism of its elevation have not been firmly established, including chronic kidney disease (CKD)8, 9, 10 and inflammatory maladies such as inflammatory bowel disease.11 For example, FGF23 levels increase in early stages of renal failure and become gradually elevated during CKD progression in parallel with increased serum phosphate levels.12 Elevated FGF23 levels in CKD have also been associated with increased cardiovascular disease and mortality.8, 13, 14 Although these studies suggest that FGF23 is an important component in CKD, the causes, sources, and the molecular mechanisms through which FGF23 is increased are largely unknown.
Injections of pro‐inflammatory cytokines and sepsis have also been shown to be linked to hypophosphatemia,15, 16, 17 suggesting a potential interplay between phosphate, inflammation, and FGF23. Consistent with these findings, studies by Atkins and colleagues have shown that inflammation increases transcription of both mouse Fgf23 and human FGF23 genes in both the murine osteocytic cell line (IDG‐SW3) and in human bone chips, respectively.18 Moreover, in vivo studies by Wolf and colleagues have shown that both acute and chronic inflammation induced by single or repeated injections of bacteria or interleukin‐1‐beta (IL‐1β) increase bone Fgf23 mRNA production.19 Because osteocytes produce the highest levels of basal Fgf23, both of these studies have focused on osseous sources of FGF23.1 Despite this, Fgf23 is also expressed at lower levels in other tissues and cell types, including the thymus, spleen, brain, activated macrophages, and dendritic cells.1, 20, 21, 22 Masuda and colleagues has shown that inflammatory stimuli can increase Fgf23 expression of activated dendritic cells and macrophages.21 Consistent with this, Fanti and colleagues recently reported that upon acute or chronic exposure to low doses of lipopolysaccharides (LPS), spleen FGF23 production increased significantly.23 Although these studies establish that inflammation increases osseous and spleen‐derived FGF23 production, the contribution of non‐osseous tissues to FGF23 production and its pathophysiological role in inflammatory diseases are unknown.
FGF23 is expressed in many cell types21, 22, 24, 25 and is regulated by hormones such as 1,25(OH)2D3 26, 27, 28 and cytokines such as IL‐1β.18 Studies to date that have explored the underlying mechanisms of this regulation have utilized a combination of (i) transient transfection techniques that are limited to an evaluation of the activity of promoter‐proximal regions attached to a reporter and/or (ii) the use of selected pathway inhibitors. These methods have revealed roles for nuclear factor kappa β (NF‐κB)18, 21 and hypoxia‐inducible factor 1α (HIF1α)19, 29 binding at the Fgf23 promoter region, suggesting that they might mediate inflammation‐induction of Fgf23 transcription. In both cases, however, inhibitors of these transcriptional activators failed to prevent the induction in Fgf23 mRNA levels by inflammation and do not provide any in vivo evidence for the gene's direct regulation, highlighting the need for unbiased identification of Fgf23 transcriptional regulatory regions.
Advances in genome‐wide techniques such as chromatin immunoprecipitation followed by DNA sequencing (ChIP‐seq) have revealed that the transcriptional regulation of many genes, including receptor activator of NF‐κB ligand (Tnfsf11), vitamin D receptor (Vdr), or matrix metallopeptidase 13 (Mmp13), are mediated through a complex network of enhancers each providing distinct factor‐ and cell‐type specificity.30, 31, 32, 33, 34, 35, 36 More importantly, these studies have shown that although promoter‐proximal regions may play a role in regulation, many if not most regulatory components (enhancers) reside distal to the promoter.31, 33, 34, 35, 36 To determine the locations of regulatory regions of the Fgf23 gene, we utilized prior genome‐wide ChIP‐seq analyses of CTCF occupied sites, histone enrichment marks associated with chromatin structure, and enhancers to identify potential enhancers of Fgf23. We deleted one of the identified putative enhancers, which was located in an intergenic region 16kb upstream of Fgf23, from the mouse genome using clustered regularly interspaced short palindromic repeats (CRISPR)‐CRISPR‐associated protein‐9 nuclease (Cas9) (CRISPR‐Cas9) technology and explored the consequence of this deletion in the newly developed mouse strain. Lack of this putative enhancer decreased basal Fgf23 mRNA levels in bone, spleen, and thymus, but did not alter the levels of circulating intact FGF23 (iFGF23) nor did it affect the expression of FGF23 target genes in kidney, bone, or distinct parameters of mineral homeostasis. However, mice lacking the −16kb enhancer showed a significantly blunted FGF23 response to LPS and the inflammatory cytokines IL‐1β and tumor necrosis factor‐alpha (TNFα) with evident tissue specificity. Deletion of the −16kb enhancer also prevented the early onset induction of renal, thymic, and osseous Fgf23 mRNA levels and blunted the increase in circulating iFGF23 in an oxalate‐induced model of CKD. Because both inflammation and parathyroid hormone (PTH) are potential causes of increased FGF23 levels in early stages of CKD, we also show that PTH mediates bone Fgf23 transcription via the −16kb enhancer. These results suggest that a novel distal Fgf23 enhancer located −16kb upstream of the gene's transcriptional start site (TSS) mediates inflammatory and PTH induction of FGF23 and is required for FGF23 upregulation in early phases of CKD.
Materials and Methods
Reagents
The following reagents were used for in vivo injections: LPS (Sigma, St. Louis, MO, USA), IL‐1β (Cell Signaling Technology, Danvers, MA, USA), and TNFα (R&D Systems, Minneapolis, MN, USA). PTH (1–84) used for the in vivo injections was purchased from Bachem California Inc. (Torrance, CA, USA). For CRISPR‐Cas9‐mediated production of the murine model, Cas9 protein was purchased from PNA Bio (Newbury Park, CA, USA). For diet‐induced CKD model, 0.67% Na Oxalate Diet (<0.01% Ca, 0.4% P; Cat. No. TD.110105) and Control Diet (0.6% Ca, 0.4% P; Cat. No. TD.97191) were purchased from Envigo (Indianapolis, IN).
Generation of mutant mice
CRISPR mice were generated at the University of Wisconsin Biotechnology Center Transgenic Animal Facility. The guides used for CRISPR‐Cas9‐mediated genome editing were optimized for the least number of potential off‐target sites and fewest sites within coding exons using the Zhang Laboratory CRISPR Design tool (http://crispr.mit.edu). The following guides: m Fgf23 −16kb Guide 1 GGAGGCGTAAACATCTGATC‐AGG (oligos F 5′‐CACC‐GGAGGCGTAAACATCTGATC‐3′ and R 5′‐AAAC‐GATCAGATGTTTACGCCTCC‐3′) and m Fgf23 −16kb Guide 2 CCATGGGCTAGGGTGCGGAA‐TGG (oligos F 5′‐CACC‐GCCATGGGCTAGGGTGCGGAA‐3′ and R 5′‐AAAC‐TTCCGCACCCTAGCCCATGGC‐3′) were annealed and cloned into plasmids pX330 or pX458 obtained from the Zhang Laboratory via Addgene (Cambridge, MA, USA) as recently described.35, 36 RNA guides to be transcribed were produced by PCR amplification of the “target guide sequence and tracrRNA sequence” included in the pX330 and pX458 plasmids with primers containing the T7 promoter sequence (5′‐TTAATACGACTCACTATAGG‐) appended to the 5′ end of the original guide sequences with reverse primer 5′‐CAAAAAAGCACCGACTCGGTGCCACTTTTTCAAG‐3′, thus producing DNA PCR template of “T7 promoter − guide sequence − tracrRNA sequence.” These PCR products were then in vitro transcribed utilizing T7 MEGAshortscript (Life Technologies, Grand Island, NY, USA) kit.37 A mixture of 50 ng/μL of the produced RNA guides and 40 ng/μL of Cas9 protein in injection buffer (5 mM Trizma Base, 5 mM Tris HCL, 0.1 mM EDTA, pH 7.4) was injected into the pronucleus of 1 day fertilized embryos isolated from hyperovulating female C57Bl6 mice as described previously38 and implanted into recipient females. The resultant pups were genotyped with spanning primers, cloned, and sequenced for validation of the deletion. One mouse that contained the desired deletion of the −16kb region was identified (Fig. 2B) and was bred to produce the murine strain lacking the −16kb region. From here on, we will refer to this genetically modified mouse strain as the Fgf23 −16KO mouse model.
Animal studies
Mice were housed in high‐density ventilated caging in the Animal Research Facility of University of Wisconsin‐Madison under 12‐hour light/dark cycles, 72°F temperature, and 45% humidity. To induce FGF23 expression in vivo, animals were injected intraperitoneally (ip) with 10 mg/kg of body weight (bw) LPS, 2 μg recombinant TNFα, 50 ng/g bw recombinant IL1‐β, or phosphate‐buffered saline (PBS) as vehicle. These single doses are well within the range utilized by other investigators and were chosen to induce maximal response. Animals were euthanized and tissues collected 3 hours after Tnfα and 6 hours after IL‐1β or LPS injection. To test PTH regulation of FGF23 expression in vivo, animals were injected ip with 230 ng/g bw PTH (1–84) or vehicle (PBS) and euthanized for experiments 1 hour later. PTH and Tnfα effects on Fgf23 transcription were previously determined by time‐course experiments to optimize for the effect examined (data not shown). For all the injections, the animals were stratified into groups according to their body weight. To induce CKD, 8‐week‐old mice were fed calcium‐deficient oxalate diet (0.67% Na Oxalate, <0.01% Ca, 0.4% P) or control diet (0.6% Ca, 0.4% P) for 7 days, at which time animals were euthanized and tissues collected. For each animal experiment, sex and age of the animals studied are indicated in each figure legend. All experiments and tissue collections were performed in the procedure rooms in the Research Animal Facility of University of Wisconsin‐Madison. All animal studies were reviewed and approved by the Research Animal Care and Use Committee of University of Wisconsin‐Madison.
ChIP followed by sequencing (ChIP‐seq) and homology comparisons
In our prior work, we performed ChIP‐seq analysis on osteocyte‐differentiated IDG‐SW3 cells, as well as MSC‐derived osteoblasts and MC‐3T3‐E1‐ and UAMS‐derived osteoblasts.35, 39, 40 With regard to the IDG‐SW3 osteocyte line, cells were cultured for 35 days on collagen‐coated plates at 33°C with osteogenic media (αMEM with 10% heat‐inactivated FBS, 100 U/mL penicillin/streptomycin, 50 μg/mL ascorbic acid, and 4 mM glycerophosphate). Cells were then fixed with 1% formaldehyde and the calcified matrix was depleted through three 15‐minute 300 mM EDTA washes. ChIP‐seq analysis was performed in triplicate using a control antibody (IgG) or experimental antibodies (CTCF, H3K9ac, H4K5ac, H3K4me1, H3K4me2, and H3K27ac) as indicated.41 ChIP‐seq data processing and statistical evaluations was as described previously.42 All sequencing data have been publicly available in the Gene Expression Omnibus database: GSE54784.41 Epigenetic histone profiles obtained from MSCs, MC3T3‐E1, and UAMS cell lines were similar, suggesting enhancer profiles across the FGF23 gene locus are common within the mesenchymal cell lineage. Conservation analysis was performed with VISTA Point comparing mouse (mm9) to human (hg19) exclusively at the FGF23 locus.43
De novo transcription factor motif analysis
Genomatix MatInspector software was used for analysis of potential de novo transcription factor binding motifs. Matrix Library 8.4 versions of the transcription factor binding sites (Weight Matrices) were used. The −16kb region (chr6:127,006,796‐127,007,720) was evaluated for binding motifs of matrix families that have been suggested to play a role in inflammatory signaling (nuclear factor kappa B/c‐rel, hypoxia inducible factor‐ bHLH/PAS protein family members, and signal transducer and activator of transcription). Matrix information is supplied in Supplemental Table S1. The search was performed with a core similarity and optimal matrix similarity of 0.75 and only matches that exhibit a matrix similarity greater than 0.95 are shown in the Table 1.
Table 1.
Transcription Factor Binding Motifs Identified by Genomatix MatIspector Analysis of Fgf23 −16kb Element
| Detailed matrix information | Strand | Core similarity | Matrix similarity | Sequence |
|---|---|---|---|---|
| Hypoxia‐response elements | – | 1 | 0.985 | tcttggtaCGTGagtgt |
| Binding site of Clock/BMAL1 and NPAS2/BMAL1 heterodimers | + | 1 | 0.983 | aagagtCACGtgtgctc |
| NF‐κB (p65) | – | 1 | 0.974 | ccaggactTTCCttt |
| STAT5 | – | 0.945 | 0.97 | tagaTTCCtagaagtgaaa |
| STAT3 | – | 1 | 0.966 | gctcTTCCagggaagtggt |
| AhR nuclear translocator homodimers | – | 1 | 0.963 | tgagcacaCGTGactct |
| STAT6 | + | 1 | 0.951 | ccacTTCCctggaagagct |
Bioinformatic analysis of the −16kb region using the Genomatix software tool MatInspector reveals the presence of several transcription factor binding motifs associated with inflammatory signaling and corresponding sequences along with highest calculated matrix similarity scores (greater than 0.95).
Bone mineral density (BMD) analysis
At age 8 weeks, BMDs of Fgf23 −16KO and their wild‐type littermates were measured and analyzed by dual‐energy X‐ray absorptiometry (DXA) with a PIXImus densitometer (GE Lunar Corp, Madison, WI, USA) as previously described.30
Gene expression
Dissected tissues were frozen immediately in liquid nitrogen and stored at −80°C. Unless otherwise indicated, the bone gene expression analyses in this article were obtained from lumbar vertebra 5 (L5). Frozen tissues were homogenized in Trizol Reagent (Life Technologies) and RNA was isolated according to the manufacturer's instructions. One microgram of RNA was used as a template to synthesize cDNA using the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). RNA isolation and cDNA production were performed in blinded fashion. Relative mRNA levels were determined via multiplex TaqMan quantitative reverse transcription‐PCR (RT‐PCR) using VIC‐labeled Mouse ACTB and FAM‐labeled TaqMan gene expression assays (Applied Biosystems). The following TaqMan Gene Expression probes (Applied Biosystems) were used for quantitative RT‐PCR: Fgf23 (Mm00445621), Il1b (Mm00434228), Il6 (Mm00446190), Tnf (Mm00443258), Cyp27b1 (Mm01165918), Cyp24a1 (Mm00487244), Tnfsf11 (Mm00441906), Fgf6 (Mm01183111), Tigar (Mm00621530), Ccnd2 (Mm00438070), Slc34a1 (Mm00441450), Slc34a3 (Mm00551746), and mouse Actb (Cat. No. 4352341E). Relative mRNA levels were calculated using the ΔCt method.30
Blood chemistry
Maxillary blood was collected during oxalate diet feeding at days 0, 4, and 7. For the remaining experiments, cardiac blood was collected at the time of death. Collected blood was incubated at room temperature for 30 minutes followed by duplicate centrifugation at 6000 rpm for 12 minutes to obtain serum or EDTA plasma. Serum calcium and phosphate levels were measured using QuantiChrom Calcium Assay Kit (Cat. No. DICA‐500, BioAssay Systems, Hayward, CA, USA) and QuantiChrom Phosphate Assay Kit (Cat. No. DIPI‐500, BioAssay Systems). Circulating intact FGF23 was measured in EDTA plasma via Mouse/Rat FGF‐23 (Intact) ELISA Kit (Cat. No. 60‐6800, Immutopics, San Clemente, CA, USA).
Statistical evaluation
All values represent the mean ± standard deviation (SD). Differences between group means were evaluated using Student's t test (Figs. 1 and 4) or one‐way ANOVA (Fig. 8 A) using GraphPad Prism 7 software (GraphPad Software, Inc., La Jolla, CA, USA). The data of Figs. 5, 6, 7, 8, 9 were analyzed by two‐way ANOVA with multiple comparison test using Benjamini‐Hochberg procedure as indicated in the figure legends. In two‐way ANOVA analysis of the data that did not pass normalization test in the original form, normalization was achieved by one of square root, log10, reciprocal, or rank transformations. If data did not achieve normality and equal variance after transformations, ranks were applied for nonparametric analysis. For two‐way ANOVA analysis of data obtained from tissues that normally do not express Fgf23 mRNA, statistical analysis was performed by imputing minimum detectable levels in place of undetectable levels followed by permutation test analysis of a 2×2 ANOVA model.
Figure 1.
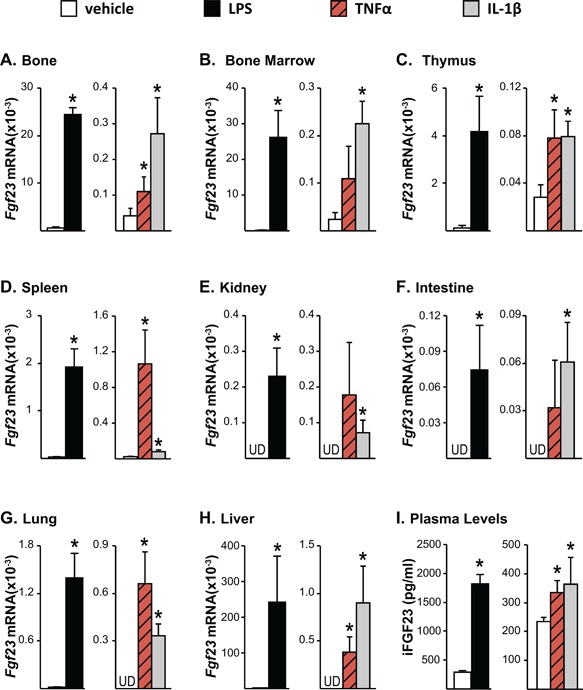
Inflammation increases Fgf23 mRNA expression in bone and non‐osseous tissues. Twelve‐month‐old female mice were injected intraperitoneally (ip) with 10 mg/kg of LPS or PBS. Eight‐week‐old female mice were injected with 2 μg of recombinant TNFα, 50 ng/g of IL‐1β, or PBS. The tissues and blood were collected 3 hours after TNFα and 6 hours after LPS or IL‐1β injection. (A–H) Fgf23 mRNA levels were measured by quantitative RT‐PCR and normalized to beta‐actin mRNA levels. (I) Blood was collected by cardiac puncture, and circulating intact FGF23 (iFGF23) levels were measured in EDTA plasma. (A–I) All values represent mean ± SD of 3 to 4 mice/group. All statistical comparisons were performed using Student's t test. *p < 0.05 compared with vehicle controls. UD indicates that Fgf23 levels were undetectable by RT‐PCR.
Figure 4.
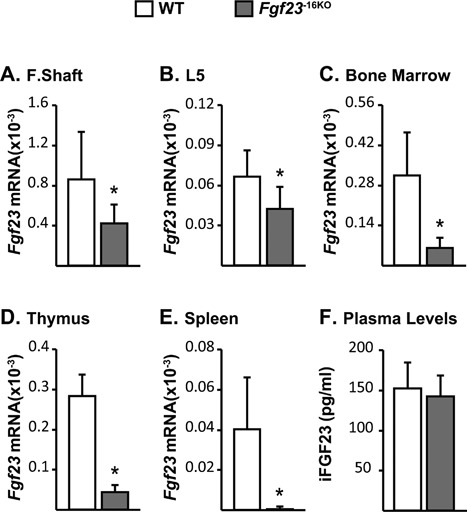
Lack of the −16kb region alters Fgf23 mRNA levels but does not alter circulating iFGF23 levels. Tissues and cardiac blood of wild‐type (WT) and Fgf23 −16KO male mice were collected at 8 weeks of age. (A–E) Femur shaft (F. Shaft) (A), lumbar vertebra 5 (L5) (B), and non‐osseous tissue (C–E) Fgf23 mRNA levels were measured by quantitative RT‐PCR and normalized to beta‐actin mRNA levels. (F) Blood was collected by cardiac puncture, and circulating intact FGF23 (iFGF23) levels were measured in EDTA plasma. (A–F) Bars represent mean ± SD of 6 to 8 mice/group. All statistical comparisons were performed using Student's t test. *p < 0.05 compared with WT controls.
Figure 8.
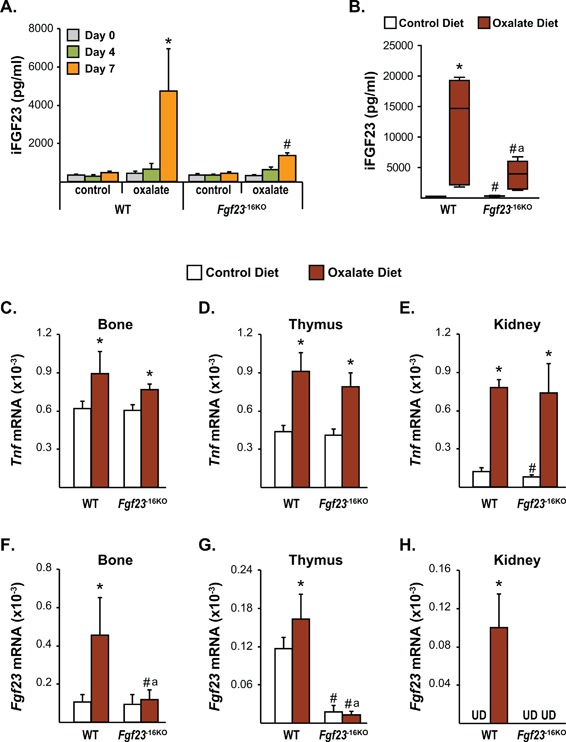
Lack of the −16kb enhancer of Fgf23 prevents kidney disease‐induced increase in Fgf23 levels. Eight‐week‐old female (A) and male (B–H) Fgf23 −16KO mice and their wild‐type (WT) littermates were fed control or oxalate diet for 1 week. (A, B) Circulating intact FGF23 (iFGF23) levels were measured at day 0 (A), day 4 (A), and day 7 (A, B) of the diet feeding. (C–H) After 7 days of diet feeding, Tnfα (C–E) and Fgf23 (F–H) mRNA levels of the male Fgf23 −16KO and WT mice were measured by RT‐PCR. (A) Bars represent the mean ± SD of 3 to 6 mice/group and statistical analysis were performed using one‐way ANOVA. For this analysis, *p < 0.05 effect of oxalate diet compared with day 0 value of the same genotype; #p < 0.05 effect of genotype within the same diet at the same time point. (B) In this box and whisker plot of iFGF23, whiskers represent the range of the values, the line located in the boxes represent the median, and the boxes represent the second and third quartiles of 5 to 7 mice per group and the statistical analysis between the groups were performed using two‐way ANOVA with Tukey's multiple comparison test. (C–H) Bars represent the mean ± SD of 5 to 7 mice/group and statistical analysis were performed using two‐way ANOVA with multiple comparison test using the Benjamini‐Hochberg procedure. For all analysis of B–H, *p < 0.05 effect of diet within the same genotype; #p < 0.05 effect of genotype within the same diet and “a”, p < 0.05 magnitude of the difference (control diet‐oxalate diet) is less in Fgf23 −16KO mice compared with WT mice. UD indicates that Fgf23 levels were undetectable by RT‐PCR.
Figure 5.
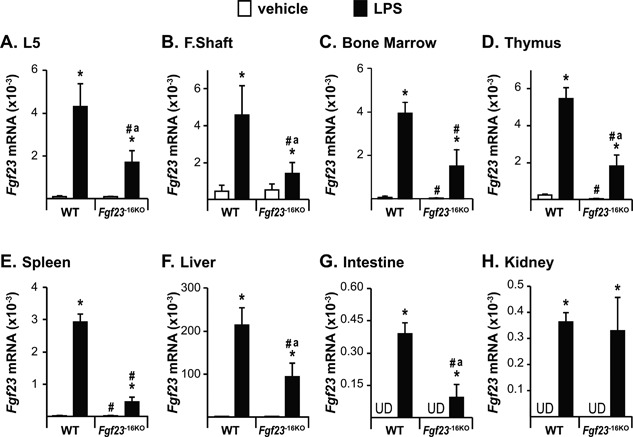
Lack of the −16kb enhancer of Fgf23 blunts the LPS‐induced increase in Fgf23 levels. Twelve‐week‐old wild‐type (WT) and Fgf23 −16KO female mice were injected with 10 mg/kg of LPS or vehicle (PBS). Tissues were collected 6 hours after injection. (A–H) Lumbar vertebra 5 (L5) (A), femur shaft (F. Shaft) (B), and non‐osseous tissue (C–H) Fgf23 mRNA levels were measured by RT‐PCR. Bars represent the mean ± SD of 4 to 6 mice/group. Statistical comparisons were performed using Two‐Way ANOVA with multiple comparison test using the Benjamini‐Hochberg procedure. *, p<0.05 effect of treatment within the same genotype; #, p<0.05 effect of genotype within the same treatment, and “a”, p<0.05 magnitude of the difference (vehicle‐injection) is less in Fgf23 −16KO mice compared to WT mice. UD indicates that Fgf23 levels were undetectable by RT‐PCR.
Figure 6.
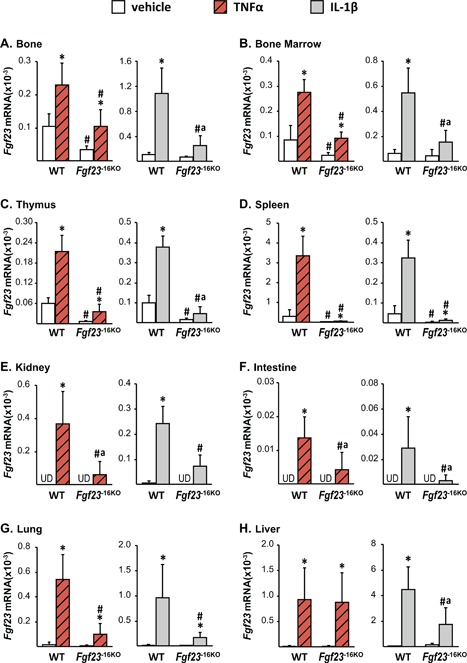
Lack of the −16kb enhancer of Fgf23 blunts the Tnfα‐ and IL‐1β‐induced increase in Fgf23 levels. Eight‐week‐old wild‐type (WT) and Fgf23 −16KO female mice were injected with 2 μg of recombinant Tnfα or vehicle, while their male littermates were injected with 50 ng/g of IL‐1β or PBS. The tissues were collected 3 hours after TNFα and 6 hours after IL‐1β injection. The Fgf23 mRNA levels were measured by RT‐PCR and represented here as the mean ± SD (n = 5 to 9 mice/group). Statistical comparisons were performed using two‐way ANOVA with multiple comparison test using the Benjamini‐Hochberg procedure. *p < 0.05 effect of treatment within the same genotype; #p < 0.05 effect of genotype within the same treatment, and “a”, p < 0.05 magnitude of the difference (vehicle‐injection) is less in Fgf23 −16KO mice compared with WT mice. UD indicates that Fgf23 levels were undetectable by RT‐PCR.
Figure 7.
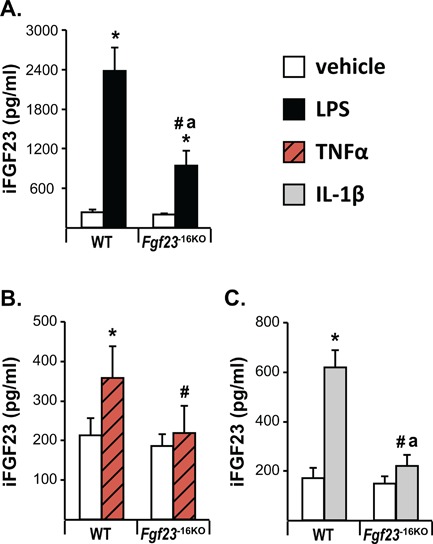
The −16kb enhancer of Fgf23 mediates the inflammation‐induced increase in circulating intact FGF23 levels. Eight‐ to 12‐week‐old wild‐type (WT) and Fgf23 −16KO mice were injected with 10 mg/kg of LPS (female mice) (A), 2 μg of recombinant TNFα (female mice) (B), 50 ng/g of IL‐1β (male mice) (C), or vehicle. Cardiac blood was collected at time of death, which was 6 hours after LPS and IL‐1β injection and 3 hours after TNFα injection. Circulating intact FGF23 (iFGF23) levels were measured by ELISA. The bars represent the mean ± SD of 4 to 9 mice/group. Statistical comparisons were performed using two‐way ANOVA with multiple comparison test using the Benjamini‐Hochberg procedure. *p < 0.05 effect of treatment within the same genotype; #p < 0.05 effect of genotype within the same treatment and “a”, p < 0.05 magnitude of the difference (vehicle injection) is less in Fgf23 −16KO mice compared with WT mice.
Figure 9.
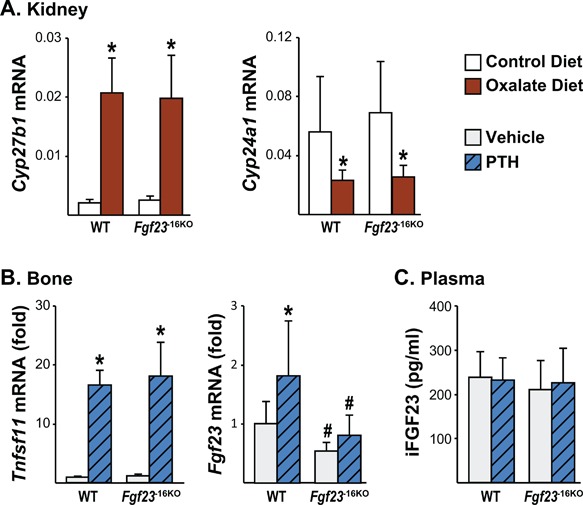
The −16kb enhancer of Fgf23 mediates the PTH‐induced increase in Fgf23 transcription. (A) Eight‐week‐old male Fgf23 −16KO mice and their wild‐type (WT) littermates were fed control or oxalate diet for 1 week. Kidney Cyp27b1 and Cyp24a1 mRNA levels were measured by RT‐PCR. Bars represent the mean ± SD of 5 to 7 mice/group. (B, C) Eight‐week‐old wild‐type (WT) and Fgf23 −16KO male mice were injected ip with 230 ng/g bw of PTH. Tissues were collected 1 hour after injection. (B) Tnfsf11 (RANKL) and Fgf23 mRNA levels were measured by RT‐PCR. Bars represent the mean ± SD of 8 to 9 mice/group as fold by comparison to mean of WT vehicle group. (C) Circulating intact FGF23 (iFGF23) levels were measured 1 hour after PTH injection. Bars represent the mean ± SD of 8 to 9 mice/group. All statistical analysis between the groups were performed using two‐way ANOVA with multiple comparison test using the Benjamini‐Hochberg procedure and “a”, p < 0.05 magnitude of the difference (vehicle‐PTH injection) is less in Fgf23 −16KO mice compared with WT mice. *p < 0.05 effect of treatment within the same genotype; #p < 0.05 effect of genotype within same treatment.
Results
Acute inflammation induces Ffg23 expression in multiple osseous and non‐osseous tissues
It has been previously shown that LPS induces Fgf23 transcription in osteocytic cell lines and bone chips.18 However, Fgf23 is expressed in non‐osseous tissues at low levels as well.1, 20, 21, 22 To address the scope of Fgf23 regulation by acute inflammation in bone and non‐osseous tissues, we injected 8‐week‐old wild‐type (WT) mice with LPS (10 mg/kg bw). As indicated in Fig. 1 A–D, LPS increased Fgf23 mRNA levels significantly in tissues that were shown previously to express Fgf23 such as bone, bone marrow, thymus, and spleen. Interestingly, LPS injection also led to the enhanced expression of Fgf23 in tissues such as kidney, intestine, lung, and liver that exhibit almost undetectable basal levels of Fgf23 (Fig. 1 E–H).
Because LPS is known to significantly increase the production of multiple pro‐inflammatory cytokines such as IL‐1β, TNFα, and IL‐6 (as confirmed in Supplemental Fig. S1), LPS induction of Fgf23 may be secondary to the actions of increased IL‐1β or TNFα. To explore this, we injected 8‐week‐old WT mice with recombinant IL‐1β (50 ng/g bw) or TNFα (2 μg). Although to a lesser extent when compared with LPS, both IL‐1β and TNFα induced Fgf23 expression in all tissues examined (Fig. 1 A–H). In line with the Fgf23 mRNA levels, IL‐1β and TNFα also increased circulating iFGF23 levels that were lower than those upregulated in response to LPS (Fig. 1 I). It is well recognized that LPS exerts effects that extend beyond those of an inflammatory nature. However, because the inflammatory cytokines show a similar response and because we see similar inductions of Fgf23 mRNA levels in ex vivo calvarial cultures (data not shown), these results suggest that the predominant effect of LPS is to modulate Fgf23 expression via its inflammatory effects, whether it be directly or in indirectly through the induction of additional inflammatory regulators. It is this latter mechanism wherein LPS induces multiple inflammatory cytokines that increase Fgf23 in an additive fashion that may be responsible for the robust nature of LPS on Fgf23 expression.
A distal enhancer located 16kb upstream of Fgf23 TSS mediates Fgf23 expression
We next sought to identify the transcriptional regulatory regions that mediate the induction of Fgf23 in response to inflammatory stimuli. Inflammatory signaling involves multiple transcription factors including NF‐κB, HIF1α, signal transducers and activators of transcription (STATs), and perhaps others such as the nuclear receptor NURR1. Many of these have also been suggested to play a role in the regulation of Fgf23,(18,19) although virtually all of these studies have focused on the promoter proximal region of the Fgf23 gene and were conducted in vitro. Given this complexity, we took the alternative approach to examine data from our previous ChIP‐seq analysis of IDG‐SW3 osteocyte cells across the extended Fgf23 gene locus in search of sites with overlapping H3K4me1, H3K4me2, H4K5ac, H3K9ac, and H3K27ac enrichment. These posttranslational histone marks are considered indicative of active regulatory regions and have been used by us previously to identify active regions in the Tnfsf11 44, 45 and Mmp13 genes.35, 36 This search yielded three potential enhancers of Fgf23 located −38kb, −16kb, and +7kb of the Fgf23 TSS (Fig. 2 A), two of which (‐38kb and +7kb) were also occupied by CTCF (Fig. 2 A), a factor with multiple pleiotropic regulatory roles. We therefore focused on the −16kb region because this segment was also highly conserved between human and mouse (Fig. 3). Previous studies utilizing pathway inhibitors have shown that HIF1α and NF‐κB play a role in inflammatory induction of Fgf23. Bioinformatics analysis of these factors and STATs and showed that the −16kb region contained an array of in silico motifs for transcription factors involved in inflammation‐induced transcription (Table 1). Modest H3K4me1 activity was also found at the promoter region of Fgf23, suggesting potential regulatory activity within this region in vivo as well.
Figure 2.
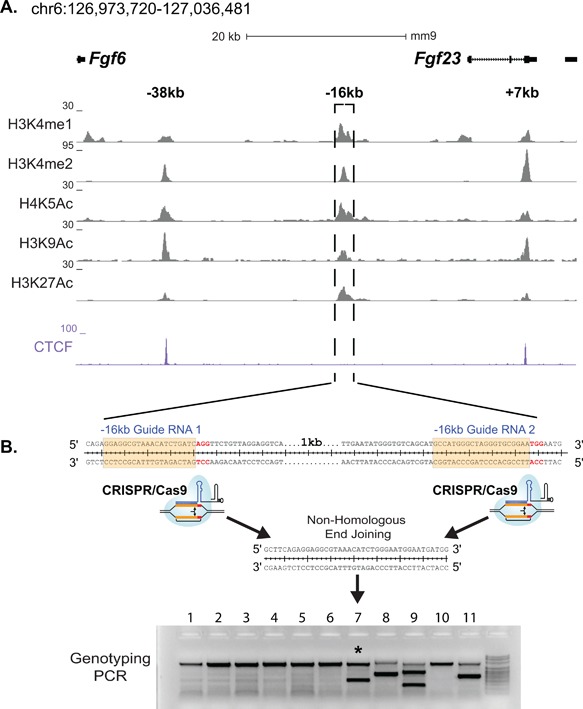
Identification of potential Fgf23 enhancers and production of Fgf23 −16KO mice. (A) ChIP‐seq tag density (normalized to 107 tags) for histone modifications and CTCF binding across the Fgf23 gene locus in differentiated IDG‐SW3 osteocytes. (B) Schematic describing the deletion of the −16kb region from the mouse genome via CRSIPR‐Cas9 genome editing.
Figure 3.
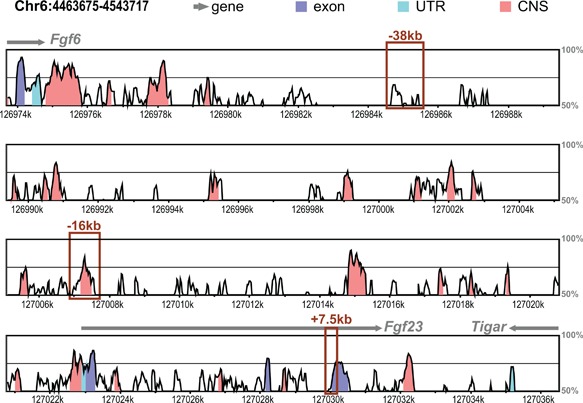
Comparative homology analysis of human and mouse Fgf23 locus. Vista‐Point Tool was used for conservation analysis comparing the mouse (mm9) to human (hg19) Fgf23 locus. Conservation score reaches significance at 70% (indicated by black middle line) for exons (dark blue), conserved untranslated region (UTR, cyan), and conserved noncoding sequence (CNS, coral). Other genomic features are indicated in legend. ChIP‐seq‐identified potential enhancers of Fgf23 are shown with boxes.
To determine if the potential enhancer at −16kb mediated Fgf23 transcription, we deleted this region from the mouse genome utilizing the CRISPR‐Cas9 genome editing technique (Fig. 2 B) and created the Fgf23 −16KO enhancer‐deleted mouse. Lack of the 16kb segment led to a ∼40% decrease in Fgf23 mRNA levels in femur shafts and whole bones (Fig. 4 A, B) and to a more than 80% decrease in Fgf23 mRNA levels in bone marrow, thymus, and spleen (Fig. 4 C–E). Moreover, deletion of the −16kb region did not alter the expression of the neighboring genes Fgf6, Tigar, or Ccnd2 (Supplemental Fig. S2), suggesting that the −16kb region likely represents a bona fide enhancer that is linked directly to the regulation of Fgf23 in vivo.
Despite the lower Fgf23 mRNA levels, circulating iFGF23 levels of Fgf23 −16KO mice were not different from their littermate controls (Fig. 4 F). This lack of change was supported by the observation that both Fgf23 −16KO mice and their WT littermates displayed similar levels of expression of the renal FGF23 target genes that encode sodium‐dependent phosphate co‐transporter genes and the enzymes involved in 1,25(OH)2D3 metabolism (Supplemental Fig. S3). Similarly, Fgf23 −16KO mice also maintain normal levels of serum calcium and phosphate, PTH levels, and bone mineral density (BMD) relative to their WT controls (Table 2). Taken together, these data suggest that lower Fgf23 mRNA levels observed in Fgf23 −16KO mice did not alter the overall biological activity of FGF23 in the blood. It is unclear why basal iFGF23 levels in the blood remain unchanged upon deletion of the −16kb Fgf23 enhancer, although there are a number of examples of this phenomenon. It is worth noting that the basal levels of Fgf23 are not only variable across the tissues examined but also extremely low and in some cases almost undetectable in non‐osseous tissues. Among all FGF23‐expressing tissues, bone expresses the highest basal amounts of FGF23 and the effect of the −16kb enhancer deletion in bone FGF23 expression is less compared with that in the non‐osseous tissues. Thus, it is possible that the impact on blood iFGF23 levels is cumulatively lost. However, tissue size is also of importance. It is also well known that FGF23 is dynamically processed posttranslationally through the complex actions of several different enzymes including Furin and Galnt 3 and that the differential activation of this processing event in tissues may be responsible for the discordance between mRNA and circulating iFGF23 levels. Further investigation of this phenomenon, however, is outside the objectives of this study of the transcriptional regulation of Fgf23.
Table 2.
Bone and Mineral Homeostasis in Fgf23 −16KO Mice
| Calcium (mg/dL) | Phosphate (mM) | PTH (pg/mL) | |
|---|---|---|---|
| WT | 10.7 ± 0.7 | 2.2 ± 0.5 | 55.2 ± 32.1 |
| Fgf23 −16KO | 10.3 ± 0.8 | 2.3 ± 0.3 | 50.9 ± 17.8 |
| Bone Mineral Density (BMD) | |||
|---|---|---|---|
| Femur | Spine | Global | |
| WT | 0.060 ± 0.005 | 0.045 ± 0.001 | 0.044 ± 0.002 |
| Fgf23 −16KO | 0.058 ± 0.005 | 0.046 ± 0.002 | 0.043 ± 0.002 |
Analysis of serum calcium and phosphate and plasma PTH were performed on 8‐week‐old wild‐type (WT) and Fgf23 −16KO male mice as explained in Materials and Methods. BMD of these mice were measured by DXA. The values represent mean ± SD of 6 to 8 mice/group. All statistical comparisons were performed using Student's t test. *p < 0.05 compared with WT controls.
The −16kb enhancer of Fgf23 mediates inflammatory induction of Fgf23
To assess the role of the −16kb Fgf23 enhancer further, we injected Fgf23 −16KO mice and their WT littermates with LPS. LPS injection induced IL‐1β, TNF, and IL‐6 expression similarly in both genotypes (Supplemental Fig. S1), suggesting equivalent inflammatory response in both genotypes. Similar to our observations shown in Fig. 1, LPS injection led to a significant induction of Fgf23 mRNA in all tissues examined (Fig. 5 A–H). Lack of the −16kb Fgf23 enhancer, however, blunted the LPS‐induced increase in Fgf23 mRNA levels in bone, bone marrow, thymus, spleen, liver, and intestine (Fig. 5 A–G) but did not alter Fgf23 induction by LPS in kidney (Fig. 5 H). These results suggest that this inflammatory modulator plays a distinct but perhaps tissue‐differential role at the genomic level in the transcriptional regulation of Fgf23.
To determine whether the −16kb enhancer provided tissue‐specific regulation of Fgf23 among the inflammatory cytokines, we injected Fgf23 −16KO mice and their WT littermates with recombinant TNFα or IL‐1β. Both cytokines increased control gene expression similarly in all genotypes, confirming equivalent injection and inflammatory response (Supplemental Fig. S4). Lack of the −16kb enhancer blunted TNFα and IL‐1β induction of Fgf23 in bone, bone marrow, thymus, spleen, kidney, intestine, and lung (Fig. 6 A–G). However, induction of Fgf23 transcription by IL‐1β was different from that of TNFα in liver. Liver Fgf23 mRNA levels increased to a lesser extent in Fgf23 −16KO mice injected with IL‐1β compared with their littermate controls, suggesting control by the −16kb enhancer. In contrast, TNFα was fully active for Fgf23 induction in the liver in Fgf23 −16KO mice, suggesting that alternative regulatory elements or factors may be involved in Fgf23 transcription in this organ. Moreover, although lack of the −16kb Fgf23 enhancer did not alter LPS induction of Fgf23 in kidney (Fig. 5 H), it blunted TNFα and IL‐1β induction of renal Fgf23 production (Fig. 6 E), suggesting that LPS upregulation may be mediated via IL‐6 operating though other transcriptional control regions for Fgf23. These results point directly to the role of additional factors such as IL‐1β and Tnfα in the LPS regulation of Fgf23 and that their induction by LPS may underlie the more complex response to this inflammatory inducer and may account for differential tissue‐specific activities as suggested earlier.
Despite the fact that basal changes in Fgf23 mRNA levels of Fgf23 −16KO mice did not alter circulating iFGF23 levels (Fig. 4 F), lack of the −16kb enhancer significantly blunted, or in the case of TNFα injections prevented, the inflammation‐induced increases found in circulating iFGF23 levels (Fig. 7 A–C). As summarized in Table 3, these results collectively suggest that the −16kb enhancer mediates Fgf23 expression in response to inflammation in a tissue‐specific manner. In contrast, lack of the −16kb region did not alter expression or transcriptional regulation of the nearby genes Ccnd2 (data not shown), Fgf6, and Tigar (Supplemental Table S2), suggesting that transcriptional regulation mediated by the −16kb enhancer was restricted to the Fgf23 gene.
Table 3.
Summary of the Role of the −16kb Enhancer in Fgf23 Regulation in Response to Acute Inflammation and Kidney Disease
| Fgf23 mRNA levels | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bone | Bone marrow | Thymus | Spleen | Kidney | Intestine | Lung | Liver | iFGF23 Plasma | |
| IL‐1β | + | + | + | + | + | + | + | + | + |
| Tnfα | + | + | + | + | + | + | + | – | + |
| LPS | + | + | + | + | – | + | + | + | + |
| Oxalate diet | + | UD | + | x | + | UD | UD | x | + |
A comparison of the results of Fgf23 mRNA regulation by inflammatory cytokines and CKD in different tissues with levels in the circulation as observed in Figs. 3, 4, 5, 6, 7.
+ = −16kb enhancer plays a role in the Fgf23 induction; − = −16kb enhancer does not play a role in Fgf23 induction; X = Fgf23 is not regulated; UD = Fgf23 levels were not measured.
The −16kb distal enhancer of Fgf23 mediates the early onset induction of Fgf23 in a kidney disease model
Recent studies have shown a positive correlation between elevations in inflammatory markers and FGF23 levels in CKD patients beginning at very early stages of the disease,46, 47 raising the possibility that inflammation may cause or contribute to the increase in FGF23 production in CKD. Because the −16kb enhancer mediates inflammatory regulation of Fgf23 transcription, we next utilized a diet‐induced CKD model to address whether the −16kb enhancer could play a role in mediating the CKD‐induced increase in Fgf23 expression.48 It has been shown previously that oxalate diet feeding initiates altered kidney function as early as 7 days post feeding, whereas 3 weeks of oxalate diet feeding is required to obtain the majority of features of CKD in this model.48 Although this is not a perfect model of CKD because of the rapid rather than chronic progression of the disease, it allowed us to test whether diet‐induced Fgf23 upregulation was mediated by the −16kb enhancer. To determine the time‐course of iFGF23 induction in the oxalate feeding‐induced CKD, we fed 8‐week‐old female and male Fgf23 −16KO mice and their littermate controls a control or oxalate diet and demonstrated that circulating iFGF23 increases after 7 days of oxalate diet feeding in WT mice (Fig. 8 A, B). Although 1 week of oxalate diet increased WT iFGF23 to a greater level in male mice, both female and male mice lacking the −16kb enhancer exhibited a blunted increase in circulating iFGF23 levels at this time point (Fig. 8 A, B).
In the genetically induced murine Col4a3 CKD knock‐out model, increases in circulating iFGF23 levels have been shown to precede an increase in Fgf23 mRNA levels in bone,49 suggesting the possibility that FGF23 production in non‐osseous tissues may contribute to the CKD‐induced increase in circulating iFGF23 levels. Because we observed that inflammation can induce Fgf23 mRNA expression in multiple non‐osseous tissues, we next sought to determine potential sources of the CKD‐induced increase in iFGF23 levels and to assess whether the upregulation of these levels of iFGF23 were mediated by the −16kb enhancer of Fgf23. Gene expression analysis performed 1 week after oxalate diet feeding showed similar increases in Tnfα expression of osseous and non‐osseous tissues of both WT and Fgf23 −16KO mice, suggesting that the oxalate diet increased inflammation in both genotypes similarly (Fig. 8 C–E). In accordance with the increased circulating iFGF23 levels, 1 week of oxalate diet feeding of WT mice increased Fgf23 mRNA levels in bones, thymus, and kidney (Fig. 8 F–H) but not in liver or spleen (data not shown). Despite a blunted, but significant, increase in iFGF23 levels (Fig. 8 B), Fgf23 −16KO mice did not show a significant increase in Fgf23 mRNA levels in any of the tissues examined (Fig. 8 F–H). Taken together, these results suggest that −16kb enhancer of Fgf23 mediates the early onset of CKD‐induced increase in bone, thymus, and kidney Fgf23 transcription but that this upregulation contributes only partially to changes in circulating iFGF23 levels.
The −16kb distal enhancer of Fgf23 mediates PTH‐induced increase in Fgf23 transcription
In addition to inflammation, PTH levels also progressively increase during CKD. Indeed, high PTH levels have also been proposed to induce Fgf23 transcription.(50) Consistent with this concept, 1 week of oxalate diet increased PTH levels, as indicated by the increase in Cyp27b1 and decrease in Cyp24a1 mRNA levels (Fig. 9 A). PTH‐PTH 1 Receptor (PTH1R) signaling activates the protein kinase A (PKA) pathway, among others, that leads to the phosphorylation of cAMP response element binding protein (CREB) followed by phospho‐CREB (pCREB) translocation to the nucleus, where it drives PTH‐mediated gene expression. However, previous in vitro studies utilizing the UMR106 cell line have suggested that PTH upregulates Fgf23 transcription and that this action may be secondary to and mediated by induction of the orphan nuclear receptor NURR1.51 Although we did not observe pCREB binding in response to PTH in the −16kb region in our previous ChIP‐seq analysis,39 there are NURR1 binding motifs in the −16kb region. To test whether PTH mediates Fgf23 expression via the −16kb region, we injected Fgf23 −16KO mice and their WT littermates with 230 ng/g bw PTH. As expected, PTH increased Tnfsf11 (RANKL) mRNA levels similarly in both genotypes, confirming equivalent treatment of all mice (Fig. 9 B). WT mice injected with PTH showed a twofold increase in Fgf23 mRNA levels in bones (Fig. 9 B) but not in other tissues such as kidney (data not shown). Moreover, lack of the −16kb distal enhancer of Fgf23 prevented this PTH‐induced increase in Fgf23 mRNA levels (Fig. 9 B). However, in line with a recent report from Knab and colleagues,52 in our experiment acute single PTH injection did not increase plasma iFGF23 levels in WT or Fgf23 −16KO mice (Fig. 9 C), providing another example of discordance between transcriptional regulation and processing. Collectively, our results suggest that although both PTH and inflammation mediate bone Fgf23 transcription via the −16kb region, the effects of PTH on Fgf23 levels are restricted to bone and are much milder compared with that of inflammation.
Discussion
Previous studies have shown that inflammation and calciotropic hormones increase Fgf23 transcription.18, 21, 23, 51, 52, 53, 54 However, the molecular mechanisms underpinning the transcriptional regulation of Fgf23 in response to inflammation and PTH in bone or non‐osseous tissues are unknown. Herein, we utilized unbiased genome‐wide analysis of epigenetic marks to identify several potential enhancers for Fgf23 and by deletion of this enhancer in vivo showed that a distal enhancer located −16kb upstream of the Fgf23 gene's TSS mediates basal‐, inflammation‐, and PTH‐induced expression of Fgf23 in a tissue‐specific manner. Because both inflammation and PTH are suggested to mediate FGF23 levels in CKD, we employed a diet‐induced CKD model and showed that the −16kb enhancer mediates induction of Fgf23 in thymus, kidney, and bone and that this led to a blunting of the increase in iFGF23 levels in circulation at early onset of this kidney disease model. Collectively, our findings support a complex role of the −16 kb Fgf23 enhancer in mediating inflammatory and other effects of Fgf23 regulation.
Importantly, this study describes the initial phases of a new approach whereby the locations of potential enhancers are identified first through unbiased genetic and/or epigenetic ChIP‐seq analyses and then explored subsequently through loss‐of‐function studies directly in the mouse in vivo. The advantages of this approach are extensive, as they enabled us to 1) link putative enhancers directly to their target genes via genomic and RNA output activity in vivo; 2) establish primary functions for enhancers; 3) identify new and unsuspected roles for individual enhancers; 4) assess activity across multiple tissues, thereby enabling the study of selective activity; and 5) avoid detailed analyses of mechanism (definition of transcription factor participation, activation, and upstream signaling processes) until such time as the regulatory regions are fully validated in vivo. This new approach is necessary because recent studies have now shown that most genes are regulated by multiple enhancers, many of which are located at sites distal to the genes they regulate and because promoter proximal studies utilized for the past several decades are now known to be highly biased and more frequently unreliable. As outlined here, we have now established that the −16kb enhancer represents a bone fide regulatory segment of the Fgf23 gene and that at least one of its activities is to facilitate the upregulatory activity of inflammatory mediators such as LPS, IL‐1β, and Tnfα, as well as the calciotropic hormone PTH at multiple tissues, many of which were not suspected to contribute to FGF23 production until now. This approach has also enabled us to define a disease role for the −16kb enhancer as a mediator of Fgf23 upregulation in a mouse model of the early stages of CKD. The results hint at a direct primary role for inflammation in that upregulation, but more importantly, now provide an experimental approach for defining the molecular details of that upregulation in vivo. Further studies are now focused on the activities of the additional putative regulatory regions within and surrounding the Fgf23 gene to understand their potential roles in mediating the activities of additional systemic regulators such as phosphate and 1,25(OH)2D3. Once these are established, studies can focus on the mechanistic details that relate to the identities of the transcription factors that are involved, the recruitment of epigenetic factors that may be involved in modulating Fgf23 output, and delineation of the upstream signaling pathways that transduce environmental information as to the physiologic or pathophysiologic state of the organism.
Because the highest concentration of FGF23 is produced by osteocytes, the regulation of FGF23 synthesized in non‐osseous tissues has been largely unexplored. However, elevations in the non‐osseous expression of Fgf23 have been observed in multiple diseases and in animal models of disease. For example, studies by Zanchi and colleagues have recently shown increases in renal Fgf23 expression in a model of progressive kidney injury induced by diabetes.55 In more recent studies, Bansal and colleagues and Wongdee and colleagues have shown elevations in splenic23 and intestinal56 expression of Fgf23. Herein, we provide additional evidence that acute inflammation can induce Fgf23 expression in several non‐osseous tissues such as thymus, spleen, bone marrow, kidney, intestine, lung, and liver and show that Fgf23 is increased even at early stages in the oxalate diet‐induced CKD model, not only in bone but also in non‐osseous tissues such as thymus and kidney. Moreover, although we show that both PTH and inflammation can increase Fgf23 levels in bone, only inflammation appears capable of inducing non‐osseous Fgf23 expression, suggesting that the higher Fgf23 levels observed in CKD bones may be due to elevated PTH or inflammation, whereas the increased non‐osseous Fgf23 levels are likely due to increased inflammation. Taken together, these results suggest that Fgf23 elevations are not limited to osseous tissues in inflammatory disorders and point out the need for further research on the pathological roles of these non‐osseous sources of FGF23.
Osteocytes are the major source of FGF23 in bones under physiological conditions and in response to inflammatory stimuli.18, 19, 57 In contrast, macrophages21 and dendritic cells21, 22 have been suggested to be the main source of FGF23 in spleen and bone marrow under normal or inflammatory conditions. Because the majority of the tissues contain tissue‐resident macrophages that play many roles including iron processing and immune response,58 macrophages may represent a potential source of non‐osseous FGF23 induced by inflammation. Importantly, macrophages from various tissue sources are not identical, and indeed the properties of resident macrophages can take on genetic and epigenetic features as determined by the host tissue. Thus, whether the inflammation‐induced Fgf23 elevations in non‐osseous tissues are due to migration of Fgf23‐expressing immune cells into these tissues or to induction of Fgf23 expression in cell types residing in these tissues is unknown and will require further investigation.
Several studies have shown that Fgf23 expression is mediated by inflammatory cytokines; however, the molecular mechanisms underpinning this regulation are unclear. Herein, we utilized ChIP‐seq analysis of epigenetic marks and identified the −16kb enhancer, which contains multiple binding motifs for transcription factors, including HIF1α, NF‐κB, and STATs that have been suggested to mediate Fgf23 transcription in response to inflammation.(18,19,21) Deletion of the −16kb enhancer of Fgf23 significantly reduced but did not completely prevent inflammatory induction of Fgf23 in bone or in other tissues, suggesting the presence of additional regulatory elements that control the output of this gene. Moreover, our results also show that the overall role of the −16kb region in controlling inflammation‐induced Fgf23 expression may be different in some tissues. Although this is likely to be due to the differential interaction of residual transcription factors that bind this enhancer, it is possible that additional enhancers may be present in other tissues that impact this differential activity. ChIP‐seq analysis of additional cell lineage types is in progress to evaluate this possibility. Given the rather ubiquitous ability of inflammatory signals to regulate FGF23 expression, we would predict that the −16 kb enhancer will likely be present in most tissues.
Our previous studies have also shown that deletion of a single enhancer can alter the transcriptional contribution of other enhancers linked to the same gene.35 Thus, although we have observed a blunting of the inflammatory response of Fgf23 expression in Fgf23 −16KO mice and identified possible motifs for several candidate transcription factors at this site, we cannot rule out the possibility that the −16kb enhancer may also function by altering the activity of additional regulatory regions playing a role in Fgf23 gene regulation. This type of interrelationship could also explain the variable contribution of the −16kb enhancer to Fgf23 expression in different tissues, although this may also be due to the actions of LPS, IL‐1β, and/or Tnfα to modulate different collections of transcription factors at the Fgf23 gene in the individual tissues. It is for these reasons that we anticipate delineating the activities of each of the additional regulatory regions of the Fgf23 gene before conducting detailed ChIP‐seq analysis to identify the multiple transcription factor candidates that are likely to play roles in mediating these tissue‐specific physiological as well as inflammatory responses. As stated earlier, this represents a largely new paradigm for studying transcriptional regulation that is based upon functional validation of enhancers in tissues in vivo before in‐depth analysis of the transcription factors and signaling pathways that may be involved.
Both inflammation and PTH have been shown to increase Fgf23 production18, 19, 50, 52 and therefore may be potential inducers of Fgf23 expression in CKD. Herein, we show that lack of the −16kb enhancer of Fgf23 prevented early onset kidney disease‐induced increases in renal, thymic, and skeletal Fgf23 expression in a diet‐induced CKD model. Although the expression of inflammatory cytokines such as TNFα and IL‐1β are induced in multiple tissues in our oxalate diet‐induced disease model, circulating PTH was also increased. In the experimental setting of an acute single injection, inflammation increases both Fgf23 mRNA levels and circulating iFGF23 in the blood at higher magnitudes compared with induction of Fgf23 by a single injection of PTH. Nevertheless, PTH levels in CKD are much higher and are sustained for a longer duration compared with those resulting from an acute single PTH injection. They also occur in a setting of uremia; therefore, PTH cannot be ruled out as a potential regulator of the osseous production of Fgf23 in CKD. Because both inflammation and PTH utilize the same −16kb region to mediate Fgf23 transcription, further refined dissection of the −16kb enhancer will be necessary to identify which of these Fgf23 mediators represents the primary inducer of FGF23 in CKD.
In summary, our data indicate that inflammation‐induced Fgf23 expression is not limited to osseous tissues, and although induction of both skeletal and non‐osseous tissue sources of Fgf23 is mediated via the −16kb distal enhancer, transcriptional regulation of Fgf23 gene may also be controlled by additional enhancers operating in concert. Moreover, whereas PTH regulation of Fgf23 levels is less robust compared with inflammatory regulation, it is also mediated by the −16kb region. Finally, transcriptional regulation of Fgf23, as exemplified here by the oxalate diet‐induced CKD model, is similarly controlled by the −16kb enhancer. Thus, this novel enhancer‐deleted murine model may be useful in future studies for examining the role of FGF23 elevations in CKD and other inflammatory diseases.
Disclosures
All authors state that they have no conflicts of interest.
Supporting information
Supporting Figure S1.
Supporting Figure S2.
Supporting Figure S3.
Supporting Figure S4.
Supporting Table S1.
Supporting Table S2.
Acknowledgments
We thank members of the UW Biochemistry Animal Support Staff for their contributions.
This work was supported by NIH grant AR064424 awarded to JWP.
Authors’ roles: Study design: JWP, MO, and AHC. Study conduct: all authors with the lead of MO and AHC. Data collection: all authors with the lead of MO and AHC. Data analysis: MO and AHC. Statistical analysis: MO and JDT. Data interpretation: all authors. Drafting manuscript: JWP and MO. Revising manuscript content: JWP and MO. Approving final version of manuscript: all authors. JWP and MO take responsibility for the integrity of the data analysis.
References
- 1. Liu S, Guo R, Simpson LG, Xiao ZS, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003; 278 (39):37419–26. [DOI] [PubMed] [Google Scholar]
- 2. Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF23 as a causative factor of tumor‐induced osteomalacia. Proc Natl Acad Sci U S A. 2001; 98 (11):6500–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. White KE, Carn G, Lorenz‐Depiereux B, Benet‐Pages A, Strom TM, Econs MJ. Autosomal‐dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF‐23. Kidney Int. 2001; 60 (6):2079–86. [DOI] [PubMed] [Google Scholar]
- 4. Fukumoto S. Post‐translational modification of fibroblast growth factor 23. Ther Apher Dial. 2005; 9 (4):319–22. [DOI] [PubMed] [Google Scholar]
- 5. Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor‐23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. 2005; 90 (3):1519–24. [DOI] [PubMed] [Google Scholar]
- 6. Larsson T, Marsell R, Schipani E, et al. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004; 145 (7):3087–94. [DOI] [PubMed] [Google Scholar]
- 7. Benet‐Pagès A, Orlik P, Strom TM, Lorenz‐Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005; 14 (3):385–90. [DOI] [PubMed] [Google Scholar]
- 8. Isakova T, Xie H, Yang W, et al. Fibroblast growth factor 23 and risks of mortality and end‐stage renal disease in patients with chronic kidney disease. JAMA. 2011; 305 (23):2432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chathoth S, Al‐Mueilo S, Cyrus C, et al. Elevated fibroblast growth factor 23 concentration: prediction of mortality among chronic kidney disease patients. Cardiorenal Med. 2015; 6 (1):73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wolf M, Molnar MZ, Amaral AP, et al. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J Am Soc Nephrol. 2011; 22 (5):956–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. El‐Hodhod MA, Hamdy AM, Abbas AA, Moftah SG, Ramadan AA. Fibroblast growth factor 23 contributes to diminished bone mineral density in childhood inflammatory bowel disease. BMC Gastroenterol. 2012; 12:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011; 79 (12):1370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mehta R, Cai X, Lee J, et al. Association of fibroblast growth factor 23 with atrial fibrillation in chronic kidney disease, from the Chronic Renal Insufficiency Cohort Study. JAMA Cardiol. 2016; 1 (5):548–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011; 121 (11):4393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chesney PJ, Davis JP, Purdy WK, Wand PJ, Chesney RW. Clinical manifestations of toxic shock syndrome. JAMA. 1981; 246(7):741–8. [PubMed] [Google Scholar]
- 16. Riedler GF, Scheitlin WA. Hypophosphataemia in septicaemia: higher incidence in gram‐negative than in gram‐positive infections. Br Med J. 1969; 1(5646):753–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barak V, Schwartz A, Kalickman I, Nisman B, Gurman G, Shoenfeld Y. Prevalence of hypophosphatemia in sepsis and infection: the role of cytokines. Am J Med. 1998; 104 (1):40–7. [DOI] [PubMed] [Google Scholar]
- 18. Ito N, Wijenayaka AR, Prideaux M, et al. Regulation of FGF23 expression in IDG‐SW3 osteocytes and human bone by pro‐inflammatory stimuli. Mol Cell Endocrinol. 2015; 399:208–18. [DOI] [PubMed] [Google Scholar]
- 19. David V, Martin A, Isakova T, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016; 89 (1):135–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF‐23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000; 277 (2):494–8. [DOI] [PubMed] [Google Scholar]
- 21. Masuda Y, Ohta H, Morita Y, et al. Expression of Fgf23 in activated dendritic cells and macrophages in response to immunological stimuli in mice. Biol Pharm Bull. 2015; 38 (5):687–93. [DOI] [PubMed] [Google Scholar]
- 22. Nakashima Y, Mima T, Yashiro M, et al. Expression and localization of fibroblast growth factor (FGF)23 and Klotho in the spleen: its physiological and functional implications. Growth Factors. 2016; 34(5–6):196–202. [DOI] [PubMed] [Google Scholar]
- 23. Bansal S, Friedrichs WE, Velagapudi C, et al. Spleen contributes significantly to increased circulating levels of fibroblast growth factor23 in response to lipopolysaccharide‐induced inflammation. Nephrol Dial Transplant. 2017; 32 (3):583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Riminucci M, Collins MT, Fedarko NS, et al. FGF‐23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003; 112 (5):683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaneko I, Saini RK, Griffin KP, Whitfield GK, Haussler MR, Jurutka PW. FGF23 gene regulation by 1,25‐dihydroxyvitamin D: opposing effects in adipocytes and osteocytes. J Endocrinol. 2015; 226 (3):155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saito H, Maeda A, Ohtomo S, et al. Circulating FGF‐23 is regulated by 1alpha,25‐dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. 2005; 280 (4):2543–9. [DOI] [PubMed] [Google Scholar]
- 27. Kolek OI, Hines ER, Jones MD, et al. 1alpha,25‐Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal‐gastrointestinal‐skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol. 2005; 289 (6):G1036–42. [DOI] [PubMed] [Google Scholar]
- 28. Yu X, Sabbagh Y, Davis SI, Demay MB, White KE. Genetic dissection of phosphate‐ and vitamin D‐mediated regulation of circulating Fgf23 concentrations. Bone. 2005; 36 (6):971–7. [DOI] [PubMed] [Google Scholar]
- 29. Zhang Q, Doucet M, Tomlinson RE, et al. The hypoxia‐inducible factor‐1α activates ectopic production of fibroblast growth factor 23 in tumor‐induced osteomalacia. Bone Res. 2016; 4:16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Onal M, St John HC, Danielson AL, Pike JW. Deletion of the distal Tnfsf11 RL‐D2 enhancer that contributes to PTH‐mediated RANKL expression in osteoblast lineage cells results in a high bone mass phenotype in mice. J Bone Miner Res. 2016; 31 (2):416–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Onal M, St John HC, Danielson AL, Markert JW, Riley EM, Pike JW. Unique distal enhancers linked to the mouse Tnfsf11 gene direct tissue‐specific and inflammation‐induced expression of RANKL. Endocrinology. 2016; 157 (2):482–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Galli C, Zella LA, Fretz JA, et al. Targeted deletion of a distant transcriptional enhancer of the receptor activator of nuclear factor‐kappaB ligand gene reduces bone remodeling and increases bone mass. Endocrinology. 2008; 149 (1):146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bishop KA, Wang X, Coy HM, Meyer MB, Gumperz JE, Pike JW. Transcriptional regulation of the human TNFSF11 gene in T cells via a cell type‐selective set of distal enhancers. J Cell Biochem. 2015; 116 (2):320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee SM, Bishop KA, Goellner JJ, O'Brien CA, Pike JW. Mouse and human BAC transgenes recapitulate tissue‐specific expression of the vitamin D receptor in mice and rescue the VDR‐null phenotype. Endocrinology 2011;2064 –76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meyer MB, Benkusky NA, Pike JW. Selective distal enhancer control of the Mmp13 gene identified through clustered regularly interspaced short palindromic repeat (CRISPR) genomic deletions. J Biol Chem. 2015; 290 (17):11093–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meyer MB, Benkusky NA, Onal M, Pike JW. Selective regulation of Mmp13 by 1,25(OH)2D3, PTH, and Osterix through distal enhancers. J Steroid Biochem Mol Biol. 2016; 164:258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang H, Yang H, Shivalila CS, et al. One‐step generation of mice carrying mutations in multiple genes by CRISPR/Cas‐mediated genome engineering. Cell. 2013; 153 (4):910–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meyer M, de Angelis MH, Wurst W, Kühn R. Gene targeting by homologous recombination in mouse zygotes mediated by zinc‐finger nucleases. Proc Natl Acad Sci U S A. 2010; 107(34):15022–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. St John HC, Meyer MB, Benkusky NA, et al. The parathyroid hormone‐regulated transcriptome in osteocytes: parallel actions with 1,25‐dihydroxyvitamin D3 to oppose gene expression changes during differentiation and to promote mature cell function. Bone. 2015; 72:81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meyer MB, Benkusky NA, Sen B, Rubin J, Pike JW. Epigenetic plasticity drives adipogenic and osteogenic differentiation of marrow‐derived mesenchymal stem cells. J Biol Chem. 2016; 291 (34):17829–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. St John HC, Bishop KA, Meyer MB, et al. The osteoblast to osteocyte transition: epigenetic changes and response to the vitamin D3 hormone. Mol Endocrinol. 2014; 28 (7):1150–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meyer MB, Goetsch PD, Pike JW. VDR/RXR and TCF4/β‐catenin cistromes in colonic cells of colorectal tumor origin: impact on c‐FOS and c‐MYC gene expression. Mol Endocrinol. 2012; 26 (1):37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Frazer K, Pachter L, Poliakov A, Rubin E, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004; 32(Web Server issue):W273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martowicz ML, Meyer MB, Pike JW. The mouse RANKL gene locus is defined by a broad pattern of histone H4 acetylation and regulated through distinct distal enhancers. J Cell Biochem. 2011; 112 (8):2030–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bishop KA, Coy HM, Nerenz RD, Meyer MB, Pike JW. Mouse Rankl expression is regulated in T cells by c‐Fos through a cluster of distal regulatory enhancers designated the T cell control region. J Biol Chem. 2011; 286 (23):20880–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Munoz Mendoza J, Isakova T, Cai X, et al. Inflammation and elevated levels of fibroblast growth factor 23 are independent risk factors for death in chronic kidney disease. Kidney Int. 2017; 91(3):711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Munoz Mendoza J, Isakova T, Ricardo AC, et al. Fibroblast growth factor 23 and inflammation in CKD. Clin J Am Soc Nephrol. 2012; 7(7):1155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mulay SR, Eberhard JN, Pfann V, et al. Oxalate‐induced chronic kidney disease with its uremic and cardiovascular complications in C57BL/6 mice. Am J Physiol Renal Physiol. 2016; 10 (8):F785–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stubbs JR, He N, Idiculla A, et al. Longitudinal evaluation of FGF23 changes and mineral metabolism abnormalities in a mouse model of chronic kidney disease. J Bone Miner Res. 2012; 27 (1):38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lavi‐Moshayoff V, Wasserman G, Meir T, Silver J, Naveh‐Many T. PTH increases FGF23 gene expression and mediates the high‐FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol. 2010; 299 (4):F882–9. [DOI] [PubMed] [Google Scholar]
- 51. Meir T, Durlacher K, Pan Z, et al. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014; 86 (6):1106–15. [DOI] [PubMed] [Google Scholar]
- 52. Knab VM, Corbin B, Andrukhova O, et al. Acute parathyroid hormone injection increases c‐terminal, but not intact fibroblast growth factor 23 levels. Endocrinology. 2017; 158 (5):1130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Katoh Y, Katoh M. Comparative genomics on mammalian Fgf6‐Fgf23 locus. Int J Mol Med. 2005; 16 (2):355–8. [PubMed] [Google Scholar]
- 54. Egli‐Spichtig D, Imenez Silva PH, Gehring N, et al. TNFα triggers renal FGF23 expression and elevates systemic FGF23 levels in mouse models of chronic kidney disease. J Bone Miner Res. 2016; 31 (Suppl 1). [Google Scholar]
- 55. Zanchi C, Locatelli M, Benigni A, et al. Renal expression of FGF23 in progressive renal disease of diabetes and the effect of ACE inhibitor. PLoS One. 2013; 8 (8):e70775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wongdee K, Teerapornpuntakit J, Sripong C, et al. Intestinal mucosal changes and upregulated calcium transporter and FGF‐23 expression during lactation: contribution of lactogenic hormone prolactin. Arch Biochem Biophys. 2016; 590:109–17. [DOI] [PubMed] [Google Scholar]
- 57. Bonewald LF, Wacker MJ. FGF23 production by osteocytes. Pediatr Nephrol. 2013; 28 (4):563–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue‐resident macrophages. Nat Immunol. 2013; 14 (10):986–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Figure S1.
Supporting Figure S2.
Supporting Figure S3.
Supporting Figure S4.
Supporting Table S1.
Supporting Table S2.