

ABSTRACT
Bacterial viruses (bacteriophages) play a significant role in microbial community dynamics. Within the human gastrointestinal tract, for instance, associations among bacteriophages (phages), microbiota stability, and human health have been discovered. In contrast to the gastrointestinal tract, the phages associated with the urinary microbiota are largely unknown. Preliminary metagenomic surveys of the urinary virome indicate a rich diversity of novel lytic phage sequences at an abundance far outnumbering that of eukaryotic viruses. These surveys, however, exclude the lysogenic phages residing within the bacteria of the bladder. To characterize this phage population, we examined 181 genomes representative of the phylogenetic diversity of bacterial species within the female urinary microbiota and found 457 phage sequences, 226 of which were predicted with high confidence. Phages were prevalent within the bladder bacteria: 86% of the genomes examined contained at least one phage sequence. Most of these phages are novel, exhibiting no discernible sequence homology to sequences in public data repositories. The presence of phages with substantial sequence similarity within the microbiota of different women supports the existence of a core community of phages within the bladder. Furthermore, the observed variation between the phage populations of women with and without overactive bladder symptoms suggests that phages may contribute to urinary health. To complement our bioinformatic analyses, viable phages were cultivated from the bacterial isolates for characterization; a novel coliphage was isolated, which is obligately lytic in the laboratory strain Escherichia coli C. Sequencing of bacterial genomes facilitates a comprehensive cataloguing of the urinary virome and reveals phage-host interactions.
IMPORTANCE Bacteriophages are abundant within the human body. However, while some niches have been well surveyed, the phage population within the urinary microbiome is largely unknown. Our study is the first survey of the lysogenic phage population within the urinary microbiota. Most notably, the abundance of prophage exceeds that of the bacteria. Furthermore, many of the prophage sequences identified exhibited no recognizable sequence homology to sequences in data repositories. This suggests a rich diversity of uncharacterized phage species present in the bladder. Additionally, we observed a variation in the abundances of phages between bacteria isolated from asymptomatic “healthy” individuals and those with urinary symptoms, thus suggesting that, like phages within the gut, phages within the bladder may contribute to urinary health.
KEYWORDS: bacteria, bacteriophages, microbiome, prophage, urinary tract, virus
INTRODUCTION
Surveys of the bacteria inhabiting the human body have led to the discovery of new taxa (1), as well as a greater diversity among “healthy” individuals than originally hypothesized (for a review, see reference 2). Studies have identified associations between the presence, absence, and/or prevalence of bacterial taxa and an individual's health and response to treatment (for reviews, see references 3–5). For instance, Lactobacillus gasseri within the bladder is strongly associated with urinary urgency incontinence (UUI) in women; this contrasts with Lactobacillus crispatus, which is associated with asymptomatic controls (6). An imbalance of the microbiota (dysbiosis) also can be connected with an individual's health, as reported within the gastrointestinal tract (7). In addition to bacteria, the microbial communities that colonize the human body include other cellular organisms (archaea and fungi) and viruses, all of which are involved in complex interactions with each other and surrounding human cells. Of particular note are the interactions between bacteria and their natural predators: bacteriophages (viruses that infect bacteria). Bacteriophages (phages) are crucial members of microbial communities, shaping bacterial metabolism and community structure (8, 9). Within the gut, for instance, this influence extends from infancy through adulthood (10, 11). Similar observations have been made within the microbiota of the oral cavity (see a review in reference 12).
Human microbiome surveys have identified phages within every community examined (13). In fact, they are the most abundant members of the human virome, far exceeding eukaryotic viruses (13). Despite their prevalence, phages are significantly understudied in comparison to their hosts. As such, phages detected within the human microbiome are often novel, with their sequences exhibiting no recognizable similarity to those of characterized species (13, 14). While phages can exploit a variety of infection strategies, lytic and lysogenic phages are most frequently found. Lytic phages are exclusively predatory, killing their host cell. Lysogenic phages have two lifestyle choices: they can be lytic immediately, or they can incorporate their genome into the host's bacterial genome or remain in the cytoplasm as a plasmid. The phage genome (now called a prophage) generally replicates in synchrony with the host chromosome until some signal induces the phage to enter the lytic path. Within the gut microbiota, this shift from lysogeny to the lytic path has been correlated with disease (14).
In comparison to the sites of the body investigated by the Human Microbiome Project (HMP), the urinary bladder is understudied. While new urinary microbiome research has primarily focused on the bacterial constituents of this community (5), a few studies have shown that the urinary microbiota also includes viruses. First, viruses have been isolated from urine, including several eukaryotic viruses (15–21) and phages (22, 23). Second, two metagenomic sequencing studies of the urinary virome (eukaryotic viruses and phages in the lytic cycle) have been conducted (24, 25). Although these virome studies found that phages vastly outnumber (>99%) eukaryotic viruses, they have sampled but a fraction of the phage community; they did not capture the diversity of prophages present.
While prophages can account for up to 20% of a bacterial genome (26), they are not simply passengers along for the ride; they can be selectively advantageous or disadvantageous to the host cell (26). For example, harboring a prophage can provide the bacterial host with protection from superinfection (27, 28). The presence of a prophage also can increase bacterial fitness through phage genes; for instance, the λ phage genes bor and lom increase the Escherichia coli host's survival in macrophages (29). Furthermore, a prophage can encode a critical virulence factor. For example, the botulinum, Shiga, and cholera toxins, among others, are encoded by prophages (30). In the event that a prophage reduces its host's fitness, selection should lead to inactivation and/or deletion (31). In fact, the Shiga toxin in Shigella dysenteriae 1 is the functional relic of a once-functional prophage (32).
Based upon prior sequencing of bacterial isolates from the bladder and the abundance of prophage sequences detected (33, 34), we hypothesize that the prophage population in the urinary microbiota is large. This is a reasonable assumption, as lysogenic phages have been found to outnumber obligately lytic phages within the microbiota of the gut, particularly in the microbiota of healthy individuals (14). Because the urinary microbiota (35–37) exists at a substantially lower biomass than the gut (38) and prior research has found that the frequency of lysogeny is inversely related to susceptible host density (39–42), we anticipate that the abundance of lysogenic phages may exceed that of the gut. To explore the lysogenic phage population of the urinary microbiome, we sequenced 181 genomes that represent the phylogenetic diversity of bacterial species isolated from the bladder (K. Thomas-White, S. C. Forster, N. Kumar, M. Van Kuiken, C. Putonti, M. Stares, E. E. Hilt, T. K. Price, A. J. Wolfe, and T. D. Lawley, unpublished data). Within these sequenced genomes, which represent 51 bacterial genera and 102 species, phage sequences were identified and characterized. Whereas seeking phage sequences within bacterial genomes would preclude the detection of most lytic phages, culture-dependent methods, such as those employed here, provide a key piece of information about the phages identified: each phage's native host. The result is the first catalogue of the rich diversity of uncharacterized phage species present in the bladder microbiota.
RESULTS
The genome assemblies of 181 bacterial isolates from the bladder were examined for the presence of phage sequences. These bacterial isolates were collected from women both with and without lower urinary tract symptoms (LUTS) and were selected to represent the phylogenetic diversity of bacteria within the urinary microbiota (see Data Set S1 in the supplemental material) (Thomas-White et al., unpublished). The taxonomy of each isolate was determined by 16S rRNA gene analysis (Data Set S2). Clustered regularly interspaced short palindromic repeat (CRISPR) arrays were identified within 109 of the 181 strains sequenced (Data Set S1). The tool VirSorter (43) was used to detect the phage sequences of both prophages that are integrated into the bacterial chromosome (categories 4 to 6) and phages that are not, including lytic phages and prophages existing as an extrachromosomal plasmid (categories 1 to 3). Given that this tool can detect both lytic and lysogenic phages, we refer to categories 1 to 3 as “unintegrated phages” and categories 4 to 6 as “integrated prophages.” Category designations indicate the confidence of the prediction defined by VirSorter (43). Categories 1 and 4 are the most confident predictions, while 3 and 6 are the least. Here, we define categories 1, 2, 4, and 5 as “high-confidence” predictions and categories 3 and 6 as “lower-confidence” predictions. In total, 226 high-confidence phage sequences and 231 lower-confidence phage sequences were identified (Table 1 and Data Set S3). The nucleotide sequences for these phages are included in Data Set S4. Each phage sequence was annotated; the presence/absence of a terminase-coding region was used as a marker to assess if the phage sequence identified by VirSorter was representative of a complete phage genome. The majority of sequences in the high-confidence group (85%) contained a recognizable terminase-coding region. In contrast, many of the phage sequences predicted with lower confidence lacked an identifiable terminase-coding region. Upon further investigation, most (∼71%) of the phage sequences within this lower-confidence group are partial phage genome sequences, which is either a residual of the assemblies themselves (as genomes are in scaffolds) or defunct integrated phages.
TABLE 1.
Number of phage sequences identified within the 181 genomes for each confidence category
| Phage type | Category | No. of phage sequences | No. of bacterial isolates |
|---|---|---|---|
| Unintegrated | 1 | 7 | 3 |
| 2 | 41 | 28 | |
| 3 | 67 | 47 | |
| Integrated | 4 | 8 | 8 |
| 5 | 170 | 105 | |
| 6 | 164 | 95 |
Phage sequences were abundant among the bacterial isolates from the bladder; 86% of these isolates had one or more phages associated with their genome sequence (Fig. 1). The number of phage sequences found within a genome had a low correlation with the genome's size (Spearman's ρ = 0.09 for high-confidence predictions and ρ = 0.21 for all predicted phages, with P values of 0.26 and 0.005, respectively). Additionally, no significant difference was found between the average length of the genomes of bacterial isolates containing phage and those lacking phage (with P values of 0.43 and 0.13 for high-confidence and lower-confidence predictions, respectively).
FIG 1.

Phage presence within bacteria of the bladder. Each phage sequence identified at a high confidence is represented by a dot on the isolate's branch in the phylogenetic tree, which was built using the 16S rRNA gene sequences of the isolates. The phyla for the isolates in the phylogenetic tree are indicated in the outer circle, with “B” indicating the single isolate from the phylum Bacteroidetes. Red and blue circles represent unintegrated phages of categories 1 and 2, respectively. Green and yellow circles represent integrated phages of categories 4 and 5, respectively. It is important to note that branches without dots may include lower-quality predictions (categories 3 and 6).
In characterizing the phage population within the bladder, we first focused on the 226 phage sequences predicted with high confidence, as these were found to resemble intact viable phages. These sequences were compared to all bacterial sequences in public repositories, as well as all publicly available viral genomic sequences. Eighty-five of the 226 high-confidence phages exhibited sequence similarity to prophage sequences within previously sequenced bacterial genomes (Data Set S5; see Materials and Methods for further details), many from the sequencing efforts that established the human microbiome reference set. In contrast, only six phages resembled previously characterized isolated phages at a strict threshold (>50% phage genome query coverage), while an additional six phages were identified at a more relaxed threshold (>25% phage genome query coverage). The majority of high-confidence phage sequences (129/226 [57%]), however, exhibited no sequence similarity to any known phages. Thus, we have uncovered 129 novel phages from the urinary microbiota. A similar examination of the lower-confidence phage sequences (Data Set S6) revealed 91 hits to previously sequenced bacterial genomes. Again, the majority of sequences in this second lower-confidence set (140/231 [61%]) appear to be novel.
The phage sequences identified within the bladder's bacterial isolates, including both the high- and lower-confidence sets, were next compared to each other. Significant sequence homology was detected within the phages found in isolates of the bacterial family Actinomycetaceae, including five Actinomyces strains and one Varibaculum cambriense strain (Fig. 2), each isolated from a different patient. The first five phages in Fig. 2 were predicted within the higher-confidence set; in contrast, the prophage from Actinomyces europaeus strain UMB0652 was predicted to be in category 6 and thus in the lower-confidence set. Next, these six phage sequences were annotated via RAST (44), revealing several phage and phage-associated coding regions (Fig. 2). The ∼60-kbp region conserved among five of the six Actinomycetaceae phages discovered in this study showed greatest similarity to a prophage within the genome of Corynebacterium atypicum strain R2070 (family Corynebacteriaceae) with 65% coverage and 90% nucleotide identity (megablast). Homologous regions between the Corynebacterium prophage and these Actinomycetaceae phages included “hallmark” viral gene sequences. We thus turned our attention to the hallmark viral genes of the capsid and terminase large subunit. blastx analysis of these genes to viral genomes in GenBank identified sequence homology (E value, 0) to Streptococcus phages, indicative of a common, albeit distant, ecological or evolutionary history between the Streptococcus and Actinomycetaceae phages. In addition to these Actinomycetaceae phages, several other phage sequences were found to include syntenic blocks across strains of bacteria from the bladder, including species from the genera Gardnerella, Aerococcus, Streptococcus, Lactobacillus, Bifidobacterium, and Escherichia (Table S1).
FIG 2.

Phage conserved among Actinomycetaceae strains isolated from different patients. Syntenic regions are shown between the strains. The annotations for coding regions within each syntenic block are listed in the corresponding colors. ECF, energy-coupling factor.
The Actinomycetaceae phages shown in Fig. 2 were all found in strains isolated from women with overactive bladder (OAB), whereas the V. cambriense strain was obtained from a woman with stress urinary incontinence (SUI). In contrast, these Actinomycetaceae phages were not identified within any of the Actinomyces strains isolated from asymptomatic controls. While the purpose of this study was to explore phage diversity and prevalence within the bacteria of the bladder, the Actinomycetaceae phages motivated us to examine the phage populations relative to the patient condition. Looking more broadly at the data as “condition−” versus “condition+,” a subset of the data was selected in which the patient had not received any treatment for urinary conditions. Thus, the condition− group included 69 genomes, and the condition+ group included 73 genomes (Data Set S1). The probability distributions of the number of high-confidence phage sequences between the two groups were not found to be statistically different, as assessed by calculating the generalized likelihood ratio test for comparing two negative binomial distribution parameters. Next, the phage populations were examined between the two cohorts by genus. A Wilcoxon test was performed; although there appears to be variation in the mean number of phage sequences within several genera, a marginally statistically significant difference was only detected within the genus Lactobacillus (P = 0.06647). It is important to note that not all Lactobacillus species chosen to be sequenced were collected from both populations; for example, the Lactobacillus iners genomes examined here were only from women in the control population, although L. iners is commonly found in the urinary microbiome of both condition− and condition+ women (6, 36). Figure 3 illustrates the number of high-confidence phages per isolate found within the condition− (control) population and the largest subset of condition+ patients, individuals with OAB symptoms (n = 63). (Similar analysis of the lower-confidence phages can be found in Fig. S1.) The mean count between the control and OAB populations was not statistically different for either category (high-confidence phages, P = 0.2057; lower-confidence phages, P = 0.2084).
FIG 3.
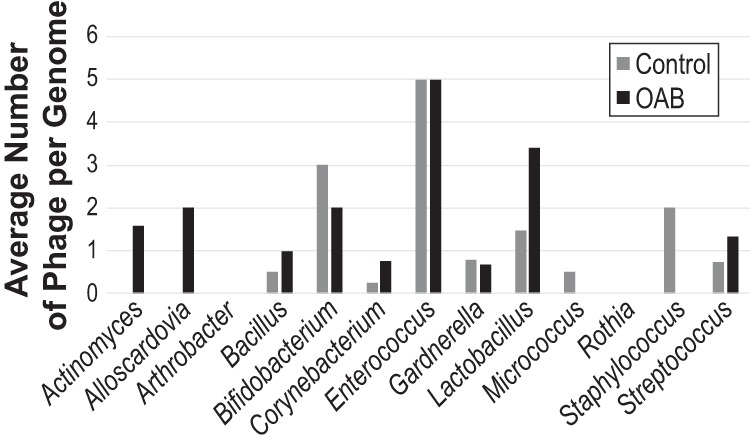
Average number of high-confidence phage sequences per genome isolated from control and OAB patients. Genera with representatives from both patient populations are shown.
Whereas we identified with high confidence an abundance of phage sequences within the bladder bacteria, some prophages integrated into the bacterial chromosome (categories 4 to 6) may be unable to enter the lytic cycle (i.e., defunct or defective prophages). For instance, eight phages were identified within the genome of E. coli strain UMB0901, including four predicted with high confidence. All four of these phage sequences are integrated prophages (categories 4 and 5) and exhibit the greatest sequence similarity to annotated prophages within E. coli genomes from the human microbiota (45) (Table 2). To determine if these prophages can enter the lytic cycle, E. coli UMB0901 was selected for further analysis in the lab. One phage was isolated (see Materials and Methods). While lysogenic within its UMB0901 host, this phage was capable of lysing laboratory strains of E. coli C and E. coli K-12 under both aerobic and anaerobic conditions; infection of the laboratory strain E. coli B was observed on several occasions, although not consistently. We also tested the phage's lytic activity on the other E. coli isolates from this study and several other E. coli bladder isolates in our collection (see Materials and Methods); lysis was not observed for any of these clinical isolates (data not shown). High-titer stocks of the purified phage were used to examine the phage's morphology via transmission electron microscopy (TEM) (Fig. 4). The presence of a contractile tail indicates that this phage belongs to the family Myoviridae of the order Caudovirales.
TABLE 2.
Sequence examination of high-confidence phages within the genome of E. coli UMB0901
| Category | Length (bp) | Best blast hit | Accession no. | Identity (%) | Query cover (%) |
|---|---|---|---|---|---|
| 4 | 38,132 | Escherichia coli K-15W01 | CP016358 | 98 | 84 |
| Salmonella phage STYP2 | KX833213 | 97 | 58 | ||
| 5 | 10,493 | E. coli ECONIH2 | CP014667 | 99 | 100 |
| E. coli K-15KW01 | CP016358 | 99 | 99 | ||
| 5 | 63,683 | E. coli 536 | CP000247 | 99 | 100 |
| E. coli ECONIH2 | CP014667 | 99 | 100 | ||
| 5 | 69,863 | E. coli 542 | CP018970 | 99 | 99 |
| E. coli ECONIH2 | CP014667 | 99 | 88 |
FIG 4.

TEM of phage isolated from bladder strain E. coli UMB0901.
DISCUSSION
The bacteria isolated from the female urinary bladder include an abundance of bacteriophages. Considering just those phage sequences predicted with high confidence, the number of phage sequences exceeded that of the number of bacterial genomes examined. The abundance of prophage sequences within the urinary bacterial strains may be the result of the bladder itself; lysogeny may serve as a survival strategy when the bacterial community density is low or in the presence of other environmental parameters, e.g., temperature and metabolic community composition (39–42). Nevertheless, not all genomes examined in this study were found to contain phage sequences; in fact, 13.8% of the bacterial isolates from the bladder did not include any detectable phage sequence, and an additional 22% only contained phage sequences predicted at the lower-confidence level (see Data Set S3 in the supplemental material). Taxa that did not show any evidence of phage infection included all three Dolosicoccus paucivorans strains examined and several, but not all, isolates of Gardnerella vaginalis and Micrococcus luteus. At present, a lytic phage from either D. paucivorans or G. vaginalis has yet to be characterized, although prophage sequences within Gardnerella spp. have previously been described (33). In contrast, a few phages capable of lysing Micrococcus strains were isolated and studied in the 1960s and 70s (see, e.g., references 46 to 48). The 226 phages predicted with high confidence were exclusive to just 116 of the genomes examined. No correspondence between the presence/absence of CRISPR arrays and prophage was observed (Data Set S1). While prior studies of the microbiomes of other niches have found a correlation between bacterial genome size and the number of prophages in genomes (49, 50), the strains from the bladder did not fit this expectation.
Whereas the number of phage sequences per genome varied, Lactobacillus strains from the bladder frequently harbored more than one phage. For example, Lactobacillus jensenii strain UMB0077 was the most phage-rich genome examined, with 10 phages predicted at high confidence and another three phages at lower confidence (Data Set S3). Surveys of the urinary microbiome have routinely found Lactobacillus species and provided evidence of their association with urinary symptoms (6, 35, 51–54). Lactobacilli also are dominant organisms within other niches, most notably the vagina (4, 55), and some species have direct correlations to health outcomes (56–58). Microbiome studies, particularly of the vaginal community, have discovered lytic phages of human-associated Lactobacillus species (see, e.g., references 59 to 61). While some of the Lactobacillus phages found in our study showed sequence homology to the previously characterized Lactobacillus phage Lv1 (60), 15 Lactobacillus phage sequences predicted with high confidence exhibited no sequence similarity to any known lytic or lysogenic phage. The fact that these urinary Lactobacillus genomes include prophages that have yet to be found within vaginal lactobacilli leads us to posit that a subset of the microbiota of these two niches may be isolated.
In addition to these novel Lactobacillus phages, novel phage genomes were also identified within other bacteria common to the urinary microbiome (5, 6, 36, 51, 62), including species of Actinomyces and Varibaculum (Fig. 2), Bifidobacterium, Gardnerella, and Streptococcus, as well as the uropathogen Proteus mirabilis (Data Set S3). This suggests a rich diversity of uncharacterized phage species present in the bladder, confirming prior conclusions of viral metagenomic analyses of urine samples (24, 25). This observation is not unique to the bladder virome, as approximately 50% of the phages within the gut virome cannot be classified (14). The phages isolated in prior studies (22, 25) and the single phage reported here (Fig. 4) are limited to two of the 51 bacterial genera found within the bladder. For many of the species within the bladder, there are no characterized lytic phages, and the ability for prophages to enter the lytic cycle is unknown. Thus, to understand intracommunity interactions, it is imperative to isolate and characterize phages from the bladder.
Further investigation into the diversity of phages within the bladder could provide critical information concerning shifts in bladder community structure. Recent studies have found that changes in the gut's phage population may be associated with dysbiosis and disease (11, 14, 63–65). Similar observations have been made from studies of the vaginal microbiota (4). Furthermore, prior studies have suggested that Lactobacillus phages could play a role in the shift in the vaginal community structure and contribute to bacterial vaginosis (for a review, see reference 66). The bacterial communities within the gut, vagina, and other areas targeted in the HMP are, however, significantly better characterized than those within the urinary bladder. The abundance of lysogenic phages found in the bladder suggests that phages also may contribute to urinary microbiota stability. Furthermore, the variation observed between the phage populations within the strains selected of individuals with and without OAB symptoms (Fig. 3), although not found to be statistically significant, suggests that phages may contribute to urinary health, thus warranting further study, including sequencing of multiple strains and species of genera found within both populations.
While metagenomic studies can comprehensively sample microbial communities, culture-dependent studies provide a key piece of information when investigating phages, namely, information regarding the bacterial host. For example, in this study, we can associate each phage with at least one of its native host species. This includes the 129 novel and 140 putative phages that exhibit no sequence homology to any known annotated (pro)phage sequence in public sequence repositories (Data Sets S5 and S6). For those phages that do resemble a previously sequenced phage, the bacterial and/or phage sequences recognized are typically annotated as the same bacterial host, e.g., the prophages of E. coli strain UMB0901 most closely resemble prophages within other E. coli strains (Table 2). However, the sequence homology observed for the Actinomycetaceae phages (Fig. 2) suggests that their host range might extend across genera and even across taxonomic families. Similarly, analysis of two prophage sequences from Actinomyces turicensis strain UMB0250 and Corynebacterium sp. strain UMB0763 suggested that these phages may also be capable of infecting bacterial species of different families (Data Set S5). These findings suggest that some phages within the microbiota of the bladder may have a broad host range and should guide subsequent experimental work to test this hypothesis. While the notion of broad-host-range phages is controversial within the phage research community (67), bioinformatic and experimental studies have identified such phages (68–70). Host range itself is likely transient, as ecological factors, including drug treatment, may enable a phage's ability to infect a new bacterial host (71).
Our survey of lysogenic phages provides the first step in the process of unraveling the complex dynamics of phage-bacterium interactions within the urinary microbiota. Additional sequencing of the bacterial population within the bladder, coupled with further metagenomic sequencing of the urinary virome, is needed to both determine if a core “bladder phageome” exists and whether phages play a role in urinary health and disease.
MATERIALS AND METHODS
Bacterial isolates.
The genome sequences examined in this study were of bacterial isolates isolated from urine collected during the conduct of several institutional review board (IRB)-approved studies. In the majority of cases, urine was collected aseptically via transurethral catheter. In a few cases, urine was collected by midstream void. In either case, the urine sample was placed in a BD Vacutainer Plus C&S preservative tube for culturing. Expanded quantitative urine culture (EQUC) was performed as described previously (36). Matrix-assisted laser desorption ionization–time of flight mass spectrophotometry (MALDI-TOF MS) with the MALDI Biotyper 3.0 software program (Bruker Daltonics, Billerica, MA) was used to identify the bacterial strains selected for sequencing, as described elsewhere (36).
Isolates were collected from individuals with or without lower urinary tract symptoms (LUTS). LUTS included overactive bladder (OAB; n = 101), interstitial cystitis/painful bladder syndrome (IC/PBS; n = 1), depression with diabetes (DD; n = 1), stress urinary incontinence (SUI; n = 2), or urinary tract infection (UTI; n = 6). One isolate was from a patient presenting with a kidney stone. Individuals without LUTS (n = 69) included a small cohort of pregnant women (n = 16). Thirty-nine of the samples collected were from individuals after LUTS treatment. The patient symptom status using these codes is indicated for each isolate in Data Set S1 in the supplemental material.
Genome sequencing and assembly.
Genome sequencing and assembly are described in full by Thomas-White et al. (unpublished). Briefly, the bacterial isolates were grown in their preferred medium and pelleted. Genomic DNA was extracted using a phenol-chloroform method (72). Genome sequencing was completed at the Wellcome Trust Genome Campus (Hinxton, UK). The Illumina Nextera kit was used for whole-genome library preparation with fragment sizes of 200 to 300 bp. Sequencing was conducted on the Illumina HiSeq 2000 platform, producing paired-end 2 × 100-bp reads, and annotated assemblies were produced using the pipeline described previously (73). Sequence reads were used to create multiple assemblies using VelvetOptimiser version 2.2.5 (74) and Velvet version 1.2 (75). The assembly with the best N50 score was selected, and an assembly improvement step was performed using the tools SSPACE (76) and GapFiller (77). Additionally, we computed the genome coverage using BBMap (78). All reads and assemblies are publicly available via EBI and GenBank; accession numbers are listed in Data Set S1.
Phylogenetic analysis.
Taxonomic classification of isolates was performed based upon the genome's 16S rRNA gene sequence. 16S rRNA gene sequences were detected through blastn queries to a local copy of the 16S rRNA gene sequences from the RNAmmer server (79). The bacterial isolates' sequences were aligned via CLUSTALW through Geneious version 9.1.4 (Auckland, New Zealand). A phylogenetic tree was derived using FastTree (80) through Geneious using the Jukes-Cantor model. The 16S rRNA gene sequences used for construction of this tree are included in Data Set S2. The tree was visualized using PhyloWidget (81).
CRISPR analysis of bacterial genomes.
CRISPR spacer arrays were detected using the software minCED (82). Arrays containing three or more spacers were identified.
Phage identification and characterization.
Contigs were examined using the tool VirSorter (43). Briefly, VirSorter examines sequences, looking for hallmark viral genes (e.g., capsid protein, terminase large subunit, and tail) and enrichment in virus-like genes. VirSorter predictions include confidence categories. Viral sequences, either prophages that remain in the cytoplasm in plasmid form or as lytic phages, are identified as category 1, 2, and 3 phages, corresponding to predictions that are “most confident,” “likely,” and “possible,” respectively. Categories 4, 5, and 6 are reserved for prophage sequences detected within a cellular contig. Category 4 refers to the most confident predictions, whereas categories 5 and 6 correspond to likely and possible predictions, respectively. Further details regarding these categories can be found online (https://pods.iplantcollaborative.org/wiki/display/DEapps/VIRSorter+1.0.2).
All VirSorter-predicted sequences were compared via blastn to all bacterial nucleotide sequences (query: “bacteria[filter] AND ddbj_embl_genbank[filter]”) and viral RefSeq (83) genomic sequences, collected in June 2017, and run locally. Phage sequences were also compared locally via blastx to annotated terminase protein sequences within the RefSeq viral collection. Predicted phage sequences of interest were further examined using the RAST annotation server (44), with blast queries of the complete nr/nt database via NCBI and by manual inspection. Upon manual curation, no sequences were found to be obvious misclassifications of viral sequences. VirSorter-predicted sequences were compared using reciprocal blastn queries locally to identify syntenic regions between the phage sequences. Phage sequences identified with a query coverage of ≥50% were clustered and examined further in Geneious via sequence alignment using Mauve (84).
Statistical analysis.
Two negative binomial distributions with different parameters were used to model the probability mass functions of the number of phage sequences observed in the two samples: patients with and without LUTS. Then, a generalized likelihood ratio test was performed to test the hypothesis that their centrality parameters are equal, assuming they have the same dispersion (85). This means that we were testing whether the overall probability distribution of the number of phage sequences is the same in the two groups. A Wilcoxon test was performed to test for differences in the mean number of phage sequence counts between the control and disorder groups for each bacterial genus.
Phage isolation and characterization.
E. coli isolates from the bladder were retrieved from −80°C storage, streaked on 1.7% LB agar plates, and incubated overnight at 37°C. Individual colonies were selected to inoculate flasks of LB medium and grown overnight with shaking at 120 rpm and 37°C. Cultures were then centrifuged, and the supernatant was filtered using 0.2-μm-pore-size cellulose acetate filters. Pour plates of the supernatant were made using laboratory strains of E. coli, including E. coli C (obtained from C. Burch, University of North Carolina), E. coli B (Ward's, VWR), and E. coli K-12 (ATCC 25404). These plates were prepared as follows: 3 ml of LB soft agar (0.7% agar), 500 μl of the respective E. coli culture (C, B, or K-12), and 100 μl of supernatant were poured atop a 1.7% LB agar plate. Plating was performed with E. coli cultures suspended during exponential growth or stationary phase.
Plaques were identified from the filtrate of the E. coli strain UMB0901 plated on E. coli C plates. The LB soft agar overlay was harvested and resuspended in LB. Phage stock was grown in high titer and triple filtered (as described above) for microscopy. Purified viral lysate was applied to Pioloform-coated copper grids and left to dry at room temperature. Samples were positively stained with 2% (wt/vol) uranyl acetate (86) and observed at 80 kV using a Hitachi H-600 transmission electron microscope (TEM).
Accession number(s).
Raw sequencing reads are available through the NCBI under BioProject no. PRJNA316969 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA316969) and EMBL-EBI under accession no. PRJEB8104 (http://www.ebi.ac.uk/ena/data/view/PRJEB8104). Individual accession numbers for each isolate sequenced are listed in Data Set S1. Assembled genomes can be accessed via the PRJNA316969 BioProject link. The accession numbers for each individual assembled genome are also listed in Data Set S1.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH (grant R01 DK104718 to A.J.W.). T.M.-E. was supported by a Loyola University Chicago Interdisciplinary Research Fellowship. A.G. is supported by the Carbon Research Fellowship at Loyola University Chicago and a CREU fellowship (Computing Research Association).
We thank Linda Brubaker and Jason Shapiro for critical reading of the manuscript and Abdul Zakkar for assistance in annotating CRISPR regions within the assembled genomes. For prior patient recruitment, we acknowledge the Loyola Urinary Education and Research Collaborative (LUEREC), specifically Mary Tulke, Linda Brubaker, Elizabeth Mueller, Cynthia Brincat, Susanne Taege, and Tanaka Dune, and the patients who provided the samples for this study.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00738-17.
REFERENCES
- 1.Wylie KM, Truty RM, Sharpton TJ, Mihindukulasuriya KA, Zhou Y, Gao H, Sodergren E, Weinstock GM, Pollard KS. 2012. Novel bacterial taxa in the human microbiome. PLoS One 7:e35294. doi: 10.1371/journal.pone.0035294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lloyd-Price J, Abu-Ali G, Huttenhower C. 2016. The healthy human microbiome. Genome Med 8:51. doi: 10.1186/s13073-016-0307-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knights D, Lassen KG, Xavier RJ. 2013. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut 62:1505–1510. doi: 10.1136/gutjnl-2012-303954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van de Wijgert JHHM, Borgdorff H, Verhelst R, Crucitti T, Francis S, Verstraelen H, Jespers V. 2014. The vaginal microbiota: what have we learned after a decade of molecular characterization? PLoS One 9:e105998. doi: 10.1371/journal.pone.0105998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brubaker L, Wolfe AJ. 2017. The female urinary microbiota, urinary health and common urinary disorders. Ann Transl Med 5:34. doi: 10.21037/atm.2016.11.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearce MM, Hilt EE, Rosenfeld AB, Zilliox MJ, Thomas-White K, Fok C, Kliethermes S, Schreckenberger PC, Brubaker L, Gai X, Wolfe AJ. 2014. The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. mBio 5:e01283-14. doi: 10.1128/mBio.01283-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sommer F, Anderson JM, Bharti R, Raes J, Rosenstiel P. 2017. The resilience of the intestinal microbiota influences health and disease. Nat Rev Microbiol 15:630–638. doi: 10.1038/nrmicro.2017.58. [DOI] [PubMed] [Google Scholar]
- 8.Weinbauer MG, Rassoulzadegan F. 2004. Are viruses driving microbial diversification and diversity? Environ Microbiol 6:1–11. doi: 10.1046/j.1462-2920.2003.00539.x. [DOI] [PubMed] [Google Scholar]
- 9.Koskella B, Brockhurst MA. 2014. Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol Rev 38:916–931. doi: 10.1111/1574-6976.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lugli GA, Milani C, Turroni F, Tremblay D, Ferrario C, Mancabelli L, Duranti S, Ward DV, Ossiprandi MC, Moineau S, van Sinderen D, Ventura M. 2016. Prophages of the genus Bifidobacterium as modulating agents of the infant gut microbiota. Environ Microbiol 18:2196–2213. doi: 10.1111/1462-2920.13154. [DOI] [PubMed] [Google Scholar]
- 11.Manrique P, Bolduc B, Walk ST, van der Oost J, de Vos WM, Young MJ. 2016. Healthy human gut phageome. Proc Natl Acad Sci U S A 113:10400–10405. doi: 10.1073/pnas.1601060113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edlund A, Santiago-Rodriguez TM, Boehm TK, Pride DT. 2015. Bacteriophage and their potential roles in the human oral cavity. J Oral Microbiol 7:27423. doi: 10.3402/jom.v7.27423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navarro F, Muniesa M. 2017. Phages in the human body. Front Microbiol 8:566. doi: 10.3389/fmicb.2017.00566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manrique P, Dills M, Young MJ. 2017. The human gut phage community and its implications for health and disease. Viruses 9:141. doi: 10.3390/v9060141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwasawa A, Kumamoto Y, Maruta H, Fukushima M, Tsukamoto T, Fujinaga K, Fujisawa Y, Kodama N. 1992. Presence of human papillomavirus 6/11 DNA in condyloma acuminatum of the urinary bladder. Urol Int 48:235–238. doi: 10.1159/000282342. [DOI] [PubMed] [Google Scholar]
- 16.Echavarria M, Forman M, Ticehurst J, Dumler JS, Charache P. 1998. PCR method for detection of adenovirus in urine of healthy and human immunodeficiency virus-infected individuals. J Clin Microbiol 36:3323–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karim RZ, Rose BR, Brammah S, Scolyer RA. 2005. Condylomata acuminata of the urinary bladder with HPV 11. Pathology 37:176–178. doi: 10.1080/00313020500058615. [DOI] [PubMed] [Google Scholar]
- 18.Burián Z, Szabó H, Székely G, Gyurkovits K, Pankovics P, Farkas T, Reuter G. 2011. Detection and follow-up of torque teno midi virus (“small anelloviruses”) in nasopharyngeal aspirates and three other human body fluids in children. Arch Virol 156:1537–1541. doi: 10.1007/s00705-011-1021-0. [DOI] [PubMed] [Google Scholar]
- 19.Hirsch HH, Kardas P, Kranz D, Leboeuf C. 2013. The human JC polyomavirus (JCPyV): virological background and clinical implications. APMIS 121:685–727. doi: 10.1111/apm.12128. [DOI] [PubMed] [Google Scholar]
- 20.Rinaldo CH, Tylden GD, Sharma BN. 2013. The human polyomavirus BK (BKPyV): virological background and clinical implications. APMIS 121:728–745. doi: 10.1111/apm.12134. [DOI] [PubMed] [Google Scholar]
- 21.Assetta B, Atwood WJ. 2017. The biology of JC polyomavirus. Biol Chem 398:839–855. doi: 10.1515/hsz-2016-0345. [DOI] [PubMed] [Google Scholar]
- 22.Brown-Jaque M, Muniesa M, Navarro F. 2016. Bacteriophages in clinical samples can interfere with microbiological diagnostic tools. Sci Rep 6:33000. doi: 10.1038/srep33000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malki K, Sible E, Cooper A, Garretto A, Bruder K, Watkins SC, Putonti C. 2016. Seven bacteriophages isolated from the female urinary microbiota. Genome Announc 4:e01003-16. doi: 10.1128/genomeA.01003-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santiago-Rodriguez TM, Ly M, Bonilla N, Pride DT. 2015. The human urine virome in association with urinary tract infections. Front Microbiol 6:14. doi: 10.3389/fmicb.2015.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rani A, Ranjan R, McGee HS, Metwally A, Hajjiri Z, Brennan DC, Finn PW, Perkins DL. 2016. A diverse virome in kidney transplant patients contains multiple viral subtypes with distinct polymorphisms. Sci Rep 6:33327. doi: 10.1038/srep33327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nanda AM, Thormann K, Frunzke J. 2015. Impact of spontaneous prophage induction on the fitness of bacterial populations and host-microbe interactions. J Bacteriol 197:410–419. doi: 10.1128/JB.02230-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bondy-Denomy J, Qian J, Westra ER, Buckling A, Guttman DS, Davidson AR, Maxwell KL. 2016. Prophages mediate defense against phage infection through diverse mechanisms. ISME J 10:2854–2866. doi: 10.1038/ismej.2016.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dedrick RM, Jacobs-Sera D, Bustamante CAG, Garlena RA, Mavrich TN, Pope WH, Reyes JC, Russell DA, Adair T, Alvey R, Bonilla JA, Bricker JS, Brown BR, Byrnes D, Cresawn SG, Davis WB, Dickson LA, Edgington NP, Findley AM, Golebiewska U, Grose JH, Hayes CF, Hughes LE, Hutchison KW, Isern S, Johnson AA, Kenna MA, Klyczek KK, Mageeney CM, Michael SF, Molloy SD, Montgomery MT, Neitzel J, Page ST, Pizzorno MC, Poxleitner MK, Rinehart CA, Robinson CJ, Rubin MR, Teyim JN, Vazquez E, Ware VC, Washington J, Hatfull GF. 2017. Prophage-mediated defence against viral attack and viral counter-defence. Nat Microbiol 2:16251. doi: 10.1038/nmicrobiol.2016.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barondess JJ, Beckwith J. 1990. A bacterial virulence determinant encoded by lysogenic coliphage lambda. Nature 346:871–874. doi: 10.1038/346871a0. [DOI] [PubMed] [Google Scholar]
- 30.Wagner PL, Waldor MK. 2002. Bacteriophage control of bacterial virulence. Infect Immun 70:3985–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Canchaya C, Proux C, Fournous G, Bruttin A, Brussow H. 2003. Prophage genomics. Microbiol Mol Biol Rev 67:238–276. doi: 10.1128/MMBR.67.2.238-276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDonough MA, Butterton JR. 1999. Spontaneous tandem amplification and deletion of the Shiga toxin operon in Shigella dysenteriae 1. Mol Microbiol 34:1058–1069. doi: 10.1046/j.1365-2958.1999.01669.x. [DOI] [PubMed] [Google Scholar]
- 33.Malki K, Shapiro JW, Price TK, Hilt EE, Thomas-White K, Sircar T, Rosenfeld AB, Kuffel G, Zilliox MJ, Wolfe AJ, Putonti C. 2016. Genomes of Gardnerella strains reveal an abundance of prophages within the bladder microbiome. PLoS One 11:e0166757. doi: 10.1371/journal.pone.0166757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Price TK, Mehrtash A, Kalesinskas L, Malki K, Hilt EE, Putonti C, Wolfe AJ. 2016. Genome sequences and annotation of two urinary isolates of E. coli. Stand Genomic Sci 11:79. doi: 10.1186/s40793-016-0202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khasriya R, Sathiananthamoorthy S, Ismail S, Kelsey M, Wilson M, Rohn JL, Malone-Lee J. 2013. Spectrum of bacterial colonization associated with urothelial cells from patients with chronic lower urinary tract symptoms. J Clin Microbiol 51:2054–2062. doi: 10.1128/JCM.03314-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hilt EE, McKinley K, Pearce MM, Rosenfeld AB, Zilliox MJ, Mueller ER, Brubaker L, Gai X, Wolfe AJ, Schreckenberger PC. 2014. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J Clin Microbiol 52:871–876. doi: 10.1128/JCM.02876-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Price TK, Dune T, Hilt EE, Thomas-White KJ, Kliethermes S, Brincat C, Brubaker L, Wolfe AJ, Mueller ER, Schreckenberger PC. 2016. The clinical urine culture: enhanced techniques improve detection of clinically relevant microorganisms. J Clin Microbiol 54:1216–1222. doi: 10.1128/JCM.00044-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sender R, Fuchs S, Milo R. 2016. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 14:e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paul JH. 2008. Prophages in marine bacteria: dangerous molecular time bombs or the key to survival in the seas? ISME J 2:579–589. doi: 10.1038/ismej.2008.35. [DOI] [PubMed] [Google Scholar]
- 40.Maurice CF, Bouvier T, Comte J, Guillemette F, Del Giorgio PA. 2010. Seasonal variations of phage life strategies and bacterial physiological states in three northern temperate lakes. Environ Microbiol 12:628–641. doi: 10.1111/j.1462-2920.2009.02103.x. [DOI] [PubMed] [Google Scholar]
- 41.Evans C, Brussaard CPD. 2012. Regional variation in lytic and lysogenic viral infection in the Southern Ocean and its contribution to biogeochemical cycling. Appl Environ Microbiol 78:6741–6748. doi: 10.1128/AEM.01388-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Payet JP, Suttle CA. 2013. To kill or not to kill: the balance between lytic and lysogenic viral infection is driven by trophic status. Limnol Oceanogr 58:465–474. doi: 10.4319/lo.2013.58.2.0465. [DOI] [Google Scholar]
- 43.Roux S, Enault F, Hurwitz BL, Sullivan MB. 2015. VirSorter: mining viral signal from microbial genomic data. PeerJ 3:e985. doi: 10.7717/peerj.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. 2014. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zurfluh K, Tasara T, Stephan R. 2016. Full-genome sequence of Escherichia coli K-15KW01, a uropathogenic E. coli B2 sequence type 127 isolate harboring a chromosomally carried blaCTX-M-15 gene. Genome Announc 4:e00927-16. doi: 10.1128/genomeA.00927-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Field AK, Naylor HB. 1962. Induction of lysogenic Micrococcus lysodeikticus. J Bacteriol 84:1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lovett PS, Shockman GD. 1970. Interaction of bacteriophage N1 with cell walls of Micrococcus lysodeikticus. J Virol 6:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sozzi T, Maret R, Cerise L. 1973. Isolation and some characteristics of two Micrococcus phages from Italian salami, type Varzi. Arch Mikrobiol 92:313–320. doi: 10.1007/BF00409284. [DOI] [PubMed] [Google Scholar]
- 49.Casjens S. 2003. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol 49:277–300. doi: 10.1046/j.1365-2958.2003.03580.x. [DOI] [PubMed] [Google Scholar]
- 50.Touchon M, Bernheim A, Rocha EP. 2016. Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J 10:2744–2754. doi: 10.1038/ismej.2016.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pearce MM, Zilliox MJ, Rosenfeld AB, Thomas-White KJ, Richter HE, Nager CW, Visco AG, Nygaard IE, Barber MD, Schaffer J, Moalli P, Sung VW, Smith AL, Rogers R, Nolen TL, Wallace D, Meikle SF, Gai X, Wolfe AJ, Brubaker L, Pelvic Floor Disorders Network . 2015. The female urinary microbiome in urgency urinary incontinence. Am J Obstet Gynecol 213:347.e1–347.e11. doi: 10.1016/j.ajog.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karstens L, Asquith M, Davin S, Stauffer P, Fair D, Gregory WT, Rosenbaum JT, McWeeney SK, Nardos R. 2016. Does the urinary microbiome play a role in urgency urinary incontinence and its severity? Front Cell Infect Microbiol 6:78. doi: 10.3389/fcimb.2016.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomas-White K, Brady M, Wolfe AJ, Mueller ER. 2016. The bladder is not sterile: history and current discoveries on the urinary microbiome. Curr Bladder Dysfunct Rep 11:18–24. doi: 10.1007/s11884-016-0345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas-White KJ, Hilt EE, Fok C, Pearce MM, Mueller ER, Kliethermes S, Jacobs K, Zilliox MJ, Brincat C, Price TK, Kuffel G, Schreckenberger P, Gai X, Brubaker L, Wolfe AJ. 2016. Incontinence medication response relates to the female urinary microbiota. Int Urogynecol J 27:723–733. doi: 10.1007/s00192-015-2847-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nunn KL, Forney LJ. 2016. Unraveling the dynamics of the human vaginal microbiome. Yale J Biol Med 89:331–337. [PMC free article] [PubMed] [Google Scholar]
- 56.Spurbeck RR, Arvidson CG. 2008. Inhibition of Neisseria gonorrhoeae epithelial cell interactions by vaginal Lactobacillus species. Infect Immun 76:3124–3130. doi: 10.1128/IAI.00101-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marrazzo JM. 2011. Interpreting the epidemiology and natural history of bacterial vaginosis: are we still confused? Anaerobe 17:186–190. doi: 10.1016/j.anaerobe.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gorgos LM, Sycuro LK, Srinivasan S, Fiedler TL, Morgan MT, Balkus JE, McClelland RS, Fredricks DN, Marrazzo JM. 2015. Relationship of specific bacteria in the cervical and vaginal microbiotas with cervicitis. Sex Transm Dis 42:475–481. doi: 10.1097/OLQ.0000000000000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kiliç AO, Pavlova SI, Alpay S, Kiliç SS, Tao L. 2001. Comparative study of vaginal Lactobacillus phages isolated from women in the United States and Turkey: prevalence, morphology, host range, and DNA homology. Clin Diagn Lab Immunol 8:31–39. doi: 10.1128/CDLI.8.1.31-39.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martín R, Escobedo S, Suárez JE. 2010. Induction, structural characterization, and genome sequence of Lv1, a prophage from a human vaginal Lactobacillus jensenii strain. Int Microbiol 13:113–121. [DOI] [PubMed] [Google Scholar]
- 61.Damelin LH, Paximadis M, Mavri-Damelin D, Birkhead M, Lewis DA, Tiemessen CT. 2011. Identification of predominant culturable vaginal Lactobacillus species and associated bacteriophages from women with and without vaginal discharge syndrome in South Africa. J Med Microbiol 60:180–183. doi: 10.1099/jmm.0.024463-0. [DOI] [PubMed] [Google Scholar]
- 62.Curtiss N, Balachandran A, Krska L, Peppiatt-Wildman C, Wildman S, Duckett J. 2017. A case controlled study examining the bladder microbiome in women with overactive bladder (OAB) and healthy controls. Eur J Obstet Gynecol Reprod Biol 214:31–35. doi: 10.1016/j.ejogrb.2017.04.040. [DOI] [PubMed] [Google Scholar]
- 63.Pérez-Brocal V, García-López R, Vázquez-Castellanos JF, Nos P, Beltrán B, Latorre A, Moya A. 2013. Study of the viral and microbial communities associated with Crohn's disease: a metagenomic approach. Clin Transl Gastroenterol 4:e36. doi: 10.1038/ctg.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, Kambal A, Monaco CL, Zhao G, Fleshner P, Stappenbeck TS, McGovern DP, Keshavarzian A, Mutlu EA, Sauk J, Gevers D, Xavier RJ, Wang D, Parkes M, Virgin HW. 2015. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160:447–460. doi: 10.1016/j.cell.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tetz G, Tetz V. 2016. Bacteriophage infections of microbiota can lead to leaky gut in an experimental rodent model. Gut Pathog 8:33. doi: 10.1186/s13099-016-0109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Turovskiy Y, Sutyak Noll K, Chikindas ML. 2011. The aetiology of bacterial vaginosis: aetiology of bacterial vaginosis. J Appl Microbiol 110:1105–1128. doi: 10.1111/j.1365-2672.2011.04977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ross A, Ward S, Hyman P. 2016. More is better: selecting for broad host range bacteriophages. Front Microbiol 7:1352. doi: 10.3389/fmicb.2016.01352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Malki K, Kula A, Bruder K, Sible E, Hatzopoulos T, Steidel S, Watkins SC, Putonti C. 2015. Bacteriophages isolated from Lake Michigan demonstrate broad host-range across several bacterial phyla. Virol J 12:164. doi: 10.1186/s12985-015-0395-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peters DL, Lynch KH, Stothard P, Dennis JJ. 2015. The isolation and characterization of two Stenotrophomonas maltophilia bacteriophages capable of cross-taxonomic order infectivity. BMC Genomics 16:664. doi: 10.1186/s12864-015-1848-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paez-Espino D, Eloe-Fadrosh EA, Pavlopoulos GA, Thomas AD, Huntemann M, Mikhailova N, Rubin E, Ivanova NN, Kyrpides NC. 2016. Uncovering Earth's virome. Nature 536:425–430. doi: 10.1038/nature19094. [DOI] [PubMed] [Google Scholar]
- 71.Modi SR, Lee HH, Spina CS, Collins JJ. 2013. Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature 499:219–222. doi: 10.1038/nature12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Green MR, Sambrook J. 2012. Molecular cloning: a laboratory manual, 4th ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 73.Page AJ, De Silva N, Hunt M, Quail MA, Parkhill J, Harris SR, Otto TD, Keane JA. 2016. Robust high-throughput prokaryote de novo assembly and improvement pipeline for Illumina data. Microb Genom 2:e000083. doi: 10.1099/mgen.0.000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gladman S, Seemann T. 2017. VelvetOptimiser. https://github.com/tseemann/VelvetOptimiser.
- 75.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. 2011. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 77.Nadalin F, Vezzi F, Policriti A. 2013. GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13(Suppl 14):S8. doi: 10.1186/1471-2105-14-S14-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bushnell B. 2017. BBTools. https://jgi.doe.gov/data-and-tools/bbtools/.
- 79.Lagesesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jordan GE, Piel WH. 2008. PhyloWidget: web-based visualizations for the tree of life. Bioinformatics 24:1641–1642. doi: 10.1093/bioinformatics/btn235. [DOI] [PubMed] [Google Scholar]
- 82.Angly F, Skennerton C. 2015. MinCED. https://github.com/ctSkennerton/minced.
- 83.O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A, Badretdin A, Bao Y, Blinkova O, Brover V, Chetvernin V, Choi J, Cox E, Ermolaeva O, Farrell CM, Goldfarb T, Gupta T, Haft D, Hatcher E, Hlavina W, Joardar VS, Kodali VK, Li W, Maglott D, Masterson P, McGarvey KM, Murphy MR, O'Neill K, Pujar S, Rangwala SH, Rausch D, Riddick LD, Schoch C, Shkeda A, Storz SS, Sun H, Thibaud-Nissen F, Tolstoy I, Tully RE, Vatsan AR, Wallin C, Webb D, Wu W, Landrumy MJ, Kimchi A, et al. 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44:D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Darling ACE, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aban IB, Cutter GR, Mavinga N. 2009. Inferences and power analysis concerning two negative binomial distributions with an application to MRI lesion counts data. Comput Stat Data Anal 53:820–833. doi: 10.1016/j.csda.2008.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ackermann HW. 2009. Basic phage electron microscopy. Methods Mol Biol 501:113–126. doi: 10.1007/978-1-60327-164-6_12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.