

Abstract
Spasmolytic polypeptide-expressing metaplasia (SPEM) develops in patients with chronic atrophic gastritis due to infection with Helicobacter pylori; it might be a precursor to intestinal metaplasia and gastric adenocarcinoma. Lineage tracing experiments of the gastric corpus in mice have not established whether SPEM derives from proliferating stem cells or differentiated, post-mitotic zymogenic chief cells in the gland base. We investigated whether differentiated cells can give rise to SPEM using a non-genetic approach in mice. Mice were given intraperitoneal injections of 5-fluorouracil, which blocked gastric cell proliferation, plus tamoxifen to induce SPEM. Based on analyses of molecular and histological markers, we found SPEM developed even in the absence of cell proliferation. SPEM therefore did not arise from stem cells. In histologic analyses of gastric resection specimens from 10 patients with adenocarcinoma, we found normal zymogenic chief cells that were transitioning into SPEM cells only in gland bases, rather than the proliferative stem cell zone. Our findings indicate that SPEM can arise by direct reprogramming of existing cells—mainly of chief cells.
Keywords: development, differentiation, exocrine cell, cancer
Spasmolytic Polypeptide-Expressing Metaplasia (SPEM) arises in the setting of chronic atrophic gastritis (CAG) in humans infected with Helicobacter pylori and may be a precursor to intestinal metaplasia and gastric adenocarcinoma1,2. CAG is characterized by chronic inflammation, parietal cell death, and metaplastic expansion of cells coexpressing spasmolytic polypeptide (TFF2) and zymogenic chief cell (ZC) markers. Genetic lineage tracing studies have not definitively identified the cell-of-origin for SPEM cells. Tracing from Mist1CreERT2-expressing cells indicated ZCs at the gland base gave rise to SPEM3–7. Another report disputed this because occasional cells in the upper, isthmal stem cell region of the gland also expressed Mist1CreERT2, indicating SPEM might be derived isthmally8. We use a new approach to determine if differentiated cells can give rise to SPEM.
For an isthmal stem cell to generate a gland full of SPEM cells, its progeny must proliferate to reach the gland base. We inhibited proliferation with the anti-mitotic drug 5-Fluorouracil (5FU) and found one 150mg/kg4 intraperitoneal injection of 5FU was sufficient to block proliferation for 24h with return to near-normal levels by 48h (Figure 1A,B).
Figure 1. SPEM can occur without proliferation.
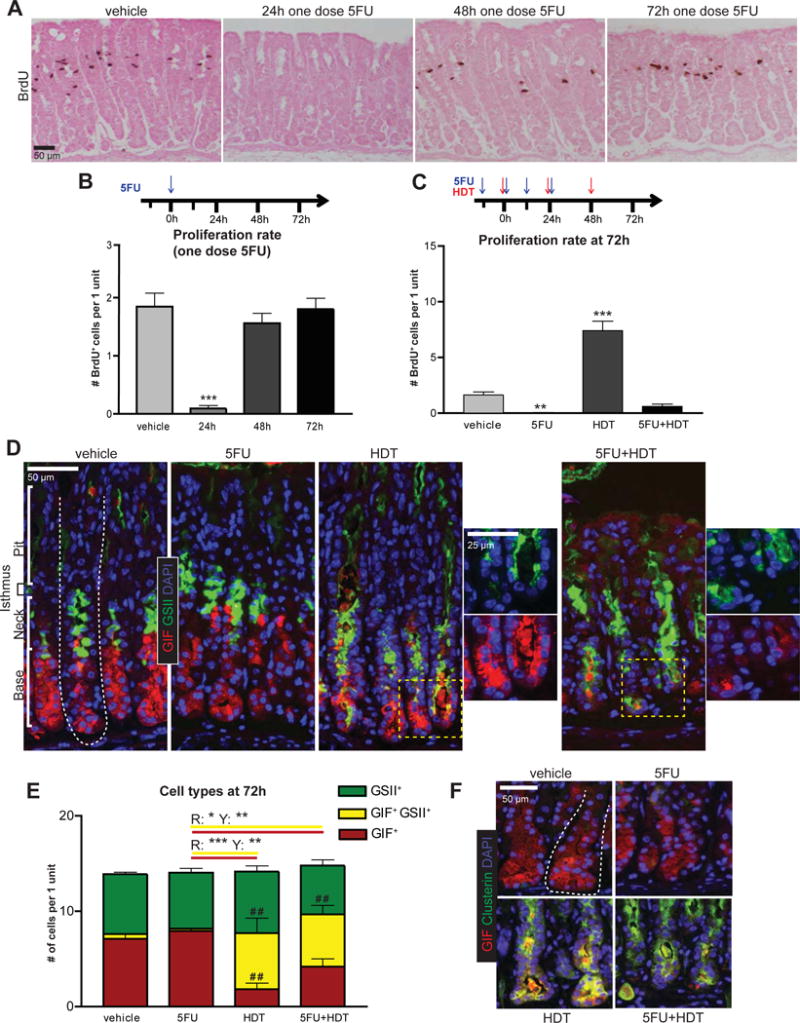
A) IHC staining for BrdU after one dose of 5FU (150mg/kg; intraperitoneal injection). Eosin Y counterstain. B) Experimental timeline and proliferation quantification. C) 5FU+HDT experimental timeline and proliferation quantification. D) Immunofluorescence of stomachs after 72h vehicle, 5FU, HDT, or 5FU+HDT treatment (red, GIF; green, GSII; blue, DAPI). Dotted line: gastric unit. E) Cell populations quantified, # indicates significance compared to vehicle, * indicates significance between treatment groups. R is significance between red columns, Y between yellow columns. F) Immunofluorescence of stomachs (red, GIF; green, Clusterin; blue, DAPI). *p≤0.05, **p≤0.01 ***p≤0.001, variance analyzed with ANOVA/Tukey (N ≥ 3 mice/group)
Long-term 5FU is toxic beyond 4–5 days, thus we used a SPEM induction method shown by multiple labs to cause maximal, synchronous SPEM within three days: high dose tamoxifen (HDT)9,10. Our selected 5FU+HDT regimen caused no mouse mortality, but blocked nearly all proliferation at the peak SPEM stage: <0.5 in 5FU+HDT vs. >8 cells/unit in HDT alone (Figure 1C).
We monitored SPEM by immunofluorescence for the ZC marker, gastric intrinsic factor (GIF; red), and a lectin (GSII; green), which co-labels with TFF2. In control mice, GIF was located in ZCs at the gland base, separated from the GSII+ mucous neck cells in the middle of the gland. Consistent with previous reports9,10, HDT caused SPEM, defined by pathognomonic GSII+GIF+ co-staining at the gland base (Figure 1D), yet few SPEM cells appeared at 24h (Supplementary Figure 1). 5FU alone, or with HDT, did not significantly alter outcomes at any timepoint. At 72h, similar numbers of SPEM cells formed in HDT and HDT+5FU: 6.0±1.5 SPEM cells in HDT, 5.6±1.0 in HDT+5FU (Figure 1D,E). Additionally, 5FU vs. HDT+5FU mice displayed a similar magnitude of GIF+ cell loss and GIF+GSII+ SPEM cell increase (Δ −3.7±1.3 GIF+ cells/unit, Δ +5.2±0.8 GIF+GSII+ cells/unit), while GSII+ neck cells decreased only slightly (ΔG: −0.76 ±0.75), implicating ZCs as the principal source for SPEM cells when proliferation is blocked.
Equivalent SPEM was confirmed in HDT±5FU mice by qRT-PCR for SPEM-associated transcripts Mist1, Gif, He4, and Clusterin11 (Supplementary Figure 2) and immunofluorescence for Clusterin (Figure 1F).
We examined the differentiation pattern and location of occasional cells escaping the 5FU blockade at 72h. Proliferating cells in control mice lacked neck/ZC markers, indicating proliferation primarily in isthmal stem cells (Figure 2A,B). HDT mice harbored proliferating isthmal, neck, SPEM, and occasional BrdU+ ZCs (Figure 2A,B). The 5FU+HDT regimen was designed such that cells escaping 5FU blockade at 72h would be in their first cell cycle, as gastric epithelial cells average one division per day12. As expected, <1 cell/unit was BrdU+ at 72h 5FU+HDT, and units with >1 BrdU+ cell were exceedingly rare (Figure 2A). Proliferating cells could not be found prior to 72h (Supplementary Figure 3). Proliferating cells at 72h expressed neck, SPEM, or ZC markers (Figure 2B). Interestingly, the largest cohort did not label with GSII or GIF. We suspect these cells are parietal cell progenitors based on cells, at 96h following HDT, expressing BrdU and beginning to show the characteristic, thick apical staining of parietal cell marker, ezrin13 (Supplementary Figure 4). Figure 2C plots zones in which proliferating cells emerged from isthmus to base, excluding upper pit/foveolar cells (Supplementary Figure 5A). In control mice, almost all proliferation was in the isthmus. In HDT and 5FU+HDT, proliferating cells appeared throughout the gland. Thus, multiple lines of evidence presented here indicate, in addition to isthmal stem cells, multiple cell types throughout the gland exhibit plasticity and can proliferate in response to damage.
Figure 2. Metaplastic cells arise below the isthmus.
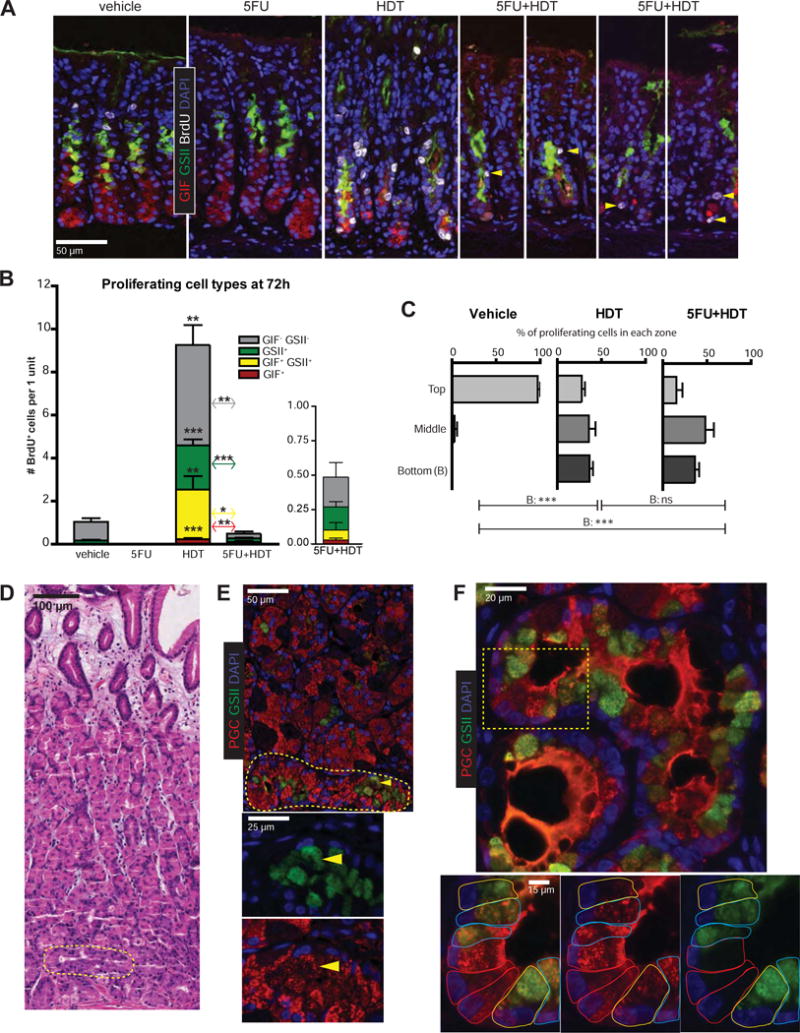
A) Immunofluorescence of stomachs after 72h vehicle, 5FU, HDT, or 5FU+HDT (green, GSII; red, GIF; white, BrdU; blue, DAPI; arrowheads, rare BrdU+ cells in 5FU+HDT). B) Proliferating cell populations quantified. C) Location of BrdU+ cells below the bottom-most AAA+ pit cell. D) Hematoxylin & eosin of early human SPEM (yellow circle) E) Immunofluorescence on serial section from 2D (red, PGC; green, GSII; blue, DAPI; arrowhead: SPEM cell). Yellow circle is SPEM area. F) Human SPEM within gland base showing transitional ZC➔SPEM forms. Dotted box indicates area shown at higher magnification. Cells outlined in colors according to cell type (red, normal ZCs; blue, hybrid SPEM; yellow, full SPEM). *p≤0.05 **p≤0.01 ***p≤0.001, variance analyzed with ANOVA/Tukey (N ≥ 3 mice/group)
We stained for proliferative SPEM with CD44v (CD44v9 in humans)11. HDT and 5FU+HDT mice exhibited CD44v staining at the gland base (Supplementary Figure 5B) and increased expression of proliferation-associated markers Check2 and Ccnb2 by qRT-PCR, indicating basally-located cells are poised to divide in 5FU+HDT stomachs (Supplementary Figure 5C).
Our data suggest stem cells are responsible for pit- and neck cell-localized proliferation following HDT; however, SPEM itself is largely derived from ZCs, supporting previous work suggesting two foci of proliferation in the stomach14. Our mouse experiments reflect ZC to SPEM transitions observed in humans previously extensively characterized from a large dataset of adenocarcinoma and gastritis biopsy and resection specimens15. Figures 2D–F and Supplementary Figure 6 depict representative stomach glands from 10 additional patients with gastric adenocarcinoma. We again observe hybrid cells (cells with features of mature ZCs and SPEM cells) occurred predominantly in the base. Specifically, we see apparent transition among a variety of cell phenotypes from mature ZCs (expressing PGC in large apical granules) to cells expressing abundant PGC with scant GSII (likely indicating early SPEM change) to phenotypically complete SPEM conversion (scant PGC with abundant GSII) (Figure 2F). A stem cell-origin of SPEM in humans would mean cell fate changes to SPEM cells should occur at the isthmus, and SPEM cells would have to expand downward and replace mature ZCs to populate the base. Thus, the interpretation that SPEM originates exclusively from the isthmus would be inconsistent with the presence of cells transitioning to SPEM consistently found in the base.
Differentiated cells are long-lived and may accumulate mutations that become unmasked upon cell cycle re-entry after injury. For example, Kras mutations in ZCs or pancreatic acinar cells can fuel tumorigenic transformation6. Therefore, our current work, highlighting how differentiated cells are called back into the cell cycle in an acute injury model, should interest those studying tumorigenesis and/or regeneration.
Materials and Methods
Animals and Injections
All experiments involving animals were performed according to protocols approved by the Washington University School of Medicine Animal Studies Committee. Mice were maintained in a specified pathogen-free barrier facility under a 12-hour light cycle. Wild-type C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). All mice used in experiments were females 6–8 weeks old. To induce SPEM, tamoxifen (250mg/kg body weight; Toronto Research Chemicals, Inc, Toronto, Canada) was administered daily for 3 days by intraperitoneal injection. Tamoxifen was dissolved in a vehicle of 10% ethanol and 90% sunflower oil (Sigma). Previously described1. To block proliferation, 5-Fluorouracil (150mg/kg body weight; Sigma F6627) was given by intraperitoneal injection twice daily for 2 days. 5-Fluorouracil was dissolved in a solution containing 10% DMSO and 0.9% sodium chloride. All mice were given an intraperitoneal injection containing 5-bromo-2′-deoxyuridine (120mg/kg) and 5-Fluoro-2′ deoxyuridine (12mg/kg) 90 minutes prior to sacrifice.
Patient Samples
Human gastric tissue was obtained with approval from the Institutional Review Board of Washington University School of Medicine. The database of metaplastic samples showing hybrid SPEM forms has been previously described2–4.
Immunofluorescence and Immunohistochemistry
Stomachs were excised immediately, flushed with PBS via the duodenal stub and then inflated with freshly prepared 4% paraformaldehyde. The stub was clamped by hemostat, and the stomach suspended in fixative in a 50 ml conical for 8–12 hours, followed by 3 rinses in 70% ETOH, arrangement in 2% agar in a tissue cassette, and routine paraffin processing. Sections (5 m) were cut, deparaffinized and rehydrated with graded xylenes, alcohols and water, then antigen-retrieved in Sodium Citrate Buffer (2.94g Sodium citrate, 500ul Tween 20, pH 6.0) using a pressure cooker. Slides were blocked in 5% Normal Goat Serum, 0.2% Triton-X 100, in PBS. Slides were incubated overnight at 4°C in primary antibodies, then rinsed in PBS, incubated 1 hour at RT in secondary antibodies and/or fluorescently labeled lectin (Alexafluor647 made by directly conjugating E–Y Labs lectin to Molecular Probes Alexauor647), rinsed in PBS, mounted in ProLong Gold Antifade Mountant with DAPI (Molecular Probes). For immunohistochemistry, steps were identical except the following. An extra quenching step was performed for 15 min in a methanol solution containing 1.5% H2O2 following antigen retrieval. Substrate reaction and detection was performed using ImmPACT VIP Peroxifase (HRP) Substrate Kit (Vector Laboratories) as detailed per the manufacture’s protocol and slides were mounted in Permount Mounting Medium.
Imaging
Fluorescence microscopy was performed using a Zeiss Axiovert 200 microscope with an Axiocam MRM camera and Apotome II instrument for grid-based optical sectioning. Post-imaging adjustments including contrast, fluorescent channel overlay, and pseudo-coloring, were performed with Axiovision and Adobe Photoshop CS6. Histology of stomach and immunohistochemistry were imaged using an Olympus BX51 light microscope and Olympus SZX12 dissecting microscope w/12 MPixel Olympus DP70 camera. Images were analyzed and post-imaging adjustments were performed with Adobe Photoshop CS6.
Antibodies
Primary antibodies used in this study were as follows: Fluorescently conjugated Anguilla anguilla lectin (AAA; Alexa Fluor 647; 1:1000; EY Labs), goat anti–5-bromo-2′-deoxyuridine (1:20,000; a gift from Dr Jeff Gordon, Washington University, St Louis, MO), rat anti-CD44 v10-e16, ortholog of human v9 (1:200; Cosmo Bio, Tokyo, Japan), goat anti-Clusterin (1:100; Santa Cruz Biotechnology), mouse anti-ezrin (1:250; Santa Cruz Biotechnology), fluorescently conjugated Griffonia simplicifolia-II lectin (GSII; Alexa Fluor 647; 1:1,000; Invitrogen), rabbit anti-human gastric intrinsic factor (1:10,000; a gift from Dr David Alpers, Washington University, St Louis, MO), sheep anti-Pepsinogen C (PGC; 1:10,000; Abcam). Secondary antibodies included AlexaFluor (488, 594, or 647) conjugated donkey anti-goat, anti-rabbit, or anti-mouse (1:500; Molecular Probes).
Immunofluorescence and Immunohistochemistry Quantification
For counts from units expressing neck, chief, and proliferative markers all time points were quantified with at least 3 mice. Stomach corpus slides were stained for 5-bromo-2′-deoxyuridine (BrdU), the neck cell marker GSII lectin, and zymogenic cell marker GIF. Images were captured as TIFF files from a Zeiss Axiovert 200 microscope with an Axiocam MRM camera with an Apotome optical sectioning filter (Carl Zeiss, Jena, Germany). Each stomach had at least five randomly distrubuted 20× images taken, which contained 10 or more well-oriented gastric units. Units were counted using the neck staining, and total quantifications of proliferating cells (5-bromo-2′-deoxyuridine) were averaged over the total unit numbers per mouse. For total proliferation counts from immunohistochemistry at least 3 mice per treatment group were quantified. Stomach corpus slides were stained with an antibody for 5-bromo-2′-deoxyuridine (BrdU). Whole slides were scanned using a NanoZoomer 2.0 HT microscope and analyzed with NanoZoomer Digital Pathology software (Hamamatsu; Hamamatsu City, Japan). At least 40 corpus units were analyzed and counted per stomach. Proliferation rate for each mouse was an average of all units counted.
qRT-PCR
RNA was isolated using RNeasy (Qiagen) per the manufacturer’s protocol. The quality of the mRNA was verified with a BioTek Take3 spectrophotometer and electrophoresis on a 2% agarose gel. RNA was treated with DNase I (Invitrogen), and 1 μg of RNA was reverse-transcribed with SuperScript III (Invitrogen) following the manufacture’s protocol. Measurements of cDNA abundance were performed by qRT–PCR using either a Stratagene MX3000P detection system or a Bio-Rad CFX Connect system. Power SYBR Green master mix (Thermo Scientific) fluorescence was used to quantify the relative amplicon amounts of each gene (normalizing gene was TATA box binding protein). Primer design and sequence in Table 1.
Table 1.
qRT-PCR primer sequences:
| Clusterin Forward | CCAGCCTTTCTTTGAGATGA |
| Clusterin Reverse | CTCCTGGCACTTTTCACACT |
| GIF Forward | GAAAAGTGGATCTGTGCTACTTGCT |
| GIF Reverse | AGACAATAAGGCCCCAGGATG |
| HE4 Forward | TGCCTGCCTGTCGCCTCTG |
| HE4 Reverse | TGTCCGCACAGTCCTTGTCCA |
| Mist1 Forward | GAGCGAGAGAGGCAGCGGATG |
| Mist1 Reverse | AGTAAGTATGGTGGCGGTCAG |
| TATA Binding Protein Forward | CAAACCCAGAATTGTTCTCCTT |
| TATA Binding Protein Reverse | ATGTGGTCTTCCTGAATCCCT |
Graphing and statistics
All graphs and statistics were completed in GraphPad Prism (La Jolla, CA), using 1-way analysis of variance with the Tukey post hoc multiple comparison test to determine significance. Sample sizes were determined based on statistical significance and practicality. A P-value of ≤0.05 was considered significant.
Supplementary Material
Supplemental Figure 1: No SPEM occurs 24h after HDT treatment
A) Immunofluorescence of stomachs after 24h of vehicle, 5FU, HDT, or 5FU+HDT treatment (red, GIF; green, GSII; blue, DAPI). B) Quantification of cell populations, # indicates significance of treatment compared to vehicle, * indicates significance between specified treatment groups. G is significance between green columns. *p≤0.05, variance analyzed with ANOVA/Tukey (N Ł 3 mice/group)
Supplemental Figure 2: qRT-PCR for SPEM-associated genes
A) Transcripts of zymogenic cell markers analyzed from RNA isolated from the whole gastric corpus of mice treated with vehicle, 5FU, HDT, or 5FU+HDT for 24 hours. B) Same RNA described from A analyzed for markers known to increase in SPEM. C) Transcripts of zymogenic cell markers analyzed from RNA isolated from the whole gastric corpus of mice treated with vehicle, 5FU, HDT, or 5FU+HDT for 72 hours. D) Same RNA described from C analyzed for markers known to increase in SPEM. *p≤0.05 **p≤0.01 ***p≤0.001, variance analyzed with ANOVA/Tukey (N Ł 3 mice/group)
Supplemental Figure 3: Proliferation is sufficiently blocked over the course of injury in 5FU+HDT mice
A) Immunofluorescence of stomachs after 24h of vehicle, 5FU, HDT, or 5FU+HDT (green, GSII; red, GIF; white, BrdU; blue, DAPI). B) Quantification of proliferating cell populations. # # p≤0.01, variance analyzed with ANOVA/Tukey (N ≥ 3 mice/group)
Supplemental Figure 4: Proliferating pre-parietal cells begin to appear 96h after HDT treatment.
Timeline of treatment scheme. Immunofluorescence of stomachs after 96h of vehicle, 5FU, HDT, or 5FU+HDT (green, GSII; red, ezrin; white, BrdU; blue, DAPI). Yellow boxes indicate BrdU+ ezrin+ cells with basolateral surfaces of each cell outlined in dashed white line.
Supplemental Figure 5: Proliferation after SPEM induction
A) Immunofluorescence of stomachs after 72h of vehicle, HDT, or 5FU+HDT treatment (red, GIF; purple, AAA (Anguilla anguilla lectin); white, BrdU; blue, DAPI). Yellow arrowheads indicate cells that were not counted in quantification because they are in the pit zone and, thus, above the bottom-most AAA+ cell. B) Immunofluorescence of stomachs treated with vehicle, 5FU, HDT, or 5FU+HDT for 72h (red, CD44v; green, GSII; blue, DAPI). Dotted line: outlined unit bases. C) Transcripts of proliferative markers analyzed from RNA isolated from the whole gastric corpus of mice treated with vehicle, 5FU, HDT, or 5FU+HDT for 72 hours.
Supplemental Figure 6: Human SPEM in gastric cancer patients
A) H&E of human corpus glands with early atrophic gastritis exhibiting focal gland bases transitioning to SPEM. Arrows refer to parietal cells in healthy glands. SPEM gland outlined in yellow has a wide lumen and lacks parietal cells. B) Same as panel A but a different patient with more active gastritis. Early SPEM gland outlined in yellow shows opening of lumen at gland base (yellow arrow) and contains degenerating parietal cells (black arrows). C) Immunofluorescence of human SPEM (red, PGC; green, GSII; blue, DAPI). Note many hybrid forms in gland bases ranging from cells with only red PGC (mature, normal ZCs) to those showing mature SPEM (prominent green GSII, scant red PGC). D) Immunofluorescence of the base of yet another patient’s corpus. A mature SPEM gland is located next to a normal gland with mature ZCs. A third gland shows hybrid forms wherein ZCs express focal, apical GSII (yellow arrowheads). E) Table detailing the demographic aspects and pathological diagnoses of the 10 patients analyzed in this manuscript.
Acknowledgments
Grant Support: This work was supported by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases (DK094989, DK105129, DK110406), by the Alvin J. Siteman Cancer Center/Barnes Jewish Hospital Foundation Cancer Frontier Fund, NIH NCI P30 CA091842, and The Barnard Trust to JCM; by the National Institutes of Health training grant 5T32GM007067-43 to MDR; by the National Institute of General Medical Sciences Cell and Molecular Biology training grant GM007067 and the Philip and Sima Needleman Student Fellowship in Regenerative Medicine to JB; and by the National Cancer Institute training grant CA00954731 to SGW. Histology was performed by the Digestive Disease Research Core Centers (P30DK052574). This work was also supported by the Alafi Neuroimaging Laboratory, the Hope Center for Neurological Disorders, and NIH Shared Instrumentation Grant (S10 RR0227552, Advanced Imaging and Tissue Analysis Core) to Washington University.
Abbreviations
- 5FU
5-Fluorouracil
- AAA
Anguilla anguilla lectin
- BrdU
5-bromo-2′-deoxyuridine
- CAG
chronic atrophic gastritis
- GIF
gastric intrinsic factor
- GSII
Griffonia simplicifolia lectin II
- HDT
high-dose tamoxifen
- PGC
pepsinogen c
- SPEM
spasmolytic polypeptide-expressing metaplasia
- TFF2
trefoil factor 2, spasmolytic polypeptide
- ZC
zymogenic chief cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
The authors disclose no conflicts.
Author Contributions:
Megan D. Radyk was responsible for the study concept, design, data acquisition, analysis, and interpretation, and drafting the manuscript. Joseph Burclaff was responsible for data acquisition, analysis and interpretation, and critical revision of the manuscript. Spencer G. Willet was responsible for data acquisition and analysis. Jason C. Mills was responsible for the study concept and design, analysis and interpretation of data, funding, supervision, and revision of the manuscript.
References
- 1.Petersen CP, et al. Cell Mol Gastroenterol Hepatol. 2017;3:11–26. doi: 10.1016/j.jcmgh.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt P, et al. Lab Investig. 1999;79:639–46. [PMC free article] [PubMed] [Google Scholar]
- 3.Nam KT, et al. Gastroenterology. 2010;139:2028–2037. doi: 10.1053/j.gastro.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stange DE, et al. Cell. 2013;155:357–368. doi: 10.1016/j.cell.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsuo J, et al. Gastroenterology. 2017;152:218–231.e14. doi: 10.1053/j.gastro.2016.09.018. [DOI] [PubMed] [Google Scholar]
- 6.Leushacke M, et al. Nat Cell Biol. 2017;19:774–786. doi: 10.1038/ncb3541. [DOI] [PubMed] [Google Scholar]
- 7.Choi E, et al. Gastroenterology. 2016;150:918–930.e13. doi: 10.1053/j.gastro.2015.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayakawa Y, Ariyama H, et al. Cancer Cell. 2015;28:800–814. doi: 10.1016/j.ccell.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huh WJ, Khurana SS, et al. Gastroenterology. 2012;142:21–24.e7. doi: 10.1053/j.gastro.2011.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saenz JB, et al. Methods Mol Biol. 2016;1422:329–39. doi: 10.1007/978-1-4939-3603-8_28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engevik AC, et al. Cell Mol Gastroenterol Hepatol. 2016;2:605–624. doi: 10.1016/j.jcmgh.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Potten CS. Philos Trans R Soc Lond B Biol Sci. 1998;353:821–830. doi: 10.1098/rstb.1998.0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lo HYG, et al. Genes Dev. 2017;31:154–171. doi: 10.1101/gad.285684.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burclaff J, et al. Gastroenterology. 2017;152:762–766.e7. doi: 10.1053/j.gastro.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lennerz JKM, et al. Am J Pathol. 2010;177:1514–33. doi: 10.2353/ajpath.2010.100328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1.Saenz JB, et al. Methods Mol Biol. 2016;1422:329–39. doi: 10.1007/978-1-4939-3603-8_28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lennerz JKM, et al. Am J Pathol. 2010;177:1514–33. doi: 10.2353/ajpath.2010.100328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capoccia BJ, et al. J Clin Invest. 2013;123:1475–91. doi: 10.1172/JCI65703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khurana SS, et al. J Biol Chem. 2013;288:16085–97. doi: 10.1074/jbc.M112.445551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: No SPEM occurs 24h after HDT treatment
A) Immunofluorescence of stomachs after 24h of vehicle, 5FU, HDT, or 5FU+HDT treatment (red, GIF; green, GSII; blue, DAPI). B) Quantification of cell populations, # indicates significance of treatment compared to vehicle, * indicates significance between specified treatment groups. G is significance between green columns. *p≤0.05, variance analyzed with ANOVA/Tukey (N Ł 3 mice/group)
Supplemental Figure 2: qRT-PCR for SPEM-associated genes
A) Transcripts of zymogenic cell markers analyzed from RNA isolated from the whole gastric corpus of mice treated with vehicle, 5FU, HDT, or 5FU+HDT for 24 hours. B) Same RNA described from A analyzed for markers known to increase in SPEM. C) Transcripts of zymogenic cell markers analyzed from RNA isolated from the whole gastric corpus of mice treated with vehicle, 5FU, HDT, or 5FU+HDT for 72 hours. D) Same RNA described from C analyzed for markers known to increase in SPEM. *p≤0.05 **p≤0.01 ***p≤0.001, variance analyzed with ANOVA/Tukey (N Ł 3 mice/group)
Supplemental Figure 3: Proliferation is sufficiently blocked over the course of injury in 5FU+HDT mice
A) Immunofluorescence of stomachs after 24h of vehicle, 5FU, HDT, or 5FU+HDT (green, GSII; red, GIF; white, BrdU; blue, DAPI). B) Quantification of proliferating cell populations. # # p≤0.01, variance analyzed with ANOVA/Tukey (N ≥ 3 mice/group)
Supplemental Figure 4: Proliferating pre-parietal cells begin to appear 96h after HDT treatment.
Timeline of treatment scheme. Immunofluorescence of stomachs after 96h of vehicle, 5FU, HDT, or 5FU+HDT (green, GSII; red, ezrin; white, BrdU; blue, DAPI). Yellow boxes indicate BrdU+ ezrin+ cells with basolateral surfaces of each cell outlined in dashed white line.
Supplemental Figure 5: Proliferation after SPEM induction
A) Immunofluorescence of stomachs after 72h of vehicle, HDT, or 5FU+HDT treatment (red, GIF; purple, AAA (Anguilla anguilla lectin); white, BrdU; blue, DAPI). Yellow arrowheads indicate cells that were not counted in quantification because they are in the pit zone and, thus, above the bottom-most AAA+ cell. B) Immunofluorescence of stomachs treated with vehicle, 5FU, HDT, or 5FU+HDT for 72h (red, CD44v; green, GSII; blue, DAPI). Dotted line: outlined unit bases. C) Transcripts of proliferative markers analyzed from RNA isolated from the whole gastric corpus of mice treated with vehicle, 5FU, HDT, or 5FU+HDT for 72 hours.
Supplemental Figure 6: Human SPEM in gastric cancer patients
A) H&E of human corpus glands with early atrophic gastritis exhibiting focal gland bases transitioning to SPEM. Arrows refer to parietal cells in healthy glands. SPEM gland outlined in yellow has a wide lumen and lacks parietal cells. B) Same as panel A but a different patient with more active gastritis. Early SPEM gland outlined in yellow shows opening of lumen at gland base (yellow arrow) and contains degenerating parietal cells (black arrows). C) Immunofluorescence of human SPEM (red, PGC; green, GSII; blue, DAPI). Note many hybrid forms in gland bases ranging from cells with only red PGC (mature, normal ZCs) to those showing mature SPEM (prominent green GSII, scant red PGC). D) Immunofluorescence of the base of yet another patient’s corpus. A mature SPEM gland is located next to a normal gland with mature ZCs. A third gland shows hybrid forms wherein ZCs express focal, apical GSII (yellow arrowheads). E) Table detailing the demographic aspects and pathological diagnoses of the 10 patients analyzed in this manuscript.