

Abstract
Bacterial O-antigens are synthesized on lipid carriers before being transferred to lipopolysaccharide core structures. Rhizobium etli CE3 lipopolysaccharide is a model for understanding O-antigen biological function. CE3 O-antigen structure and genetics are known. However, proposed enzymology for CE3 O-antigen synthesis has been examined very little in vitro, and even the sugar added to begin the synthesis is uncertain. A model based on mutagenesis studies predicts that 2-acetamido-2,6-dideoxy-d-glucose (QuiNAc) is the first O-antigen sugar and that genes wreV, wreQ and wreU direct QuiNAc synthesis and O-antigen initiation. Previously, synthesis of UDP-QuiNAc was shown to occur in vitro with a WreV orthologue (4,6-hexose dehydratase) and WreQ (4-reductase), but the WreQ catalysis in this conventional deoxyhexose-synthesis pathway was very slow. This seeming deficiency was explained in the present study after WreU transferase activity was examined in vitro. Results fit the prediction that WreU transfers sugar-1-phosphate to bactoprenyl phosphate (BpP) to initiate O-antigen synthesis. Interestingly, WreU demonstrated much higher activity using the product of the WreV catalysis [UDP-4-keto-6-deoxy-GlcNAc (UDP-KdgNAc)] as the sugar-phosphate donor than using UDP-QuiNAc. Furthermore, the WreQ catalysis with WreU-generated BpPP-KdgNAc as the substrate was orders of magnitude faster than with UDP-KdgNAc. The inferred product BpPP-QuiNAc reacted as an acceptor substrate in an in vitro assay for addition of the second O-antigen sugar, mannose. These results imply a novel pathway for 6-deoxyhexose synthesis that may be commonly utilized by bacteria when QuiNAc is the first sugar of a polysaccharide or oligosaccharide repeat unit: UDP-GlcNAc → UDP-KdgNAc → BpPP-KdgNAc → BpPP-QuiNAc.
Keywords: Rhizobium, quinovosamine, O-antigen, biosynthesis, bactoprenyl-phosphate, deoxysugar
Introduction
O polysaccharide, or O-antigen, is the outermost component of the lipopolysaccharide (LPS) that is the major constituent of the outer leaflet of the outer membrane in bacteria [1]. Bacterial mutants lacking O-antigen have deficiencies that reveal the profound physiological and ecological importance of this portion of LPS [2–8]. For instance, complete and abundant O-antigen (Fig. 1a) of the model bacterium of this study, Rhizobium etli strain CE3, is indispensable for infection and development of nitrogen-fixing root nodules on its legume host, Phaseolus vulgaris [9–11].
Fig. 1.

(a) Structure of Rhizobium etli CE3 O-antigen. The O-antigen structure of R. etli LPS is shown linked to the lipid A core. Abbreviations for the sugars: QuiNAc, N-acetyl-d-quinovosamine; Man, mannose; Fuc, fucose; MeGlcA, methyl-glucuronate; 3OMe6dTal, 3-O-methyl-6-deoxytalose; terminal residue, TOMFuc, 2,3,4-tri-O-methylfucose or DOMFuc, 2,3-di-O-methylfucose. The proposed first O-antigen sugar, QuiNAc, is highlighted. (b) R. etli CE3 O-antigen genetic clusters. Upper panel: the chromosomal wre gene cluster (previously called lps region α) spanning nucleotides 784 527 to 812 262 of the genome sequence consists of 25 predicted ORFs. Another chromosomal ORF (wreQ) spanning nucleotides 2 969 313 to 2 970 242 is required for QuiNAc synthesis [27]. Lower panel: a 4-kilobase cluster on plasmid pCFN42b consists of three predicted ORFs. In each panel the predicted GTase-encoding genes are in grey. Genes encoding enzymes studied in the current work, wreG, wreQ, wreU and wreV, are specifically labelled. (c) Two hypotheses of O-antigen initiation in R. etli CE3. Reactions in each hypothesis and the enzyme that catalyses each reaction are indicated. In both hypotheses, the first reaction (1) is the same, the conversion of UDP-GlcNAc to UDP-KdgNAc catalysed by the predicted 4,6-dehydratase WreV. The two hypotheses differ in reactions (2) and (3). In hypothesis 1, KdgNAc is reduced to QuiNAc on the UDP linkage by WreQ and then QuiNAc-1-P is transferred by WreU. In hypothesis 2, KdgNAc-1-P is transferred by WreU first and then KdgNAc is reduced to QuiNAc by WreQ on the Und-PP linkage.
The CE3 O-antigen is also an intriguing model for studying polysaccharide biosynthesis. It has features, such as its precisely controlled number of repeat units [12], which are not readily explained by known mechanisms. Making it attractive is also the fact that all 29 genes considered necessary specifically for its synthesis have been mutated. For instance, it has been possible to identify nine genes encoding glycosyltransferases (GTs), and, by biochemical analysis of truncated LPS from each GT mutant, to explain which sugar linkages are catalysed by each of the nine GTs [13] (Fig. 1b). However, these assignments have not been confirmed by investigation in vitro with purified enzymes and defined substrates. The initial step of the biosynthesis is a logical first reaction to investigate.
The biosynthesis of all characterized O-antigens is believed to share a conserved initial type of reaction catalysed by a family of GTs that are integral membrane proteins. The initiating GTs catalyse transfer of a sugar-1-phosphate moiety from a nucleotide-sugar donor to the membrane lipid carrier bactoprenyl phosphate (BpP), resulting in bactoprenyl-pyrophosphoryl-sugar (BpPP-sugar) [14, 15]. Due to the difficulty in obtaining purified enzymes and the limited availability of substrates, this initial step in the synthesis of an O-antigen has been demonstrated in only a few cases [16–18]. In R. etli CE3 O-antigen (Fig. 1a), the proposed first sugar is 2-acetamido-2,6-dideoxy-d-glucose (d-QuiNAc, hereafter referred to as QuiNAc) [12, 13, 19]. Although QuiNAc is found in a number of bacterial polysaccharides, the mechanism of its incorporation into a polysaccharide, in particular as the initiating sugar, has not been reported. The predicted initiating GT for R. etli CE3 O-antigen synthesis is encoded by the wreU gene (Fig. 1b) [13]. The LPS of a wreU null mutant lacks all O-antigen-specific sugars including QuiNAc [13].
QuiNAc is derived from the central metabolite UDP-GlcNAc (UDP-N-acetyl-d-glucosamine [UDP-2-acetamido-2-deoxy-d-glucose]) [19, 20]. Besides WreU, two additional enzyme activities are expected in a pathway from UDP-GlcNAc to BpPP-QuiNAc (Fig. 1c). The first is a 4,6-dehydratase that catalyses conversion of UDP-GlcNAc to UDP-2-acetamido-2,6-dideoxy-d-xylo-4-hexulose (also known as UDP-4-keto-6-deoxyGlcNAc and hereafter referred to as UDP-KdgNAc). R. etli gene wreV (Fig. 1b) encodes a protein whose predicted sequence aligns with enzymes known to catalyse this reaction in vitro [19, 21–25]. The gene for one of these characterized enzymes, Pseudomonas aeruginosa wbpM, complements R. etli wreV mutants [19].
The other expected enzyme activity is a 4-reductase that catalyses the reduction of the KdgNAc moiety to QuiNAc (Fig. 1c). Analysis of the R. etli CFN42 total nucleotide sequence assigns this type of activity to the protein encoded by wreQ (Fig. 1b), and mutation of this gene has previously been shown to cause the absence of QuiNAc from the R. etli CE3 LPS [26]. Recently, it was shown that WreQ catalyses the conversion of UDP-KdgNAc to UDP-QuiNAc in vitro [19]. However, the catalysis by WreQ was relatively very slow, raising the question of whether UDP-KdgNAc is the natural substrate of WreQ in vivo. Also relevant is the fact that in the small amount of LPS O-antigen produced by a wreQ null mutant, the QuiNAc residue is replaced by KdgNAc [27]. This result raises the possibility that WreU acts on either the QuiNAc or KdgNAc moiety, or, considering the observed slow WreQ catalysis with UDP-KdgNAc as the substrate, the normal route to QuiNAc in vivo may be the one shown in Fig. 1(c) as hypothesis 2.
In the present study, the alternative hypothetical pathways of Fig. 1(c) were tested by an in vitro biochemical approach using enzymes expressed from hybrid cloned genes in Escherichia coli. In addition, BpPP-QuiNAc was shown to be a functional acceptor substrate for the next step in CE3 O-antigen synthesis in an assay in vitro using the predicted sugar donor and the predicted transferase encoded by gene wreG.
Results
Recombinant R. etli WreU was expressed in E. coli
The wreU gene of R. etli CE3 was cloned into a pET15b vector, yielding a genetic construct from which the expressed WreU protein included an amino-terminal six-histidine (His6) tag. When this ORF was subcloned into a vector that replicates in R. etli, its expression complemented the LPS-deficient phenotype of R. etli wreU-null mutant strain CE566 (Fig. S1, available with the online version of this article). After overexpression in E. coli, His6-WreU was found exclusively in the cell membrane fraction. Attempts to purify His6-WreU free of membrane were not successful despite trying various detergents, various expression conditions, and making other types of WreU constructs. Thus, the E. coli membrane fraction containing His6-WreU was used in the in vitro studies of WreU.
WreU possessed GT activity with preference for UDP-KdgNAc as the nucleotide-sugar substrate
For testing WreU enzymatic activity, the lipid carrier substrate, undecaprenyl phosphate (Und-P), was synthesized in situ from undecaprenol and ATP with an enzyme having polyprenol kinase activity as described by [28]. UDP-KdgNAc or UDP-QuiNAc, each enzymatically synthesized as described previously [19], or UDP-GlcNAc was added as a possible nucleotide-sugar substrate. The reactions were started by adding WreU-containing membranes, or control membranes lacking WreU, and terminated by chloroform-methanol extraction. Und-PP-sugars, such as the predicted products of WreU catalysis, partition into the organic phase in this type of extraction thereby separating them from the nucleotide-sugar substrates [29, 30].
To facilitate visualization of the product after TLC separation, the lipid substrate Und-P was labelled with 32P by using ATP (γ-32P) in its synthesis. The result of the WreU assay is shown in Fig. 2. An abundant product corresponding to an undecaprenyl pyrophosphate-linked sugar (Und-PP-sugar) candidate (compound I) was detected in the reaction only with UDP-KdgNAc as the nucleotide-sugar substrate (Fig. 2, lane 4). In the reaction with an equal concentration of UDP-QuiNAc, a faint spot representing a different Und-PP-sugar candidate (compound II) was observed (Fig. 2, lane 5), and no product was detected in the reaction with UDP-GlcNAc (Fig. 2, lane 3). When quantified with a phosphorimager, compound I in lane 4 had a 30-fold higher intensity than compound II in lane 5. Importantly, the production of compounds I and II required both the lipid substrate Und-P and the enzyme WreU (Fig. 2, lane 1, 2). These results provided evidence that WreU is an initiating GT and UDP-KdgNAc is the preferred sugar-P-donor substrate. UDP-QuiNAc was much less favoured, and UDP-GlcNAc led to no detectable reaction.
Fig. 2.
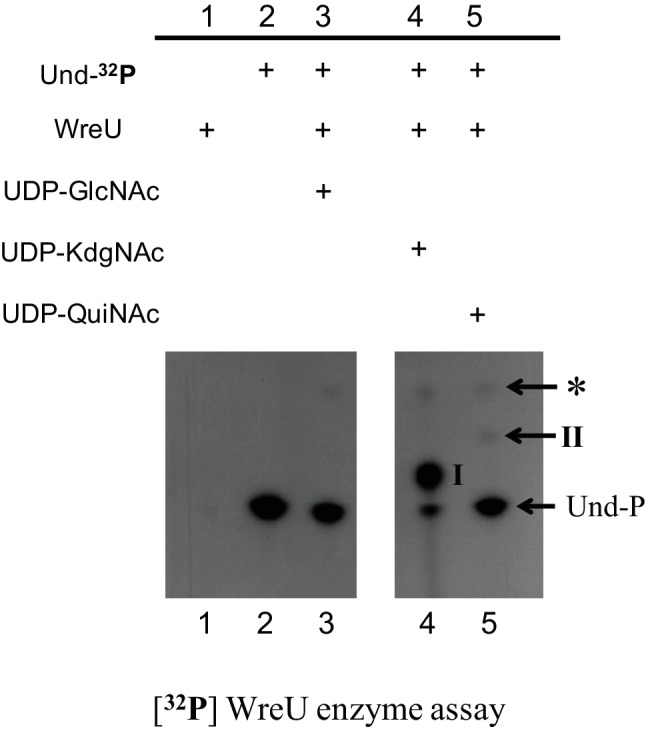
Nucleotide sugar substrate specificity of R. etli WreU and TLC analysis of the products of its activity. The images show TLC separation of products extracted into the organic phase after generation in WreU reactions that included 32P-labelled lipid substrate Und-P. Two portions of one autoradiogram from one experiment are shown. Lanes: lane 1, by omitting the polyprenyl kinase for its synthesis, lipid substrate Und-P was absent (negative control 1); lane 2, no WreU crude enzyme was added to the reaction (negative control 2); lanes 3–5, different nucleotide sugar substrates were added, as indicated in the table above the TLC images. Inferred compounds: I, Und-PP-KdgNAc; II, Und-PP-QuiNAc; *, a compound derived from Und-P in the presence of E. coli membrane (its Rf value with a different TLC solvent (not shown) matches that of Und-PP [44]).
WreQ catalysed the reduction of KdgNAc to QuiNAc on Und-PP linkage
The foregoing results with WreU were consistent with the second step of hypothesis 2 (Fig. 1c). Hence, the next step of this hypothesis was tested: does WreQ catalyse the reduction of Und-PP-KdgNAc to Und-PP-QuiNAc? His6-WreQ had been produced and purified in a previous study, in which it had been shown to catalyse the reduction of d-KdgNAc stereospecifically to d-QuiNAc [19].
A WreU-WreQ-coupled assay was carried out with 32P-radiolabelling (Fig. 3). The Und-32PP-KdgNAc (compound I) produced in the WreU reaction (Fig. 3, lane 1) served as a substrate for WreQ. NADH was chosen as the reducing substrate. When both WreQ and NADH were added to the WreU reaction mixture, compound I was completely converted to a faster-moving compound (compound II) (Fig. 3, lane 4). When NADH alone was added without WreQ, no conversion occurred (Fig. 3, lane 3). When WreQ was added but NADH was omitted, a very small amount of compound II was produced (Fig. 3, lane 2), possibly due to contaminating NADH from the crude WreU enzyme (membrane).
Fig. 3.
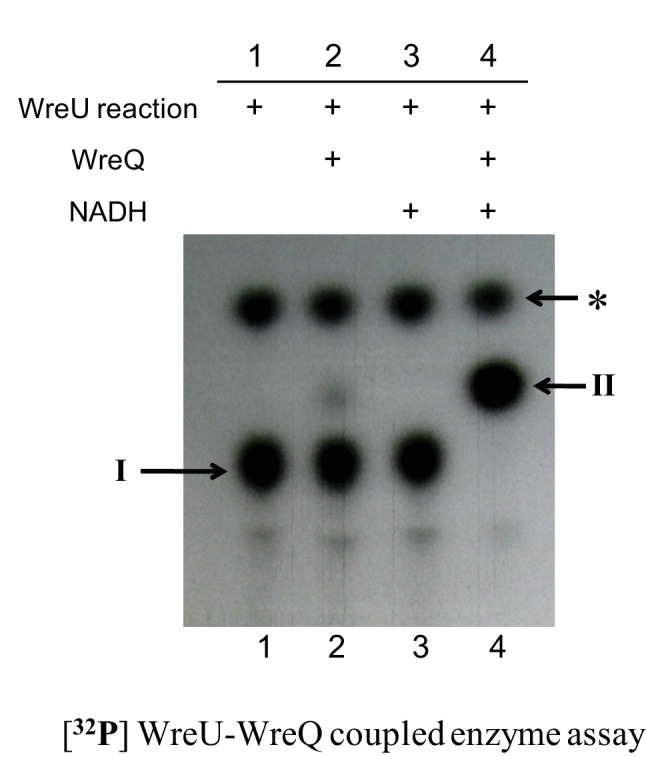
Alteration of WreU reaction product by addition of WreQ and NADH. The autoradiogram shows TLC separation of products extracted into the organic phase after generation in WreU-WreQ-coupled enzyme assays. All reactions were set up as noted for lane 4 of Fig. 2, with UDP-KdgNAc as the nucleotide substrate for WreU, and the reactions were allowed to proceed for 1 h. The reactions then varied by whether WreQ and NADH were added at this point, and incubation continued for another hour. Lanes: lane 1, WreU reaction only; lane 2, WreQ added to the WreU reaction; lane 3, NADH added to the WreU reaction; lane 4, both WreQ and NADH added to the WreU reaction. Inferred compounds: I, Und-PP-KdgNAc; II, Und-PP-QuiNAc; *, Und-PP (see Fig. 2 legend).
WreQ catalysis was much faster when KdgNAc was linked to Und-PP rather than UDP
The result of the WreU-WreQ-coupled assay suggested that WreQ has Und-PP-KdgNAc (compound I) reductase activity, leading to Und-PP-QuiNAc (compound II) as the product. WreQ can also catalyse UDP-KdgNAc reduction to UDP-QuiNAc in vitro, but that reaction is relatively slow [19]. The rates of catalysis with Und-PP-KdgNAc as the substrate versus UDP-KdgNAc as the substrate were compared by TLC and autoradiography in the following experiments.
To provide conditions for estimating the rate of a WreQ-catalysed reduction of the lipidated substrate Und-PP-KdgNAc, the WreU-WreQ-coupled reactions were performed with serially diluted WreQ concentrations, and the WreQ reaction was allowed to proceed for only 1 min (instead of 1 h in the experiment shown in Fig. 3). The conversion of Und-PP-KdgNAc (compound I) to Und-PP-QuiNAc (compound II) gradually increased with decreasing dilution of WreQ (from 10−6 to 10−2) (Fig. 4a, lanes 2–6). The negative control (0 min) indicated that the method to stop the reaction was effective (Fig. 4a, lane 1). At 10−2 dilution, the conversion was almost complete in 1 min (Fig. 4a, lane 6), whereas the reactions with 10−3 and 10−4 diluted WreQ enzyme were slow enough that reaction rates/enzyme concentration (V/[E]) could be estimated (Table 1).
Fig. 4.

Comparison of the WreQ kinetic activity with lipid-linked versus nucleotide-linked substrates. Autoradiograms show products separated on TLCs after controlled times of incubation and concentrations of WreQ. (a) WreQ catalysis with the lipidated substrate. First, [32P] WreU transferase reactions with the UDP-KdgNAc substrate were allowed to proceed for 1 h (i.e. under the same conditions as for Fig. 2, lane 4). Then, WreQ and NADH were added. After 1 min, the reactions were terminated by adding and rapidly mixing with chloroform-methnol/3 : 2 (solvent I). Lanes: lane 1 is a 0 min control in which solvent I was added before WreQ; lanes 2–8 are groups that contain serially diluted WreQ, from 106 to 1 (undiluted). Inferred compounds: I, Und-PP-KdgNAc; II, Und-PP-QuiNAc. (b) WreQ catalysis with UDP-KdgNAc. WreQ and NADH were added to reactions in which UDP-[3H]KdgNAc was produced by complete conversion from UDP-[3H]GlcNAc catalysed by WbpM in a 30 min reaction [19]. WreQ concentration was the same as the undiluted concentration used in panel (a) (10 µg His6-WreQ per 100 µl reaction). Catalysis was allowed to proceed for different times before being terminated by boiling. Lanes: lane 1, 0 min; lane 2, 1 min; lane 3, 5 min; lane 4, 30 min.
Table 1. Comparing the estimated enzymatic activities of WreQ with the two substrates, UDP-KdgNAc and Und-PP-KdgNAc.
Substrate conversion was calculated from the TLC result shown in Fig. 4. The intensities of spots were measured by ImageQuantTL software for 32P spots in Fig. 4(a) and by ImageJ software for 3H spots in Fig. 4(b).
| Substrate | Substrate concentration [S] (µM) | WreQ concentration [E] (µM) | Substrate conversion (%) | Product concentration [P] (µM) | Reaction time t (min) | Reaction rate V=[P]/t (µM min−1) | Substrate conversion per active site per min V/[E] (min−1) |
|---|---|---|---|---|---|---|---|
| UDP-KdgNAc | 500 | 28 | 45 | 225 | 30 | 7.5 | 0.27 |
| Und-PP-KdgNAc | 0.33 | 0.0028 | 62.5 | 0.206 | 1 | 0.206 | 74 |
| 0.3 | 0.00028 | 14 | 0.042 | 1 | 0.042 | 150 |
For visual comparison, the WreQ catalysis with the nucleotide substrate UDP-KdgNAc was carried out with a single (much higher) WreQ concentration and terminated at different time points (Fig. 4b). The reaction showed near linear progression and the data obtained at 30 min reaction time (Fig. 4b, lane 2) was used for calculation of the reaction rate (Table 1). The V/[E] calculated from the results in Fig. 4 indicated that the WreQ catalysis was at least two orders of magnitude faster when KdgNAc was linked to Und-PP rather than UDP (Table 1), even though the lipidated substrate was presented at 1000-fold lower concentration.
WreG catalysed the addition of the second O-antigen sugar (mannose) to Und-PP-QuiNAc
A previous study based on mutant phenotypes proposed that WreG is the GT that transfers the second O-antigen sugar mannose (Man) to QuiNAc [13]. The in vitro assay system developed in the present study provided a means to obtain biochemical evidence for the role of WreG and to confirm that Und-PP-QuiNAc is the precursor for further O-antigen synthesis. As a first step, the wreG gene of R. etli CE3 was cloned into vector pET21b such that the expressed WreG protein in E. coli included a carboxy-terminal His6 tag. When this ORF was subcloned into a vector that replicates in R. etli, its expression complemented the LPS-deficient phenotype of R. etli wreG-null mutant strain CE358 (Fig. S2). To test the enzymatic activity of WreG-His6, two potential acceptor substrates, Und-PP-KdgNAc (compound I) and Und-PP-QuiNAc (compound II), were produced by the WreU reaction and the WreU-WreQ-coupled reaction, respectively (Fig. 5, lanes 2 and 3). GDP-Man was added as the donor of Man, and WreG was added as a crude membrane fraction (Fig. 5, lanes 4 and 5) or as purified enzyme (Fig. 5, lane 6).
Fig. 5.

The product generated by the combined action of WreU, WreQ and predicted Bp-PP-QuiNAc mannosyl transferase WreG. 32P-labelled compounds were extracted into the organic phase after reactions with six varied combinations of WreU, WreQ, WreG and substrates. The two panels show autoradiograms of TLCs carried out with two different solvents, 2-propanol/ammonium hydroxide/water, 6 : 3 : 1 (a) and chloroform/methanol/water, 65 : 25 : 4 (b). All reaction mixtures contained substrates UDP-KdgNAc, Und-32P and GDP-Man. They varied by lacking added NADH or enzyme as follows: lane 1, no WreU; lane 2, no WreQ, WreG or NADH; lane 3, no WreG; lane 4, no WreQ or NADH; lane 5, WreG activity provided by crude membrane fraction and all other reagents and enzymes added; lane 6, all reagents and enzymes added, including purified WreG enzyme. Inferred compounds: I, Und-PP-KdgNAc; compound II, Und-PP-QuiNAc; compound III, Und-PP-QuiNAc-Man; *, Und-PP (see Fig. 2 legend).
Und-PP-KdgNAc (compound I) remained almost unchanged when crude WreG and GDP-Man were included in the WreU reaction (compare lane 4 to lane 2 in Fig. 5). In contrast, the Und-PP-QuiNAc (compound II) produced when WreQ was added to the WreU reaction mixture was converted to a slower-moving compound (compound III) when WreG and GDP-Man were also added (as shown by lanes 5 and 6 compared to lane 3 in Fig. 5). When WreG was added as a crude membrane preparation, compound II was almost completely replaced by compound III (Fig. 5, lane 5), whereas the purified WreG was less active, as revealed by partial conversion of compound II to compound III in lane 6 of Fig. 5.
Based on the substrate requirements for its formation, compound III was inferred to be Und-PP-QuiNAc-Man, the predicted lipid-linked disaccharide resulting from the GT activity of WreG. Because compound III migration was nearly identical to Und-P in TLC solvent A (Fig. 5a), a different solvent (solvent B) was also used. This second solvent system separated compound III from Und-P and other radiolabelled compounds (Fig. 5b).
The result of this assay provided in vitro evidence that WreG is the mannosyltransferase for adding the second O-antigen sugar (Man) in R. etli CE3. Furthermore, Und-PP-QuiNAc was utilized much more readily than Und-PP-KdgNAc as the acceptor of Man in vitro, suggesting that normally the reduction of KdgNAc to QuiNAc by WreQ would occur before Man addition.
Discussion
Results in this study lead to the following inferences regarding QuiNAc and O-antigen synthesis in R. etli, as discussed further in succeeding paragraphs: (1) the conversion of d-GlcNAc to d-QuiNAc in R. etli occurs in an unconventional manner compared with other characterized deoxysugar syntheses (i.e. it follows hypothesis 2 of Fig. 1c). As with biosynthesis of other 6-deoxysugars, it proceeds with formation of a 4-keto-6-deoxyhexose intermediate (KdgNAc). However, the KdgNAc-P intermediate is first transferred from nucleotide linkage to a bactoprenyl phosphate carrier before undergoing 4-reduction to the 6-deoxy product, QuiNAc. (2) The switch from nucleotide linkage to lipid carrier is directed by WreU, whose reaction requirements confirm the prediction that it is the initiating GT for R. etli O-antigen biosynthesis. The specificity of WreU for KdgNAc dictates that bactoprenyl-PP-KdgNAc is the first lipid-linked O-antigen intermediate. (3) The unconventional lipidated-substrate specificity of 4-reductase WreQ is responsible for delaying conversion of KdgNAc to QuiNAc until KdgNAc is attached to bactoprenol-PP. Without WreQ, KdgNAc would be the predicted proximal sugar of the final O-antigen, and, in fact such is the case in the small amount of O-antigen produced in wreQ-null mutant strain CE166 [27]. (4) The acceptor-substrate specificity of mannosyltransferase WreG ensures that QuiNAc predominately replaces KdgNAc before O-antigen synthesis can proceed. WreG operates fastest with bactoprenyl-PP-QuiNAc and thereby dictates that QuiNAc and WreQ are needed for efficient synthesis of the R. etli CE3 O-antigen.
WreU is a UDP-KdgNAc:bactoprenyl-P KdgNAc-1-P transferase (reaction 2 of hypothesis 2 in Fig. 1c). This conclusion is based on (1) the substrate requirements of the reaction it catalyses, (2) physical and chemical properties of the inferred product, and (3) the degree of sequence alignment with other characterized initiating GTs. The lipid substrate of WreU in the in vitro reactions was undecaprenyl phosphate (Und-P or C55-P). However, the exact form of bactoprenol lipid carrier in R. etli CE3 is not known. It is likely dodecaprenol-P (C60-P) as reported for Rhizobium leguminosarum 3841 and Sinorhizhobium meliloti 1021 [31]. Of the two hypothetical sugar-donor substrates (Fig. 1c, hypothesis 1 vs hypothesis 2), UDP-KdgNAc yielded 30-fold greater activity than UDP-QuiNAc in vitro. The lack of activity with UDP-GlcNAc as the donor substrate indicates that WreU requires the 6-deoxy moiety for activity. The products, compounds I (from UDP-KdgNAc) and II (from UDP-QuiNAc), behaved in solvent extraction and relative TLC migration as would be predicted. They also carried the input 32P of the lipid substrate. The WreU enzyme assay was also carried out with different radioisotope labelling in which the UDP-KdgNAc carried tritium [3H] in the sugar moiety. After extraction into the organic solvent, the 3H-labelled product showed the same relative migration on TLC as the 32P-compound I (data not shown). Hence, radiolabelling provided additional evidence of both the input lipid and the sugar being present in the product.
A final argument for the enzymatic identity of WreU is based on the predicted amino-acid sequence of translated wreU. The predicted topology and sequence alignment of WreU (Fig. S3) places it within a large subgroup of the superfamily of initiating GTs represented by the active carboxy-terminal portion of Salmonella WbaP [18], WecP from Aeromonas hydrophila AH-3 [17], and PglC from Campylobacter jejuni NCTC 11168 [32]. Like other members of this subgroup, WreU is predicted to have a single transmembrane segment near the amino-terminus followed by a cytoplasmic catalytic domain that constitutes the rest of the polypeptide of these ‘small’ phospho-GTs [33].
WreQ catalysed the inferred 4-reduction of KdgNAc to QuiNAc orders of magnitude faster when KdgNAc was attached to Und-PP than when it was attached to UDP. The very slow reduction of UDP-KdgNAc by the WreQ catalysis reported in a previous study [19] is thereby explained. The study of that slower reaction, however, had the advantage that it was chemically very clean and allowed definitive demonstration that the QuiNAc produced by the WreQ catalysis has the d-stereo configuration [19].
Although this may be the first report of 6-deoxyhexose biosynthesized in this way, a conceptually analogous precedent is N-acetylgalactosamine (GalNAc) synthesis in E. coli by the Gnu pathway, in which the WecA-initiating GT first transfers GlcNAc-1-P to Und-P and the Und-PP-GlcNAc product is converted to Und-PP-GalNAc via a Gnu epimerase [34, 35]. The overall outcome is to generate a primer for synthesis of a polysaccharide or oligosaccharide repeat that will have GalNAc at its reducing terminus. This is exactly analogous to the apparent metabolic role of the WreV-WreU-WreQ pathway and its product bactoprenyl-PP-QuiNAc in R. etli.
Another pathway for d-QuiNAc synthesis was reported recently [20]. It proceeds by the first two steps as outlined in hypothesis 1 of Fig. 1c, i.e. the path not followed by WreV-WreU-WreQ in R. etli. Discovered in Bacillus cereus strain ATCC 14579, it is catalysed by a 4,6-dehydratase and a 4-reductase that are not homologous with WreV and WreQ [20]. Whereas the bactoprenyl-P-coupled pathway of the Proteobacteria seems suited to provide QuiNAc only to begin polysaccharides and oligosaccharide repeat units, this pathway in the bacilli conceivably could be used to provide QuiNAc either for interior glycosyl positions or the initial position in a growing chain. Surprisingly, though, this more conventional pathway may be very limited phylogenetically. The 4-reductase, Preq, shows high sequence similarity only with proteins in other bacilli and perhaps certain closely related firmicutes. An extensive database search did not find it in Proteobacteria, where the bactoprenyl-P-coupled pathway catalysed by WreV-WreU-WreQ orthologues is widely distributed (Table S1).
Results obtained with WreG validated the functionality of the in vitro product of WreU and WreQ activity for CE3 O-antigen synthesis. Based on its structure and the responsible wre genes, the CE3 O-antigen synthesis has been deduced [13] to follow the lesser known of the two common overall mechanisms of O-antigen synthesis [1], in which the complete O-antigen with all repeat units is made on the cytoplasmic face of the inner membrane and then transported across the membrane. The model for CE3 O-antigen [13] proposes that QuiNAc is the primer residue [1, 36] for the remainder of O-antigen synthesis, with Man being the next ‘adaptor’ sugar added (Fig. 1a). WreG is the predicted transferase that catalyses this addition and GDP-Man is the donor substrate for Man addition. The results of the in vitro assay of WreG activity (Fig. 5) supported both predictions of the model and Und-PP-QuiNAc as the product from the WreU and WreQ catalysis. The existence of WreQ and the specificity of WreG are coupled. Selectivity for the 4-OH of QuiNAc by WreG [i.e. much slower catalysis with bactoprenyl-PP-KdgNAc (Fig. 5)] is the key reason that WreQ-null mutants have low abundance of O-antigen [26]. However, WreG in vivo apparently has enough activity with KdgNAc as the Man acceptor that such mutants have a low amount of O-antigen that is identical to the normal O-antigen except for substitution of KdgNAc for QuiNAc [27]. A faint spot visible in lane 4 of Fig. 5, (circled in Fig. S4a) may be due to this lower activity of WreG with Und-PP-KdgNAc as the acceptor substrate in vitro. This logic leads to the prediction that greatly increasing the specific concentration of just the WreG enzyme will lead to higher O-antigen production in a wreQ-minus genetic background. Fig. S4(b) shows results that confirm this prediction. This result explains the basis of the genetic suppression of the WreQ-minus phenotype by multiple copies of the main wre cluster [26]. Importantly, it also supports hypothesis 2 over hypothesis 1 of Fig. 1(c) by means of in vivo results that are independent of the in vitro assays.
The phylogenetic distribution of this pathway of QuiNAc synthesis was investigated by blast searches of the sequenced protein database (Table S1). At least 40 genera had at least one strain that carried orthologues of all three genes–wreV, wreU and wreQ. Two genera of green-sulfur bacteria had strong matches, but almost all of the rest were in the Proteobacteriaceae, with all its subphyla being represented (Table S1). blast e-values were less than e−30 for all three homologues in all strains.
It should be noted that WreQ orthologues are the genes needed specifically for QuiNAc synthesis by the bactoprenyl pathway. As stated above, WreV-WreQ are often linked with WreU orthologues, but, at lower frequency, they are found with orthologues of WbpL, another initiating GT. In a limited search of WreQ hits with e-values below e−89, 211 were linked to a WreU homologue (Table S1) and 63 to a WbpL homologue (Table S2). WreU and WbpL are not homologous; they represent the two very different types of initiating GT structures. It is reasonable to suppose that other initiating GT subtypes are coupled with WreVQ in a strain, depending on how the genetic cluster has evolved.
Recently, Colwellia psychrerythraea 34 h, was found to make an ‘antifreeze’ polysaccharide that has a repeat unit containing QuiNAc [37]. Its ability to synthesize QuiNAc had been predicted the previous year [19] because it had WreV and WreQ sequence matches with very low e-values. A gene whose encoded protein has a significant match with WreU is adjacent to the wreQ orthologue on the genome, and the wreV orthologue is separated by three genes (Fig. S5).
In summary, results in this study strongly support the second of the two alternative hypotheses of Fig. 1(c). A main conclusion is that biosynthesis of QuiNAc in R. etli CE3 (and probably in many other bacteria) is tightly coupled to initiation of the synthesis of a polysaccharide on which it is ultimately the first (reducing-end) sugar. Fig. 6 depicts this coupling, the steps in the pathway, and its association with the membrane. Phylogenomic searches suggest that this pathway is distributed widely among the Proteobacteria. The outcome is Bp-PP-QuiNAc, which in R. etli CE3 becomes the platform for the rest of O-antigen synthesis, the next step of which was also demonstrated in this study and is depicted in Fig. 6 as well.
Fig. 6.

Model of QuiNAc synthesis coordinated with O-antigen initiation. The three phases of QuiNAc synthesis in R. etli CE3 are indicated with numbers: first, UDP-GlcNAc is converted to intermediate UDP-KdgNAc by the dehydratase WreV (phase 1); second, KdgNAc-1-P is transferred by WreU to the bactoprenyl phosphate (BpP) lipid carrier (phase 2); third and last, the KdgNAc moiety is reduced to QuiNAc by WreQ (phase 3). The final product of QuiNAc synthesis serves as the platform for further O-antigen synthesis to which a Man was transferred by WreG. Compounds: I, Bp-PP-KdgNAc; II, Bp-PP-QuiNAc; III, Bp-PP-QuiNAc-Man.
Methods
Bacterial strains and growth conditions
Rhizobium etli CE3 was derived from R. etli wild-type strain CFN42 by a spontaneous mutation conferring resistance to streptomycin [38]. As in almost all past studies of the LPS of R. etli CFN42, strain CE3 was the wild-type source of DNA and genotype for strain constructions. All R. etli strains were grown to stationary phase at 30 °C in TY liquid medium [0.5 % tryptone (Difco Laboratories), 0.3 % yeast extract (Difco) and 10 mM CaCl2]. All Escherichia coli strains were grown to stationary phase at 37 °C in Luria–Bertani (LB) liquid medium (1.0 % tryptone, 0.5 % yeast extract and 0.5 % NaCl). Agar medium contained 1.5 % Bacto Agar (Difco).
DNA techniques
Genomic DNA was isolated using GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich) and plasmid DNA was isolated using QIAprep Spin Miniprep Kit (Qiagen). DNA extraction from agarose gels was performed using Gel/PCR DNA Fragments Extraction Kit (IBI Scientific). DNA amplification by PCR was performed using Expand High Fidelity PCR System (Roche Applied Science). Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs (NEB).
Cloning of R. etli wreU and wreG for overexpression
The R. etli CE3 wreU gene was amplified from R. etli CE3 genomic DNA using primers 5′-CCGGCATATGGGCTTGAAACGGGCG-3′ (forward) and 5′-GGCCGGATCCCTAGTGCTTTATTCC-3′ (reverse). The PCR product was cloned into the pET15b vector (Novagen) using NdeI and BamHI sites, generating plasmid pLS5. It encodes the WreU protein with additional amino acids at the amino-terminus, MGSSHHHHHHSSGLVPRGSH (the 6xHis tag is underlined). This WreU construct is referred to as His6-WreU in this work.
The R. etli CE3 wreG gene was amplified from R. etli CE3 genomic DNA using primers 5′- GCGCTAGCATGAGAGTCCTTCATTT-3′ (forward) and 5′-TTCTCGAGGCGGGAACCGGCCACGT-3′ (reverse). The PCR product was cloned into the pET21b vector (Novagen) using NheI and BamHI sites, generating plasmid pTL59. It encodes the WreG protein with amino-terminal additional amino acids MAS, and carboxy-terminal additional amino acids LEHHHHHH (the 6xHis tag is underlined). This WreG construct is referred to as WreG-His6 in this work.
Overexpression of His6-WreU, WreG-His6 and the polyprenyl kinase (DGK)
Plasmid pLS5 (His6-WreU) and pTL59 (WreG-His6) were separately transformed into E. coli BL21(DE3) cells by electroporation. The polyprenol kinase used in these studies is expressed from cloned dgk DNA from Streptococcus mutans. Although homologous to E. coli dgk, the protein encoded by Streptococcus mutans dgk has higher [28], or much higher [39], kinase activity with undecaprenol as the substrate than with diacylglycerols. BL21 cells carrying a pET vector construct encoding this protein with a carboxy-terminal His6-tag [28] was provided by Dr Barbara Imperiali, Massachusetts Institute of Technology, Cambridge, MA. Hereafter in this section, it will be referred to as DGK, to conform with the extant abbreviation in the literature.
Expression of His6-WreU, WreG-His6 and DGK followed the same procedure: a flask of 1 l LB medium containing appropriate antibiotics (ampicillin 100 µg ml−1 for His6-WreU and WreG-His6, kanamycin 50 µg ml−1 for DGK) was inoculated with a 5 ml overnight start culture and shaken at 37 °C until an optical density between 0.6 and 0.8 was reached. Then the flask was chilled for 1 h. Protein expression was induced by adding IPTG to the culture (1 mM for DGK, 0.01 mM for His6-WreU and 0.1 mM for WreG-His6), and the culture was shaken for a further 20 h at 16 °C. Cells were harvested by centrifugation at 5000 g for 15 min at 4 °C, and the pellets was stored at −80 °C until needed.
Complementation of R. etli CE3 mutants with the respective His-tagged constructs
The DNA sequence-encoding His6-WreU together with the RBS sequence was amplified from plasmid pLS5 with primers 5′-GCCGAATTCATACCCACGCCGAAACAAG-3′ (forward) and 5′-GCCGGTACCAGTTCCTCCTTTCAGCAAA-3′ (reverse). The PCR product was cloned into plasmid pFAJ1708 [40] with EcoRI and KpnI sites, generating plasmid pLS22.
The DNA sequence-encoding WreG-His6 together with the RBS sequence was amplified with primers 5′-GCCGAA TTCATACCCACGCCGAAACAAG-3′ (forward) and 5′-GCCGGTACCAGTTCCTCCTTTCAGCAAA-3′ (reverse). The PCR product was cloned into plasmid pFAJ1708 [40] with XbaI and KpnI sites, generating plasmid pTL63.
Separately, pLS22 (His6-wreU) was transferred into CE566 (wreU::Km) and pTL63 (wreG-His6) was transferred into CE358 (wreG::Tn5) by triparental mating [41] with plasmid-mobilizer strain MT616 [42], as described previously [13]. Strains containing these constructs were selected on TY agar plates supplemented with 200 µg of streptomycin ml−1, 30 µg of nalidixic acid ml−1, 5 µg tetracycline ml−1, 30 µg of kanamycin ml−1. Single colonies were purified and analysed by SDS-PAGE.
Preparation of membrane fractions
Membrane fractions were prepared from E. coli cells expressing DGK, His6-WreU and WreG-His6 for use as crude enzyme or for purification of membrane-located proteins. Frozen cell pellets from 500 ml culture were thawed with lysis buffer (buffer A for DGK, 50 mM Tris, 1 mM ethylenediaminetetraacetic acid; buffer B for His6-WreU, 20 mM Tris, 300 mM NaCl, pH 8.5; buffer C for WreG-His6, 20 mM sodium phosphate, 300 mM NaCl, 5 mM imidazole, pH=7.0, with 14.3 mM 2-mercaptoethanol), and lysed by sonication. The lysate was centrifuged first at a low speed (6000 g, 20 min at 4 °C) to remove most of the cellular debris and then followed by a high speed spin (65 000 g, 120 min at 4 °C) to pellet the cell membrane fraction (stored at −80 °C if not used). The pellet of His6-WreU and WreG-His6 was homogenized in the respective lysis buffer and aliquoted into 100 µl fractions for storage at −80 °C.
Purification of DGK from membrane fractions
Frozen cell membrane was thawed and resuspended in 0.5 ml buffer D (20 mM sodium phosphate, 300 mM NaCl, 5 mM imidazole, pH 8.0) and incubated with 1 % CHAPS for 1 h at 4 °C to solubilize membrane proteins. Then the sample was incubated with 250 µl Ni2+-profinity IMAC resin (Bio-Rad) for 30 min at 4 °C. The resin was placed in a 0.2 µm filter in a microcentrifuge tube for the subsequent wash and elution steps. The resin was washed twice with 375 µl of buffer D containing 1 % CHAPS, and twice with 375 µl of the same buffer with 45 mM imidazole. The protein was eluted three times in 200 µl of the same buffer containing 300 mM imidazole. The combined elution fraction was dialysed and concentrated with an Amicon Ultra-0.5 (nominal molecular weight limit: 10 kDa) filter device. The concentrated protein was aliquoted into smaller fractions for storage at −80 °C.
Purification of WreG-His6 from membrane fractions
One tube of 0.5 ml frozen cell membrane fraction was thawed and incubated with 1 % Triton X-100 for 2.5 h at 4 °C. Then the sample was incubated with 200 µl Ni2+-profinity IMAC resin (Bio-Rad) for 30 min at 4 °C. The resin was placed in a 0.2 µm filter in a microcentrifuge tube for the subsequent wash and elution steps. The resin was washed twice with 250 µl of buffer C containing 0.1 % Triton X-100, and twice with 250 µl of the same buffer with 20 mM imidazole. The protein was eluted twice in 250 µl of the same buffer containing 300 mM imidazole. Protein sample dialysis and concentration were performed exactly as described for DGK above.
In vitro enzyme assays
WreU GT assay – the lipid substrate Und-P was prepared according to [28] with modification. Briefly, 3 µl DMSO and 10 µl 10 % Triton X-100 were mixed with 13 nmol of dried undecaprenol (American Radiolabeled Chemicals). The tube was vortexed to ensure solubilization of the lipid. To the same tube, 5 µM [γ-32P]-ATP (2000 mCi mmol−1) (PerkinElmer), 1 µl of purified DGK (~50 ng), 50 mM Tris buffer, pH 8.0, 40 mM MgCl2 were added to a total volume of 100 µl. The DGK reaction was incubated at 30 °C for 1 h. To start the WreU enzyme assay, 1 µl (~2 µg) His6-WreU membrane fraction was added to the DGK reaction. In the [32P]-WreU assay, nucleotide sugar substrates tested were: UDP-GlcNAc (Sigma), purified UDP-KdgNAc and UDP-QuiNAc. The concentration of each nucleotide sugar substrate was 0.05 mM.
The WreU reactions were incubated at 30 °C for 1 h, then quenched into 500 µl of solvent I (chloroform-methanol/3 : 2) and extracted with 400 µl PSUP (chloroform-methanol-1M MgCl2-water/18 : 294 : 293 : 1) [43]. The organic layers were dried with lyophilization and re-dissolved in 20 µl solvent I. 1 µl of each sample was spotted on an aluminum-backed precoated Silica gel 60 plate (EMD Chemicals) and developed in TLC solvent A (2-propanol/ammonium hydroxide/water, 6 : 3 : 1). Dried TLC plates were exposed to films or photostimulable phosphor (PSP) plates and viewed by autoradiogram.
WreU-WreQ-coupled assay – the WreU reactions with UDP-KdgNAc as the substrate were incubated for 1 h at 30 °C. To the WreU reactions, 0.1 mM NADH and 1 µl (~10 µg) WreQ enzyme were added. The reactions were allowed to proceed for 1 h at 30 °C after WreQ addition. Then they were quenched and prepared for analysis as described above for WreU enzyme assay.
WreU-WreQ-WreG-coupled reaction – in reactions that aimed to test the GT activity of WreG, 10 µl crude (~10 µg) or purified WreG enzyme (~40 µg) and 0.1 mM GDP-Man were added to [32P]-WreU reactions together with (or without) 10 µg WreQ and 0.1 mM NADH. Reactions were allowed for 1 h after adding WreG and then quenched and prepared for TLC analysis. For analysis of the reaction products, two TLC solvents were used: solvent A and solvent B (choloform/methanol/water, 65 : 25 : 4).
Rate comparison of WreQ-catalysed reaction with different substrates
To estimate the rate of the WreQ reaction with nucleotide sugar substrate, 10 µg His6-WreQ protein and 1 mM NADH were added to the WbpM reaction which was incubated for 30 min to generate product UDP-KdgNAc [19] and the WreQ reactions were quenched at 1, 5 and 30 min. Reducing the WreQ enzyme concentration by a factor of 10 was attempted which led to a very slow reaction, thus only one concentration of WreQ was used for this reaction.
To estimate the rate of the WreQ reaction with the lipidated substrate, firstly [32P] WreU transferase reactions with the UDP-KdgNAc substrate for 1 h. Then 0.1 mM NADH and 1 µl serially diluted WreQ enzyme (10−6, 10−5, 10−4, 10−3, 10−2, 10−1 and undiluted) was added and the WreQ reactions were allowed for only 1 min. In one reaction, solvent I was added before the addition of the WreQ enzyme as a 0 min control, to show that the method of quenching the reactions was effective. The organic phases of these reactions were analysed by TLC. Radioactive (32P) spots were quantified by phosphorimager and used for estimating reaction kinetics.
Funding information
This work was supported by National Institutes of Health Grant 1 R15 GM087699-01A1.
Acknowledgements
We thank Dr Barbara Imperiali for the gift of an expression vector construct encoding the Streptococcus mutans undecaprenol kinase with a carboxy-terminal His6-tag and Dr J. S. Lam for providing the WbpM-His-S262 expression vector.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Supplementary Data
Footnotes
Abbreviations: BpP, bactoprenyl phosphate; BpPP, bactoprenyl pyrophosphate; DGK, kinase encoded by Streptococcus mutans dgk, with preference for polyprenol substrates; GlcNAc, 2-acetamido-2-deoxy-d-glucose, also known as N-acetyl-d-glucosamine; GT, glycosyltransferase; KdgNAc, 2-acetamido-2,6-dideoxy-d-xylo-4-hexulose, also known as 4-keto-6-deoxy-GlcNAc; LPS, lipopolysaccharide; Man, mannose; QuiNAc, 2-acetamido-2,6-dideoxy-d-glucose, also known as N-acetyl-d-quinovosamine; Und-P, undecaprenyl phosphate; Und-PP, undecaprenyl pyrophosphate.
Two supplementary tables and five supplementary figures are available with the online version of this article.
Edited by: I. J. Oresnik and G. H. Thomas
References
- 1.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank MM, Joiner K, Hammer C. The function of antibody and complement in the lysis of bacteria. Rev Infect Dis. 1987;9:S537–S545. doi: 10.1093/clinids/9.Supplement_5.S537. [DOI] [PubMed] [Google Scholar]
- 3.Bowden MG, Kaplan HB. The Myxococcus xanthus lipopolysaccharide O-antigen is required for social motility and multicellular development. Mol Microbiol. 1998;30:275–284. doi: 10.1046/j.1365-2958.1998.01060.x. [DOI] [PubMed] [Google Scholar]
- 4.Toguchi A, Siano M, Burkart M, Harshey RM. Genetics of swarming motility in Salmonella enterica serovar typhimurium: critical role for lipopolysaccharide. J Bacteriol. 2000;182:6308–6321. doi: 10.1128/JB.182.22.6308-6321.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kierek K, Watnick PI. The Vibrio cholerae O139 O-antigen polysaccharide is essential for Ca2+-dependent biofilm development in sea water. Proc Natl Acad Sci USA. 2003;100:14357–14362. doi: 10.1073/pnas.2334614100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hölzer SU, Schlumberger MC, Jäckel D, Hensel M. Effect of the O-antigen length of lipopolysaccharide on the functions of type III secretion systems in Salmonella enterica. Infect Immun. 2009;77:5458–5470. doi: 10.1128/IAI.00871-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgenstein RM, Clemmer KM, Rather PN. Loss of the waaL O-antigen ligase prevents surface activation of the flagellar gene cascade in Proteus mirabilis. J Bacteriol. 2010;192:3213–3221. doi: 10.1128/JB.00196-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Post DM, Yu L, Krasity BC, Choudhury B, Mandel MJ, et al. O-antigen and core carbohydrate of Vibrio fischeri lipopolysaccharide: composition and analysis of their role in Euprymna scolopes light organ colonization. J Biol Chem. 2012;287:8515–8530. doi: 10.1074/jbc.M111.324012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noel KD, Vandenbosch KA, Kulpaca B. Mutations in Rhizobium phaseoli that lead to arrested development of infection threads. J Bacteriol. 1986;168:1392–1401. doi: 10.1128/jb.168.3.1392-1401.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carlson RW, Kalembasa S, Turowski D, Pachori P, Noel KD. Characterization of the lipopolysaccharide from a Rhizobium phaseoli mutant that is defective in infection thread development. J Bacteriol. 1987;169:4923–4928. doi: 10.1128/jb.169.11.4923-4928.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cava JR, Elias PM, Turowski DA, Noel KD. Rhizobium leguminosarum CFN42 genetic regions encoding lipopolysaccharide structures essential for complete nodule development on bean plants. J Bacteriol. 1989;171:8–15. doi: 10.1128/jb.171.1.8-15.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forsberg LS, Bhat UR, Carlson RW. Structural characterization of the O-antigenic polysaccharide of the lipopolysaccharide from Rhizobium etli strain CE3. A unique O-acetylated glycan of discrete size, containing 3-O-methyl-6-deoxy-l-talose and 2,3,4-tri-O-methyl-l-fucose. J Biol Chem. 2000;275:18851–18863. doi: 10.1074/jbc.M001090200. [DOI] [PubMed] [Google Scholar]
- 13.Ojeda KJ, Simonds L, Noel KD. Roles of predicted glycosyltransferases in the biosynthesis of the Rhizobium etli CE3 O antigen. J Bacteriol. 2013;195:1949–1958. doi: 10.1128/JB.02080-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valvano MA. Export of O-specific lipopolysaccharide. Front Biosci. 2003;8:s452–s471. doi: 10.2741/1079. [DOI] [PubMed] [Google Scholar]
- 15.Price NP, Momany FA. Modeling bacterial UDP-HexNAc: polyprenol-P HexNAc-1-P transferases. Glycobiology. 2005;15:29R–42R. doi: 10.1093/glycob/cwi065. [DOI] [PubMed] [Google Scholar]
- 16.Al-Dabbagh B, Mengin-Lecreulx D, Bouhss A. Purification and characterization of the bacterial UDP-GlcNAc:undecaprenyl-phosphate GlcNAc-1-phosphate transferase WecA. J Bacteriol. 2008;190:7141–7146. doi: 10.1128/JB.00676-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merino S, Jimenez N, Molero R, Bouamama L, Regué M, et al. A UDP-HexNAc:polyprenol-P GalNAc-1-P transferase (WecP) representing a new subgroup of the enzyme family. J Bacteriol. 2011;193:1943–1952. doi: 10.1128/JB.01441-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel KB, Ciepichal E, Swiezewska E, Valvano MA. The C-terminal domain of the Salmonella enterica WbaP (UDP-galactose:Und-P galactose-1-phosphate transferase) is sufficient for catalytic activity and specificity for undecaprenyl monophosphate. Glycobiology. 2012;22:116–122. doi: 10.1093/glycob/cwr114. [DOI] [PubMed] [Google Scholar]
- 19.Li T, Simonds L, Kovrigin EL, Noel KD. In vitro biosynthesis and chemical identification of UDP-N-acetyl-d-quinovosamine (UDP-d-QuiNAc) J Biol Chem. 2014;289:18110–18120. doi: 10.1074/jbc.M114.555862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang S, Aronov A, Bar-Peled M. The Biosynthesis of UDP-d-QuiNAc in Bacillus cereus ATCC 14579. PLoS One. 2015;10:e0133790. doi: 10.1371/journal.pone.0133790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Creuzenet C, Schur MJ, Li J, Wakarchuk WW, Lam JS. FlaA1, a new bifunctional UDP-GlcNAc C6 Dehydratase/ C4 reductase from Helicobacter pylori. J Biol Chem. 2000;275:34873–34880. doi: 10.1074/jbc.M006369200. [DOI] [PubMed] [Google Scholar]
- 22.Creuzenet C, Lam JS. Topological and functional characterization of WbpM, an inner membrane UDP-GlcNAc C6 dehydratase essential for lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Mol Microbiol. 2001;41:1295–1310. doi: 10.1046/j.1365-2958.2001.02589.x. [DOI] [PubMed] [Google Scholar]
- 23.Olivier NB, Chen MM, Behr JR, Imperiali B. In vitro biosynthesis of UDP-N,N'-diacetylbacillosamine by enzymes of the Campylobacter jejuni general protein glycosylation system. Biochemistry. 2006;45:13659–13669. doi: 10.1021/bi061456h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schoenhofen IC, Mcnally DJ, Vinogradov E, Whitfield D, Young NM, et al. Functional characterization of dehydratase/aminotransferase pairs from Helicobacter and Campylobacter: enzymes distinguishing the pseudaminic acid and bacillosamine biosynthetic pathways. J Biol Chem. 2006;281:723–732. doi: 10.1074/jbc.M511021200. [DOI] [PubMed] [Google Scholar]
- 25.Pinta E, Duda KA, Hanuszkiewicz A, Kaczyński Z, Lindner B, et al. Identification and role of a 6-deoxy-4-keto-hexosamine in the lipopolysaccharide outer core of Yersinia enterocolitica serotype O:3. Chemistry. 2009;15:9747–9754. doi: 10.1002/chem.200901255. [DOI] [PubMed] [Google Scholar]
- 26.Noel KD, Forsberg LS, Carlson RW. Varying the abundance of O antigen in Rhizobium etli and its effect on symbiosis with Phaseolus vulgaris. J Bacteriol. 2000;182:5317–5324. doi: 10.1128/JB.182.19.5317-5324.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forsberg LS, Noel KD, Box J, Carlson RW. Genetic locus and structural characterization of the biochemical defect in the O-antigenic polysaccharide of the symbiotically deficient Rhizobium etli mutant, CE166. Replacement of N-acetylquinovosamine with its hexosyl-4-ulose precursor. J Biol Chem. 2003;278:51347–51359. doi: 10.1074/jbc.M309016200. [DOI] [PubMed] [Google Scholar]
- 28.Hartley MD, Larkin A, Imperiali B. Chemoenzymatic synthesis of polyprenyl phosphates. Bioorg Med Chem. 2008;16:5149–5156. doi: 10.1016/j.bmc.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Osborn MJ, Cynkin MA, Gilbert JM, Müller L, Singh M, et al. Methods in enzymology. Academic Press; 1972. Synthesis of bacterial O-antigens; pp. 583–601. [Google Scholar]
- 30.Schäffer C, Wugeditsch T, Messner P, Whitfield C. Functional expression of enterobacterial O-polysaccharide biosynthesis enzymes in Bacillus subtilis. Appl Environ Microbiol. 2002;68:4722–4730. doi: 10.1128/AEM.68.10.4722-4730.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanjilal-Kolar S, Basu SS, Kanipes MI, Guan Z, Garrett TA, et al. Expression cloning of three Rhizobium leguminosarum lipopolysaccharide core galacturonosyltransferases. J Biol Chem. 2006;281:12865–12878. doi: 10.1074/jbc.M513864200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glover KJ, Weerapana E, Chen MM, Imperiali B. Direct biochemical evidence for the utilization of UDP-bacillosamine by PglC, an essential glycosyl-1-phosphate transferase in the Campylobacter jejuni N-linked glycosylation pathway. Biochemistry. 2006;45:5343–5350. doi: 10.1021/bi0602056. [DOI] [PubMed] [Google Scholar]
- 33.Lukose V, Luo L, Kozakov D, Vajda S, Allen KN, et al. Conservation and covariance in small bacterial phosphoglycosyltransferases identify the functional catalytic core. Biochemistry. 2015;54:7326–7334. doi: 10.1021/acs.biochem.5b01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rush JS, Alaimo C, Robbiani R, Wacker M, Waechter CJ. A novel epimerase that converts GlcNAc-P-P-undecaprenol to GalNAc-P-P-undecaprenol in Escherichia coli O157. J Biol Chem. 2010;285:1671–1680. doi: 10.1074/jbc.M109.061630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cunneen MM, Liu B, Wang L, Reeves PR. Biosynthesis of UDP-GlcNAc, UndPP-GlcNAc and UDP-GlcNAcA involves three easily distinguished 4-epimerase enzymes, Gne, Gnu and GnaB. PLoS One. 2013;8:e67646. doi: 10.1371/journal.pone.0067646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vinogradov E, Frirdich E, Maclean LL, Perry MB, Petersen BO, et al. Structures of lipopolysaccharides from Klebsiella pneumoniae. Eluicidation of the structure of the linkage region between core and polysaccharide O chain and identification of the residues at the non-reducing termini of the O chains. J Biol Chem. 2002;277:25070–25081. doi: 10.1074/jbc.M202683200. [DOI] [PubMed] [Google Scholar]
- 37.Casillo A, Parrilli E, Sannino F, Mitchell DE, Gibson MI, et al. Structure-activity relationship of the exopolysaccharide from a psychrophilic bacterium: a strategy for cryoprotection. Carbohydr Polym. 2017;156:364–371. doi: 10.1016/j.carbpol.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noel KD, Sanchez A, Fernandez L, Leemans J, Cevallos MA. Rhizobium phaseoli symbiotic mutants with transposon Tn5 insertions. J Bacteriol. 1984;158:148–155. doi: 10.1128/jb.158.1.148-155.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lis M, Kuramitsu HK. The stress-responsive dgk gene from Streptococcus mutans encodes a putative undecaprenol kinase activity. Infect Immun. 2003;71:1938–1943. doi: 10.1128/IAI.71.4.1938-1943.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dombrecht B, Vanderleyden J, Michiels J. Stable RK2-derived cloning vectors for the analysis of gene expression and gene function in gram-negative bacteria. Mol Plant Microbe Interact. 2001;14:426–430. doi: 10.1094/MPMI.2001.14.3.426. [DOI] [PubMed] [Google Scholar]
- 41.Glazebrook J, Walker GC. Genetic techniques in Rhizobium meliloti. Methods Enzymol. 1991;204:398–418. doi: 10.1016/0076-6879(91)04021-f. [DOI] [PubMed] [Google Scholar]
- 42.Finan TM, Kunkel B, de Vos GF, Signer ER. Second symbiotic megaplasmid in Rhizobium meliloti carrying exopolysaccharide and thiamine synthesis genes. J Bacteriol. 1986;167:66–72. doi: 10.1128/jb.167.1.66-72.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patel KB, Valvano MA. In vitro UDP-sugar:undecaprenyl-phosphate sugar-1-phosphate transferase assay and product detection by thin layer chromatography. Methods Mol Biol. 2013;1022:173–183. doi: 10.1007/978-1-62703-465-4_14. [DOI] [PubMed] [Google Scholar]
- 44.El Ghachi M, Bouhss A, Blanot D, Mengin-Lecreulx D. The bacA gene of Escherichia coli encodes an undecaprenyl pyrophosphate phosphatase activity. J Biol Chem. 2004;279:30106–30113. doi: 10.1074/jbc.M401701200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.