

ABSTRACT
OPTN (optineurin), a ubiquitin-binding scaffold protein, functions as an important macroautophagy/autophagy receptor in selective autophagy processes. Mutations in OPTN have been linked with human neurodegenerative diseases including ALS and glaucoma. However, the mechanistic basis underlying the recognition of ubiquitin by OPTN and its regulation by TBK1-mediated phosphorylation are still elusive. Here, we demonstrate that the UBAN domain of OPTN preferentially recognizes linear ubiquitin chain and forms an asymmetric 2:1 stoichiometry complex with the linear diubiquitin. In addition, our results provide new mechanistic insights into how phosphorylation of UBAN would regulate the ubiquitin-binding ability of OPTN and how disease-associated mutations in the OPTN UBAN domain disrupt its interaction with ubiquitin. Finally, we show that defects in ubiquitin-binding may affect the recruitment of OPTN to linear ubiquitin-decorated mutant Huntington protein aggregates. Taken together, our findings clarify the interaction mode between UBAN and linear ubiquitin chain in general, and expand our knowledge of the molecular mechanism of ubiquitin-decorated substrates recognition by OPTN as well as the pathogenesis of neurodegenerative diseases caused by OPTN mutations.
KEYWORDS: autophagy receptor, linear ubiquitin chain, optineurin, TBK1, UBAN domain
Abbreviations
- ALS
amyotrophic lateral sclerosis
- Atg8
autophagy-related protein 8
- DAPI
4',6-diamidino-2-phenylindole
- DiUb
diubiquitin
- GFP
green fluorescent protein
- HsOPTN
Homo sapiens/human optineurin
- HSQC
heteronuclear single-quantum coherence
- HTT-polyQ
polyQ proteins derived from human huntingtin
- IKBKG
inhibitor of nuclear factor kappa B kinase subunit gamma
- ITC
isothermal titration calorimetry
- JOAG
juvenile open-angle glaucoma
- LIR
LC3-interacting region
- MALS
multi-angle light scattering
- mCherry
mCherry fluorescent protein
- MM
molecular mass
- MmOPTN
Mus musculus/mouse optineurin
- NMR
nuclear magnetic resonance
- OPTN
optineurin
- TBK1
TANK-binding kinase 1
- UBAN
ubiquitin binding in TNIP/ABIN and IKBKG/NEMO proteins.
Introduction
Autophagy, a highly conserved and lysosome-dependent intracellular catabolic process in eukaryotes, plays essential roles in maintaining cellular homeostasis and/or adapting to various cellular stresses.1–3 Although autophagy was thought to be a nonselective process that indiscriminately engulfs and sequesters cellular components for autophagic degradation, increasing evidences suggest that at least a subset of autophagy-mediated degradations, including bulk protein aggregates, dysfunctional organelles and cytosol-invading pathogens, are highly selective, and require specific substrate-recognition and targeting by the autophagy machinery.2,4–8 During these selective autophagy processes, a panel of unique adaptor proteins termed autophagy receptors plays crucial roles.7,9,10 In mammals, the currently known autophagy receptors including SQSTM1/p62, NBR1 (NBR1, autophagy cargo receptor), OPTN (optineurin), CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), TAX1BP1 (Tax1 binding protein 1), FUNDC1 (FUN14 domain containing 1), BNIP3L (BCL2 interacting protein 3 like), STBD1 (starch binding domain 1), TRIM5/TRIM5α, NCOA4 (nuclear receptor coactivator 4), LMNB1 (lamin B1), and RETREG1/FAM134B (reticulophagy regulator 1), can be mainly divided into 2 categories, soluble autophagy receptors and membrane-associated autophagy receptors, all of which contain an LC3-interacting region (LIR) that can specifically recruit the Atg8-family proteins and a cargo-associating motif, which can associate with certain types of autophagic cargoes.9–19 Accordingly, autophagy receptors can function as bridging adaptors to specifically link the cargoes to the autophagy machinery for their subsequent autophagic degradation. In order to timely and efficiently remove the deleterious substrates during selective autophagy, the functions of autophagy receptors are subject to tight regulation.6,7,9 In this regard, the phosphorylation of autophagy receptors in higher eukaryotes regulates autophagy in response to diverse cellular stresses by not only tuning the associations of autophagy receptors with cargoes but also affecting the interactions between the autophagy receptors and Atg8-family proteins.20–24 However, the molecular mechanisms of those phosphorylation-dependent regulations of autophagy receptors are still not well understood.
Similar to that K48 ubiquitination of degradative substrates for proteasomes, the ubiquitation of substrates is also thought to mark substrates for autophagic degradation.5,7,19,23,25 Many substrates of selective autophagy are decorated with ubiquitin proteins before they can be discriminated by the ubiquitin-binding autophagy receptors.6,7,9,25 For instance, in aggrephagy (the selective autophagy of bulk aggregated proteins), the bulk protein aggregates are marked with polyubiquitin chains which serve as recognition signals for the recruitment of autophagy receptors together with downstream autophagic machinery.25,26 Ubiquitin has an extreme N-terminal methionine residue and 7 lysine residues (K6, K11, K27, K29, K33, K48, and K63), all of which can be used to form distinct linkage types of ubiquitin chains.27,28 Different linkage types of ubiquitin chains can be selectively recognized by specific ubiquitin-binding proteins and are endowed with distinct cellular functions in mammalian cells.27,29,30 So far, the M1-linked, K48-linked, and K63-linked polyubiquitin chains are found in selective autophagy processes, and are implicated as specific degradation signals.25,31–33 However, given that there are many different ubiquitin-binding autophagy receptors, how the ubiquitin-modified substrates are selectively and efficiently recognized by certain types of autophagy receptors is an important question that needs to be understood.
OPTN is a ubiquitin-binding autophagy receptor and participates in xenophagy (the selective autophagy of pathogens),20 aggrephagy,34 and recently is reported to participate in depolarization-dependent mitophagy (the selective autophagy of mitochondria).21,35,36 The N-terminal part of OPTN mainly contains 2 coiled-coil domains sandwiching a LIR motif, whereas its C-terminal part has a ubiquitin-binding UBAN (ubiquitin binding domain in IKBKG/NEMO and TNIPs/ABINs) domain followed by a zinc finger (Fig. 1A). The UBAN domain of OPTN is homologous to that of IKBKG,37 a crucial regulatory subunit of the IKK complex involved in the NFKB1/NF-κB pathway, and binds preferentially to the M1-linked or linear ubiquitin chain as well as the K63-linked chain.38–40 The N-terminal coiled-coil domain of OPTN directly interacts with the C-terminal domain of TBK1 to mediate the stable OPTN-TBK1 hetero-tetramer complex formation.41,42 Interestingly, the functions of OPTN in selective autophagy are tightly regulated by TBK1. Particularly, TBK1 can directly phosphorylate the S172 residue adjacent to the LIR motif of OPTN to enhance the binding affinities of OPTN to Atg8-family proteins,20 and the S473 residue located within the UBAN region to increase the ability of OPTN to recognize ubiquitin proteins,21,22 thereby promoting the efficiency of OPTN-involved selective autophagy processes. However, the structural mechanisms governing the selective recognition of ubiquitin proteins by the OPTN UBAN domain and its regulation by TBK1-mediated phosphorylation are still poorly understood. Many missense and truncation disease-causing mutations in the UBAN region of OPTN are found in patients with amyotrophic lateral sclerosis (ALS), frontal temporal dementia and glaucoma,43–50 supporting the functional importance of this domain. However, it is not clear how such mutations lead to these devastating human diseases, and the underlying disease mechanism caused by these UBAN mutations remains to be elucidated.
Figure 1.
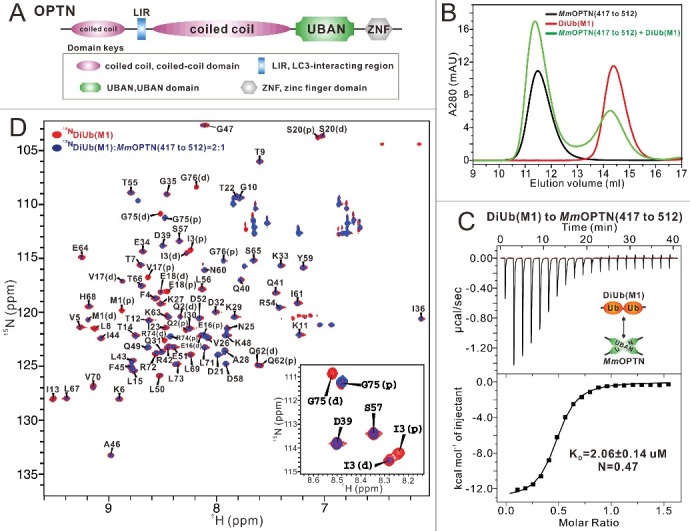
Biochemical characterization of the OPTN and DiUb(M1) interaction. (A) A schematic diagram showing the domain arrangements of OPTN. (B) Analytical gel filtration chromatography analyses of the interaction between MmOPTN(417 to 512) fragment and DiUb(M1). mAU, milli absorbance unit. (C) ITC-based measurement of the binding affinity of the MmOPTN UBAN region with DiUb(M1). The KD error is the fitted error obtained from the data analysis software, when using the one-site binding model to fit the ITC data. The ITC-measured N value, which is related to the binding stoichiometry, indicates that the binding stoichiometry of MmOPTN UBAN and DiUb(M1) interaction is 2:1. (D) Superposition plot of the 1H-15N HSQC spectra of DiUb(M1) titrated with unlabeled MmOPTN(417 to 512) proteins at different molar ratios. In this drawing, the residues of distal and proximal ubiquitin in the DiUb(M1) were labeled with “D” and “P”, respectively. For clarity, the insert shows the enlarged view of a representative region of the overlaid 1H-15N HSQC spectra.
To address the molecular basis of ubiquitin recognition by OPTN as well as its regulation by TBK1, we biochemically and structurally characterized the interactions of the UBAN region of OPTN with different ubiquitin proteins, and discovered that the UBAN domain of OPTN preferentially recognizes linear ubiquitin chain and forms a stable 2:1 stoichiometry complex with the linear diubiquitin. We provide the first atomic structure of the OPTN UBAN domain in complex with linear diubiquitin, demonstrating the molecular mechanism by which a UBAN dimer asymmetrically binds to a linear diubiquitin. We reveal mechanistic insights as to how phosphorylation of the OPTN Ser473 residue would promote the ability of the UBAN domain of OPTN to bind ubiquitin proteins. Furthermore, our study also provides structural explanations as to the functional significance of several disease-associated mutations found in the OPTN UBAN region in autophagic degradation. In all, our findings expand our knowledge on the molecular mechanism of ubiquitin-decorated autophagic substrates recognition by OPTN as well as the mutual regulation between OPTN and TBK1 in selective autophagy. Our results have implication in our understanding the pathogenesis of neurodegenerative diseases caused by OPTN defects.
Results
The UBAN domain of OPTN selectively recognizes the linear ubiquitin chain and forms a 2:1 stoichiometry complex with the linear diubiquitin
As an IKBKG/NEMO-like ubiquitin-binding autophagy receptor, OPTN relies on its UBAN domain to specifically recognize ubiquitinated cargoes for autophagic degradation.37,45 To elucidate the molecular interaction mechanism between OPTN UBAN and ubiquitin proteins, we first carried out a sequence alignment of OPTN from different species, which showed that the UBAN region of OPTN (residues 399 to 509 of human OPTN, hereafter referred to as HsOPTN(399 to 509)) is highly conserved throughout evolution (Fig. S1A), in line with its crucial roles in mediating the autophagic function of OPTN. Next, using purified recombinant proteins, we biochemically characterized the interactions of the OPTN UBAN region (residues 417 to 512 of mouse OPTN, hereafter referred to as MmOPTN(417 to 512)) with monoubiquitin and different linkage types of ubiquitin chains implicated in the OPTN-involved selective autophagy processes including M1-linked diubiquitin (hereafter referred to as DiUb[M1]), K48-linked diubiquitin (hereafter referred to as DiUb[K48]), and K63-linked diubiquitin (hereafter referred to as DiUb[K63]). Analytical gel filtration chromatography assays showed that the OPTN UBAN domain can specifically bind to the DiUb(M1) and DiUb(K63), but is unable to interact with the monoubiquitin or DiUb(K48) (Fig. 1B; Fig. S1B to D). In addition to confirming that there is no detectable interaction between the OPTN UBAN and monoubiquitin or DiUb(K48) (Fig. S1E and F), quantitative ITC analyses also revealed that the OPTN UBAN efficiently binds to DiUb(M1) and DiUb(K63), but with distinct binding affinity KD values, about 2.06 μM and 60.70 μM, respectively (Fig. 1C; Fig. S1G). Thus, our data clearly demonstrated that the OPTN UBAN domain preferentially binds to a linear ubiquitin chain. Notably, the ITC-measured N value of MmOPTN(417 to 512) and DiUb(M1) interaction is 0.47, very close to 0.5 (Fig. 1C), indicating that the stoichiometry of the OPTN UBAN binding to DiUb(M1) is 2:1. Furthermore, MALS-based measurement demonstrated that the OPTN UBAN is a dimer in solution and the binding stoichiometry between the OPTN UBAN and DiUb(M1) in solution is 2:1, as the measured molecular mass (MM) of HsOPTN(399 to 509) is about 24.4 kD, roughly 2-fold of its theoretical MM, and the measured MM of purified HsOPTN(399 to 509)-DiUb(M1) complex is about 35.9 kD, close to the theoretical MM of one OPTN UBAN dimer in complex with one DiUb(M1) molecule (Fig. S2A). To further validate that the binding stoichiometry between the OPTN UBAN and DiUb(M1) is 2:1 rather than 2:2, we also designed a biochemical assay using the purified HsOPTN(399 to 509) fused with a DiUb(M1) through a TEV enzyme cutting site, and then digested by TEV enzyme. The following FPLC combined with SDS-PAGE analyses revealed that a large amount of DiUb(M1) proteins were disassociated from the HsOPTN(399 to 509)-DiUb(M1) complex (Fig. S2B, C), indicating that the binding stoichiometry of OPTN UBAN to DiUb(M1) is unlikely to be 2:2.
We also used NMR spectroscopy to further characterize the interaction between DiUb(M1) and the OPTN UBAN domain. Titration of 15N-labeled DiUb(M1) with MmOPTN(417 to 512) fragment showed that a select set of peaks in the 1H-15N HSQC spectrum of DiUb(M1) underwent significant peak-broadening (Fig. 1D), confirming that the OPTN UBAN region can specifically bind to the linear ubiquitin chain. After backbone assignments, we found that majority of residues on the distal and proximal ubiquitin of DiUb(M1) only showed one set of peaks in the 1H-15N HSQC spectrum, however, some residues displayed 2 distinct sets of peaks in the spectrum due to the different chemical environments of those residues in the distal and proximal ubiquitin, such as M1, Q2, I3, V17, E18, S20, R74, G75 and G76 (Fig. 1D). Importantly, further analyses revealed that these 2-peak residues showed very different peak-broadening profiles when titrated with the MmOPTN(417 to 512) protein (Fig. 1D). For instance, in the presence of the 2:1 molar ratio of the OPTN fragment, the peaks corresponding to the I3 and G75 residues in the distal ubiquitin of DiUb(M1) completely disappeared, but their counterparts in the proximal ubiquitin were still well detectable (Fig. 1D), suggesting that the OPTN UBAN binds to different surfaces of 2 ubiquitin moieties in the DiUb(M1). Altogether, our systematically biochemical analyses clearly demonstrated that the OPTN UBAN domain selectively recognizes linear diubiquitin to form a 2:1 stoichiometry complex.
Overall structure of the OPTN UBAN domain in complex with the linear diubiquitin
To uncover the mechanistic basis underlying the specific interaction between the OPTN UBAN domain and DiUb(M1), we sought to determine their complex structure. Fortunately, after more than 2 y of extensive crystal screening and optimization, we were finally able to obtain good crystals that diffracted to 3.0 Å using the purified complex protein formed by MmOPTN(417 to 512) and DiUb(M1). The complex structure was determined by the molecular replacement method using the previously solved structure of the IKBKG UBAN dimer (PDB ID: 2ZVO),38 and a ubiquitin monomer structure as the search modes (Table S1). In the final refined structural model, each asymmetric unit contains 4 OPTN molecules and 2 DiUb(M1), to form 2 2:1 stoichiometry complexes (Fig. 2A), in line with our aforementioned ITC and MALS results that 2 OPTN UBAN molecules bind to one DiUb(M1) (Fig. 1C; Fig. S2A). In the complex structure, each MmOPTN UBAN region adopts a crescent-shaped continuous α-helix, and packs together to form a parallel coiled-coil homodimer (Fig. 2A). Intriguingly, the DiUb(M1) molecule asymmetrically binds to one surface side of the OPTN coiled-coil dimer in the complex structure (Fig. 2A), and packs extensively with a solvent-exposed elongated groove formed by 2 OPTN helices, burying a total surface area of ∼940 Å2 (Fig. 2A, B). Further structural comparison revealed that the binding mode of the OPTN UBAN domain to DiUb(M1) is very similar to that of the interaction of the IKBKG UBAN domain and DiUb(M1) (Fig. S3), however, the binding stoichiometry of the 2 complexes is totally different. In contrast to the 2:1 stoichiometry of OPTN-DiUb(M1) complex, in the crystal structure of the IKBKG-DiUb(M1) complex (PDB ID: 2ZVO), the 2 DiUb(M1) molecules symmetrically accommodate 2 surface sides of the IKBKG dimer,38 resulting in a 2:2 stoichiometry complex (Fig. S3B, C).
Figure 2.
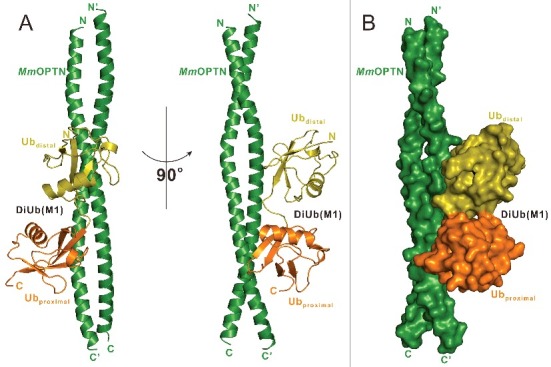
Overall structure of MmOPTN(417 to 512) in complex with the DiUb(M1). (A) Ribbon diagram showing the overall structure of MmOPTN(417-512)-DiUb(M1) complex. In this drawing, MmOPTN(417 to 512) is shown in forest green, distal- and proximal- ubiquitin of DiUb(M1) in olive and orange, respectively. (B) The surface representation showing the overall architecture of MmOPTN(417 to 512)-DiUb(M1) complex with the same color plan as in panel (A).
The molecular interface of the OPTN and linear diubiquitin complex
Consistent with our NMR data, in the MmOPTN-DiUb(M1) complex structure, 2 ubiquitin moieties in the DiUb(M1) bind differentially to MmOPTN (Fig. 2). Further structural analysis revealed that the distal ubiquitin of DiUb(M1) interacts with both chains of the OPTN dimer through its canonical Ile44 hydrophobic patch together with the C-terminal tail, while the proximal ubiquitin contacts only one chain of OPTN dimer via a solvent-exposed surface, which is composed of polar residues and adjacent to the Ile44 hydrophobic site (Fig. 3A). Specifically, the interaction between OPTN and distal ubiquitin is primarily mediated by hydrophobic and polar interactions (Fig. 3A, B). In particular, the hydrophobic side chains of Val466, Ala469 and Val473 from one chain of the OPTN dimer pack against the canonic ubiquitin hydrophobic patch formed by the side chains of Ile44 and Val70 from the distal ubiquitin, and the side chain of Leu73 located at the C-terminal tail of the distal ubiquitin is embedded in the hydrophobic pocket formed by the side chains of Tyr474 from one chain of the OPTN dimer and Cys475, Phe478 from the other chain of the OPTN dimer (Fig. 3B). Furthermore, the polar side chain groups of OPTN Glu462, Ser476 and Asp477 residues form 3 hydrogen bonds with the side chains of His68 and Arg72 as well as the backbone amide of Leu73 from the distal ubiquitin, respectively (Fig. 3B). In addition, 2 pairs of salt bridges between the negatively charged Asp477, Glu481 residues of OPTN and the positively charged Arg72, Arg74 residues of distal ubiquitin further strengthen the OPTN and distal ubiquitin interaction (Fig. 3B). In contrast, the interaction between proximal ubiquitin and the OPTN UBAN domain is highly specific and only mediated by polar interactions (Fig. 3A, C). Particularly, the side chains of OPTN Arg482, Arg485 and Glu490 residues interact with the side chains of Glu92, Glu140 and Lys87 in the proximal ubiquitin to form 3 salt bridges, respectively, and the Arg482, Arg485, Glu486, His489 residues of OPTN coupled with the Thr90, Gln78, Thr88 and Thr142 residues of the proximal ubiquitin to form 5 hydrogen bonds (Fig. 3C). Notably, the residues of OPTN involved in the proximal ubiquitin-binding and which mediated the interaction with the canonical Ile44 hydrophobic patch of distal ubiquitin are located at the different chains of OPTN dimer, and Leu73 is the only residue of DiUb(M1), which simultaneously binds to both chains of OPTN dimer (Fig. 3). Importantly, all the key residues involved in the binding interface either from OPTN or ubiquitin are highly conserved throughout evolution (Fig. S1A; Fig. S4). Using quantitative ITC analysis, we further verified the specific interaction between OPTN and DiUb(M1) observed in the complex structure (Fig. S5). In agreement with the structural data, individual point mutations of the key residues involved in the binding interface either from OPTN or DiUb(M1), such as the V466S, D477R, F478A, E486A mutations of OPTN (Fig. S5A to D) or the I44A, L73S, R74E, T90R mutations of DiUb(M1) (Fig. S5E to H), all largely attenuated or essentially abolished the specific interaction between OPTN and DiUb(M1) (Fig. S5).
Figure 3.
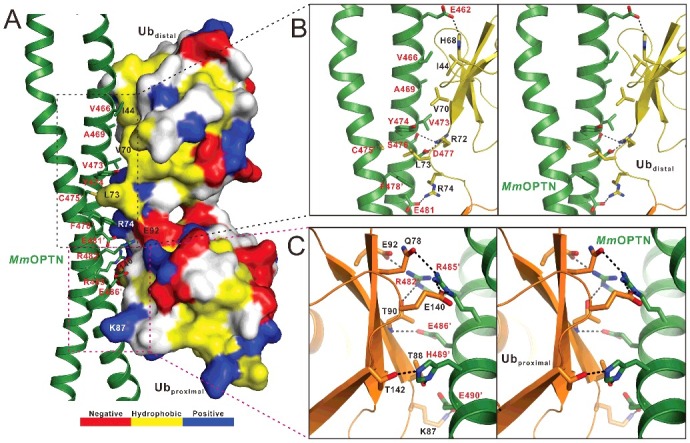
The molecular interface of the MmOPTN(417 to 512)-DiUb(M1) complex. (A) The combined surface representation and the ribbon-stick model showing the interaction interface between the MmOPTN UBAN region and DiUb(M1). In this presentation, the DiUb(M1) molecule is shown in the surface model and MmOPTN in the ribbon-stick model. The hydrophobic amino acid residues in the surface model of DiUb(M1) are drawn in yellow, the positively charged residues in blue, the negatively charged residues in red, and the uncharged polar residues in gray. The key residues of MmOPTN and DiUb(M1) involved in the interaction were labeled in red and black, respectively. (B) Stereo view of ribbon-stick model showing the detailed interactions between MmOPTN and the distal ubiquitin of DiUb(M1). The hydrogen bonds and salt bridges involved in the binding are shown as dotted lines. (C) Stereo view of ribbon-stick model showing the detailed interactions between MmOPTN and the proximal ubiquitin of DiUb(M1).
Phosphorylation of MmOPTN S476 changes the binding affinities and selectivity of UBAN to ubiquitin proteins
Recent studies reveal that the TBK1 kinase can directly phosphorylate the Ser473 residue of human OPTN (corresponding to Ser476 in the mouse OPTN) to promote the ubiquitin-binding ability of OPTN.21,22 In our MmOPTN-DiUb(M1) complex structure, one of the 2 Ser476 residues of MmOPTN dimer is in close proximity to a highly positively charged surface of the distal ubiquitin in the bound DiUb(M1) (Fig. 4A), and its side chain forms a weak hydrogen bond with the positively-charged Arg72 residue of the distal ubiquitin (Fig. 4B). Thus, once the Ser476 residue was phosphorylated, the negatively charged phosphate group would enhance the interaction between OPTN and distal ubiquitin, which provides a mechanistic explanation for phosphorylation-mediated enhancement of its interaction with ubiquitin.
Figure 4.
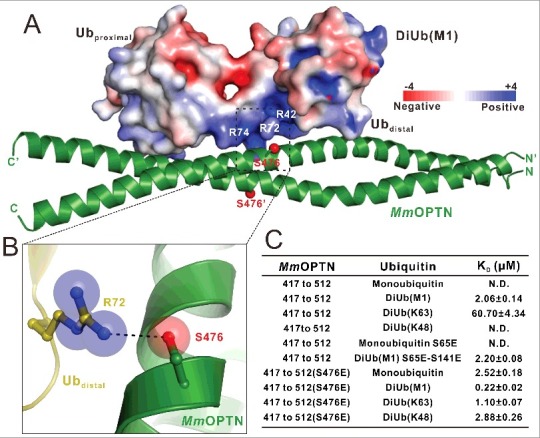
Phosphorylation of the OPTN UBAN domain by TBK1 promotes the ability of UBAN to bind to different ubiquitin proteins. (A) The combined surface charge representation and the ribbon-stick model showing the overall position of the critical Ser476 residues in the MmOPTN-DiUb(M1) complex. In this presentation, the side chains of S476 on the MmOPTN UBAN domain are highlighted and shown in the stick-ball model. (B) The combined ribbon and stick-sphere representations showing the detailed roles of the MmOPTN S476 residue in the OPTN and DiUb(M1) interaction. In this drawing, the side chains of the key residues are shown in the stick mode, and the hydrogen bond is indicated by a black dashed line. (C) The measured binding affinities between different MmOPTN and ubiquitin proteins by ITC-based assays. N.D. indicates that the KD value is not detectable. The KD errors are the fitted errors obtained from the data analysis software, when using the one-site binding model to fit the ITC data.
To further examine the effects induced by the S476-phosphorylation on the binding of the MmOPTN UBAN region to ubiquitin proteins, we purified the recombinant MmOPTN(417 to 512) protein with the S476E mutation that mimics the S476-phosphorylation state of the MmOPTN UBAN, and analyzed its interactions with different ubiquitin proteins. The ITC-based measurements revealed that the phosphomimetic S476E mutant of MmOPTN showed a stronger binding to DiUb(M1) or DiUb(K63) than that of wild-type MmOPTN protein (Fig. S6A and B; Fig. 4C). Strikingly, in contrast to the wild-type MmOPTN that only selectively recognized DiUb(M1) and DiUb(K63), the S476E mutant of the MmOPTN UBAN domain has lost this selectivity and interacted with the monoubiquitin and DiUb(K48) with very similar binding affinity KD values of about 2.52 μM and 2.88 μM, respectively (Fig. S6C and D; Fig. 4C). Since the phosphomimetic S476E mutant can effectively recognize monoubiquitin, therefore any type of polyubiquitin chain was likely to be recognized by the MmOPTN UBAN once its S476 residue was phosphorylated by TBK1. Thus, the phosphorylation of S476 on the MmOPTN UBAN region, by TBK1 could dramatically change the binding affinities and selectivity of OPTN to ubiquitin proteins, thereby expanding the capability of OPTN to recognize different types of ubiquitinated cargoes for autophagic degradation.
The phosphorylation of the Ser65 residue of ubiquitin by the PINK1 kinase during the mitophagy affects the interactions between OPTN and ubiquitin proteins.21,35 However, our structural analysis showed that the 2 corresponding serine residues in DiUb(M1), Ser65 (the Ser65 residue in the distal ubiquitin) and Ser141 (the Ser65 residue in the proximal ubiquitin), are far away from the MmOPTN dimer and are not directly involved in the binding interface of the MmOPTN-DiUb(M1) complex (Fig. S7A and B; Fig. 3). In addition, further quantitative ITC analyses showed that the phosphomimetic S65E mutant of monoubiquitin was unable to interact with the MmOPTN UBAN domain (Fig. 4C; Fig. S6E), and the binding affinity of the phosphomimetic S65E and S141E double mutant of DiUb(M1) towards MmOPTN is very similar to that of the wild-type DiUb(M1) protein (Fig. 4C; Fig. S6F). Accordingly, the phosphorylation of ubiquitin on Ser65 residue may be unlikely to regulate the interactions between the OPTN UBAN and ubiquitin proteins.
Mechanistic insights into the disease-causing mutations in the OPTN UBAN region
Previous studies have identified 3 disease-associated missense mutations located at the UBAN region of OPTN from human patients,45,46 of which Q454E and E478G are associated with ALS,48 and the H486R mutation is linked with juvenile open-angle glaucoma (JOAG).51 In addition, the D474N mutation of human OPTN has been a subject of functional studies.35,52,53 According to our determined MmOPTN-DiUb(M1) complex structure, although the Q457 residue of MmOPTN (corresponding to Q454 in the human OPTN) does not directly participate in the ubiquitin-binding, the Q454E mutation is likely to alter the dimeric structure of OPTN and thus, might indirectly affect the binding with ubiquitin (Fig. 3). However, we could not directly investigate this possibility as the recombinant protein of the Q454E mutation of the OPTN UBAN domain, expressed in E. coli cells is in inclusion bodies. In contrast, the other 3 residues that were associated with human ALS or JOAG, D474, E478 and H486 (corresponding to D477, E481 and H489 in the mouse OPTN), are all directly involved in the DiUb(M1)-binding of OPTN (Fig. 5A–C). Particularly, the side chain of MmOPTN D477 forms a salt bridge with the side chain of R72 and a hydrogen bond with the backbone amine group of L73 from the distal ubiquitin (Fig. 5A), the negatively charged E481 residue forms a charge-charge interaction network with the positively charged R74 residue from distal ubiquitin as well as the adjacent R482 and R485 residues from the neighbor chain of the MmOPTN dimer (Fig. 5B), and the side chain of H489 forms 2 pairs of hydrogen bonds with the side chain hydroxide groups of T88 and T142 residues from the proximal ubiquitin (Fig. 5C). Thus, the D477N, E481G and H489R mutations of OPTN are predicted to disrupt or disturb those interactions seen in the wild-type MmOPTN-DiUb(M1) complex (Fig. 5A–C). Consistent with the structural predictions, ITC analyses revealed that the E478G mutation of HsOPTN totally abolished the interaction between OPTN and DiUb(M1), while the D474N and H486R mutations significantly attenuated the binding affinity of OPTN to DiUb(M1) (Fig. 5D to F). Collectively, our data demonstrate that all the currently known disease-causing OPTN mutations in the UBAN region are likely to directly or indirectly affect the binding ability of the OPTN UBAN domain to ubiquitin, and thereby interfere with the functions of OPTN in selective autophagy or other signaling pathways.
Figure 5.
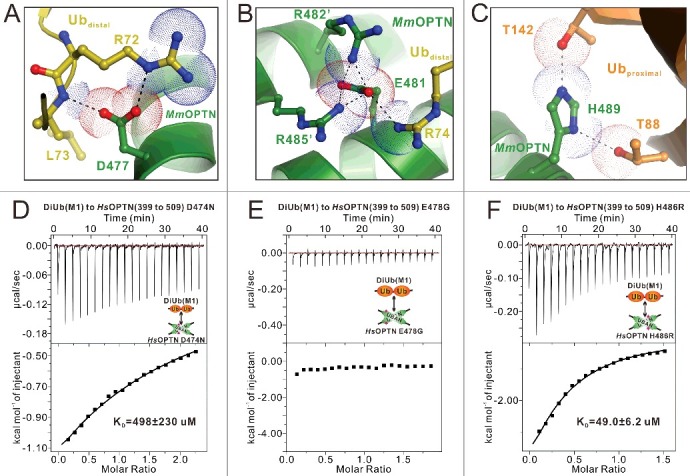
Mechanistic insights into the disease-causing OPTN mutations in the UBAN domain. (A to C) The combined ribbon and stick-dot representations showing the detailed roles of MmOPTN D477 (A), E481 (B) and H489 (C) residues in the OPTN UBAN domain and DiUb(M1) interaction. In this drawing, the side chains of key residues are shown in the stick model, and the salt bridges, hydrogen bonds and charge-charge interactions are indicated by black dashed lines. (D to F) The ITC-based measurements of the affinities of the D474N (D), E478G (E) and H486R (F) mutants of HsOPTN(399 to 509) to bind with DiUb(M1). The KD errors are the fitted errors obtained from the data analysis software, when using the one-site binding model to fit the ITC data.
Cellular colocalization of OPTN and HTT-polyQ aggregates required the specific interaction between the OPTN UBAN domain and ubiquitin
OPTN was found to be a common component in neuronal aggregates associated with many neurodegenerative diseases,43 such as ALS and Huntington disease.48,54 To mimic the Huntington disease condition, we sought to test the cellular localization of OPTN with HTT-polyQ aggregates in HeLa cells. In order to exclude the potential interference by endogenously expressed OPTN, we first generated an OPTN-knockout HeLa cell line using CRISPR-Cas9 technology (Fig. S8; Fig. 6A). As expected, the GFP-HTT140Q forms bulk cellular aggregates in transfected OPTN-knockout cell (Fig. S9A and B). Interestingly, using an antibody that can specifically recognize M1-linked ubiquitin chains, we found that those GFP-HTT140Q aggregates were well decorated with M1-linked ubiquitin proteins (Fig. S9A), and the mCherry-tagged human OPTN were readily recruited to those M1-linked ubiquitin chain-positive HTT140Q aggregates in cotransfected cells (Fig. S9B). In addition, our image data showed that overexpression of OTUB1, a deubiquitinating enzyme that can specifically suppress the K63-linked ubiquitin chain formation,55,56 did not affect the colocalization of OPTN with HTT140Q aggregates (Fig. S9C and D). Therefore, our data strongly implied that the recruitment of OPTN to HTT140Q aggregates is mainly driven by the presence of M1-linked ubiquitin in Huntingtin protein aggregates. Next, we investigated the roles of the interaction of the OPTN UBAN domain and ubiquitin, as well as the disease-associated E478G and H486R mutations of OPTN, on the cellular colocalizations of OPTN with HTT-polyQ aggregates in HeLa cells. We cotransfected different mCherry-tagged human OPTN variants with the GFP-tagged HTT140Q into those OPTN-knockout cells, and analyzed the colocalization of those OPTN variants with the HTT-polyQ aggregates. Consistent with the data reported earlier,34 when cotransfected, the exogenously expressed wild-type OPTN displayed a diffused localization pattern, but formed some large puncta that colocalized well with the bulk HTT140Q inclusions in the cytoplasm of transfected cells (Fig. 6B, J). In contrast, the I463S mutation of human OPTN (corresponding to the V466S mutant of mouse OPTN) displayed an attenuated ability to colocalize with the HTT-polyQ puncta (Fig. 6C, J), in line with the aforementioned structural and biochemical results (Fig. 3B; Fig. S5A). Interestingly, the S473A mutant that is unable to be phosphorylated on the UBAN domain by TBK1 and the phosphomimetic S473E mutant of OPTN both colocalized with the HTT-polyQ aggregates in the cotransfected HeLa cells (Fig. 6D, E and J), indicating that the recruitment of OPTN to HTT140Q aggregates is not mediated by TBK1-mediated phosphorylation of the UBAN domain. On the other hand, the D474R, F475A and ALS-related E478G mutations of OPTN, all of which essentially abolished the interaction between the OPTN UBAN region and DiUb(M1) in vitro as shown by our biochemical and structural analyses, significantly abrogated the colocalization of OPTN with the HTT140Q puncta (Fig. 6F–H and J). In addition, the JOAG-associated H486R mutant of OPTN, which showed a decreased binding affinity to DiUb(M1), exhibited an reduced colocalization with the HTT140Q inclusions (Fig. 6I, J). As control, when expressed individually, mCherry-tagged OPTN proteins mainly displayed a diffused localization, and the OPTN wild type together with some mutants (S473A and S473E) that still can strongly bind to ubiquitin, also formed many small puncta in the cytosol of transfected cells (Fig. S10). However, all of them were unable to form very large puncta as found in the coexpression system with HTT140Q. Therefore, all the above data demonstrated that the ubiquitin-binding ability of the OPTN UBAN domain is crucial for OPTN to colocalize with HTT140Q protein aggregates in transfected cells.
Figure 6.
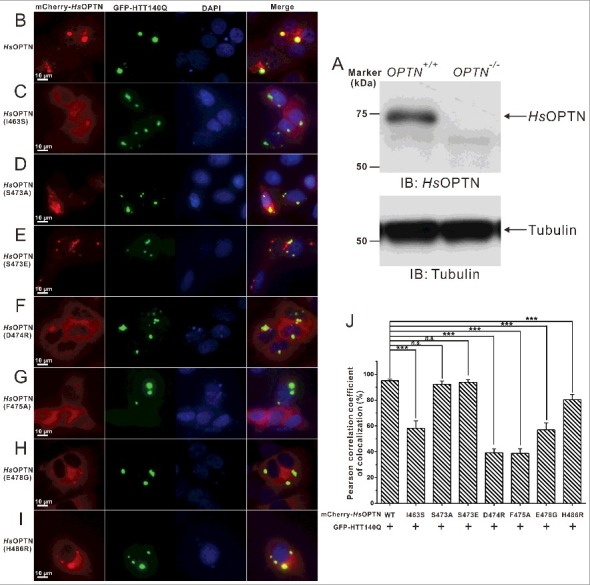
The specific interaction between the OPTN UBAN domain and ubiquitin is required for the cellular colocalization of OPTN and HTT-polyQ aggregates in transfected HeLa cells. (A) Western blot analyses of OPTN expression in the wild-type HeLa cells and OPTN−/− Hela cells generated by the CRISPR/Cas9 system. The anti-tubulin analysis is used as the input assay. (B to I) Cotransfection of GFP-HTT140Q with different OPTN variants in OPTN−/− HeLa cells, after 48 h, the cell images containing GFP-positive puncta were captured using a Leica microscope equipped with LAS X software system, and the nuclei were shown by staining with DAPI. Scale bar: 10 µm. In this assay, when cotransfected, the wild-type OPTN colocalizes very well with the GFP-HTT140Q aggregate clusters in the cytosol (B). The I463S mutant of OPTN, which displays a decreased binding ability with DiUb(M1) in vitro, shows a dramatically reduced colocalization with the HTT140Q puncta in cells (C). The S473A mutant that is unable to be phosphorylated on the UBAN domain colocalizes well with the HTT140Q aggregates (D). The phosphomimetic S473E mutant of OPTN that displays an enhanced binding ability to different ubiquitin proteins in vitro, also colocalizes very well with the HTT140Q clusters (E). The D474R (F), F475A (G) and the ALS-related E478G mutations of OPTN (H), which would totally abolish the interaction between OPTN UBAN and DiUb(M1) in vitro, largely abrogate the colocalization of OPTN with the HTT140Q puncta. The JOAG-associated H486R mutant of OPTN, which shows a decreased binding affinity to DiUb(M1), exhibits an obviously reduced colocalization with HTT140Q clusters (I). (J) Statistical results related to the colocalizations of the GFP-HTT140Q aggregates and the mCherry-tagged OPTN variants in the cotransfected OPTN−/− HeLa cells shown as a Pearson correlation. The Pearson correlation coefficient analysis was performed using the LAS X software based on a randomly selected region that roughly contains one cotransfected HeLa cell. The data represent mean±s.d. of >50 analyzed cells (selected regions) from 3 independent experiments. The unpaired Student t test analysis was used to define a statistically significant difference, and the stars indicate the significant differences between the indicated bars (***P<0.001) and n.s. stands for not significant.
Discussion
Previously, the interaction between the IKBKG UBAN domain and ubiquitin proteins has been extensively characterized, however, the exact interaction mode is still elusive.38,39,57,58 In particular, biochemical analyses demonstrates that the binding stoichiometry of IKBKG to DiUb(M1) is 2:1,39 while the solved crystal structure of the IKBKG UBAN-DiUb(M1) complex (PDB ID: 2ZVO) shows a 1:1 stoichiometry ratio.38 In this study, we demonstrated that the binding stoichiometry between the OPTN UBAN and DiUb(M1) is 2:1. Given that the OPTN UBAN forms a symmetric dimer containing 2 binding sites for the DiUb(M1), it is puzzling why the OPTN UBAN dimer only asymmetrically binds to one DiUb(M1) molecule. Based on the dimeric nature of OPTN UBAN structure, one potential explanation is that once a DiUb(M1) molecule bound to one of the binding sites on the OPTN dimer, it would induce conformation changes on the other site, thereby to preclude its ability to interact with other DiUb(M1) molecules. Indeed, detailed structural comparison of the ubiquitin-bound site with the corresponding ubiquitin-free site in the MmOPTN-DiUb(M1) complex structure revealed that when DiUb(M1) binds to the OPTN UBAN dimer on one binding site, the dimeric UBAN helices on the other site would exhibit an significant “inward” rotation (Fig. S11), thereby the inter-residue arrangements of some critical residues for the DiUb(M1) binding in the 2 sites are quite different (Fig. S11). Notably, the space among the Y474, C475 and F478 in the ubiquitin-free site is much smaller than that of the ubiquitin-bound site, and thus, due to the increasing steric hindrance, the bulk hydrophobic side chain of L73 from distal ubiquitin is unable to occupy this distorted hydrophobic pocket in the ubiquitin-free site (Fig. S11). In addition, the assumed distances between the positive-charged side chains of R72, R74 on distal ubiquitin and the negative-charged side chains of D477, E481 on the ubiquitin-free site of UBAN are much larger than that of on the ubiquitin-bound site (Fig. S11). Given that all these hydrophobic and charge-charge interactions are essential for the interaction of the OPTN UBAN domain and DiUb(M1), based on our aforementioned data (Fig. 3; Fig. S5), these allosteric changes on the ubiquitin-free site of the OPTN UBAN region are likely to prohibit the interaction between UBAN dimer and other DiUb(M1), thereby the OPTN UBAN dimer can only asymmetrically bind to one DiUb(M1) molecule.
It is also worthwhile to mention that based on our structural modeling analyses, the OPTN-DiUb(M1) complex with a 2:1 stoichiometry may make it possible for the TBK1-mediated phosphorylation of OPTN dimer at the UBAN region, as one of the Ser473 residues is unoccupied by the ubiquitin proteins and is exposed to the solvent (Fig. S12A). In contrast, in the assumed 2:2 stoichiometry complex, both Ser473 residues of OPTN UBAN dimers are directly involved in the ubiquitin binding and are unlikely to be phosphorylated by TBK1 due to the potential steric exclusion induced by the bound ubiquitin proteins (Fig. S12B). Thus, this unique asymmetric interaction between the linear ubiquitin chain and the dimeric OPTN may have functional significance for the downstream TBK1-mediated phosphorylation of OPTN. The interaction between UBAN and linear ubiquitin proteins is still in debate in the literature,38,39 and all the previously reported crystal structures of the UBAN and linear ubiquitin complex are obtained by just mixing 2 proteins together rather than using the purified protein complex.38,59 Presumably due to crystal packing, the UBAN dimer of IKBKG or OPTN in those determined complex structures all symmetrically binds to 2 linear ubiquitin molecules.38,59 Therefore, our structural study clarified a long-standing question, how a UBAN domain exactly interacts with the linear ubiquitin proteins.
In addition to binding linear ubiquitin chain, our data together with other people's results,39,58 also showed that the OPTN UBAN domain can bind to K63-linked polyubiquitin chains (Fig. S1D and G). Interestingly, the OPTN UBAN binds to DiUb(K63) also with a 2:1 stoichiometry as indicated by our ITC-based analysis (Fig. S1G). Further NMR-based titration assay showed that the 2 ubiquitin moieties of DiUb(K63) can directly interact with the OPTN UBAN region (Fig. S13A and C), but using different interaction surfaces (Fig. S13B and D). Due to the poor homogeneity of the 1H-15N HSQC spectrum of DiUb(K63) in complex with UBAN (Fig. S13A and C), we sought to use X-ray crystallography to solve this complex structure. Unfortunately, after numerous trials, we failed to obtain high quality crystals for structural determination. Therefore, further studies are required to elucidate the detailed molecular mechanism governing the specific interaction between OPTN and K63-linked ubiquitin chain.
As a highly specific ubiquitin-binding module, the UBAN domain only selectively recognizes the M1- and K63-liked polyubiquitin chains.21,22,39,40,60 However, in contrast to the UBAN domains of IKBKG and TNIP-family proteins, the OPTN UBAN contains a unique serine residue (Fig. S7C), phosphorylation of which mediated by TBK1 kinase could promote the binding affinities and change the binding selectivity of OPTN to ubiquitin proteins (Fig. 4).21,22 Notably, monoubiquitin and any linkage types of polyubiquitin chains are likely to be recognized by the phosphorylated OPTN (Fig. 4). Thus, the ubiquitin-binding diversity of OPTN could be subject to tuning by TBK1. Interestingly, the activation of TBK1 kinase in OPTN-involved selective autophagy has been proven to rely on the polyubiquitin binding ability of the OPTN UBAN domain.21,22 Therefore, our data imply that different linkage types of ubiquitin chains might play distinct roles in the OPTN-mediated selective autophagy, and the M1- and K63-liked polyubiquitin chains are likely essential for the initial recruitment of OPTN-TBK1 complex to the ubiquitin-coated autophagic substrates. However, the exact functions of different linkage types of ubiquitin chains in OPTN-mediated selective autophagy remain to be elucidated.
In all, we proposed a model depicting the recognition and recruitment of ubiquitin-coated protein aggregates by OPTN as well as the regulation of OPTN by TBK1 in aggrephagy (Fig. S14). In this model, once the protein aggregates are modified by polyubiquitin chains, the stable OPTN-TBK1 hetero-tetrameric complexes were recruited to ubiquitinated aggregates through the UBAN domain of OPTN, which could form a unique dimeric structure and specifically recognize the M1- and K63-linked polyubiquitin chains decorated on the protein aggregates through one of its ubiquitin-binding site. Then, activated TBK1 molecules may directly phosphorylate the OPTN dimer on a critical serine residue (Ser473 of human OPTN) located in the ubiquitin-free site of the dimeric UBAN to promote the binding ability of OPTN to further recognize and recruit ubiquitin-decorated protein aggregates even without M1- and K63-linked polyubiquitin chains. In parallel, the activated TBK1 could also directly phosphorylate the Ser172 residue of OPTN, which is close to the OPTN LIR motif, to enhance the binding affinities of OPTN to Atg8-family proteins, thereby promoting the recruitment of downstream autophagic machinery and amplifying the aggrephagy process to timely and efficiently clear the deleterious protein aggregates.
Materials and methods
Protein expression and purification
The different DNA fragments encoding human OPTN (residues 399 to 509), mouse OPTN (residues 417 to 512), and human ubiquitin (residues 1 to 76) were PCR amplified from the full-length human OPTN, mouse Optn and human UBA52 cDNA, respectively. All these fragments were either cloned into the pET-32M vector (a modified version of the pET32a vector (Novagen, 69015-3) containing a N-terminal Trx-tag and His6-tag) or the pET-GST vector (a modified version of pET32a vector containing a N-terminal GST-tag) for recombinant protein expressions. For fluorescence imaging experiment, the DNA fragments encoding human OPTN (HsOPTN) and HsHTT-polyQ (140Q) were cloned into pmCherry-C1 and pEGFP-C1 vectors (Clontech Laboratories, 6084-1), respectively. All the point mutations of OPTN and ubiquitin used in this study were created using the standard PCR-based mutagenesis method, further checked by PCR screen using Taq Master mix (Vazyme Biotech Co., P112-01) enzyme and confirmed by DNA sequencing.
Recombinant proteins were expressed in BL21 (DE3) E. coli cells induced by 200 μM IPTG (Merck Millipore, 420322) at 16°C. The bacterial cell pellets were resuspended in binding buffer (50 mM Tris, 500 mM NaCl, 5 mM imidazole, pH 7.9), and then lysed using the ultrahigh pressure homogenizer FB-110XNANO homogenizer machine (Shanghai Litu Machinery Equipment Engineering Co., Ltd., Shanghai, China). Then the lysate was spun down by centrifugation at 35000 g for 30 min to remove the pellets. His6-tagged proteins were purified with Ni2+-NTA agarose (GE Healthcare, 17–5318–03) affinity chromatography, while GST-tagged proteins were purified with glutathione sepharose 4B (GE Healthcare, 17–0756–04) affinity chromatography. Each recombinant protein was further purified by size-exclusion chromatography. The N-terminal tag of each recombinant protein was cleaved with 3C protease (purified in-house) and further removed by size-exclusion chromatography. Uniformly 15N or 15N, 13C-labeled DiUb(M1) or DiUb(K63) proteins were prepared by growing bacteria in M9 minimal medium (In 1 liter M9 minimal medium, containing 6.8 g Na2HPO4, 3.0 g KH2PO4, 0.5 g NaCl, 1.0 g NH4Cl, 4.0 g D-glucose, 2 mM MgSO4 and 0.1 mM CaCl2) using 15NH4Cl (Cambridge Isotope Laboratories Inc., NLM-467) as the sole nitrogen source or 15NH4Cl and 13C6-glucose (Cambridge Isotope Laboratories Inc., CLM-1396) as the sole nitrogen and carbon sources, respectively.
Preparation of K48-linked, K63-linked, and M1-linked diubiquitin proteins
The K48R, K63R (unlabeled or 15N-labeled) and D77 (unlabeled or 15N-labeled) monoubiquitin mutants were cloned into the pET-M-3C vector (a modified version of the pET32a vector containing a N-terminal His6-tag), and expressed and purified as monoubiquitin for enzymatic synthesis of K48-linked diubiquitin or K63-linked diubiquitin according to published protocols.61,62 For M1-linked diubiquitin, 2 repeats of a DNA fragment encoding the human ubiquitin (residues 1 to 76) were directly fused together as one extended DNA fragment, which was cloned into pET-32M-3C vectors. The M1-linked diubiquitin proteins were expressed and purified following the same procedure for monoubiquitin.
Analytical gel filtration chromatography
Analytical gel filtration chromatography was carried out on an AKTA FPLC system (GE Healthcare, Pittsburgh, PA, USA). Protein samples were loaded on to a Superose 12 10/300 GL column (GE Healthcare, 17–5173–01) equilibrated with a buffer containing 20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM DTT.
NMR spectroscopy
The labeled protein samples for NMR studies were concentrated to ∼0.1 mM for titration experiments and ∼0.6 mM for backbone resonance assignment experiments in 50 mM potassium phosphate buffer containing 50 mM NaCl, 1 mM DTT, pH 6.5. NMR spectra were acquired at 25 °C on an Agilent 800 MHz spectrometer (Agilent Technologies, CA, USA) at the Shanghai Institute of Organic Chemistry equipped with an actively z gradient shielded triple resonance cold probe. Backbone resonance assignments were achieved by combination of the standard heteronuclear correlation experiments.63
Protein crystallization and structural elucidation
Crystals of the MmOPTN(417 to 512)-DiUb(M1) complex were obtained by mixing the freshly purified complex protein (10 or 20 mg/ml in 50 mM Tris-HCl, 100 mM NaCl, 1 mM DTT, 1 mM EDTA, pH 7.5) with equal volumes of reservoir solution containing 0.1 M HEPES, pH 7.5, 30% w/v PEG 3350 (Hampton research, HR2-144) using the sitting-drop vapor-diffusion method at 16°C.64 Before diffraction experiments, glycerol (10%; Sigma-Aldrich, G5516) was added as the cryoprotectant. A 3.0 Å resolution X-ray data set for MmOPTN(417 to 512)-DiUb(M1) complex were collected at the beamline BL19U1 of the Shanghai Synchrotron Radiation Facility. The diffraction data were processed and scaled using HKL2000.65
The phase problem of the MmOPTN(417 to 512)-DiUb(M1) complex was solved by molecular replacement method using the structure of the IKBKG-DiUb(M1) complex with PHASER.66 The initial model was rebuilt manually using COOT,67 and then refined using REFMAC.68 Further manual model building and adjustment were completed using COOT. The qualities of the final model were validated by MolProbity.69 In the final stage, an additional TLS refinement was performed in PHENIX.70 The final refinement statistics of solved structures in this study are listed in Table S1. Structural diagrams were prepared using the program PyMOL (http://www.pymol.org/).
Isothermal titration calorimetry assay
ITC measurements were carried out on an ITC200 (GE Healthcare, Pittsburgh, PA, USA) or Microcal PEAQ-ITC (Malvern, Enigma Business Park, UK) calorimeter at 25°C. All protein samples were in the same buffer containing 50 mM Tris, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT. The concentrated 0.1 mM MmOPTN fragments and 1 mM of various ubiquitin proteins were loaded into the cell and the syringe, respectively. The titration processes were performed by injecting 40-μl aliquots of the ubiquitin proteins into the MmOPTN fragments at time intervals of 2 min to ensure that the titration peak returned to the baseline. The titration data were analyzed using the program Origin7.0 from Micro Cal and fitted using the one-site binding model.
Multi-angle light scattering
For multi-angle light scattering measurement, HsOPTN(399 to 509) and HsOPTN(399 to 509)-DiUb(M1) complex samples (100 μL at a concentration of 20 μM) were injected into an AKTA FPLC system (GE Healthcare, Pittsburgh, PA, USA) with a Superose 12 10/300 GL column (GE Healthcare, Pittsburgh, PA, USA) with the column buffer containing 50 mM Tris-HCl, 100 mM NaCl, 1 mM DTT, 1 mM EDTA, pH 7.5. The chromatography system was coupled to a static light-scattering detector (miniDawn, Wyatt Technology, Santa Barbara, CA, USA) and a differential refractive index detector (Optilab, Wyatt Technology, Santa Barbara, CA, USA). Data were collected every 0.5 second with a flow rate of 0.5 mL/min. Data were analyzed using ASTRA 6 (Wyatt Technology, Santa Barbara, CA, USA).
Generation of OPTN knockout cell line
The OPTN gene was knocked out from HeLa cells using the CRISPR/Cas9 system,71 with a guide RNA spanning the exon 1 of OPTN gene. Guide RNA sequence: 5’-GTAAACGTGTCCAGGTTTGGG-3’. The guide RNA was individually cloned into the pX330 vector and transfected into HeLa cells. Transfected cells were sorted by fluorescence-activated cell sorting base on the green fluorescent protein. Single colonies were screened by western blot using a specific OPTN antibody (Santa Cruz biotechnology, sc-166576, 1:1000) to confirm the loss of OPTN protein expression and detecting the expression of tubulin with anti-tubulin antibody as the loading control (1:10,000 dilution; MBL, PM054).
Cell culture, transfection and fluorescence imaging
OPTN-knockout HeLa cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen, 11995–065) supplemented with 10% fetal bovine serum (Invitrogen, 10099–141). Transfections of GFP-HTT140Q and mCherry-HsOPTN alone, or Cotransfections of GFP-HTT140Q with mCherry-HsOPTN or related plasmids were performed using Lipofectamine 2000 (Invitrogen, 11668–019) according to the manufacturer's instructions. After 48 h, cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 (Sangon Biotech, A110694) in PBS, and the nuclei were visualized by staining with DAPI. The cell images were captured and analyzed using a TCS SP5 microscope equipped with LAS X software (Leica, Inc., Thornwood, NY, USA). Particularly, the M1-linked ubiquitin chains were stained using a commercial antibody (Sigma, MABS199) that can specifically recognize M1-linked ubiquitin proteins, and the Pearson correlation coefficient analysis was performed using the LAS X software based on a randomly selected region that roughly contains 1 cotransfected HeLa cell. The statistical data represent mean±s.d. of >50 analyzed cells (selected regions) from 3 independent experiments. The unpaired Student t test analysis was used to define a statistically significant difference.
Coordinates
The coordinate and structure factor of the MmOPTN(417 to 512)-DiUb(M1) complex has been deposited in the Protein Data Bank under the accession code 5WQ4.
Supplementary Material
Funding Statement
This work was supported by grants from the National Key R&D Program of China [grant number 2016YFA0501903, 2013CB836900], National Natural Science Foundation of China [grant number 31470749, 21621002], Science and Technology Commission of Shanghai Municipality [grant number 15JC1400400], the Strategic Priority Research Program of the Chinese Academy of Sciences [grant number XDB20000000], a Thousand Talents Program young investigator award, the start-up fund from State Key Laboratory of Bioorganic and Natural Products Chemistry and Chinese Academy of Sciences (to L.P.); and grants from the Chinese Academy of Sciences, the China Ministry of Science and Technology Program [grant number 2014ZX09102001-002], and National Natural Science Foundation of China [grant number 31530041], (to J.Y.).
Disclosure of potential conflicts of interest
There were no potential conflicts of interest that needed to be disclosed.
Acknowledgements
We thank SSRF BL17U and NCPSS BL19U1 for X-ray beam time, Dr. Shang Yuan and Dr. Jianchao Li for help in the X-ray diffraction data collection, Dr. Cong Liu for providing the machines for ITC and MALS measurements.
References
- [1].Klionsky DJ, Emr SD. Cell biology - Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. PMID:11099404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. PMID:18006683 [DOI] [PubMed] [Google Scholar]
- [3].Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. doi: 10.1038/cr.2013.161. PMID:24323045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. doi: 10.1038/nrm2708. PMID:19491929 [DOI] [PubMed] [Google Scholar]
- [5].Feng YC, He D, Yao ZY, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. PMID:24366339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Randow F, Youle RJ. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host & Microbe. 2014;15:404–412. doi: 10.1016/j.chom.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. PMID:24875736 [DOI] [PubMed] [Google Scholar]
- [8].Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell. 2014;54:224–233. doi: 10.1016/j.molcel.2014.03.009. PMID:24766886 [DOI] [PubMed] [Google Scholar]
- [9].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011;7: 279–296. doi: 10.4161/auto.7.3.14487. PMID:21189453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rogov V, Dotsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53:167–178. doi: 10.1016/j.molcel.2013.12.014. PMID:24462201 [DOI] [PubMed] [Google Scholar]
- [11].Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. PMID:19820708 [DOI] [PubMed] [Google Scholar]
- [12].Jiang SX, Wells CD, Roach PJ. Starch-binding domain-containing protein 1 (Stbd1) and glycogen metabolism: identification of the Atg8 family interacting motif (AIM) in Stbd1 required for interaction with GABARAPL1. Biochem Biophys Res Commun. 2011;413:420–425. doi: 10.1016/j.bbrc.2011.08.106. PMID:21893048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liu L, Feng D, Chen G, Chen M, Zheng QX, Song PP, Ma Q, Zhu CZ, Wang R, Qi WJ, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. PMID:22267086 [DOI] [PubMed] [Google Scholar]
- [14].Mancias JD, Wang XX, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–109. doi: 10.1038/nature13148. PMID:24695223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105–109. doi: 10.1038/nature15548. PMID:26524528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354–358. doi: 10.1038/nature14498. PMID:26040720 [DOI] [PubMed] [Google Scholar]
- [17].Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, Nakatogawa H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015;522: 359–362. doi: 10.1038/nature14506. PMID:26040717 [DOI] [PubMed] [Google Scholar]
- [18].Mandell MA, Jain A, Arko-Mensah J, Chauhan S, Kimura T, Dinkins C, Silvestri G, Munch J, Kirchhoff F, Simonsen A, et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell. 2014;30: 394–409. doi: 10.1016/j.devcel.2014.06.013. PMID:25127057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zaffagnini G, Martens S. Mechanisms of selective autophagy. J Mol Biol. 2016;428: 1714–1724. doi: 10.1016/j.jmb.2016.02.004. PMID:26876603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. PMID:21617041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015;60:7–20. doi: 10.1016/j.molcel.2015.08.016. PMID:26365381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016;113:4039–4044. doi: 10.1073/pnas.1523926113. PMID:27035970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].McEwan DG, Dikic I. The three musketeers of autophagy: phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 2011;21:195–201. doi: 10.1016/j.tcb.2010.12.006. PMID:21277210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–289. doi: 10.1016/j.molcel.2011.07.039. PMID:22017874 [DOI] [PubMed] [Google Scholar]
- [25].Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836–841. doi: 10.1038/ncb0910-836. PMID:20811356 [DOI] [PubMed] [Google Scholar]
- [26].Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. 2012;2012:736905. doi: 10.1155/2012/736905. PMID:22518139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol. 2009;10:659–671. doi: 10.1038/nrm2767. PMID:19773779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. PMID:15571809 [DOI] [PubMed] [Google Scholar]
- [29].Hicke L, Schubert HL, Hill CP. Ubiquitin-binding domains. Nat Rev Mol Cell Biol. 2005;6:610–621. doi: 10.1038/nrm1701. PMID:16064137 [DOI] [PubMed] [Google Scholar]
- [30].Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. PMID:16753028 [DOI] [PubMed] [Google Scholar]
- [31].Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ. 2013;20:21–30. doi: 10.1038/cdd.2012.72. PMID:22722335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].van Wijk SJ, Fiskin E, Putyrski M, Pampaloni F, Hou J, Wild P, Kensche T, Grecco HE, Bastiaens P, Dikic I. Fluorescence-based sensors to monitor localization and functions of linear and K63-linked ubiquitin chains in cells. Mol Cell. 2012;47:797–809. doi: 10.1016/j.molcel.2012.06.017. PMID:22819327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fiskin E, Bionda T, Dikic I, Behrends C. Global analysis of host and bacterial ubiquitinome in response to Salmonella Typhimurium infection. Mol Cell. 2016;62:967–981. doi: 10.1016/j.molcel.2016.04.015. PMID:27211868 [DOI] [PubMed] [Google Scholar]
- [34].Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, Terzic J, Dikic I. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci. 2013;126:580–592. doi: 10.1242/jcs.114926. PMID:23178947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. PMID:26266977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wong YC, Holzbaur ELF. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111:E4439–E4448. doi: 10.1073/pnas.1405752111. PMID:25294927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ying HY, Yue BYJT. Cellular and molecular biology of optineurin. Int Rev Cell Mol Biol. 2012;294:223–258. doi: 10.1016/B978-0-12-394305-7.00005-7. PMID:22364875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136:1098–109. doi: 10.1016/j.cell.2009.03.007. PMID:19303852 [DOI] [PubMed] [Google Scholar]
- [39].Lo YC, Lin SC, Rospigliosi CC, Conze DB, Wu CJ, Ashwell JD, Eliezer D, Wu H. Structural basis for recognition of diubiquitins by NEMO. Mol Cell. 2009;33: 602–615. doi: 10.1016/j.molcel.2009.01.012. PMID:19185524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Clark K, Nanda S, Cohen P. Molecular control of the NEMO family of ubiquitin-binding proteins. Nat Rev Mol Cell Biol. 2013;14:673–685. doi: 10.1038/nrm3644. PMID:23989959 [DOI] [PubMed] [Google Scholar]
- [41].Morton S, Hesson L, Peggie M, Cohen P. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett. 2008;582:997–1002. doi: 10.1016/j.febslet.2008.02.047. PMID:18307994 [DOI] [PubMed] [Google Scholar]
- [42].Li FX, Xie XQ, Wang YL, Liu JP, Cheng XF, Guo YJ, Gong YK, Hu SC, Pan LF. Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat Commun. 2016;7. doi: 10.1038/ncomms12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Osawa T, Mizuno Y, Fujita Y, Takatama M, Nakazato Y, Okamoto K. Optineurin in neurodegenerative diseases. Neuropathology. 2011;31:569–574. doi: 10.1111/j.1440-1789.2011.01199.x. PMID:21284751 [DOI] [PubMed] [Google Scholar]
- [44].Sirohi K, Swarup G. Defects in autophagy caused by glaucoma-associated mutations in optineurin. Exp Eye Res. 2015;144:54–63. doi: 10.1016/j.exer.2015.08.020. PMID:26302410 [DOI] [PubMed] [Google Scholar]
- [45].Kachaner D, Genin P, Laplantine E, Weil R. Toward an integrative view of Optineurin functions. Cell Cycle. 2012;11:2808–2818. doi: 10.4161/cc.20946. PMID:22801549 [DOI] [PubMed] [Google Scholar]
- [46].Slowicka K, Vereecke L, van Loo G. Cellular functions of optineurin in health and disease. Trends Immunol. 2016;37:621–633. doi: 10.1016/j.it.2016.07.002. PMID:27480243 [DOI] [PubMed] [Google Scholar]
- [47].Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, Heon E, Krupin T, Ritch R, Kreutzer D, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295:1077–1079. doi: 10.1126/science.1066901. PMID:11834836 [DOI] [PubMed] [Google Scholar]
- [48].Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–U109. doi: 10.1038/nature08971. PMID:20428114 [DOI] [PubMed] [Google Scholar]
- [49].Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603–608. doi: 10.1126/science.aaf6803. PMID:27493188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441. doi: 10.1126/science.aaa3650. PMID:25700176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Willoughby CE, Chan LLY, Herd S, Billingsley G, Noordeh N, Levin AV, Buys Y, Trope G, Sarfarazi M, Heon E. Defining the pathogenicity of optineurin in juvenile open-angle glaucoma. Invest Ophthalmol Vis Sci. 2004;45:3122–3130. doi: 10.1167/iovs.04-0107. PMID:15326130 [DOI] [PubMed] [Google Scholar]
- [52].Mankouri J, Fragkoudis R, Richards KH, Wetherill LF, Harris M, Kohl A, Elliott RM, Macdonald A. Optineurin negatively regulates the induction of IFNbeta in response to RNA virus infection. PLoS Pathog. 2010;6:e1000778. doi: 10.1371/journal.ppat.1000778. PMID:20174559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nagabhushana A, Chalasani ML, Jain N, Radha V, Rangaraj N, Balasubramanian D, Swarup G. Regulation of endocytic trafficking of transferrin receptor by optineurin and its impairment by a glaucoma-associated mutant. BMC Cell Biol. 2010;11:4. doi: 10.1186/1471-2121-11-4. PMID:20085643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schwab C, Yu S, McGeer EG, McGeer PL. Optineurin in Huntington's disease intranuclear inclusions. Neurosci Lett. 2012;506:149–154. doi: 10.1016/j.neulet.2011.10.070. PMID:22085693 [DOI] [PubMed] [Google Scholar]
- [55].Wiener R, Zhang X, Wang T, Wolberger C. The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature. 2012;483:618–22. doi: 10.1038/nature10911. PMID:22367539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sato Y, Yamagata A, Goto-Ito S, Kubota K, Miyamoto R, Nakada S, Fukai S. Molecular basis of Lys-63-linked polyubiquitination inhibition by the interaction between human deubiquitinating enzyme OTUB1 and ubiquitin-conjugating enzyme UBC13. J Biol Chem. 2012;287:25860–25868. doi: 10.1074/jbc.M112.364752. PMID:22679021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kensche T, Tokunaga F, Ikeda F, Goto E, Iwai K, Dikic I. Analysis of nuclear factor-kappa B (NF-kappa B) essential modulator (NEMO) binding to linear and lysine-linked ubiquitin chains and its role in the activation of NF-kappa B. J Biol Chem. 2012;287:23626–23634. doi: 10.1074/jbc.M112.347195. PMID:22605335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yoshikawa A, Sato Y, Yamashita M, Mimura H, Yamagata A, Fukai S. Crystal structure of the NEMO ubiquitin-binding domain in complex with Lys 63-linked di-ubiquitin. FEBS Lett. 2009;583:3317–3322. doi: 10.1016/j.febslet.2009.09.028. PMID:19766637 [DOI] [PubMed] [Google Scholar]
- [59].Nakazawa S, Oikawa D, Ishii R, Ayaki T, Takahashi H, Takeda H, Ishitani R, Kamei K, Takeyoshi I, Kawakami H, et al. Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat Commun. 2016;7. doi: 10.1038/ncomms12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gleason CE, Ordureau A, Gourlay R, Arthur JSC, Cohen P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon beta. J Biol Chem. 2011;286:35663–35674. doi: 10.1074/jbc.M111.267567. PMID:21862579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hofmann RM, Pickart CM. In vitro assembly and recognition of Lys-63 polyubiquitin chains. J Biol Chem. 2001;276:27936–27943. doi: 10.1074/jbc.M103378200. PMID:11369780 [DOI] [PubMed] [Google Scholar]
- [62].Piotrowski J, Beal R, Hoffman L, Wilkinson KD, Cohen RE, Pickart CM. Inhibition of the 26 S proteasome by polyubiquitin chains synthesized to have defined lengths. J Biol Chem. 1997;272:23712–23721. doi: 10.1074/jbc.272.38.23712. PMID:9295315 [DOI] [PubMed] [Google Scholar]
- [63].Bax A, Grzesiek S. Methodological Advances in Protein Nmr. Acc Chem Res. 1993;26:131–138. doi: 10.1021/ar00028a001. [DOI] [Google Scholar]
- [64].DeLucas LJ, Bray TL, Nagy L, McCombs D, Chernov N, Hamrick D, Cosenza L, Belgovskiy A, Stoops B, Chait A. Efficient protein crystallization. J Struct Biol. 2003;142:188–206. doi: 10.1016/S1047-8477(03)00050-9. PMID:12718931 [DOI] [PubMed] [Google Scholar]
- [65].Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromol Crystallogr, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- [66].Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallog, Sect D-Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- [67].Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D-Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- [68].Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr Sect D-Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- [69].Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. PMID:17452350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr Sect D-Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- [71].Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. PMID:24157548 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.