

Abstract
Compared with other mammalian species, humans are particularly susceptible to tau-mediated neurodegenerative disorders. Differential interactions of the tau protein with other proteins are critical for mediating tau's physiological functions as well as tau-associated pathological processes. Primate tau harbors an 11-amino acid-long motif in its N-terminal region (residues 18–28), which is not present in non-primate species and whose function is unknown. Here, we used deletion mutagenesis to remove this sequence region from the longest human tau isoform, followed by glutathione S-transferase (GST) pulldown assays paired with isobaric tags for relative and absolute quantitation (iTRAQ) multiplex labeling, a quantitative method to measure protein abundance by mass spectrometry. Using this method, we found that the primate-specific N-terminal tau motif differentially mediates interactions with neuronal proteins. Among these binding partners are proteins involved in synaptic transmission (synapsin-1 and synaptotagmin-1) and signaling proteins of the 14-3-3 family. Furthermore, we identified an interaction of tau with a member of the annexin family (annexin A5) that was linked to the 11-residue motif. These results suggest that primate Tau has evolved specific residues that differentially regulate protein–protein interactions compared with tau proteins from other non-primate mammalian species. Our findings provide in vitro insights into tau's interactions with other proteins that may be relevant to human disease.
Keywords: Tau protein (Tau), tauopathy, neurochemistry, mass spectrometry (MS), protein-protein interaction
Introduction
Alzheimer's disease (AD),3 the most common form of dementia, is characterized by amyloid-β (Aβ)-containing plaques and intracellular neurofibrillary tangles, which predominantly contain hyperphosphorylated forms of the neuronal microtubule-associated protein tau (1). Both aggregated tau and soluble tau species contribute to pathomechanisms in AD (2). Tau shows largely axonal localization in neurons (3). However, a smaller pool of tau localizes to the somatodendritic compartment of tau (1). Mechanisms of neuronal dysfunction mediated through tau include mitochondrial, synaptic, and axonal transport deficits (4).
Protein–protein interactions are critical for tau's physiological functions, and contribute to its role in disease (5). Tau was shown to interact with key neuronal proteins, for example, scaffolding proteins Grb2 (6), C-Jun-amino-terminal kinase-interacting protein 1 (7), and postsynaptic density protein 95 (8), signaling molecules such as Fyn (9), phospholipase C (6), and protein phosphatase 2A (10) as well as pre-synaptic factors such as α-synuclein (11). However, the full interactome of tau remains incompletely resolved. Furthermore, localized pools of tau appear to undergo different interactions, driving diverse functions mediated by tau. Thus, classical axonal interactions of tau with microtubules affect axonal transport (12). Dendritic tau, on the other hand, interacts with post-synaptic protein complexes that mediate signals downstream of post-synaptic neurotransmitter receptors that contribute to Aβ toxicity in AD (8). In addition, tau protein interactions are dynamically regulated, likely reflecting changes in neuronal states of excitation/inhibition or other cellular processes. Post-translational modification of tau, such as phosphorylation, can affect tau complex formation with resulting changes in tau downstream signaling, imparting tau with detrimental (13, 14), but also beneficial (15) properties in relationship to Aβ toxicity. Finally, in familial forms of tauopathies (=neurological diseases with tau pathology), tau mutations affect protein interactions with tau (16). Thus, deciphering tau protein–protein interactions may be a path to understanding tau physiology and associated pathomechanisms.
Here, we speculated that primate (including human) tau has evolved to undergo species-specific interactions that depend on protein features inherent to primate tau. We report a primate-specific amino acid sequence segment between residues 18 and 28 in the N terminus of human tau that is present only in other primate species, but absent from other mammalian species. In this study, we use deletion mutagenesis in human tau to identify interaction partners using isobaric tags for relative and absolute quantitation (iTRAQ) multiplex labeling. We found that this primate-specific N-terminal feature of tau differentially mediates interactions with neuronal proteins of the vesicle-associated machinery and synaptic transmission.
Results
Human tau harbors an 11-amino acid, primate-specific N-terminal sequence
Alignment of human and murine tau primary sequences revealed 11 consecutive amino acids (aa) in the extreme N-terminal region of human tau with no sequence homology to mouse (Fig. 1A). Extending the alignment study to other mammalian tau sequences, and using the longest human tau isoform (441 aa) as a template, showed that this motif comprising aa 18 to 28 of tau was conserved in primates, and did not share sequence homology with other mammalian tau N termini, including bovine, goat, cat, and rodent tau (Fig. 1, A and B). The only notable exception was canine tau, with 54% sequence identity to human tau in these 11 aa within its N terminus. For comparison, non-mammalian tau showed no homology for the primate 11 aa sequence. To study the requirement of this 11-aa region within N-terminal tau for function and involvement in protein interactions, we used site-directed mutagenesis to delete corresponding codons from the longest isoform of human tau, resulting in a deletion variant of tau we termed tauΔ18–28 (Fig. 1C). DNA sequencing confirmed successful deletion mutagenesis. Expression of tauΔ18–28 protein in cells showed a lower molecular weight band compared with full-length tau (tauFL), as shown by SDS-PAGE and Western blotting with a tau-specific antibody (Fig. 1D).
Figure 1.
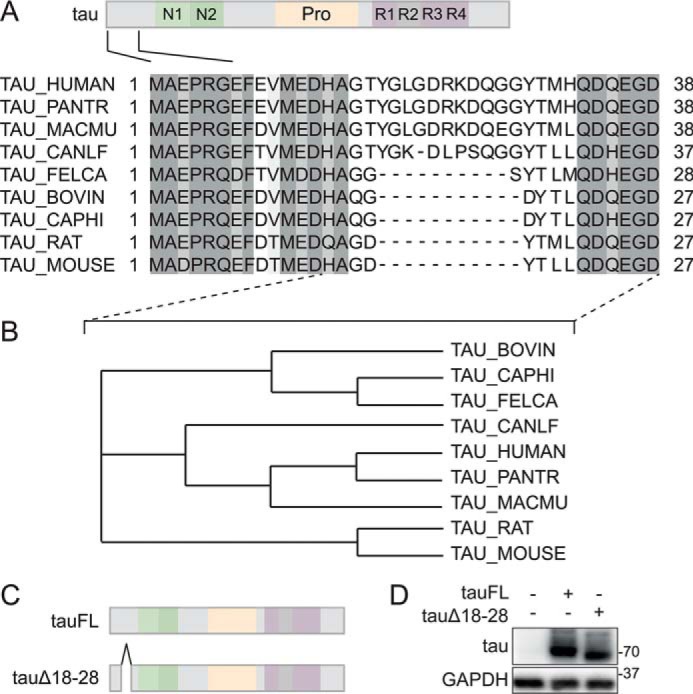
A unique 11-amino acid motif in the N-terminal region of primate tau. A, alignment of mammalian tau primary sequences. Primary sequences of N-terminal regions of human, non-human primate, macaque, bovine, goat, dog, cat, and rodent tau proteins were aligned using ClustalOmega with marked up similarities (gray shading). Note the absence of a motif between amino acids 18 and 28 of primate tau in other mammalian tau sequences. Only canine tau contains a 10-amino acid motif of lesser similarity to primate tau. B, dendrogram of N-terminal mammalian tau protein sequences. Dendrogram analysis of sequences between the indicated residues in A (dashed lines) was performed using ClustalOmega. Note that canine tau clusters with primate (human, Pan troglodytes, and Macaca mulatta) tau N-terminal sequences due to its 10-amino acid motif albeit with weaker similarity. C, amino acids 18–28 were deleted in human tau40 to generate the tauΔ18 construct. D, tauΔ18 and tau full-length (tauFL) were transfected into 293T cells. Immunoblot of cell lysates from tauFL, tauΔ18, or untransfected control was detected with anti-human tau antibody. tauΔ18 shows reduced retention, consistent with deletion of amino acids 18–28. Probing for glyceraldehyde-phosphate dehydrogenase (GAPDH) served as loading control.
Differential interaction mediated by aa 18–28 of human tau
Protein interactions involving the microtubule-binding repeats or the poly-proline region of tau have been reported before (17, 18). However, N-terminal tau interactions and its sequence-dependent requirements remain poorly understood. Presence of a unique 11-aa segment in primate tau N terminus suggested that human tau may engage in interactions distinct from murine (and other non-primate mammalian) tau. To address differential interaction in dependence of these 11 aa, we produced human tauFL or tauΔ18–28 as a glutathione S-transferase (GST) fusion protein for glutathione (GSH)-mediated pulldown experiments. GST fusion proteins were produced in Escherichia coli and purified on GSH beads (Fig. 2A). Purified GST-tauFL and GST-tauΔ18–28 fusion proteins were then used as bait protein for pulldown experiments and incubated with murine cortical lysate from tau-deficient (tau−/−) mice (Fig. 2B). Lysates from tau−/− mice were chosen to avoid competition of bait protein with endogenous murine tau for potential interaction partners. To enable unbiased quantitative comparison of proteins present in multiple samples, we used labeling of peptides with isobaric isotope tags (iTRAQ) for quantitation (19). GST pulldown samples for tauFL and tauΔ18–28 from 4 independent (biological) replicates were multiplexed with different iTRAQ tags (Fig. 2B). Cortical lysates from 4 different age- and gender-matched tau−/− mice were used for GST pulldown replicates. Supporting Table S1 summarizes the results of the multiplex comparison of pulldowns of tauFL and tauΔ18–28, including the number of proteins identified at the 1% false discovery rate. One-hundred and 33 proteins were identified in both tauΔ18–28 and tauFL samples, across the 4 biological replicates (Table S1). Only murine proteins with a minimum unused score of >1.3 (> =95% confidence in sequence identification) and at least two distinct peptides detected by mass spectrometry were included in further analysis. ProteinPilot requires a minimum of 40 counts of iTRAQ reporter ion intensity to calculate iTRAQ ratios. Proteins identified with iTRAQ tag ion intensities below this threshold were not quantified. We calculated differential detection ratios using tauFL as denominator in ProteinPilot, thus reflecting proteins differentially bound to tauΔ18–28 versus tauFL (Table 1). A list of 8 proteins fulfilled the statistical criteria for differential binding regulated by aa 18–28 of tau. Proteins that were identified without differential binding between tauΔ18–28 and tauFL included known interaction partners of tau (e.g. tubulin).
Figure 2.
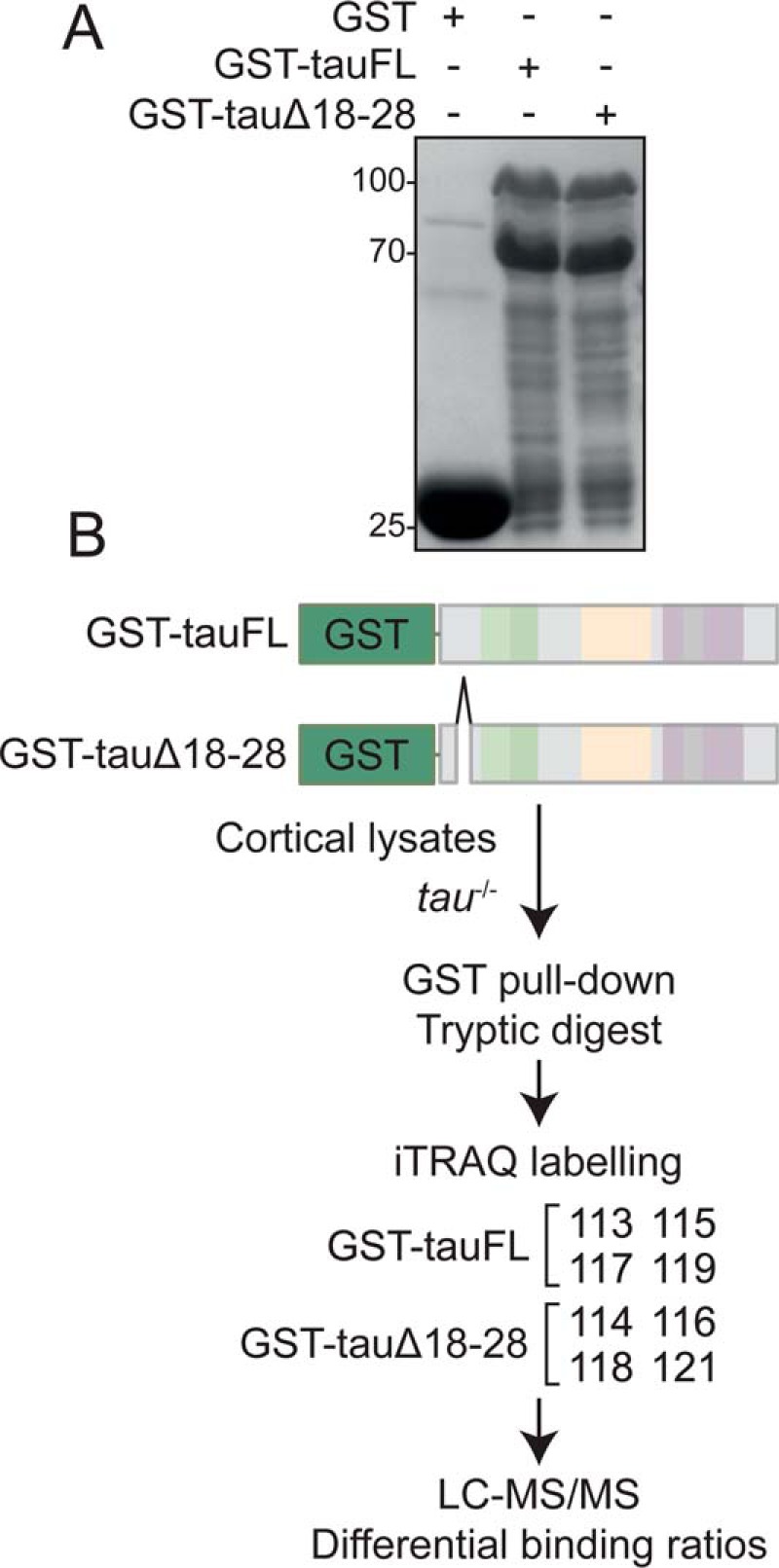
iTRAQ-based mass spectrometry to identify tau protein–protein interactions modulated by aa 18–28. A, production of recombinant tau proteins. GST-tagged tauFL and tauΔ18–28 were produced in E. coli. Samples of washed and eluted protein were separated by SDS-PAGE and visualized by Coomassie Brilliant Blue. B, experimental setup for quantitative comparison of tau protein–protein interactions in dependence of the 11-aa motif in tau's N-terminal region using 8-plex iTRAQ. Technical replicates of GST pulldown with GST-tagged tauFL and tauΔ18–28 were subjected to tryptic digest and iTRAQ labeled (labels 113, 114, 115, 116, 117, 118, 119, and 121). Peptides were detected by mass spectrometry (Triple TOF 5600+ mass spectrometer, SCIEX). Cross-comparison ratios of iTRAQ-labeled peptides were determined by ProteinPilot version 4.0 software.
Table 1.
iTRAQ-based relative ratios of peptides/proteins identified by GST pulldown
Cross-comparison ratios of iTRAQ-labeled peptides were determined by ProteinPilot software and analyzed using MATLAB. Ratios are averaged across 4 biological replicates. A protein had to show the same deregulation trend in at least 2 of the 4 replicates to be considered as deregulated with a p value <0.05. Ratios of >1.2 for increased interactions or <0.82 for reduced interactions were used as cut-off from our previous work using iTRAQ (26).
| UNIPROT ID | Protein name | Total (ID confidence) | Distinct peptides | tauΔ18–28:tauFL mean ratio (n = 4) | p value (n = 4 replicates) |
|---|---|---|---|---|---|
| 1433E_MOUSE | 14-3-3 β | 8.28 | 6 | 1.68 | 0.016 |
| 1433B_MOUSE | 14-3-3 β | 10.12 | 10 | 1.51 | 0.014 |
| NEUG_MOUSE | Neurogranin | 4.00 | 2 | 0.78 | 0.003 |
| SYT1_MOUSE | Synaptotagmin-1 | 2.85 | 2 | 0.77 | 0.025 |
| NEUM_MOUSE | Neuromodulin/growth associated protein 43 | 6.42 | 3 | 0.77 | 0.044 |
| SYN1_MOUSE | Synapsin-1 | 7.54 | 4 | 0.73 | 0.038 |
| ANXA5_MOUSE | Annexin A5/Lipocortin V | 2.09 | 2 | 0.59 | 0.014 |
Tau interactors regulated by aa 18–28 form gene ontology clusters
Performing gene ontology analysis of the differentially bound proteins specific to aa 18–28 revealed significant enrichment of several clusters. Specifically, we found enriched gene ontology term clusters included axon, dendrite, postsynaptic density, and synapse (Fig. 3A, Table S2). Next, we plotted differential interactors identified by ProteinPilot analysis of the iTRAQ data in a network map using STRING (Fig. 3B, Table S3). We also included tau to obtain information on how our list of aa 18–28-linked N-terminal binding candidates relate to previous evidence on tau interactions. Three distinct clusters emerged: one centered around tau and the phospho-protein binding factors 14-3-3β and 14-3-3η, one including synaptic vesicle membrane-associated proteins synapsin-1 (Syn1) and synaptotagmin-1 (Syt1), and a third cluster of neuromodulin/Gap43 and neurogranin. The interaction of tau and annexin A5/Lipocortin-IV had not been previously described. Taken together, these results suggest that the N-terminal human-specific residues aa 18–28 in tau contribute to interactions that may affect distinct cellular and molecular targets of synapse function.
Figure 3.
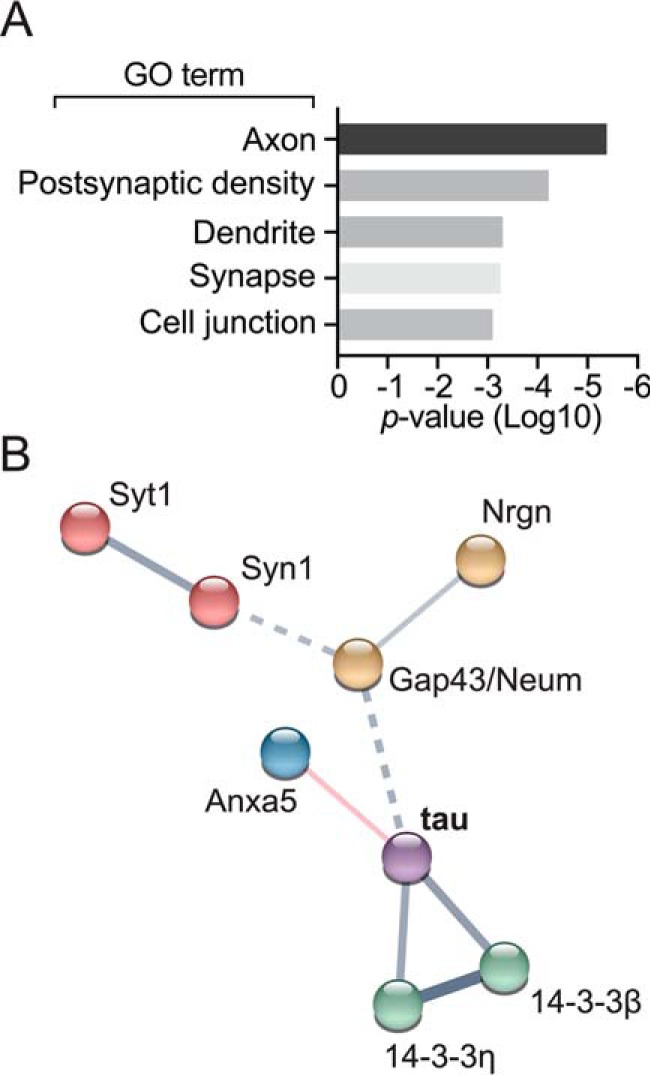
Gene ontology (GO) and protein network analysis of differential protein–protein interactions modulated by presence of the aa 18 to 28 N-terminal tau motif. A, protein ontology was analyzed using DAVID (version 6.8) gene ontology. B, protein interaction network of deregulated protein–protein interactions with tauΔ18–28 was analyzed using STRING (v10.5). Network edges are defined by confidence indicating the strength of data support by line thickness.
Co-immunoprecipitation validates differential interaction linked to aa 18–28 of tau
To address interactions of tau identified by mass spectrometry using an independent method, we selected several candidates (Table 1) for co-immunoprecipitation experiments using transiently transfected cells. Both, previously unreported interactions as well as a known interaction partner of tau (i.e. GSK3β) were chosen. Where expression constructs were not available, coding sequences were amplified from reverse transcribed murine cortical mRNA. Transiently transfecting candidates 14-3-3β, 14-3-3η, Syt1, and annexin A5/Lipocortin V in 293T cells together with V5-tagged tauFL or tauΔ18–28 followed by co-immunoprecipitation confirmed differential affinity to tau that was affected by the presence of aa 18–28 of the human tau N terminus (Fig. 4, A–E). Lower levels of annexin A5, 14-3-3β, and 14-3-3η were found bound to tauΔ18–28 as compared with tauFL (Fig. 4, A–C). Interaction of tau with Syt1 was confirmed by co-immunoprecipitation, however, with increased binding affinity to tauΔ18–28 as compared with tauFL (Fig. 4D).
Figure 4.

Validated interactions modulated by primate-specific N-terminal tau motif. A–E, selected tau protein interaction partners annexin A5/Lipocortin-IV (AnxA5) (A) 14-3-3β (B), 14-3-3η (C), synaptotagmin-1 (D), and GSK3β (E) were expressed in 293T cells together with V5-tagged human tauFL or tauΔ18–28. Protein–protein interaction was assessed by co-immunoprecipitation (IP) using a V5-specific antibody and detected by immunoblotting for HA, myc, green fluorescent protein (GFP), or GSK3. Results from 3 independent experiments are represented as mean ± S.D. (Student t test) ** p < 0.05. ns, not significant.
For comparison, co-immunoprecipitation from cells co-transfected with tauΔ18–28 or tauFL together with a previously reported tau interaction partner GSK3β showed comparable interaction of tauΔ18–28 and tauFL (Fig. 4E), suggesting that aa 18–28 regulate tau protein interactions selectively. Taken together, we could corroborate differential interactions of several binding partners identified by iTRAQ screening that are modulated by the N-terminal aa 18–28 of human tau.
Discussion
In the present study, we revealed that a 11-aa sequence unique to the extreme N terminus of primate tau is linked to differential interaction of tau with a small subset of partners implicated in synaptic function. All confirmed interaction partners showed differential interactions with a human tau variant that lacks aa 18–28, suggesting this unique sequence modulates these interactions. Our results suggest that primate tau may differ in its interactome, and thus in its molecular function as compared with non-primate tau.
Disease mechanisms involving tau as well as physiological functions of tau remain incompletely understood. Protein–protein interactions of tau have been shown to be critical for the role of tau in Aβ toxicity in AD (8). Furthermore, interactions of tau with other factors that mediate intracellular signals can be modulated through post-translational modifications on tau or intrinsic factors in tau (7, 15, 20, 21). The N-terminal region of human tau is defined by splicing-dependent inclusions of 2 primary sequence segments, which contribute to differential functions of tau regarding protein interactions or localization (20).
When aligning primary amino acid sequences of N-terminal tau from different mammalian species, we observed that primate tau (human, chimp, and macaque) contains a segment from aa 18 to 28 that is not present in other species tested. This confirms observations by others (22, 23). Furthermore, antibodies raised against this segment of human tau do not show immunoreactivity with tau from rat, mouse, and bovine brains (24), confirming human/primate-specific differences in the very N-terminal region of tau on the protein level. We postulated that this segment may affect protein interactions of tau and confirmed this hypothesis using pulldown experiments coupled to mass spectrometric binding partner identification.
Seeking to identify potential molecular determinants engaging this segment in N-terminal tau, we employed a deletion approach in the longest human isoform of tau (441 aa) to address the requirement of this segment for tau protein–protein interactions. We based our approach on the idea that deletion would lead to a change in the behavior of the entire tau molecule thus altering interactions, possibly by effects on more distal parts of the protein. This is supported by findings of intramolecular interactions of tau N- and C-terminal regions that are affected through post-translational modifications in either region (25). Thus, binding partners identified by a deletion approach in tau in comparison with full-length tau (tauFL) may reflect the binding propensity of the tau molecule in complex formation rather than direct binding via aa 18–28.
Consistent with this idea, the small N-terminal alteration to tau (=Δ18–28) did not lead to a large change in numbers of binding partners from cortical brain lysates. We and others previously identified differentially expressed proteins using iTRAQ in AD mouse and human samples (20, 26, 27). Using stringent criteria employed in our previous studies using iTRAQ, we identified a small number of dysregulated binding partners of tau with the tauΔ18–28 variant.
The identified proteins were enriched for synaptic and vesicle membrane proteins. Our iTRAQ data suggests differential interaction of synaptic vesicle proteins Syt1 and Syn1/2 with the tauΔ18–28 variant. Using co-immunoprecipitation as an independent method to address interactions, we confirmed interaction of Syt1 and tau in cells (Fig. 4D), with an increased affinity to tauΔ18. Our results suggest that the primate tau N terminus modulates tau's interaction with proteins on synaptic vesicles. Vesicular protein interactions with tau have been reported for Syn1, Syt1, and syntaxin-1B (20, 28), supporting our own findings. Pathological human tau interacts with presynaptic vesicles though its N-terminal region (28). Although physiological implications of tau interactions with vesicular proteins are unknown, human/primate tau may show different functional involvement with synaptic vesicles as compared with rodent tau due to different binding affinities, potentially differentially affecting synaptic functions across species.
We previously identified Lipocortin IV/annexin A5 among mitochondria-associated proteins that are dysregulated in the AD mouse model (26). AnxA5 is a circulating protein with anti-inflammatory and phospholipid-binding properties (29). Interestingly, evidence from cultured neurons and a mouse model suggests AnxA5 as biomarker for AD (30). We observed reduced binding of tauΔ18–28 to AnxA5, which was verified by co-immunoprecipitation. This is the first evidence of an interaction of tau and AnxA5. Virtual loss of interaction of AnxA5 with tauΔ18–28 suggests that the presence of the N-terminal aa 18–28 segment in primate tau critically regulates this interaction unique to primate tau. Interestingly, another member of the annexin family, annexin A2, was shown to interact with tau and tether it to the cell cortex in neurons (31). Together with our finding that tau interacts also with AnxA5 in the present study, it is an intriguing possibility that tau engages in different functional interactions with multiple annexin family members.
Our iTRAQ data showed a reduced affinity of tauΔ18–28 to Gap43/neuromodulin, a protein involved in axonal outgrowth and synaptic development (32, 33). In cultured neurons, Gap43 shows synaptic localization in dependence of synaptic maturation (34). Interestingly, its synaptic localization inversely correlates with the presence of synapsin/synaptotagmin during maturation of axonal synapses. Therefore, tau may contribute to axonal maturation of primate neurons by differential interaction between Syn/Syt1 and Gap43, regulated by the N-terminal aa 18 to 28.
With Nrgn, a postsynaptic protein implicated in synaptic plasticity (35), we identified another tau interaction partner whose affinity to tau is mediated by the 18–28-aa segment in the N terminus of tau. Nrgn had recently been suggested as a cerebrospinal fluid biomarker for AD (36, 37). Forced expression of Nrgn enhances local synaptic plasticity in mice (38). Nrgn associates with the post-synaptic density (39) and with neuronal exosomes (14). Other neuronal exosome-associated proteins identified in our study are synapsin-1 (14) and potentially Syt1. Synaptotagmins can be released in presynaptic exosomes to facilitate postsynaptic signals important in synaptic plasticity and development (40). Interestingly, tau pathology can propagate between neurons via a mechanism that may involve exosomes (41, 42). Increased affinity of primate tau to binding partners associated with exosomes may help explain mechanisms of tau propagation seen in human tauopathies.
Finally, we identified two members of the 14-3-3 protein family, 14-3-3β and 14-3-3η, with differential affinity to tauΔ18–28 and tauFL. In brain lysates, both were detected with iTRAQ ratios >1.2, suggesting increased abundance of these proteins bound to tauΔ18–28 compared with tauFL. In contrast, when testing complex formation with tau in cultured cells, we found lower binding of 14-3-3β/η to tauΔ18–28 compared with tauFL. Interactions with 14-3-3 proteins are dependent on the phosphorylation status of binding targets (43), and tau was previously shown to bind 14-3-3 proteins in a phosphorylation-dependent manner (44). However, tau phosphorylation status may vary depending on incubation conditions and exposure to kinases in cell culture as compared with brain tissue-derived lysates. Given that recombinant tau used for GST pulldown and mass spectrometry was not phosphorylated may explain the discrepancies in 14-3-3 protein binding between our in vitro and cell culture experiments. Nevertheless, our results indicate that aa 18–28 modulate the binding of tau to 14-3-3β and 14-3-3η. 14-3-3 binding normally depends on phosphorylation of partners at SP/TP sites in conserved motifs (43). Since the 18 to 28-aa segment does not harbor the SP/TP phosphorylation site, this sequence may modulate the interaction of tau with 14-3-3β and 14-3-3η in a non-conventional way.
It is noteworthy that canine tau is the only non-primate species to harbor a N-terminal sequence with some, but low similarity to the primate aa 18–28 sequence. Interestingly, there are reports of human-like AD and tau pathology in dogs (45, 46), whereas AD mouse models with Aβ do not develop human-like tau pathology (i.e. NFTs) unless human tau is co-expressed (47). Although we have no direct evidence that this species difference depends on the aa 18–28 motif, it is an intriguing possibility that distinct sequence features that contribute to specific functions of human/primate tau also render it susceptible to disease-driving changes.
To this end, our results define a small, but specific feature within the N terminus of human/primate tau that is linked to differential interaction with synaptic and vesicle-associated proteins, possibly providing a basis for species-specific insights into tau relevant to human disease. Further studies are required to define the specific role of this motif in vivo.
Experimental procedures
Mice
Tau knock-out mice with a targeted allele for Mapt were described previously (48). Mice were housed in individually ventilated cages with food ad libitum and a 12/12-h light-dark cycle. Genotypes of offspring were determined at postnatal day 16 by PCR using primers listed in Table S4. All animal experiments were approved by the University of New South Wales Animal Care and Ethics Committee.
Plasmid constructs
Codons encoding amino acids 18–28 were deleted from the coding sequence of human full-length tau (hTau40 or tau 441 aa) using the Q5 site-directed mutagenesis kit (New England Biolabs) thus generating the tauΔ18 construct. TauFL and tauΔ18 were cloned into pGEX-4-T-1 for recombinant protein production using conventional restriction enzyme cloning. Murine coding DNA (cDNA) encoding AnxA5 was amplified from murine mRNA isolated from cortical brain samples using the first strand cDNA synthesis kit (New England Biolabs) for reverse transcription. AnxA5 was amplified with a C-terminal HA tag and cloned into pcDNA3.1. Plasmid encoding 14-3-3η was a kind gift from Dr. Yue Xiong (Addgene plasmid number 19957), 14-3-3β was a kind gift from Dr. Michael Yaffe (Addgene plasmid number 13270). 14-3-3β and 14-3-3η were subcloned into pENTR-SD-TOPO, and then transferred into a pcDNA3.2-myc mammalian expression destination vector (kind gift from Dr. Romanic) by LR-clonase reaction (Invitrogen, ThermoFisher, Sydney, Australia). Enhanced green fluorescent protein-E-Syt1 was a kind gift from Dr. Pietro De Camilli (Addgene plasmid number 66830). Primers for cloning and mutagenesis are listed in Table S5. All constructs were confirmed by sequencing. Expression of proteins was confirmed by immunoblotting after transfection into 293T cells.
Cell culture
Human embryonic kidney 293T cells were cultured in Dulbecco's modified Eagle's medium high glucose (Sigma, Sydney, Australia) supplemented with 10% fetal bovine serum, 2 mm l-glutamine and 1% penicillin/streptomycin at 37 °C and 5% CO2. Cells were transfected using calcium precipitation of plasmid DNA as previously described (49). Briefly, cells were plated out 24 h prior to transfection and media was replaced 2 h prior to transfection. Plasmid DNA (5 μg) was used per 6-cm dish and 2.5 m calcium chloride (CaCl2) and 2× HBS (281 mm sodium chloride (NaCl), 100 mm HEPES, 1.5 mm disodium hydrogen phosphate (Na2HPO4, pH 7.05–7.09)) was used to precipitate DNA. Cells were lysed and sonicated on ice 48 h post-transfection, in RIPA buffer (1 m Tris, pH 8.0, 150 mm NaCl, 5 mm EDTA, 1% Nonidet P-40, 0.1% SDS, 10 mm sodium fluoride (NaF), 1 mm sodium vanadate (Na3VO4), 1 mm sodium pyrophosphate (NaPP), 0.1% glycerophosphate, 1× Complete Mini protease inhibitor (Roche Applied Science, Sydney, Australia)), then centrifuged (10 min, 16,000 × g, 4 °C). Protein concentration was determined using a Bradford assay (Bio-Rad, Sydney, Australia).
Antibodies
Antibodies against the following epitopes were used: glyceraldehyde-phosphate dehydrogenase (clone 6C5, 1:5,000, Invitrogen, ThermoFisher), Tau (catalog number A0024, 1:5,000, DAKO, Sydney, Australia), green fluorescent protein (catalog number ab290, 1:1,000, Abcam, Sydney, Australia), glycogen synthase kinase-3 (GSK3; catalog number ab131356, 1:1,000, Abcam, Sydney, Australia), hemagglutinin tag (clone HA-7; 1:5,000, Sigma, Sydney, Australia), myc tag-horseradish peroxidase conjugate (MYC-HRP; catalog number R951–25, 1:5,000, Invitrogen, ThermoFisher), and V5 tag (catalog number PA1–993, 1:500, Invitrogen, ThermoFisher). Horseradish peroxidase-coupled secondary antibodies donkey anti-rabbit (1:5,000, Santa Cruz, ThermoFisher).
Co-immunoprecipitation and immunoblotting
Western blotting was performed as previously described (50). Briefly, for immunoprecipitation, 500 μg of protein lysates were incubated with 0.5 μl (0.5 μg) of V5 antibody at 4 °C for 16 h with rotation, before the addition of 20 μl of RIPA-buffer equilibrated Protein G beads (New England Biolabs) and incubation with Protein G Beads for 1 h at 4 °C. Samples were washed 3 times in RIPA buffer and then resuspended in 40 μl of SDS loading buffer, then incubated 5 min at 95 °C. The supernatant was separated from beads using a magnetic holder and for input samples, 5 μg of protein was used. Samples were separated on 8% SDS-PAGE and following protein transfer, membranes were blocked with 3% BSA in Tris-buffered saline solution with 0.1% Tween 20 (TBS-T) before probing with primary antibodies overnight at 4 °C.
Preparation of recombinant proteins
pGEX-4-T-1-tauFL, pGEX-4-T-1-tauΔ18, or empty pGEX-4-T-1 were transformed into E. coli BL21DE3 pLys (Fermentas, ThermoFisher) and proteins were induced in log phase (A600 = 0.4) with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside (Sigma) for 2 h at 37 °C. Cells were pelleted by centrifugation, washed, and lysed by sonication in bacterial lysis buffer (50 mm Tris, pH 7.5, 300 mm NaCl, lysozyme (Sigma), 2 mm EDTA, 0.5% Triton X-100, 10% glycerol, 0.25 mm dithiothreitol (DTT), 1 μg/ml DNase I, 10 mm NaF, 1 mm Na3VO4, 1 mm NaPP, 0.1% glycerophosphate, 1× Complete Mini protease inhibitor (Roche Applied Science, Penzberg, Germany)). After clearing lysates by centrifugation (16,000 × g, 4 °C, 10 min), buffer-equilibrated glutathione-Sepharose beads (GE Healthcare) were used to affinity purify GST fusion proteins. Glutathione-Sepharose beads were washed 3 times with TBS buffer (10 mm Tris, 150 mm NaCl). Bacterial lysis, clearing, and affinity purification was monitored on SDS-PAGE with Coomassie Brilliant Blue staining (Sigma).
GST pulldown
Murine cortical tissue was dissected from brain after transcardial perfusion with cold phosphate-buffered saline, pH 7.4 (PBS). Tissue was homogenized in lysis buffer (50 mm Tris, 7.5, 50 mm NaCl, 5 mm EDTA, 0.3 m sucrose, 0.05% Tween 20, 10 mm NaF, 1 mm Na3VO4, 1 mm NaPP, 0.1% glycerophosphate, 1× Complete Mini protease inhibitor (Roche Applied Science, Penzberg, Germany)) using a Dounce homogenizer (Heidolph Douncer, Schwabach, Germany) on ice. Insoluble materials were removed by centrifugation (16,000 × g, 4 °C, 10 min). Total protein concentration was determined by BCA assay (Bio-Rad). Tissue extract (1 mg) and GST fusion proteins (10 μg) were incubated with buffer-equilibrated GSH beads overnight (16 h) at 4 °C with gentle agitation (6 rpm) on a rotator. Beads were recovered by gentle centrifugation at 400 × g and washed three times with GST buffer. For SDS-PAGE analysis, bound proteins were eluted from the beads by the addition of SDS loading buffer, followed by incubation (5 min, 95 °C). Supernatant proteins were then separated by molecular weight using SDS-PAGE (8%).
ITRAQ sample processing, mass spectrometry, and data analysis
Fig. 2 shows iTRAQ labels and corresponding samples: briefly labels 114, 116, 118, and 121 represent tauΔ18, whereas, 113, 115, 117, and 119 represent tauFL pulldown samples. Labeling of protein samples was performed according to the Applied Biosystems iTRAQ manual. Briefly, GST pulldown samples were desalted and buffer exchanged using 3-kDa Amicon filter units (EMD Millipore) and 50 mm sodium bicarbonate. For the 8-plex iTRAQ labeling, 100 μg of protein sample was reduced by adding 2 μl of Tris(2-carboxyethyl)phosphine (Sigma) and incubating samples at 60 °C for 1 h. Next, samples were treated with 1 μl of iodoacetamide (37 mg/ml) for alkylation/cysteine blocking (10 min, ambient temperature). Protein samples were digested using 4 μg of reconstituted trypsin (sequencing grade, low autolysis trypsin; Promega), at 37 °C for 16 h. Digested samples were briefly spun in a microcentrifuge and pH, if necessary the pH was adjusted to about 9–10 with a few microliters of sodium carbonate (500 mm Na2CO3).
iTRAQ 8-plex reagents were reconstituted in 50 μl of absolute isopropyl alcohol (Sigma, Sydney, Australia) and the whole contents of one vial was transferred to one sample (Fig. 2B shows the iTRAQ labels and corresponding samples) and incubated at ambient temperature for 1 h. The labeled samples were then combined into one sample tube, vortexed, and spun briefly. To reduce the concentration of buffer salts and organics, the sample mixture was diluted 10-fold with cation exchange load buffer (10 mm potassium phosphate in 25% acetonitrile, pH 3.0) before loading sample mixture onto a strong cation exchange cartridge (9.5 ml/h). The flow through was discarded. To elute the peptides from the cation exchange cartridge, 500 μl of cation exchange elution buffer (10 mm potassium phosphate in 25% acetonitrile, 350 mm potassium chloride, pH 3.0) was injected and the eluent was collected in a 1.5-ml polypropylene tube and then dried in a vacuum centrifuge (SpeedVac ThermoFisher Scientific, Sydney, Australia). An additional clean-up step was performed by resuspending the dried sample in 500 μl of 0.2% heptafluorobutyric acid (Sigma, Sydney, Australia), then loading onto an Oasis HLB cartridge (Waters, Sydney, Australia). The peptides were eluted in 700 μl of 0.1% formic, 50% neat acetonitrile, dried under vacuum, and dissolved in 400 μl of 0.05% heptafluorobutyric acid, 1.0% formic acid. Samples were run in triplicate (6 μl injected per run) on a tripleTOF 5600+ LC-MS-MS system (ABSciex, Foster City, CA), as previously described (51). Chromatographic separation of peptides was performed on a ∼12 cm C18 column (350 μm inner diameter, Reprosil-Pur, 1.9 μ, 200 Å, Dr. Maisch GmbH, Ammerbuch-Entringen, Germany) using a 240-min gradient (Dionex UltiMate 3000 RSLCnano pump, ThermoFisher Scientific Dionex, Waltham, MA), with buffer A (H2O:CH3CN of 98:2 containing 0.1% formic acid) to buffer B (H2O:CH3CN of 20:80 containing 0.1% formic acid) at 200 nl/min. Data were processed with ProteinPilot version 4.0 software (ABSciex, Foster City, CA). Reporter ion peak area ratios of tauΔ18 were expressed relative to tauFL for the 4 biological replicates. Cut-off for differential expression was set at a ratio of >1.2 for increased interactions and <0.82 for reduced interaction based on previous iTRAQ experiments (26). Only proteins with ≥2 annotated peptides and an unused score of >1.3 in ProteinPilot were used for further analysis. Quantitative ratios from ProteinPilot were further analyzed using a custom script using MATLAB (vR2016b) by averaging over the 4 biological replicates and comparisons applying a p value (Student's t test) criterion of p < 0.05 to determine the statistically significant differentially bound proteins.
Bioinformatics
Protein ontology was analyzed using DAVID gene ontology (version 6.8) software using gene ontology annotations for the entire murine proteome. Protein–protein interactions were analyzed using STRING (version 10.5).
Author contributions
K. S., A. V., J. B., and A. P. data curation; K. S., A. V., J. B., and A. P. formal analysis; K. S. validation; K. S., A. V., J. B., A. P., Y. D. K., and A. I. methodology; Y. D. K., L. M. I., and A. I. supervision; Y. D. K., L. M. I., and A. I. funding acquisition; Y. D. K., L. M. I., and A. I. writing-review and editing; L. M. I. and A. I. conceptualization; L. M. I. and A. I. writing-original draft; L. M. I. and A. I. project administration.
Supplementary Material
This work was supported in part by National Health and Medical Research Council (NHMRC), Australian Research Council (ARC), CurePSP, and Dementia Australia. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S5.
- AD
- Alzheimer's disease
- aa
- amino acid(s)
- GST
- glutathione S-transferase
- Syn1
- synapsin-1
- Syt1
- synaptotagmin-1
- AnxA5
- annexin A5
- Nrgn
- neurogranin
- GSK3
- glycogen synthase kinase-3
- tauFL
- tau full-length
- HA
- hemagglutinin
- iTRAQ
- isobaric tags for relative and absolute quantitation.
References
- 1. Ittner L. M., and Gotz J. (2011) Amyloid-β and tau: a toxic pas de deux in Alzheimer's disease. Nat. Rev. Neurosci. 12, 65–72 10.1038/nrn2982,10.1038/nrn2985 [DOI] [PubMed] [Google Scholar]
- 2. Ashe K. H. (2007) A tale about tau. N. Engl. J. Med. 357, 933–935 10.1056/NEJMcibr073552 [DOI] [PubMed] [Google Scholar]
- 3. Aronov S., Aranda G., Behar L., and Ginzburg I. (2001) Axonal tau mRNA localization coincides with tau protein in living neuronal cells and depends on axonal targeting signal. J. Neurosci. 21, 6577–6587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mandelkow E. M., and Mandelkow E. (2012) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harbor Perspect. Med. 2, a006247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bakota L., and Brandt R. (2016) Tau biology and tau-directed therapies for Alzheimer's disease. Drugs 76, 301–313 10.1007/s40265-015-0529-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reynolds C. H., Garwood C. J., Wray S., Price C., Kellie S., Perera T., Zvelebil M., Yang A., Sheppard P. W., Varndell I. M., Hanger D. P., and Anderton B. H. (2008) Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J. Biol. Chem. 283, 18177–18186 10.1074/jbc.M709715200 [DOI] [PubMed] [Google Scholar]
- 7. Ittner L. M., Ke Y. D., and Götz J. (2009) Phosphorylated Tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease. J. Biol. Chem. 284, 20909–20916 10.1074/jbc.M109.014472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ittner L. M., Ke Y. D., Delerue F., Bi M., Gladbach A., van Eersel J., Wölfing H., Chieng B. C., Christie M. J., Napier I. A., Eckert A., Staufenbiel M., Hardeman E., and Götz J. (2010) Dendritic function of tau mediates amyloid-β toxicity in Alzheimer's disease mouse models. Cell 142, 387–397 10.1016/j.cell.2010.06.036 [DOI] [PubMed] [Google Scholar]
- 9. Klein C., Kramer E. M., Cardine A. M., Schraven B., Brandt R., and Trotter J. (2002) Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J. Neurosci. 22, 698–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sontag J. M., Nunbhakdi-Craig V., White C. L. 3rd, Halpain S., and Sontag E. (2012) The protein phosphatase PP2A/Bα binds to the microtubule-associated proteins Tau and MAP2 at a motif also recognized by the kinase Fyn: implications for tauopathies. J. Biol. Chem. 287, 14984–14993 10.1074/jbc.M111.338681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qureshi H. Y., and Paudel H. K. (2011) Parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and α-synuclein mutations promote Tau protein phosphorylation at Ser-262 and destabilize microtubule cytoskeleton in vitro. J. Biol. Chem. 286, 5055–5068 10.1074/jbc.M110.178905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dixit R., Ross J. L., Goldman Y. E., and Holzbaur E. L. (2008) Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089 10.1126/science.1152993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoover B. R., Reed M. N., Su J., Penrod R. D., Kotilinek L. A., Grant M. K., Pitstick R., Carlson G. A., Lanier L. M., Yuan L. L., Ashe K. H., and Liao D. (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081 10.1016/j.neuron.2010.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goetzl E. J., Kapogiannis D., Schwartz J. B., Lobach I. V., Goetzl L., Abner E. L., Jicha G. A., Karydas A. M., Boxer A., and Miller B. L. (2016) Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer's disease. FASEB J. 30, 4141–4148 10.1096/fj.201600816R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ittner A., Chua S. W., Bertz J., Volkerling A., van der Hoven J., Gladbach A., Przybyla M., Bi M., van Hummel A., Stevens C. H., Ippati S., Suh L. S., Macmillan A., Sutherland G., Kril J. J., et al. (2016) Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer's mice. Science 354, 904–908 10.1126/science.aah6205 [DOI] [PubMed] [Google Scholar]
- 16. Goedert M., and Jakes R. (2005) Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta 1739, 240–250 10.1016/j.bbadis.2004.08.007 [DOI] [PubMed] [Google Scholar]
- 17. Guo T., Noble W., and Hanger D. P. (2017) Roles of tau protein in health and disease. Acta Neuropathol. 133, 665–704 10.1007/s00401-017-1707-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee G., Newman S. T., Gard D. L., Band H., and Panchamoorthy G. (1998) Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 111, 3167–3177 [DOI] [PubMed] [Google Scholar]
- 19. Latosinska A., Vougas K., Makridakis M., Klein J., Mullen W., Abbas M., Stravodimos K., Katafigiotis I., Merseburger A. S., Zoidakis J., Mischak H., Vlahou A., and Jankowski V. (2015) Comparative analysis of label-free and 8-plex iTRAQ approach for quantitative tissue proteomic analysis. PLoS ONE 10, e0137048 10.1371/journal.pone.0137048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu C., Song X., Nisbet R., and Götz J. (2016) Co-immunoprecipitation with tau isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N tau in disease. J. Biol. Chem. 291, 8173–8188 10.1074/jbc.M115.641902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mondragón-Rodriguez S., Trillaud-Doppia E., Dudilot A., Bourgeois C., Lauzon M., Leclerc N., and Boehm J. (2012) Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-d-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 287, 32040–32053 10.1074/jbc.M112.401240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holzer M., Craxton M., Jakes R., Arendt T., and Goedert M. (2004) Tau gene (MAPT) sequence variation among primates. Gene 341, 313–322 10.1016/j.gene.2004.07.013 [DOI] [PubMed] [Google Scholar]
- 23. Poorkaj P., Kas A., D'Souza I., Zhou Y., Pham Q., Stone M., Olson M. V., and Schellenberg G. D. (2001) A genomic sequence analysis of the mouse and human microtubule-associated protein tau. Mamm. Genome 12, 700–712 10.1007/s00335-001-2044-8 [DOI] [PubMed] [Google Scholar]
- 24. Crowe A., Ksiezak-Reding H., Liu W. K., Dickson D. W., and Yen S. H. (1991) The N terminal region of human tau is present in Alzheimer's disease protein A68 and is incorporated into paired helical filaments. Am. J. Pathol. 139, 1463–1470 [PMC free article] [PubMed] [Google Scholar]
- 25. Jeganathan S., Hascher A., Chinnathambi S., Biernat J., Mandelkow E. M., and Mandelkow E. (2008) Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J. Biol. Chem. 283, 32066–32076 10.1074/jbc.M805300200 [DOI] [PubMed] [Google Scholar]
- 26. Rhein V., Song X., Wiesner A., Ittner L. M., Baysang G., Meier F., Ozmen L., Bluethmann H., Dröse S., Brandt U., Savaskan E., Czech C., Götz J., and Eckert A. (2009) Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proc. Natl. Acad. Sci. U.S.A. 106, 20057–20062 10.1073/pnas.0905529106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang P., Joberty G., Buist A., Vanoosthuyse A., Stancu I. C., Vasconcelos B., Pierrot N., Faelth-Savitski M., Kienlen-Campard P., Octave J. N., Bantscheff M., Drewes G., Moechars D., and Dewachter I. (2017) Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo. Acta Neuropathol. 133, 731–749 10.1007/s00401-016-1663-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou L., McInnes J., Wierda K., Holt M., Herrmann A. G., Jackson R. J., Wang Y. C., Swerts J., Beyens J., Miskiewicz K., Vilain S., Dewachter I., Moechars D., De Strooper B., Spires-Jones T. L., De Wit J., and Verstreken P. (2017) Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun. 8, 15295 10.1038/ncomms15295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burgmaier M., Schutters K., Willems B., van der Vorst E. P., Kusters D., Chatrou M., Norling L., Biessen E. A., Cleutjens J., Perretti M., Schurgers L. J., and Reutelingsperger C. P. (2014) AnxA5 reduces plaque inflammation of advanced atherosclerotic lesions in apoE(−/−) mice. J. Cell Mol. Med. 18, 2117–2124 10.1111/jcmm.12374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamaguchi M., Kokai Y., Imai S., Utsumi K., Matsumoto K., Honda H., Mizue Y., Momma M., Maeda T., Toyomasu S., Ito Y. M., Kobayashi S., Hashimoto E., Saito T., and Sohma H. (2010) Investigation of annexin A5 as a biomarker for Alzheimer's disease using neuronal cell culture and mouse model. J. Neurosci. Res. 88, 2682–2692 [DOI] [PubMed] [Google Scholar]
- 31. Gauthier-Kemper A., Weissmann C., Golovyashkina N., Sebo-Lemke Z., Drewes G., Gerke V., Heinisch J. J., and Brandt R. (2011) The frontotemporal dementia mutation R406W blocks tau's interaction with the membrane in an annexin A2-dependent manner. J. Cell Biol. 192, 647–661 10.1083/jcb.201007161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frey D., Laux T., Xu L., Schneider C., and Caroni P. (2000) Shared and unique roles of CAP23 and GAP43 in actin regulation, neurite outgrowth, and anatomical plasticity. J. Cell Biol. 149, 1443–1454 10.1083/jcb.149.7.1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang C. Y., Lin H. C., Song Y. P., Hsu Y. T., Lin S. Y., Hsu P. C., Lin C. H., Hung C. C., Hsu M. C., Kuo Y. M., Lee Y. J., Hsu C. Y., and Lee Y. H. (2015) Protein kinase C-dependent growth-associated protein 43 phosphorylation regulates gephyrin aggregation at developing GABAergic synapses. Mol. Cell. Biol. 35, 1712–1726 10.1128/MCB.01332-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morita S., and Miyata S. (2013) Synaptic localization of growth-associated protein 43 in cultured hippocampal neurons during synaptogenesis. Cell Biochem. Funct. 31, 400–411 10.1002/cbf.2914 [DOI] [PubMed] [Google Scholar]
- 35. Díez-Guerra F. J. (2010) Neurogranin, a link between calcium/calmodulin and protein kinase C signaling in synaptic plasticity. IUBMB Life 62, 597–606 10.1002/iub.357 [DOI] [PubMed] [Google Scholar]
- 36. Portelius E., Zetterberg H., Skillbäck T., Törnqvist U., Andreasson U., Trojanowski J. Q., Weiner M. W., Shaw L. M., Mattsson N., and Blennow K. (2015) Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer's disease. Brain 138, 3373–3385 10.1093/brain/awv267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kester M. I., Teunissen C. E., Crimmins D. L., Herries E. M., Ladenson J. H., Scheltens P., van der Flier W. M., Morris J. C., Holtzman D. M., and Fagan A. M. (2015) Neurogranin as a cerebrospinal fluid biomarker for synaptic loss in symptomatic Alzheimer disease. JAMA Neurol. 72, 1275–1280 10.1001/jamaneurol.2015.1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhong L., Brown J., Kramer A., Kaleka K., Petersen A., Krueger J. N., Florence M., Muelbl M. J., Battle M., Murphy G. G., Olsen C. M., and Gerges N. Z. (2015) Increased prefrontal cortex neurogranin enhances plasticity and extinction learning. J. Neurosci. 35, 7503–7508 10.1523/JNEUROSCI.0274-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Watson J. B., Szijan I., and Coulter P. M. 2nd. (1994) Localization of RC3 (neurogranin) in rat brain subcellular fractions. Brain Res. Mol. Brain Res. 27, 323–328 10.1016/0169-328X(94)90017-5 [DOI] [PubMed] [Google Scholar]
- 40. Korkut C., Li Y., Koles K., Brewer C., Ashley J., Yoshihara M., and Budnik V. (2013) Regulation of postsynaptic retrograde signaling by presynaptic exosome release. Neuron 77, 1039–1046 10.1016/j.neuron.2013.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Asai H., Ikezu S., Tsunoda S., Medalla M., Luebke J., Haydar T., Wolozin B., Butovsky O., Kügler S., and Ikezu T. (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593 10.1038/nn.4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Y., Balaji V., Kaniyappan S., Krüger L., Irsen S., Tepper K., Chandupatla R., Maetzler W., Schneider A., Mandelkow E., and Mandelkow E. M. (2017) The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 12, 5 10.1186/s13024-016-0143-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pozuelo Rubio M., Geraghty K. M., Wong B. H., Wood N. T., Campbell D. G., Morrice N., and Mackintosh C. (2004) 14-3-3-affinity purification of over 200 human phosphoproteins reveals new links to regulation of cellular metabolism, proliferation and trafficking. Biochem. J. 379, 395–408 10.1042/bj20031797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sadik G., Tanaka T., Kato K., Yamamori H., Nessa B. N., Morihara T., and Takeda M. (2009) Phosphorylation of tau at Ser214 mediates its interaction with 14-3-3 protein: implications for the mechanism of tau aggregation. J. Neurochem. 108, 33–43 10.1111/j.1471-4159.2008.05716.x [DOI] [PubMed] [Google Scholar]
- 45. Schmidt F., Boltze J., Jager C., Hofmann S., Willems N., Seeger J., Hartig W., and Stolzing A. (2015) Detection and Quantification of β-amyloid, pyroglutamyl Abeta, and tau in aged canines. J. Neuropathol. Exp. Neurol. 74, 912–923 10.1097/NEN.0000000000000230 [DOI] [PubMed] [Google Scholar]
- 46. Smolek T., Madari A., Farbakova J., Kandrac O., Jadhav S., Cente M., Brezovakova V., Novak M., and Zilka N. (2016) Tau hyperphosphorylation in synaptosomes and neuroinflammation are associated with canine cognitive impairment. J. Comp. Neurol. 524, 874–895 10.1002/cne.23877 [DOI] [PubMed] [Google Scholar]
- 47. Oddo S., Caccamo A., Shepherd J. D., Murphy M. P., Golde T. E., Kayed R., Metherate R., Mattson M. P., Akbari Y., and LaFerla F. M. (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421 10.1016/S0896-6273(03)00434-3 [DOI] [PubMed] [Google Scholar]
- 48. Tucker K. L., Meyer M., and Barde Y. A. (2001) Neurotrophins are required for nerve growth during development. Nat. Neurosci. 4, 29–37 10.1038/82868 [DOI] [PubMed] [Google Scholar]
- 49. Ittner A. A., Bertz J., Chan T. Y., van Eersel J., Polly P., and Ittner L. M. (2014) The nucleotide exchange factor SIL1 is required for glucose-stimulated insulin secretion from mouse pancreatic beta cells in vivo. Diabetologia 57, 1410–1419 10.1007/s00125-014-3230-z [DOI] [PubMed] [Google Scholar]
- 50. Ittner A., Block H., Reichel C. A., Varjosalo M., Gehart H., Sumara G., Gstaiger M., Krombach F., Zarbock A., and Ricci R. (2012) Regulation of PTEN activity by p38Δ-PKD1 signaling in neutrophils confers inflammatory responses in the lung. J. Exp. Med. 209, 2229–2246 10.1084/jem.20120677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muenchhoff J., Poljak A., Thalamuthu A., Gupta V. B., Chatterjee P., Raftery M., Masters C. L., Morris J. C., Bateman R. J., Fagan A. M., Martins R. N., and Sachdev P. S. (2016) Changes in the plasma proteome at asymptomatic and symptomatic stages of autosomal dominant Alzheimer's disease. Sci. Rep. 6, 29078 10.1038/srep29078 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.