

Abstract
Glioblastoma multiforme are mortifying brain tumors that contain a subpopulation of tumor cells with stem-like properties, termed as glioblastoma stem-like cells (GSCs). These GSCs constitute an autonomous reservoir of aberrant cells able to initiate, maintain, and repopulate the tumor mass. A new therapeutic strategy would consist of targeting the GSC population. The GSCs are situated in perivascular niches, closely associated with brain microvascular endothelial cells thereby involved in bidirectional molecular and cellular interactions. In this scenario, the endothelium not only supplies oxygen and necessary nutrients but also seeds a protective microenvironment for tumor growth. Although GSC fate, plasticity, and survival are regulated by external cues emanating from endothelial cells, the nature of such angiocrine signals remains unknown. Our laboratory conclusively demonstrated that brain endothelial cells positively control the expansion of GSCs.1 Notably, we found that GSCs are addicted to the hormonal peptide apelin (APLN) secreted by surrounding endothelial cells, and identified the APLN/APLNR nexus as a promising druggable network in glioblastoma.
Keywords: Glioblastoma, apelin, GPCR, endothelium
Comment on: Harford-Wright E, Andre-Gregoire G, Jacobs KA, Treps L, Le Gonidec S, Leclair HM, Gonzalez-Diest S, Roux Q, Guillonneau F, Loussouarn D, Oliver L, Vallette FM, Foufelle F, Valet P, Davenport AP, Glen RC, Bidere N, Gavard J. Pharmacological targeting of apelin impairs glioblastoma growth. Brain. 2017 Nov 1;140(11):2939-2954. doi: 10.1093/brain/awx253. PubMed PMID: 29053791.
Glioblastoma is the most common primary brain tumor in adults, which is associated with an extremely poor prognosis, with a median survival following diagnosis of 15 months.2 Glioblastoma are aggressive tumors, characterized by areas of rapid cell proliferation, angiogenesis, and necrosis, reflecting the aggressive nature of this disease. To date, standard treatment for glioblastoma involves surgical resection of as much of the tumor bulk as possible, followed by radiotherapy and chemotherapy to eliminate the remaining cells. However, these treatments are palliative in nature, and tumors fatally relapse within 7 to 10 months. As such, long-term survival rates for glioblastoma patients have not significantly improved over the past decade.3
Glioblastoma are associated with significant inter- and intratumoral heterogeneity. Within glioblastoma exists a population of tumor-initiating cells also named as glioblastoma stem-like cells (GSCs) that have a proposed role in tumor initiation, resistance to current therapies, invasion, and angiogenesis.4–6 Although some debates on the origin and definition of GSCs remain, the presence of these cells within specific niches within tumors is now well accepted.4 The GSCs interact with the tumor microenvironment, residing in hypoxic niches near the endothelial vessels.7–10 The localization of GSCs in these vascular niches facilitates communication between endothelial cells and GSCs, enabling a privileged access for GSCs to tumor endothelium-secreted factors. Indeed, soluble growth factors released from the endothelium, collectively termed as “angiocrines,” have been reported in various physiological and pathological conditions.11,12 However, the specific endothelial-secreted factors involved in the maintenance of GSCs are yet to be identified. Characterization of the protein secretome involved in the expansion of GSCs thus has considerable implications in the improvement of treatment options for glioblastoma patients.
To study the molecular basis of this cross talk, we first developed an original in vitro model by co-culturing human brain endothelial cells with patient-derived GSCs.8,9,13,14 We demonstrated that factors secreted by brain endothelial cells positively control the expansion and survival of GSCs, and vice versa, GSCs modulate the endothelial behavior. Recent research from our laboratory has further suggested that the vasoactive peptide apelin (APLN) may be a central regulator of GSC maintenance.1 This short peptide is one of the endogenous ligands of the G protein–coupled receptor APJ (APLNR)15 and is widely expressed throughout various tissues, including the brain.16 Apelin was identified by mass spectrometry from the brain endothelial secretome and found expressed in glioblastoma patient tissue in close proximity to blood vessels.1 In vitro, the addition of exogenous apelin was able to sustain GSC growth via its receptor APJ. Knocking down the expression level of APJ in GSCs via small interfering RNA/short hairpin RNA (shRNA) indeed curbs the effects of endothelial cell–conditioned media and of apelin alone. Likewise, pharmacologic inhibition of APJ with a novel competitive antagonist bicyclic peptide MM5417 inhibited endothelial-mediated GSC expansion in vitro. In addition, when tested in vivo in 2 mouse models of tumor growth, MM54 was found to be safe and effective in reducing tumor growth and increasing survival of GSC-implanted mice. Notably, no obvious adverse effects on the cardiovascular parameters were noted on repeated injections. Together, these results suggest that endothelial-secreted apelin may act as a paracrine signal that sustains GSCs and therefore represents a possible novel therapeutic target for the treatment of glioblastoma.
Mechanistically, the precise molecules mediating the effect of endothelial apelin on GSCs requires further investigation. Although the in vitro data clearly demonstrate that apelin directly sustains GSC spheres, the reduction in tumor growth in vivo following administration of the apelin antagonist could be explained by either a direct or an indirect effect on GSCs. However, it does appear that glycogen synthase kinase 3β (GSK3β) signaling may play a role in this process downstream of APJ.1 Moreover, phosphorylation of GSK3β was observed both in vitro and in vivo, suggesting an inhibitory effect of the apelin antagonist on GSK3β signaling.1 Indeed, the function of GSK3β signaling has been previously reported to play varied roles in malignancy, depending on the substrate and tumor type. It, has been previously reported that GSK3β has a role in cancer stem cell self-renewal and glioblastoma tumorigenesis.18 Consistent with the proposed mechanism of apelin in GSC maintenance, administration of the GSK3β competitive inhibitor Tideglusib is effective in a preclinical model of glioblastoma, with inhibition of GSK3β found to inhibit GSC self-renewal and sensitize glioblastoma to temozolomide (TMZ), the alkylating agent used in clinics, without effecting normal astrocytes.18 Likewise, the apelin receptor antagonist MM54 reduces GSC self-renewal and aggravate the effects of the chemotherapeutic agent TMZ.1 Collectively, these data suggest that apelin may directly affect GSCs by inhibition of GSK3β signaling. Future work will be designed to precisely address the signaling complexes engaged downstream of APJ in GSCs, in vitro and in vivo.
An alternate argument is that the observed reduction in tumor volume associated with MM54 administration may be due to a reduction in angiogenesis. Apelin has previously been implicated in angiogenesis and is reported to induce vessel sprouting and stabilization of contacts between endothelial cells.15,16 Indeed, apelin has a proposed role in tumor angiogenesis and response to anti-angiogenic therapies, with apelin messenger RNA reported to be elevated in patients who do not respond to anti-angiogenic therapy.19 Moreover, apelin expression has been positively correlated with increased microvessel densities and subsequent tumor growth in human non–small-cell lung carcinoma.20 It is well established that tumors rely heavily on neo-angiogenesis to receive the nutrients they require to survive.21 Consequently, the observed effect on tumor growth by blocking apelin in vivo may also be associated with an anti-angiogenic effect rather than by directly targeting the GSCs. However, the weight of the in vitro data suggests that endothelial-derived apelin has a clear role in the maintenance of these human GSCs.1 Moreover, implantation of GSCs in which the apelin receptor has been silenced while left intact in host endothelial cells demonstrated a reduction in tumor size compared with shRNA control groups, a result which cannot be explained by apelin-mediated changes toward angiogenesis.
Although impressive results were obtained with the MM54 compound in xenografted mice, it is important to note that both genetic and pharmacological evidence for the role of APJ in glioma growth were established in immuno-compromised animals. In keeping with this idea, recent published data suggest that in melanoma, point mutations of the APLNR gene are associated with a failure of targeted immunotherapies22 indicating that the interaction between APLNR and the immune system may warrant further investigation. Nonetheless, tumor growth in vivo is a complicated and multifaceted process that is rarely due to one factor or mechanism alone, and compounds that target multiple aspects of tumorigenesis may prove extremely beneficial. Together, the results of this study highlight the potential of endothelial-derived apelin as an exciting target for glioma growth (Figure 1).
Figure 1.
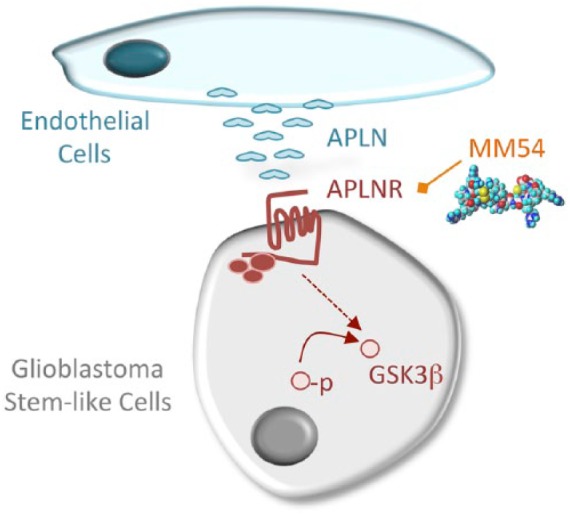
Proposed model for the angiocrine action of APLN in glioma growth. Endothelial cells produce and release the hormonal peptide apelin (APLN), which operates on GSCs, through its cognate receptor the G protein–coupled receptor APLNR. The APLN/APLNR complex allows the activation of the glycogen synthase kinase, GSK3β (nonphosphorylated on S9) and maintains in turn the stemness properties of GSCs. The competitive antagonist MM54 counteracts the effect of endothelial cells on GSCs and reduces tumor growth in mice. GSCs indicate glioblastoma stem-like cells.
Acknowledgments
The authors thank the members of the SOAP team (Nantes, France).
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Ligue nationale contre le cancer comité de Loire-Atlantique et Vendée (J.G) and Region Pays de la Loire et Nantes Metropole under Connect Talent Grant (J.G).
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: EHW wrote the manuscript; JG edited the text and prepared the figure.
ORCID iD: Julie Gavard  https://orcid.org/0000-0002-7985-9007
https://orcid.org/0000-0002-7985-9007
References
- 1. Harford-Wright E, Andre-Gregoire G, Jacobs KA, et al. Pharmacological targeting of apelin impairs glioblastoma growth. Brain. 2017;140:2939–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. [DOI] [PubMed] [Google Scholar]
- 3. Stupp R, Taillibert S, Kanner AA, et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA. 2015;314:2535–2543. [DOI] [PubMed] [Google Scholar]
- 4. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hale JS, Sinyuk M, Rich JN, Lathia JD. Decoding the cancer stem cell hypothesis in glioblastoma. CNS Oncol. 2013;2:319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yan K, Yang K, Rich JN. The evolving landscape of glioblastoma stem cells. Curr Opin Neurol. 2013;26:701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. [DOI] [PubMed] [Google Scholar]
- 8. Galan-Moya EM, Le Guelte A, Lima Fernandes E, et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep. 2011;12:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jacobs KA, Harford-Wright E, Gavard J. Neutralizing gp130 interferes with endothelial-mediated effects on glioblastoma stem-like cells. Cell Death Differ. 2017;24:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin X, Kim LJY, Wu Q, et al. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat Med. 2017;23:1352–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ding BS, Nolan DJ, Butler JM, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature. 2010;468:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cao Z, Ye T, Sun Y, et al. Targeting the vascular and perivascular niches as a regenerative therapy for lung and liver fibrosis. Sci Transl Med. 2017;9:eaai8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Treps L, Edmond S, Harford-Wright E, et al. Extracellular vesicle-transported semaphorin3A promotes vascular permeability in glioblastoma. Oncogene. 2016;35:2615–2623. [DOI] [PubMed] [Google Scholar]
- 14. Le Guelte A, Galan-Moya EM, Dwyer J, et al. Semaphorin 3A elevates endothelial cell permeability through PP2A inactivation. J Cell Sci. 2012;125:4137–4146. [DOI] [PubMed] [Google Scholar]
- 15. Pitkin SL, Maguire JJ, Kuc RE, Davenport AP. Modulation of the apelin/APJ system in heart failure and atherosclerosis in man. Br J Pharmacol. 2010;160:1785–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kalin RE, Kretz MP, Meyer AM, Kispert A, Heppner FL, Brandli AW. Paracrine and autocrine mechanisms of apelin signaling govern embryonic and tumor angiogenesis. Dev Biol. 2007;305:599–614. [DOI] [PubMed] [Google Scholar]
- 17. Macaluso NJ, Pitkin SL, Maguire JJ, Davenport AP, Glen RC. Discovery of a competitive apelin receptor (APJ) antagonist. ChemMedChem. 2011;6:1017–1023. [DOI] [PubMed] [Google Scholar]
- 18. Zhou A, Lin K, Zhang S, et al. Nuclear GSK3beta promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat Cell Biol. 2016;18:954–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zuurbier L, Rahman A, Cordes M, et al. Apelin: a putative novel predictive biomarker for bevacizumab response in colorectal cancer. Oncotarget. 2017;8:42949–42961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berta J, Kenessey I, Dobos J, et al. Apelin expression in human non-small cell lung cancer: role in angiogenesis and prognosis. J Thorac Oncol. 2010;5:1120–1129. [DOI] [PubMed] [Google Scholar]
- 21. Azzi S, Hebda JK, Gavard J. Vascular permeability and drug delivery in cancers. Front Oncol. 2013;3:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Patel SJ, Sanjana NE, Kishton RJ, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548:537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]