

SUMMARY
To identify novel targets for acute myeloid leukemia (AML) therapy, we performed genome-wide CRISPR-Cas9 screening using AML cell lines, followed by a second screen in vivo. Here, we show that the mRNA decapping enzyme scavenger (DCPS) gene is essential for AML cell survival. The DCPS enzyme interacted with components of pre-mRNA metabolic pathways, including spliceosomes, as revealed by mass spectrometry. RG3039, a DCPS inhibitor originally developed to treat spinal muscular atrophy, exhibited anti-leukemic activity via inducing pre-mRNA mis-splicing. Humans harboring germline bi-allelic DCPS loss-of-function mutations do not exhibit aberrant hematologic phenotypes, indicating that DCPS is dispensable for human hematopoiesis. Our findings shed light on a pre-mRNA metabolic pathway and identify DCPS as a target for AML therapy.
eTOC
Yamauchi et al. perform in vitro and in vivo CRISPR-Cas9 genetic screening of p53 WT AML to identify potential therapeutic targets. They find that AML relies on the DCPS decapping enzyme, and a DCPS inhibitor shows anti-leukemia activity in tumor models without impacting normal hematopoiesis.
INTRODUCTION
Acute myeloid leukemia (AML) is a devastating disease with a long-term survival rate of less than 30% (Ferrara and Schiffer, 2013). Recent progress has been made to define its mechanisms, and sequencing studies now provide a near-complete picture of the AML genome (Welch et al., 2012). Nonetheless, to devise urgently needed therapies, functional studies are necessary to assess the significance of AML-associated mutations (Boehm and Hahn, 2011; Garraway and Lander, 2013; Lawrence et al., 2014).
Successful application of the S. pyogenes-derived type 2 clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system for genome editing is transforming the landscape of genetic research in many organisms (Cho et al., 2013; Cong et al., 2013; Jinek et al., 2012; Mali et al., 2013). Furthermore, given its high efficiency and flexibility, the system is ideal for use in genome-wide recessive genetic screens and proof-of-principle studies using cancer cell lines demonstrate the potential of this technology to identify genes essential for cancer cell survival (Koike-Yusa et al., 2014; Shalem et al., 2014; Shi et al., 2015; Wang et al., 2014). Since AML cells generally exhibit low mutational burdens (Alexandrov et al., 2013) and mutation of the TP53 tumor suppressor gene has a significant impact on AML prognosis (Zhang et al., 2016), it is critical to perform a screen employing AML lines whose genetic background, namely TP53 status, are well-defined.
RESULTS
A genome-wide CRISPR-Cas9 screen identifies DCPS as an AML essential gene
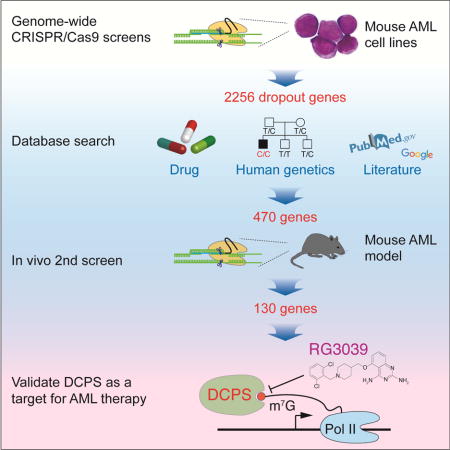
To establish AML cell lines with a relatively “clean” genetic background, we first generated AML in mice by transducing either the CALM/AF10 or MLL/AF9 leukemia oncogene into mouse bone marrow hematopoietic stem cells (HSCs) and then transferred cells to sub-lethally irradiated recipients. Primary AML cells were harvested 3-6 months later, serially transplanted three times, and then cultured in the presence of cytokines. The resultant two independent lines were then transduced with the Cas9 nuclease (Figure 1A). Cells of both lines exhibited normal karyotypes (Figure S1A). To perform the genome-wide CRISPR-Cas9 screen, we used the mouse lentivirus-based GeCKO v2 library, which contains 130,209 single-guide RNAs (sgRNAs) targeting 20,611 protein-coding genes and 1,175 miRNAs (Sanjana et al., 2014; Shalem et al., 2014). Cas9-expressing AML cells of both lines were transduced with the library (day 0) and treated with puromycin on day 1. We passaged the cells 5-6 times over a 16-day incubation period, while maintaining at least 500 cells per sgRNA throughout (Figure S1B). Genomic DNA was isolated from cells on days 3 and 18 and deep-sequenced to measure read counts of each sgRNA. Changes in abundance of each sgRNA were assessed using the MAGeCK program (Li et al., 2014; Shalem et al., 2014). We obtained over 400 million mapped reads per sample, suggesting that at least 600 cells were transduced with each sgRNA (Figure S1C). Strikingly, sgRNAs targeting Trp53 were among the most enriched after a 16-day incubation of both AML lines, indicating intact TP53 activity in both lines (Figures 1B and 1C). We identified nearly 1,700 dropout genes in each line at a false discovery rate (FDR) of 0.25, with significant overlap between the lines (Figure 1D, Figure S1D and Table S1). As expected, genes encoding components of basal cellular machineries were highly enriched in dropout genes (Figure S1E). Dropout genes were abundantly expressed in primary mouse CALM/AF10 or MLL/AF9 leukemia cells, an observation strongly suggesting those genes are functional in AML cells and that sgRNA off-target effects are negligible (Figure S2A). Overall, we identified 2256 dropout genes at a FDR of 0.25 using two AML lines (Figure 1D). sgRNAs targeting the genes with a well-defined function in leukemogenesis, among them, Kras, Nras, Bcl2l1, Jak1, Jak2, Brd4 and Brd9, were significantly depleted, confirming quality and efficiency of our dropout screens (Figure S2B).
Figure 1. Genome-wide CRISPR-Cas9 screens identify Dcps as an AML essential gene.
(A) Generation of Cas9-expressing mouse AML cell lines. Two mouse lines expressing Cas9 endonuclease (CALM/AF10-Cas9 and MLL/AF9-Cas9) were used for screens. (B) Genes significantly enriched or dropped-out after a 16-day incubation were identified using the MAGeCK program (Li et al., 2014; Shalem et al., 2014). Representative results (GeCKO library B screen in MLL/AF9 cells) of the enrichment screen are shown. A modified robust ranking aggregation (RRA) algorithm was used to rank sgRNAs based on p values (Li et al., 2014; Shalem et al., 2014). (C) Graphs show read counts of individual sgRNAs targeting Trp53 before and after a 16-day incubation. P values were calculated using a Wilcoxon matched-pairs signed rank test. (D) Experimental schema of in vitro and in vivo CRISPR-Cas9 screens. Overall, we identified 130 AML essential genes for further evaluation. (E) A representative RRA score plot showing top dropout genes (GeCKO library B screen in CALM/AF10 cells). (F) Read counts of sgRNAs targeting Dcps significantly decreased after a 16-day incubation in both AML lines. P values were calculated using a Wilcoxon matched-pairs signed rank test. (G) Read counts of single sgRNAs targeting Dcps before and 3 weeks after leukemia transfer are shown. Read counts of 7 sgRNAs are shown, as one out of 8 sgRNA (#14) was not detected on day 0. (H) Domain-saturating DCPS mutagenesis. All NGG-restricted sgRNAs (n=154) were identified within Dcps coding exons. The pool was transduced into CALM/AF10-Cas9 cells and a dropout screen was performed. Read counts from final and initial time points were normalized to non-targeting guides and log2 fold-change in guide abundance was calculated. Guides were then mapped to protein by mapping double-stranded break sites to a codon. (I) Dropout scores (log2 fold-change) for each amino acid were mapped onto a structure publicly available in the Protein Data Bank (PDB ID: 1vlr). Since DCPS forms a homodimer, one monomer is depicted in pink for visual clarity, and scores are divided into bins of 1 log2 fold-change. Red asterisk indicates HIT sequence, where the 7-methylhuaninosine (m7G) cap of mRNA binds. See also Figures S1, S2 and Tables S1, S2 and S3.
To identify potentially actionable targets, we selected genes meeting the following criteria: 1) they encoded a protein with an available inhibitor and/or 2) their germline mutation loss-of-function phenotype was relatively moderate based on the literature or the human exome-sequencing database (Lek et al., 2016; Lim et al., 2014; Narasimhan et al., 2016; Saleheen et al., 2017; Sulem et al., 2015). We excluded genes encoding components of basal cellular machineries (e.g. histones, ribosomal proteins, or polymerases), as targeting these factors would likely be deleterious to normal tissues (Figure 1D). Since AML lines used in initial screens were maintained in the presence of cytokines, it was possible that some dropout genes encoded essential effectors of cytokine signaling, but were dispensable for activity of primary AML cells. To address this issue, we performed a second in vivo validation screen targeting 470 genes (Figure 1D and Table S2). Cas9-expressing MLL/AF9 cells were transduced with a library containing 8 sgRNAs per gene plus 100 non-targeting sgRNAs and then transferred into sub-lethally irradiated recipients (Figure S2C). DNA samples from pre- and post-transplant were deep-sequenced with sgRNA abundance determined using MAGeCK software (Figure S2D). Nearly one fourth of the 470 genes tested in the second screen were dispensable in vivo and another one fourth exhibited only moderate effects (not shown). Non-targeting sgRNAs showed variable effects (Figure S2E). Overall, we identified 130 genes necessary for AML cell survival both in vitro and in vivo for further evaluation (Figure 1D).
Among genes significantly depleted in our primary screen was the mRNA decapping enzyme scavenger (Dcps), which encodes a mRNA 5′ cap binding enzyme implicated in mRNA decay (Milac et al., 2014) (Figure 1E). Read counts for each Dcps-targeted sgRNA significantly decreased in both AML lines over the 16-day incubation period (Figure 1F), which also demonstrated significant dropout in the second screen (Figure 1G). DCPS protein harbors an N-terminal domain that shares structural homology to yeast mRNA export factor Mex67 and a C-terminal domain that contains a histidine triad (HIT) sequence (His274, His276, and His278) in which His276 is critical for decapping activity (Gu et al., 2004; Han et al., 2005). To determine DCPS amino acid residues necessary for AML cell survival, we performed a negative selection CRISPR-Cas9 mutagenesis scan (Shi et al., 2015) of all Dcps coding exons using the Ctps (cytidine 5′-triphosphate synthase) gene as control (Table S3). sgRNAs targeting either the N- or C-terminal DCPS domains, namely those targeting aa 230-240 or the HIT sequence, respectively, were significantly decreased after the 16-day incubation, strongly suggesting a critical role for those residues in AML survival (Figure 1H and 1I). In contrast, 238 sgRNAs targeting all Ctps coding exons were largely dispensable (Figure S2F).
The DCPS inhibitor RG3039 slows proliferation and induces AML cell differentiation
We next asked whether DCPS inactivation inhibits proliferation of human AML cell lines. To this end, we generated lentivirus-based, GFP-tagged shRNA targeting DCPS and evaluated the proportions of GFP-positive (shRNA+) and -negative (shRNA-) cells by FACS over time (Figure 2A). Efficient DCPS knockdown in the GFP-positive fraction was confirmed by Western blot (Figure 2B). Cells that expressed shRNA-DCPS, but not those transduced with scrambled shRNA, demonstrated a proliferative disadvantage as compared to non-transduced cells, indicating a toxic effect of shRNA-DCPS in AML cells (Figure 2A). DCPS knockdown induced cell cycle arrest (Figure 2C) and apoptosis (Figure 2D). Notably, DCPS-knockdown MOLM-13 cells underwent myelo/monocytic differentiation, as evidenced by FACS analysis and morphological examination (Figure 2E and 2F).
Figure 2. Treatment of AML cells with the DCPS inhibitor RG3039 induces cell-cycle arrest, apoptosis and differentiation.
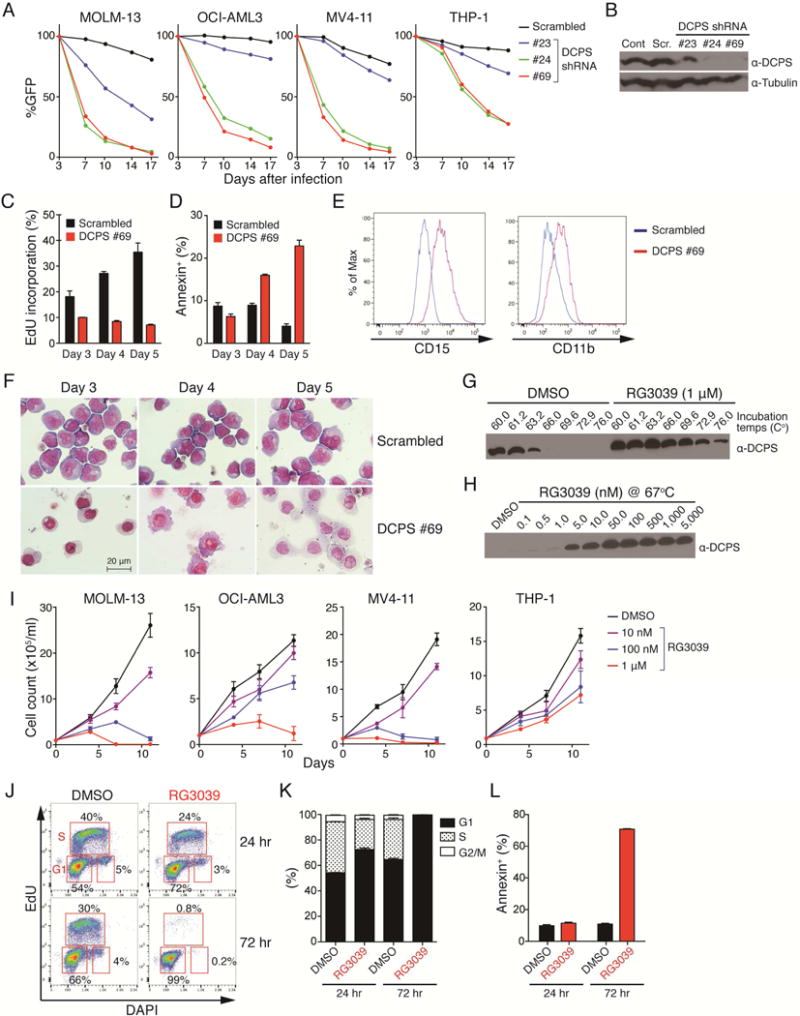
(A) Human AML cell lines were transduced with a lentivirus vector encoding an shRNA and GFP cassette, and the fraction of GFP-positive cells was measured at indicated times by FACS. At each time point that proportion was normalized to the number of GFP-positive cells present at day 3 (two days after transduction). Scrambled-shRNA served as control. The data is from one experiment. (B) shRNA knockdown efficiencies were assessed by Western blot. Cont: control sample without transduction. Scr: scrambled shRNA. (C) EdU (5-ethynyl-2′-deoxyuridine) incorporation assay. MOLM-13 cells transduced with either scrambled or DCPS shRNA clone#69 were pulsed with EdU and proportions of EdU-positive cells were assessed by FACS on indicated days after transduction. (D) Proportions of apoptotic cells were assessed by Annexin V staining. (E) Expression levels of surface CD15 and CD11b in MOLM-13 cells were assessed by FACS following shRNA-mediated DCPS knockdown (day 5). (F) May-Giemsa stain of cytospin preparations showing monocytic differentiation of MOLM-13 cells upon DCPS knockdown. (G) Representative result of a cellular thermal shift assay (CETSA). (H) DCPS protein is resistant to heat denaturation in the presence of RG3039 dose-dependently. (I) Growth curves were generated for AML lines treated with RG3039. Data are represented as means ± SD. (J) MOLM-13 cells were pulsed with EdU and treated with either DMSO or RG3039. Proportions of EdU-positive cells and their DNA contents were analyzed by FACS 24 hr and 72 hr after RG3039 treatment. (K) Bar graphs show proportions of cells at each stage of the cell cycle after DMSO or RG3039 treatment. (L) Proportions of apoptotic cells were assessed by Annexin V stain following control DMSO or RG3039 treatment. Data (n=3) are represented as means ± SD. See also Figure S3.
The DCPS inhibitor RG3039, an orally-active quinazoline derivative, was originally developed to treat spinal muscular atrophy (SMA) (Jarecki et al., 2005). We examined RG3039 binding to DCPS protein in AML cells via a cellular thermal shift assay (CETSA) (Martinez Molina et al., 2013; Xu et al., 2016). To do so, we treated MOLM-13 cells with RG3039 or control DMSO vehicle and then raised resulting cell lysates to various temperatures in order to test whether DCPS protein is protected against heat-induced denaturation in the presence of RG3039. Soluble fractions of protein lysates were collected and subjected to Western blot. While DCPS protein was not detected in lysates of DMSO control cells subjected to higher temperature, DCPS protein remained soluble in lysates of RG3039-treated cells dose-dependently and thus was detectable by Western blot (Figures 2G and 2H). Time course CETSA analysis showed that engagement of ligand with DCPS occurred immediately after RG3039 administration and then gradually decreased over 24 hours (not shown).
We next assessed anti-leukemia effects of RG3039 by treating 4 human AML lines with RG3039 for 11 days and generating growth curves. That analysis showed that RG3039 had dose-dependent anti-proliferative effects, although compound sensitivity varied among lines (Figure 2I). We also assessed the effects of RG3039 on the cell cycle via a 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay. While EdU was efficiently incorporated into DMSO-treated control cells, EdU incorporation was significantly decreased in RG3039-treated cells, and the proportion of cells in S phase was extremely low in treated versus untreated cells (Figures 2J and 2K). RG3039-treated cells also underwent apoptosis after 72 hr of drug treatment, as revealed by Annexin V stain (Figure 2L). As observed in DCPS-knockdown AML cells, RG3039 treatment induced differentiation of human (Figures S3A and S3B) and mouse (Figures S3C and S3D) AML. Importantly, observed anti-leukemic effects of RG3039 were caused via TP53-independent mechanisms, as CALM/AF10 and MLL/AF9 AML cells whose TP53 function was abrogated by CRISPR-Cas9-mediated Trp53 knockout were also sensitive to the compound (Figure S3E). Preferential DCPS dependency of leukemia cells was also validated using the DEMETER, an siRNA-based gene dependency database (Tsherniak et al., 2017). Strikingly, leukemia cells (n=28) were significantly more dependent on DCPS than were cells from non-hematological tumors (n=454) (Figure S3F). Of note, the leukemia cell line least dependent on DCPS was THP-1, which is relatively insensitive to shRNA-mediated DCPS knockdown or RG3039 treatment (Figures 2A and 2I).
DCPS binds to the nuclear RNA processing machinery
Since DCPS protein was predominantly nuclear in human primary AML cells (Figure S4A), we hypothesized that DCPS primarily functions in that compartment rather than in the cytoplasmic mRNA decay pathway. To search for nuclear proteins potentially interacting with DCPS, we undertook immunoprecipitation (IP) with an anti-DCPS antibody of lysates of MOLM-13 cells followed by mass spectrometry analysis. Analysis was done in triplicate using non-specific immunoglobulins as control. Western blot analysis of IP’d proteins indicated successful pull-down of endogenous DCPS protein in each experiment (Figure 3A). Among highly significant interactors, we identified components of pre-mRNA processing machineries including spliceosomes, the transcription-export complex (TREX), and the nuclear pore complex (NUP), as well as complexes functioning in pre-rRNA processing (Figures 3B, 3C and Table S4). DCPS also bound to NuRD (Nucleosome Remodeling Deacetylase) subunits, suggesting a potential function in transcriptional regulation. Importantly, sgRNAs targeting genes encoding DCPS-interacting proteins were generally depleted in our CRISPR-Cas9 screens, indicating that binding events are functionally significant (Figures 3D). The cap-binding complex (CBC), which consists of CBP20 (also known as NCBP2) and CBP80 (NCBP1), reportedly binds the 5′ cap of an mRNA in the nucleus and functions in RNA export and pre-mRNA splicing (Izaurralde et al., 1995; Izaurralde et al., 1994; Visa et al., 1996). We therefore used publicly available datasets to ask whether DCPS interacts with proteins that also participate in the CBC complex (Andersen et al., 2013). We found that some DCPS-interacting proteins are common to the CBC complex, including proteins also found in the TREX and NUP complex (Figure S4B).
Figure 3. DCPS protein interacts with components of pre-mRNA metabolic pathways.
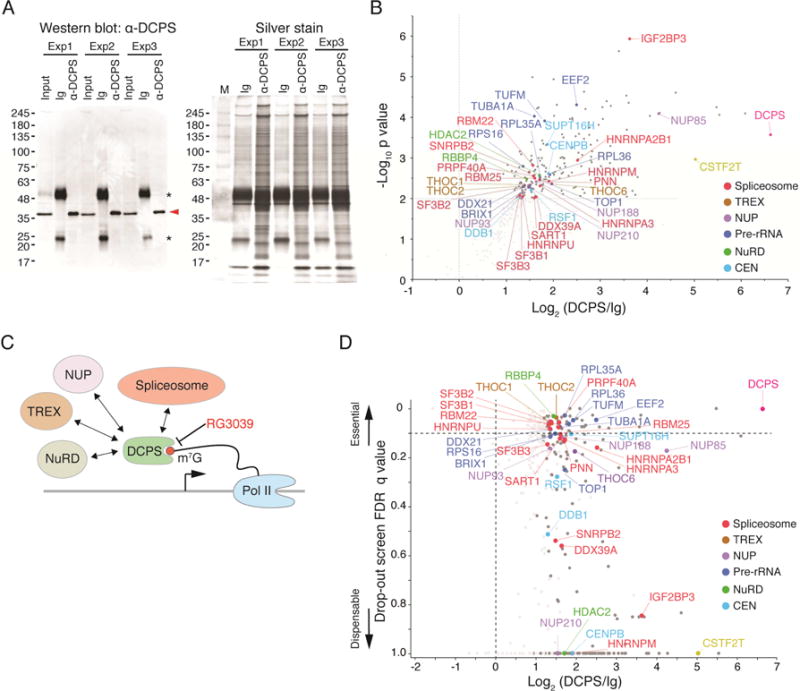
(A) Immunoprecipitation (IP) of endogenous DCPS protein was performed in triplicate. DCPS was detected (red arrowhead) by Western blot when IP was performed with anti-DCPS antibody but not with control immunoglobulins (Ig), indicative of specificity (left). Asterisks denote non-specific immunoglobulin light and heavy chain signals. Protein lysates used for mass spectrometry were analyzed by SDS-PAGE and silver stained (right). (B) Volcano plot displaying results of triplicate IP/mass spectrometry experiments. Y-axis shows negative log10 p values, which represent reproducibility of events among three independent experiments; x-axis indicates log2 ratio of normalized protein abundance between anti-DCPS antibody and control Ig IPs. (C) Proposed model for nuclear DCPS function. DCPS complexes with components of pre-mRNA processing pathways including, spliceosomes, the transcription-export complex (TREX) and the nuclear pore complex (NUP). (D) Genes encoding DCPS-interacting proteins were found essential for AML survival in CRISPR-Cas9 screens. The Y-axis shows the lowest FDR q-value of each gene among 4 dropout screens (GeCKO library A and B screens in CALM/AF10 or MLL/AF9 cells); the X-axis indicates log2 ratio of normalized protein abundance between anti-DCPS antibody and control Ig IPs. See also Figure S4 and Table S4.
DCPS inactivation causes pre-mRNA mis-splicing and induces a type I interferon response in AML cells
Given that DCPS binds the splicing machinery, we asked whether DCPS inhibition would impair pre-mRNA splicing in AML cells. To assess this possibility, we treated AML cells or granulocyte macrophage progenitors (GMPs), the normal counterpart of AML cells, with RG3039 and performed RNA-seq to determine potential transcriptome-wide splicing changes (Figure 4A). Of note, CETSA analysis showed comparable binding affinity between RG3039 and DCPS protein in cells, regardless of cell type (Figure S4C), suggesting that RG3039 inhibits DCPS enzymatic activity comparably in GMPs and AML cells. RNA-seq data were analyzed using a bioinformatic pipeline enabling quantification of exon-exon junctions without predetermining alternative splicing models or annotating splice junctions as described (Darman et al., 2015). We observed 493 and 704 mis-splicing events in AML cells at 6 hr and 10 hr after RG3039 treatment, respectively, while there were significantly fewer in GMPs (294 and 416 events at 6 hr and 10 hr, respectively) (Figure 4B and Table S5). Alternative 5′ splice site (ss) selection was most frequently observed in both AML cells and GMPs (Figure 4B), although mis-spliced genes differed between the two cell types (Figure 4C). Mis-spliced mRNAs containing premature termination codons undergo nonsense-mediated mRNA decay (NMD), preventing production of aberrant proteins (Lykke-Andersen and Jensen, 2015). Bioinformatic prediction analysis revealed that approximately 40% of genes aberrantly-spliced at either alternative 3′ or 5′ splice sites were NMD-sensitive in AML cells, but this outcome was not evident in GMPs (Figure 4D). Transcripts predicted to undergo NMD in AML cells were, in fact, significantly down-regulated after 10 hours of RG3039 treatment (Figure 4D). Notably, most aberrant splicing events observed in AML cells occurred at the first exon (or the first intron, in cases of intron retention) (Figure 4E).
Figure 4. DCPS inhibition impedes pre-mRNA processing pathways in AML cells.
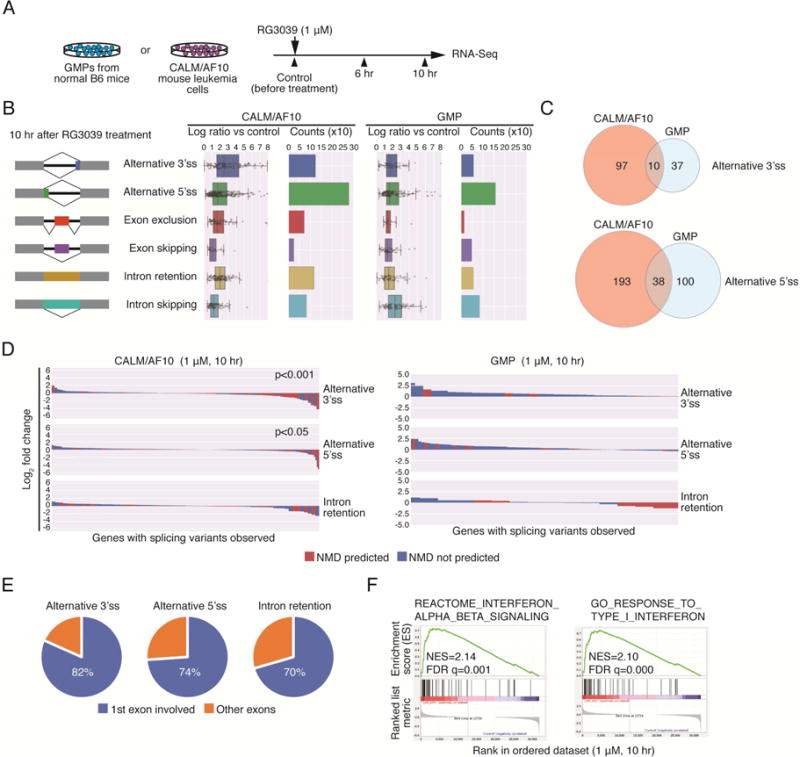
(A) RNA-Seq analysis of effects of DCPS inhibition on pre-mRNA splicing and transcriptome activity. RNA samples were prepared prior to and 6 hr and 10 hr after RG3039 treatment of CALM/AF10 mouse leukemia cells or GMPs. (B) Event counts and ratios relative to control of indicated splicing patterns (10 hr after RG3039 treatment) are shown. Data are represented as means ± SD. (C) Venn diagrams show overlap of mis-spliced genes between CALM/AF10 AML cells and GMPs. (D) Waterfall plots indicate NMD-sensitivities and expression changes of genes aberrantly-spliced following RG3039 treatment. (E) Locations of Mis-splicing events. (F) GSEA analysis of RG3039-treated AML cells. See also Table S5.
Since DCPS associates with several pre-mRNA processing machineries, its inhibition could significantly impact pre-mRNA metabolism in ways not limited to pre-mRNA splicing. In fact, a genome-wide CRISPR screen, in which RG3039 was added to the medium of Cas9-expressing MLL/AF9 cells from days 10 to 18, failed to identify sgRNAs uniquely enriched in the presence of RG3039 (Table S1), suggesting that RG3039 exerts anti-leukemic activity via multiple mechanisms rather than a single pathway. To assess overall effects of DCPS inhibition on the leukemia transcriptome, we performed Gene Set Enrichment Analysis (GSEA) and functional annotation clustering using the Metascape program (http://metascape.org/). Both analyses identified gene signatures representing a type-I interferon response in RG3039-treated AML cells but not in GMPs (Figures 4F, S4D and Table S5). This activity may account for anti-leukemic effects observed following DCPS inhibition. Of note, genes aberrantly-spliced in AML cells were not necessarily enriched for factors functioning in IFN signaling (Figure S4E).
DCPS is dispensable for steady-state hematopoiesis in mice and humans
To determine effects of RG3039 on normal mouse hematopoiesis in vivo, we treated wild-type C57BL/6J mice with RG3039 daily for 12 days and examined peripheral blood (PB) counts over a time course (Figure S5A). RG3039/DCPS engagement was confirmed via CETSA in bone marrow (BM) cells the day after the last RG3039 injection (Figure S5A). While RG3039 treatment was tolerable and did not cause gross side effects, it induced a mild leukocytopenia at higher dosage (Figure S5B). BM cellularity was also slightly reduced with no cell-type specificity in RG3039-treated mice (Figures S5C and S5D). When mice were treated with RG3039 for a longer period (28 days), we observed mild transient anemia, which was normalized within a week of drug withdrawal (Figure S5E). Nonetheless, these data indicate that mice largely tolerate pharmacological inhibition of DCPS enzymatic activity.
To evaluate effects of DCPS deficiency on human hematopoiesis, we employed a xenotransplant model, in which human cord blood-derived CD34 cells reconstitute human hematopoiesis in mice (Yamauchi et al., 2013). To do so, we initiated RG3039 treatment 6 weeks after transplant and administered drug daily for 14 days (Figure 5A). We observed comparable proportions of blood cells of human origin (hCD45+) in peripheral blood in DMSO (vehicle)- and RG3039-treated mice (Figure 5B). Mice were euthanized after 14 injections (days), at which time we examined proportions of hCD45+ cells and cellularity in BM. A small portion (2 × 106 cells) of BM mononuclear cells (BMMNCs) was transferred to irradiated recipients (6 mice per experimental group) for the 2nd transplant. While RG3039 treatment did not alter reconstitution of human hematopoiesis in recipients’ BM upon 1st transplant (Figure 5C), subsets of BMMNCs were slightly skewed toward the myeloid lineage (Figure 5D). RG3039 treatment also did not alter proportions of hematopoietic stem/progenitor cells (HSPCs) (Figure 5E).
Figure 5. DCPS is dispensable for steady-state hematopoiesis in humans.
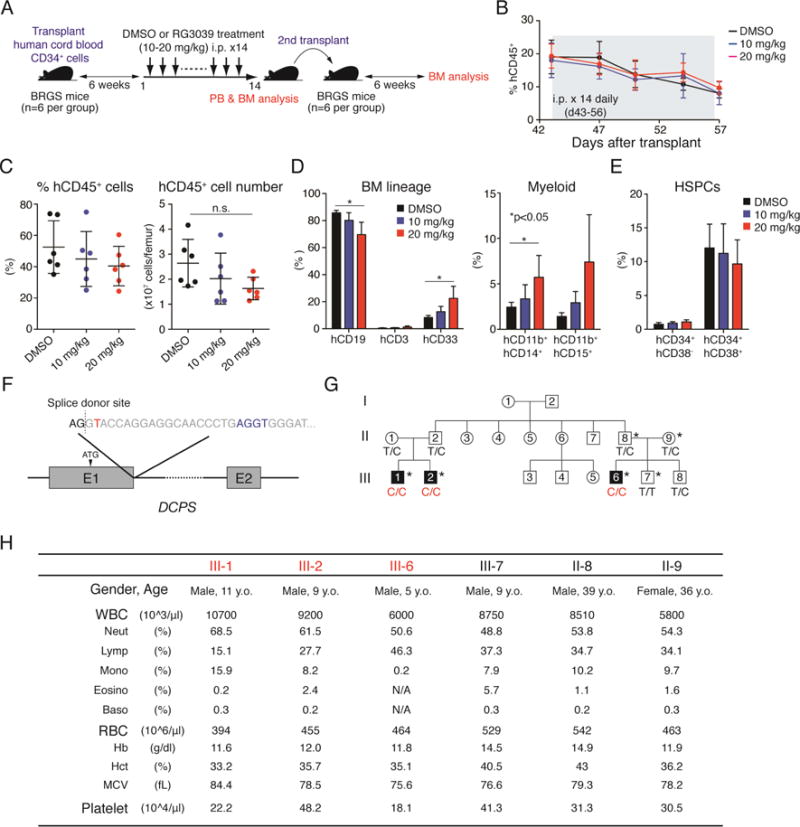
(A) Schematic representations of xenotransplant experiments. BM cells of mice treated with DMSO (vehicle) or RG3039 were transferred to secondary recipients to assess capacity to reconstitute human hematopoiesis. BRGS mice (Yamauchi et al., 2013) were used as recipients. (B) Proportions of human CD45+ cells were assessed by FACS on indicated days after the first transplant. 6 mice per condition were analyzed at each time point. (C) Dot graphs show proportions and numbers of hCD45+ cells in BM after first transplant. n.s.: not significant. Data are represented as means ± SD (n=6 per condition). (D) Bar graphs show proportions of B cells (hCD19+), T cells (hCD3+) and myeloid cells (hCD33+) in BM (n=6 per condition). Myeloid compartments were further defined using the myelo/monocytic markers CD11b, CD14 and CD15. Data are represented as means ± SD. P values were calculated using an unpaired t-test with Welch’s correction. (E) Bar graphs show proportions of hematopoietic stem cells (HSCs: CD34+CD38−) and progenitors (CD34+CD38+) in BM. Data are represented as means ± SD (n=6 per condition). (F) Schematic representations of the germline loss-of-function mutation observed in a Jordanian family (Ng et al., 2015). Thymine (red) near the splice donor site downstream of exon 1 was mutated to cytosine, creating an alternative cryptic splice site (blue) and resulting in an in-frame premature termination 40bp downstream exon 1. (G) Family tree of the affected pedigree. Asterisks denote individuals whose peripheral blood counts were assessed in this study. (H) Peripheral blood counts of affected individuals and unaffected relatives are shown. See also Figure S5.
We next assessed effects of DCPS inhibition on human HSPC function via 2nd transplantation assay (Figure 5A) and observed comparable engraftment efficiency, regardless of BM source: engraftment of human blood cells was observed in 4/6, 3/6 and 4/6 of recipient mice that were transplanted with BM cells from DMSO-, RG3039 (10 mg/kg) - and RG3039 (20 mg/kg) treated donors in consistent with our previous study (Yamauchi et al., 2013) (Figure S5F). Furthermore, proportions of hCD45+ cells in BM were comparable among experimental groups, indicating that DCPS inhibition does not grossly perturb human HSPC function (Figure S5F).
To explore effects of DCPS deficiency on normal hematopoiesis in humans, we examined PB counts of three children harboring germline homozygous loss-of-function mutations as well as heterozygous relatives in a Jordanian family reported previously (Ng et al., 2015). The variant (c.201+2T>C) at the first splice donor site of intron 1 (Figure 5F) led to complete loss of the most abundant DCPS isoform as well as its decapping activity, leaving minimal levels of a novel DCPS isoform barely detectable at the protein level (Ng et al., 2015). Three individuals homozygous for the loss-of-function variant (III-1, -2 and -6) and the two carriers (II-8 and -9) exhibited normal blood counts (Figures 5G and 5H), indicating that DCPS is dispensable for steady-state hematopoiesis in humans.
RG3039 exhibits anti-leukemia effects in human AML xenograft models
Next, we explored potential anti-leukemia effects of RG3039 in vivo in a mouse model of human AML. Specifically, we tested RG3039 efficacy using patient-derived xenograft (PDX) AML models established from three human AML lines (Townsend et al., 2016) (Figure S6A). After xenotransplantation, proportions of AML cells per total mononuclear cells in PB were examined over time by FACS, and RG3039 treatment was initiated once AML cells constituted 0.1-0.5% (Figure 6A). In the first cohort of mice, we monitored proportions of AML cells in PB until the last day of a two-week drug treatment (Figure 6B) and then euthanized mice to assess proportions of AML cells in BM (Figure 6C). RG3039 exhibited anti-leukemic activity, as evidenced by the lower leukemia burden in PB and BM, although treatment response varied by lines tested (Figures 6B and 6C). RG3039 binding to DCPS protein in AML cells in vivo was confirmed by CETSA using AML cells in BM (Figure S6B). In the second cohort, RG3039 was administered intraperitoneally for two weeks once AML cells were detected in PB as described above, and survival curves were generated. RG3039-treated mice survived significantly longer than vehicle-treated mice, indicative of therapeutic efficacy of RG3039 monotherapy against AML in vivo (Figure 6D). Of note, FHIT (fragile histidine triad), a newly-identified scavenger decapping enzyme (Taverniti and Séraphin, 2015), is differentially expressed in AML cells and normal hematopoietic cells (Figure S6C): Western blot analysis showed that human AML cells (cell lines, PDX lines and primary AML samples) express FHIT protein at significantly lower levels than do normal hematopoietic cells (cord blood and BMMNCs) (Figure S6C). Thus, lack of FHIT-mediated decapping activity may render AML cells more vulnerable to DCPS deficiency, accounting in part for leukemia-specific sensitivity to DCPS depletion or DCPS inhibition.
Figure 6. Treatment with a DCPS inhibitor has anti-leukemia effects in AML PDX models.
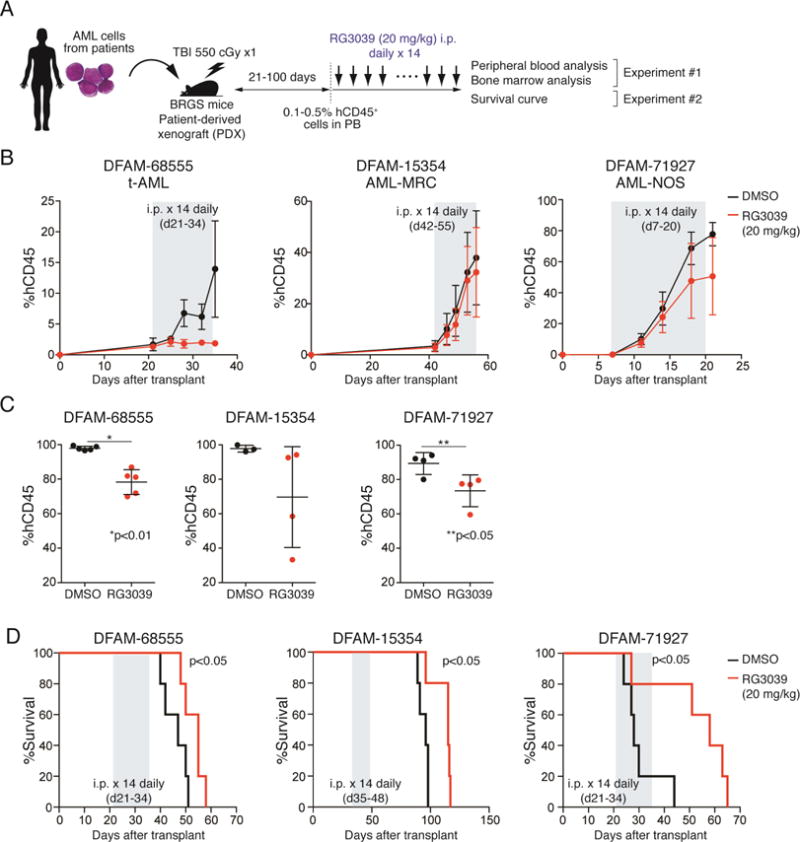
(A) Workflow of PDX experiments. (B) Proportions of hCD45+ cells in PB were examined by FACS. Duration of RG3039 treatment (20 mg/kg for 14 days) is depicted in grey. Data are represented as means ± SD (n=4-5). (C) Mice were euthanized after RG3039 treatment, and BM leukemia burden was assessed by FACS. Dot graphs show proportions of hCD45+ cells in BM. Data are represented as means ± SD (n=3-5). P values were calculated using an unpaired t-test with Welch’s correction. (D) Survival curves (n=5 per group). Duration of RG3039 treatment (20 mg/kg for 14 days) is depicted in grey. P values were calculated using a Mantel-Cox log-rank test. See also Figure S6.
DISCUSSION
Investigators have discovered genes essential for cancer using genome-wide CRISPR-Cas9 screening (Hart et al., 2015; Tzelepis et al., 2016; Wang et al., 2017). We established two mouse AML lines whose genetic backgrounds are well-defined: both exhibit a normal karyotype and harbor functionally-normal Trp53. This experimental system is particularly relevant as it mimics human primary AML cells where TP53 is less frequently mutated than in solid tumors (Welch et al., 2012). Furthermore, an in vivo validation screen controlled for potential artifacts emerging from in vitro culture conditions. Another unique feature of our screen was that we selected potentially-actionable targets using databases for chemical inhibitors (Google search and DGIdb (Wagner et al., 2016)) and human genomic databases [ExAC (Lek et al., 2016), gnomAD (http://gnomad.broadinstitute.org), OMIM, ClinVar]. This approach was inspired by recent discovery of low-frequency coding-sequence variants that alter the risk of coronary artery disease and the successful translation of that approach to drug development (Abifadel et al., 2003; Myocardial Infarction and Investigators, 2016; Sabatine et al., 2015).
DCPS was identified as an actionable target for AML after rigorous discovery processes, including unbiased genome-wide screens in vitro, “manual” selection of genes implicated in a human genetics database, and then an in vivo validation screen We focused on DCPS for the following reasons: 1) sgRNAs targeting DCPS were markedly depleted both in vitro and in vivo, with high statistical significance; 2) RG3039, a DCPS inhibitor, was available and tested safe in a phase I clinical trial (Van Meerbeke et al., 2013), suggesting it could be “re-purposed’ as AML therapy; and 3) individuals homozygous for a DCPS loss-of-function mutation survive but exhibit intellectual disability and craniofacial and neuromuscular abnormalities, suggesting DCPS is dispensable in adulthood. Domain-mapping experiments further confirmed that DCPS is essential for AML cell survival and identified amino acid residues required for that activity.
RG3039 was originally developed as a drug to treat SMA (Jarecki et al., 2005). It inhibits DCPS catalytic activity via binding to a site within the HIT sequence, interfering with DCPS binding to the 7-methylguanosine (m7G) cap of mRNA (Gu et al., 2004). RG3039 and its derivatives significantly improve symptoms and survival in SMA mouse models at similar doses that used in our in vivo studies (Gogliotti et al., 2013; Gopalsamy et al., 2017; Van Meerbeke et al., 2013). Importantly, RG3039 was granted Orphan Drug and Fast Track designations from the FDA and has been judged safe in a phase I trial in healthy volunteers (Gogliotti et al., 2013; Van Meerbeke et al., 2013), although details of the study (e.g. treatment schedule, dosage, side effects) were not currently available in publicly-available databases. Our data also indicate that RG3039-mediated DCPS inhibition does not grossly alter normal hematopoiesis. Furthermore, BM cells from RG3039-treated mice reconstituted human hematopoiesis similarly to controls in second transplant experiments, suggesting that DCPS inhibition does not perturb HSC/progenitor function. Importantly, individuals with Al-Raqad syndrome (MIM 616459) who harbor biallelic loss-of-function germline DCPS mutations exhibit normal peripheral blood counts (Ahmed et al., 2015; Ng et al., 2015). While the loss-of-function allele (c.201+2T>C) reportedly expresses an aberrant transcript, its mRNA levels are very low and the endogenous protein is barely detected in patient-derived primary fibroblasts, which also lack detectable DCPS enzymatic activity (Ng et al., 2015). Since DCPS inhibitors with more potency and specificity are being developed (Gopalsamy et al., 2017), it would be interesting to test their anti-leukemia activity.
CETSA analysis confirmed binding of RG3039 and DCPS in AML cells as previously reported (Martinez Molina et al., 2013; Xu et al., 2016). RG3039 suppressed proliferation of human AML cells in vitro and in vivo. Since amino acid residues outside the HIT sequence that bind RG3039 were necessary for AML survival in a domain-mapping experiment, we expect that pharmacological inhibition of DCPS function beyond its enzymatic activity could exert more potent anti-leukemia effects. In fact, DCPS knockout (via CRISPR/Cas9) or knockdown (via shRNA) promotes more potent anti-leukemic activity than inhibition of its enzymatic activity by RG3039 treatment. For example, following DCPS knockdown in AML cells, expression of differentiation markers was more robust than was seen following RG3039 treatment. In this regard, a Cereblon-based target degradation system (Winter et al., 2015) might be an interesting option to completely inactivate DCPS function.
It is unclear why AML cells are more vulnerable to DCPS deficiency than are cells functioning in normal human hematopoiesis. It also remains unknown exactly how DCPS inhibition causes cell cycle arrest, cellular apoptosis and differentiation in AML cells. Considering that DCPS interacts with multiple pre-mRNA processing machineries, either directly or indirectly (e.g. through RNA or chromatin), DCPS inhibition could broadly alter pre-mRNA metabolism in the nucleus, including pre-mRNA splicing.
RNA-seq experiments revealed that RG3039-induced mis-splicing is much less pronounced in GMPs, the normal counterpart of AML cells. Furthermore, genes mis-spliced to create alternative 3′ss and 5’ss vary between AML cells and GMPs and exhibit distinct NMD-prediction patterns. Importantly, an IFN signature was not evident in RG3039-treated GMPs, indicating differential effects of DCPS inhibition on pre-mRNA splicing and the overall transcriptome between cell types. RG3039 induced aberrant splicing, namely alternative splicing at 5′ and 3′ splice sites of the first exon, in marked contrast to aberrant splicing observed in the SF3B1-mutant chronic lymphocytic leukemia (CLL) cells (Darman et al., 2015), Sf3b1-mutant mouse myeloid progenitors (Obeng et al., 2016) or Srsf2-mutant mouse AML cells (Lee et al., 2016). We do not yet know how DCPS depletion leads to pre-mRNA mis-splicing. However, two explanations are plausible. The first involves the nuclear cap binding complex (CBC), which consists of a 5’cap binding protein NCBP20 (a.k.a. CBP20) and NCBP1 (a.k.a. CBP80) and is implicated in pre-mRNA splicing (Gonatopoulos-Pournatzis and Cowling, 2014). CBC reportedly interacts with components of small nuclear ribonucleoproteins (snRNPs), which are necessary for efficient co-transcriptional spliceosomal assembly (Pabis et al., 2013). Multiple lines of evidence suggest that interaction between the 5′ end m7G cap and CBC is necessary to splice the first intron (Inoue et al., 1989; Izaurralde et al., 1995; Izaurralde et al., 1994; Jiao et al., 2013). Like CBC, DCPS, which binds to the m7G cap in the nucleus, may function as a platform to recruit spliceosomes co-transcriptionally. An alternative is that in the nucleus CBC and DCPS may compete for m7G cap-containing pre-mRNAs, and DCPS depletion would favor engagement of CBC with the m7G cap, perturbing pre-mRNA splicing, as described (Shen et al., 2008).
RG3039 treatment affects splicing of more than 300 genes in AML cells, and DCPS complexes with multiple pre-mRNA processing machineries. Given that a type I interferon response was the most enriched gene expression signature following RG3039 treatment in AML cells, we postulate that DCPS inhibition broadly alters pre-mRNA metabolism, including splicing, and produces aberrant transcripts that induce a cell-intrinsic response, such as ribotoxic stress-induced apoptosis, an outcome reminiscent of that observed in RNAi-treated cells (Bridge et al., 2003; Wurtmann and Wolin, 2009).
We report that RG3039 monotherapy had anti-leukemic effects and prolonged survival of human AML PDX models. In this model, instead of treating mice immediately after AML cell transfer, we initiated RG3039 treatment once they constituted 0.1-0.5% of peripheral blood mononuclear cells, at which point AML cells constituted 70-90% of BM cells. Considering that leukemia burden at the time of treatment was very high, and that these AML cells harbor complex genomic backgrounds and are refractory to multiple chemotherapies, the observation of anti-leukemic activity in vivo by RG3039 as a monotherapy is significant. Since somatic mutations of spliceosomal genes are common in myelodysplastic syndromes (MDS) and AML (Lee et al., 2016) and mRNA splicing is reportedly impeded in those cells (Darman et al., 2015; Lee et al., 2016; Obeng et al., 2016), it would be interesting to test whether AML/MDS cells harboring a spliceosome mutation are particularly sensitive to DCPS inhibition. Along the same line, combinatorial treatment using RG3039 plus a compound targeting spliceosomal function, such as an SF3B1 inhibitor (Lee and Abdel-Wahab, 2016), might be a potential therapeutic approach worthy of investigation.
In summary, we identified DCPS as a target for AML therapy. While DCPS is implicated in the mRNA decay pathway, it is primarily localized to the nucleus and functions to maintain pre-mRNA metabolism; moreover, its loss renders leukemia cells vulnerable to cell cycle arrest and apoptosis. Since DCPS is dispensable for steady-state-hematopoiesis in humans, DCPS inhibitors, such as RG3039, warrant attention as potential therapeutic approaches for AML.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact, Takahiro Maeda (t_maeda@cancer.med.kyushu-u.ac.jp).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
12–16 weeks-old C57BL/6 male mice were purchased from Jackson Laboratory. C57BL/6.Rag2nullIl2rgnullNOD-Sirpa (BRGS) mice were described previously (Yamauchi et al., 2013). 6-10 weeks-old BRGS mice were used for experiments. Mice were bred and maintained in individual ventilated cages and fed with autoclaved food and water at Kyushu University Animal Facility. All animal experiments were approved by the Institutional Animal Care and Use Committees, according to national and institutional guidelines.
Generation of Cas9-expressing mouse AML cell lines
Mouse bone marrow HSCs were harvested from male C57BL/6 mice and transduced with a retrovirus encoding the CALM/AF10 or MLL/AF9 oncogene. Transduced cells were transplanted into lethally-irradiated recipient mice. Primary AML cells were collected 3-6 months later, serially transplanted 3 times to obtain clonally homogenous population, and harvested to establish cell lines (Figure 1A). Cells were cultured in the presence of cytokines as described above and infected with lentivirus encoding both the S. pyogenes Cas9 protein and the blasticidin resistance gene (lentiCas9-Blast, Addgene). Blasticidin (Sigma) selection was initiated 24 h later. Trp53 mutant CALM/AF10 or MLL/AF9 AML cells were established by lentivirally transducing sgRNA against Trp53 into parental AML lines, followed by Nutlin-3a, a Mdm2 inhibitor, treatment.
Cell culture
Mouse leukemia cells (CALM/AF10 and MLL/AF9) were cultured in Iscove’s Modified Dulbecco’s Media (IMDM) (Life Technologies) supplemented with 20% Fetal Bovine Serum (FBS) (Omega Scientific), 1% penicillin streptomycin (Life Technologies), and mouse SCF (20 ng/ml), mouse IL-3 (10 ng/ml) and mouse IL-6 (10 ng/ml) (all from PeproTech). Human leukemia cell lines (MOLM-13, OCI-AML3, MV4-11, and THP-1) were cultured in Roswell Park Memorial Institute (RPMI) medium (Life Technologies) with 10% FBS and 1% penicillin streptomycin.
Lentiviral transduction
Lentivirus transduction was performed as described (Shalem et al., 2014). In brief, HEK293T cells were cultured with Dulbecco’s Modified Eagle’s Medium (DMEM) (Life Technology) supplemented with 10% FBS and 1% penicillin stereptomycin. Cells were transfected with 6.7 μg psPAX2 (Addgene), 4.1 μg VSV-G (Addgene), and 10 μg lentiviral vectors using 60 μg of linear polyethylenimine (Polysciences). Lentiviral supernatants were harvested at 48 and 72 hours post-transfection and concentrated by ultracentrifugation (24,000 rpm for 2 hours at 4°C with a Beckman Coulter SW 32 Ti rotor). AML cells were plated into 12-well plates (0.5-3 × 106 cells per well) with medium supplemented with 8 μg/ml polybrene (Sigma) and spin-infected at 2,000 rpm for 2 hours at 37°C.
Xenotransplantation
Lineage (Lin)-depleted cord blood (CB) cells were obtained using magnetic beads (Lineage cell depletion kit; Miltenyi Biotec, Bergisch Gladbach, Germany). To reconstitute human hematopoiesis in mice, Lin− CB cells (2 × 104 per mouse) were injected into irradiated (550 cGy) BRGS mice via the tail vein (Yamauchi et al., 2013). Mice were euthanized 6 weeks later, and their BM cells (2 × 106 per mouse) were transferred to irradiated secondary recipients to assess HSPC reconstitution capacity. Recipients of the second transplant were euthanized 6 weeks later, and engraftment of human blood cells in BM was assessed by FACS.
To establish patient-derived xenograft (PDX) models, three PDX lines were obtained from Leukemia/Lymphoma Xenograft Core (LLX) of the Dana-Farber Cancer Institute (DFCI). Somatic mutations in cells were assessed via the Rapid Heme Panel (DFCI). 1-2 × 106 AML cells were injected into irradiated (550 cGy) BRGS mice (Yamauchi et al., 2013) via the tail vein. After transplantation, mice were given sterile water containing prophylactic enrofloxacin (Baytril; Bayer HealthCare).
Peripheral blood analysis in patients with biallelic DCPS loss-of-function
A Jordanian family consisting of three children (III-1, III-2 and III-6) exhibiting intellectual disability, craniofacial and neuromuscular abnormalities and two unaffected siblings (III-7 and III-8) were described previously (Ng et al., 2015). Peripheral blood counts of all were examined with standard methodology using an automated analyzer, with consent of patient or parent.
METHOD DETAILS
Immunohistochemistry
Immunohistochemistry was performed on paraffin sections with the conventional avidin–biotin–peroxidase method. Antigen retrieval was performed using citrate buffer. Endogenous peroxidases were quenched by incubating sections in 3% H2O2 in PBS for 5 min. Sections were blocked in PBS with 0.1% Tween-20 (PBST) for 1h. DCPS antibody (Santa Cruz, A-12) was added overnight at 4°C. Slides were incubated with biotin-conjugated secondary antibodies (1:400; Vector Laboratories) using a VECTA-STAIN Elite ABC kit (Vector Laboratories) and developed with 3,3′-diaminobenzidine (DAB). Slides were mounted with Permount medium (Thermo Fisher Scientific).
Genome-wide CRISPR screen
The GeCKO v2 mouse library (Shalem et al., 2014) was purchased from Addgene. It consists of two half-libraries (A and B), containing altogether 130,209 sgRNAs (67,405 in Library A and 62,804 in Library B) targeting 20,611 protein-coding genes and 1,175 miRNAs plus 1,000 non-targeting control sgRNAs. The plasmid pool was prepared as described (https://www.addgene.org/pooled-library/zhang-human-gecko-v2/). The library representation was validated via Miseq (Illumina). Genome-wide screens were performed as described (Shalem et al., 2014). Briefly, 1.2×108 AML cells (per half library) were transduced at a transduction efficiency of 30% with the viral pool to achieve an average coverage of more than x500. Cells were treated with puromycin (Sigma) 24 h after transduction (on day 1) and 3×106 cells were harvested two days later (day 3) to obtain input DNA. Remaining cells were passaged 5-6 times over a 16-day incubation period. At least 3×107 cells were maintained at any given time to ensure sgRNA representation (Figure S1B). On day18, cells were harvested and genomic DNA extracted using a Blood & Cell Culture DNA Midi Kit (Qiagen).
sgRNA library design for the second screen
470 genes were chosen for a secondary screen in vivo as described (Figure 1D). For each gene, eight sgRNAs were chosen from the previously published mouse genome-wide sgRNA libraries “Brie” and “Asiago” (Doench et al., 2016). If fewer than 8 sgRNAs per gene were available within these libraries, sgRNA sequences were taken from the mouse “GeCKOv2” genome-wide library until 8 sgRNAs were obtained (Sanjana et al., 2014). The final library included 3,760 gene-targeting sgRNAs and 100 non-targeting sgRNAs as negative controls. Each sgRNA oligo was synthesized as described (Canver et al., 2015) and cloned using a Gibson Assembly master mix (New England Biolabs) into lentiGuide-Puro plasmid (Addgene plasmid no. 52963), which had been BsmBI-digested, PCR-purified, and dephosphorylated. Gibson Assembly products were transformed to electrocompetent cells (E. cloni, Lucigen). Sufficient colonies were isolated to ensure ~50x coverage of the library. The plasmid library was deep-sequenced using Miseq to confirm representation.
In vivo second screen in a mouse AML model
2×107 MLL-AF9-Cas9 cells were transduced with the viral pool at transduction efficiencies of 20% to achieve over x1,000 coverage (Figures S2C and S2D). Cells were treated with puromycin 24h after transduction and cultured 2 days, at which time 2×106 puromycin-resistant cells were harvested to obtain input DNA. Remaining cells were transferred to sub-lethally irradiated recipient mice (Figure S2C). Mice were euthanized 3 weeks later, and AML cells were harvested from bone marrow to obtain output DNA samples.
Domain saturating mutagenesis
All NGG-restricted sgRNAs were identified within coding exons of Dcps (n=154) and Ctps (n=238) (Canver et al., 2017). 100 non-targeting sgRNAs were included as negative controls. Library synthesis was performed as per the second screening library, as noted above. Read counts from final and initial time points were normalized to control-nontargeting guides via the MAGeCK count function (Li et al., 2014), which was used to calculate log2 fold-changes in guide abundance. Guides were then mapped to the protein by mapping the double-stranded break site to the corresponding codon. Lastly, scores for amino acids with no assigned guide were interpolated via LOESS regression, using known guide scores and location as input. Scores for each amino acid were then mapped onto the DCPS structure publicly available in the Protein Data Bank (PDB ID: 1vlr (Han et al., 2005)). Structure sequence was aligned to the sequence of the protein isoform to which guides were originally mapped using Biopythons pairwise2 module (Cock et al., 2009) (local alignment with Blosum62 matrix, opening gap cost -10, extension -0.5). Scores from guides mapping to the same amino acid were averaged. The protein structure was recolored in PyMOL (https://www.pymol.org) based on aligned scores, by placing them in the B-factor field. For visual clarity, scores were divided into bins of 1 log2 fold-change.
Library preparation for next generation sequencing
Library preparation was performed as described (Shalem et al., 2014). sgRNA inserts were PCR-amplified from 130 μg genomic DNA using Herculase 2 fusion DNA polymerase (Agilent). Resulting PCR products were purified and sequenced on a NextSeq500 sequencer (Illumina) to assess change in abundance of each sgRNA between initial and final cell populations.
shRNA-mediated DCPS knockdown
Lentivirus vectors containing shRNA against human DCPS were purchased from Sigma Aldrich (TRCN0000335923, TRCN0000335924 and TRCN0000005569). Vector fragments containing shRNA sequence were isolated by MfeI/SpeI digestion of the pLKO vector, gel-purified and sub-cloned into PPIG (pLKO Puro-IRES-GFP) lentivirus vectors. Lentivirus transduction was performed as described above.
Cellular thermal shift assay (CETSA)
CETSA was performed to assess binding of RG3039 to DCPS protein in cells as described (Martinez Molina et al., 2013; Xu et al., 2016).
Cell cycle and apoptosis assay
For cell cycle analysis, a 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay was performed using the Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit following the manufacturer’s specifications (Life Technologies). FxCycle™ Violet (Life Technologies) was used for DNA content analysis. FITC-Annexin V (BioLegend) was used to identify apoptotic cells.
Immunoprecipitation/mass spectrometry
MOLM-13 cells were cultured in RPMI containing 10% FBS. Proteins were extracted with RIPA Buffer. DCPS complexes were pulled down using anti-DCPS antibody (Bethyl) followed by separation on SDS-PAGE. Proteins in gels were reduced using 10 mM dithiothreitol for 30 min, and alkylated using 50 mM iodoacetamide for 30 min. After overnight trypsin digestion, peptides were purified with SDB-XC StageTip. NanoLC-MS/MS were conducted using a Triple TOF 6600 (SCIEX) with an ekspert nanoLC 415 system (Eksigent Technologies). The mobile phase consisted of (A) 0.1% FA in water and (B) 0.1% FA in acetonitrile. A linear gradient of 5% B to 32% B in 60 min, 32% B to 80% B in 5 min and 80% B for 5 min was employed. Samples were injected onto a ChromXP C18CL (3 μm, 200μm × 0.5 mm) column serving as a trap column and then separated on a ChromXP C18CL (3 μm, 75 μm × 15 mm) column. For Information Dependent Acquisition, precursor ions were scanned from 400 to 1250 with an accumulation time of 50 msec. For sequential window acquisition of all theoretical mass spectra (SWATH), precursor ions ware taken by a Variable Q1 Window Acquisition from 100 to 2000. MS data were subjected to a search against the Uniprot Human database with Protein Pilot V.5.0 (AB Sciex). The false discovery rate was set below 1%. The spectral library for SWATH analysis was generated from IDA data, and peak area of peptides was calculated by PeakView (AB Sciex).
RNA-Seq
Two independent primary AML lines per oncogene (CALM/AF10 or MLL/AF9) were obtained from the bone marrow of leukemic mice (CALM/AF10 #1 and #2 and MLL/AF9 #1 and #2; Figure S2A) and RNA was extracted using an RNeasy Mini Kit (Qiagen). Libraries were generated using the Ovation Mouse RNA-Seq System following the manufacturer’s specifications (NuGEN). All libraries underwent 75 bp single-end sequencing on an Illumina NextSeq500 sequencer. For RNA-seq of RG3039-treated AML cells (Figure 4), CALM/AF10 cells or GMPs (Lin−Sca-1−c-Kit+FcγR+CD34+) were treated with RG3039 (1 μM) in the presence of cytokines, harvested over a 0, 6 and 10h time course, and RNA samples were prepared. Libraries were constructed and underwent 150 bp paired-end sequencing on an Illumina NextSeq500 sequencer.
QUANTIFICATION AND STATISTICAL ANALYSIS
Comparing dropout p values with gene expression
To investigate the relationship between dropout probability and gene expression, we used a scatter plot, as proposed by Tzelepis et al (Tzelepis et al., 2016). Dropout probability was obtained using MAGeCK software (Li et al., 2014). We used Salmon (Patro et al., 2017) with default parameters for RNA-seq quantification, and averaged TPM values across three replicates per condition.
Quantification of RNA-Seq data and identification of differential splicing events
Raw sequence reads were aligned to mouse reference mm10 using the STAR aligner (Dobin et al., 2013). We assessed individual junction counts for each cohort by summing these counts over all samples in that cohort. Estimation of intron retention counts were performed by defining a 6 nt window over each exon-exon junction (3 nt in exon and 3 nt in intron), and all reads which completely overlapped this window were considered intronic (Darman et al., 2015). Individual junction usage in each cohort was then converted to a percent spliced in (PSI) measurement by dividing the count of that junction by the total count of all junctions sharing a splice site with it in that cohort. We assessed differential junction PSI between cohorts similar to (Darman et al., 2015), however a binomial z-test of logit-transformed PSI values was used to assess the likelihood of observation, and these p values were corrected using FDR. A q-value < 0.05 was considered significant.
Gene expression analysis
Isoform quantification was performed using Kallisto (Bray et al., 2016) against GENCODE M2. Total gene counts were compiled by summing isoform counts for all isoforms belonging of that gene. Differential analysis was performed using EdgeR (Robinson et al., 2010) to assess significance of gene expression changes between cohorts. Gene expression changes corresponding to a q-value < 0.05 were considered significant.
NMD prediction
Prediction of splice variant induction of nonsense mediated decay (NMD) was performed as previously described (Darman et al., 2015). Briefly, all RefSeq annotated transcripts for each gene containing a given significant splicing event were modified to contain the novel junction (i.e., intron retention events involve the removal of annotated junctions, alternative 3′ splice sites cause the extension of downstream exons, etc). Each transcript was then translated in silico and it was determined whether a premature termination codon (PTC) had been inserted more than 50 nt from the last exon-exon junction, which is predicted to result in targeting by the NMD pathway (Rivas et al., 2015). If all transcripts associated with a given gene were predicted to result in degradation, that gene was predicted to be NMD-targeted. Similarly, if all transcripts were not predicted to contain a PTC that gene was predicted not to be targeted by the NMD pathway.
DATA AND SOFTWARE AVAILABILITY
RNA-seq data from CALM/AF10 AML or normal GMP cells were deposited in the Gene Expression Omnibus (GEO) database under accession number GSE107288.
Supplementary Material
Table S1. Summary of genome-wide screening results, Related to Figure 1.
Table S2. Summary of in vivo second screening results, Related to Figure 1.
Table S3. Summary of domain saturating mutagenesis results and a list of sgRNA sequences used, Related to Figure 1.
Table S4. Summary of mass-spectrometry results, Related to Figure 3.
Table S5. Summary of RNA-seq results, Related to Figure 4.
SIGNIFICANCE.
Genome-wide CRISPR-Cas9 knockout screening is a powerful tool for functional genomics. We performed genome-wide CRISPR-Cas9 dropout screens utilizing AML lines exhibiting a normal karyotype and harboring functionally-normal Trp53, which enabled straightforward interpretation of screening results. We then selected potentially actionable targets using databases relevant to chemical inhibitors and human genetics. We identified DCPS as a drug target for AML therapy. We report that RG3039, a DCPS inhibitor proven safe in a phase I clinical trial, exhibits anti-leukemia activity and identify a pre-mRNA metabolic pathway required for AML survival. Our work suggests a potential opportunity to “re-purpose’ DCPS inhibitors for AML therapy.
Highlights.
Genome-wide CRISPR-based screens were performed with Trp53-intact mouse AML cells.
Potentially actionable targets were identified using publicly-available databases.
DCPS, an mRNA decapping enzyme, was identified as a target for AML therapy.
RG3039, a DCPS inhibitor, showed toxicity against AML but not normal blood cells.
Acknowledgments
We thank Hai Yi, Catherine Zhu and members in the Division of Hematology at BWH and Department of Medicine and Biosystemic Science at Kyushu University for assistance, advice and helpful discussion, Zach Herbert for help and advice on sequencing and Elise Lamar for critical reading of the manuscript. This work is supported in part by a Grant-in-Aid for JSPS Fellows (15J10130) (to T.Y.), an NIDDK Predoctoral National Research Service Award for an M.D./Ph.D. Fellowship (F30DK103359-01A1) (to M.C.C.), a Strategic Positioning Fund for Genetic Orphan Diseases from the Agency for Science, Technology, and Research in Singapore (to B.R), NIH R00HG008399 (to L.P.), NIDDK (K08DK093705, R03DK109232), NHLBI (DP2OD022716), Burroughs Wellcome Fund, and American Society of Hematology (to D.E.B.), NIH P01HL032262 and P30DK049216 grants (Center of Excellence in Molecular Hematology) (to S.H.O.), a Grant-in-Aid for Scientific Research (S)(16H06391)(to K.A.) and a Grant-in-Aid for Scientific Research (A) (17H01567), American Cancer Society (RSG-13-379-01-LIB) and an American Society of Hematology Bridge Grant (to T.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
T.Y., M.C.C., L.P., D.E.B. and T.M. designed CRISPR-Cas9 screen experiments. T.Y., Y.S. and T.M reviewed CRISPR screen data. T.M., M.M., Y.I., Y.S. and M.N. executed CRISPR-CAS9 experiments, cell biology experiments, RNA-Seq, western blot analysis, immunohistochemistry and in vivo mouse studies, supervised by F.A., L.P., S.H.O., K.A., D.E.B. and T.M. T.M. performed mass-spectrometry and analyzed proteomic data. M.S., S.B. Y.S., Q.Y. and L.P. analyzed RNA-Seq data. M.S., M. A. R. and B.R. provided patient data. M.C.C., V.A.C.S., M.A.C. and C.M.T. analyzed CRISPR saturation mutagenesis data, supervised by L.P. and D.E.B. T.Y and T.M. wrote the manuscript with help from all authors.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Abifadel M, Varret M, Rabès J-P, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- Ahmed I, Buchert R, Zhou M, Jiao X, Mittal K, Sheikh TI, Scheller U, Vasli N, Rafiq MA, Brohi MQ, et al. Mutations in DCPS and EDC3 in autosomal recessive intellectual disability indicate a crucial role for mRNA decapping in neurodevelopment. Hum Mol Genet. 2015;24:3172–3180. doi: 10.1093/hmg/ddv069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen Dale A-L, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PR, Domanski M, Kristiansen MS, Storvall H, Ntini E, Verheggen C, Schein A, Bunkenborg J, Poser I, Hallais M, et al. The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat Struct Mol Biol. 2013;20:1367–1376. doi: 10.1038/nsmb.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm JS, Hahn WC. Towards systematic functional characterization of cancer genomes. Nat Rev Genet. 2011;12:487–498. doi: 10.1038/nrg3013. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Bridge AJ, Pebernard S, Ducraux A, Nicoulaz A-L, Iggo R. Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet. 2003;34:263–264. doi: 10.1038/ng1173. [DOI] [PubMed] [Google Scholar]
- Canver MC, Lessard S, Pinello L, Wu Y, Ilboudo Y, Stern EN, Needleman AJ, Galactéros F, Brugnara C, Kutlar A, et al. Variant-aware saturating mutagenesis using multiple Cas9 nucleases identifies regulatory elements at trait-associated loci. Nat Genet. 2017 doi: 10.1038/ng.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJ. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015;13:1033–1045. doi: 10.1016/j.celrep.2015.09.053. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara F, Schiffer CA. Acute myeloid leukaemia in adults. Lancet. 2013;381:484–495. doi: 10.1016/S0140-6736(12)61727-9. [DOI] [PubMed] [Google Scholar]
- Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Gogliotti RG, Cardona H, Singh J, Bail S, Emery C, Kuntz N, Jorgensen M, Durens M, Xia B, Barlow C, et al. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum Mol Genet. 2013;22:4084–4101. doi: 10.1093/hmg/ddt258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonatopoulos-Pournatzis T, Cowling VH. Cap-binding complex (CBC) Biochem J. 2014;457:231–242. doi: 10.1042/BJ20131214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalsamy A, Narayanan A, Liu S, Parikh MD, Kyne RE, Fadeyi O, Tones MA, Cherry JJ, Nabhan JF, LaRosa G, et al. Design of Potent mRNA Decapping Scavenger Enzyme (DcpS) Inhibitors with Improved Physicochemical Properties To Investigate the Mechanism of Therapeutic Benefit in Spinal Muscular Atrophy (SMA) J Med Chem. 2017;60:3094–3108. doi: 10.1021/acs.jmedchem.7b00124. [DOI] [PubMed] [Google Scholar]
- Gu M, Fabrega C, Liu S-W, Liu H, Kiledjian M, Lima CD. Insights into the structure, mechanism, and regulation of scavenger mRNA decapping activity. Mol Cell. 2004;14:67–80. doi: 10.1016/s1097-2765(04)00180-7. [DOI] [PubMed] [Google Scholar]
- Han GW, Schwarzenbacher R, McMullan D, Abdubek P, Ambing E, Axelrod H, Biorac T, Canaves JM, Chiu H-J, Dai X, et al. Crystal structure of an Apo mRNA decapping enzyme (DcpS) from Mouse at 1.83 A resolution. Proteins. 2005;60:797–802. doi: 10.1002/prot.20467. [DOI] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015 doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Inoue K, Ohno M, Sakamoto H, Shimura Y. Effect of the cap structure on pre mRNA splicing in Xenopus oocyte nuclei. Genes Dev. 1989;3:1472–1479. doi: 10.1101/gad.3.9.1472. [DOI] [PubMed] [Google Scholar]
- Izaurralde E, Lewis J, Gamberi C, Jarmolowski A, McGuigan C, Mattaj IW. A cap-binding protein complex mediating U snRNA export. Nature. 1995;376:709–712. doi: 10.1038/376709a0. [DOI] [PubMed] [Google Scholar]
- Izaurralde E, Lewis J, McGuigan C, Jankowska M, Darzynkiewicz E, Mattaj IW. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell. 1994;78:657–668. doi: 10.1016/0092-8674(94)90530-4. [DOI] [PubMed] [Google Scholar]
- Jarecki J, Chen X, Bernardino A, Coovert DD, Whitney M, Burghes A, Stack J, Pollok BA. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum Mol Genet. 2005;14:2003–2018. doi: 10.1093/hmg/ddi205. [DOI] [PubMed] [Google Scholar]
- Jiao X, Chang JH, Kilic T, Tong L, Kiledjian M. A mammalian pre-mRNA 5′ end capping quality control mechanism and an unexpected link of capping to pre-mRNA processing. Mol Cell. 2013;50:104–115. doi: 10.1016/j.molcel.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera MDC, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32:267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC-W, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22:976–986. doi: 10.1038/nm.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC-W, Dvinge H, Kim E, Cho H, Micol J-B, Chung YR, Durham BH, Yoshimi A, Kim YJ, Thomas M, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med. 2016;22:672–678. doi: 10.1038/nm.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS. MAGeCK enables robust identification of essential genes from genome scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ET, Würtz P, Havulinna AS, Palta P, Tukiainen T, Rehnström K, Esko T, Mägi R, Inouye M, Lappalainen T, et al. Distribution and medical impact-of-loss of function variants in the Finnish founder population. PLoS Genet. 2014;10:e1004494. doi: 10.1371/journal.pgen.1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. 2015;16:665–677. doi: 10.1038/nrm4063. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- Milac AL, Bojarska E, Wypijewska del Nogal A. Decapping Scavenger (DcpS) enzyme: advances in its structure, activity and roles in the cap-dependent mRNA metabolism. Biochim Biophys Acta. 2014;1839:452–462. doi: 10.1016/j.bbagrm.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Myocardial Infarction, G., and Investigators, C. A. E. C. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N Engl J Med 2016 [Google Scholar]
- Narasimhan VM, Hunt KA, Mason D, Baker CL, Karczewski KJ, Barnes MR, Barnett AH, Bates C, Bellary S, Bockett NA, et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science. 2016 doi: 10.1126/science.aac8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CKL, Shboul M, Taverniti V, Bonnard C, Lee H, Eskin A, Nelson SF, Al-Raqad M, Altawalbeh S, Séraphin B, Reversade B. Loss of the scavenger mRNA decapping enzyme DCPS causes syndromic intellectual disability with neuromuscular defects. Hum Mol Genet. 2015;24:3163–3171. doi: 10.1093/hmg/ddv067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell. 2016;30:404–417. doi: 10.1016/j.ccell.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabis M, Neufeld N, Steiner MC, Bojic T, Shav-Tal Y, Neugebauer KM. The nuclear cap-binding complex interacts with the U4/U6-U5 tri-snRNP and promotes spliceosome assembly in mammalian cells. RNA. 2013;19:1054–1063. doi: 10.1261/rna.037069.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas MA, Pirinen M, Conrad DF, Lek M, Tsang EK, Karczewski KJ, Maller JB, Kukurba KR, DeLuca DS, Fromer M, et al. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science. 2015;348:666–669. doi: 10.1126/science.1261877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–1509. doi: 10.1056/NEJMoa1500858. [DOI] [PubMed] [Google Scholar]
- Saleheen D, Natarajan P, Armean IM, Zhao W, Rasheed A, Khetarpal SA, Won H-H, Karczewski KJ, O’Donnell-Luria AH, Samocha KE, et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. 2017;544:235–239. doi: 10.1038/nature22034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nature methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen V, Liu H, Liu S-W, Jiao X, Kiledjian M. DcpS scavenger decapping enzyme can modulate pre-mRNA splicing. RNA. 2008;14:1132–1142. doi: 10.1261/rna.1008208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33:661–667. doi: 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulem P, Helgason H, Oddson A, Stefansson H, Gudjonsson SA, Zink F, Hjartarson E, Sigurdsson GT, Jonasdottir A, Jonasdottir A, et al. Identification of a large set of rare complete human knockouts. Nat Genet. 2015;47:448–452. doi: 10.1038/ng.3243. [DOI] [PubMed] [Google Scholar]
- Taverniti V, Séraphin B. Elimination of cap structures generated by mRNA decay involves the new scavenger mRNA decapping enzyme Aph1/FHIT together with DcpS. Nucleic Acids Res. 2015;43:482–492. doi: 10.1093/nar/gku1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend EC, Murakami MA, Christodoulou A, Christie AL, Köster J, DeSouza TA, Morgan EA, Kallgren SP, Liu H, Wu S-C, et al. The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell. 2016;29:574–586. doi: 10.1016/j.ccell.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. Defining a Cancer Dependency Map. Cell. 2017;170:564–576.e516. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzelepis K, Koike Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, Mupo A, Grinkevich V, Li M, Mazan M, et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016;17:1193–1205. doi: 10.1016/j.celrep.2016.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meerbeke JP, Gibbs RM, Plasterer HL, Miao W, Feng Z, Lin M-Y, Rucki AA, Wee CD, Xia B, Sharma S, et al. The DcpS inhibitor RG3039 improves motor function in SMA mice. Hum Mol Genet. 2013;22:4074–4083. doi: 10.1093/hmg/ddt257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visa N, Izaurralde E, Ferreira J, Daneholt B, Mattaj IW. A nuclear cap-binding complex binds Balbiani ring pre-mRNA cotranscriptionally and accompanies the ribonucleoprotein particle during nuclear export. J Cell Biol. 1996;133:5–14. doi: 10.1083/jcb.133.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AH, Coffman AC, Ainscough BJ, Spies NC, Skidmore ZL, Campbell KM, Krysiak K, Pan D, McMichael JF, Eldred JM, et al. DGIdb 2.0: mining clinically relevant drug-gene interactions. Nucleic Acids Res. 2016;44:D1036–1044. doi: 10.1093/nar/gkv1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, Sabatini DM. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell. 2017 doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtmann EJ, Wolin SL. RNA under attack: cellular handling of RNA damage. Crit Rev Biochem Mol Biol. 2009;44:34–49. doi: 10.1080/10409230802594043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Gopalsamy A, Hett EC, Salter S, Aulabaugh A, Kyne RE, Pierce B, Jones LH. Cellular thermal shift and clickable chemical probe assays for the determination of drug-target engagement in live cells. Org Biomol Chem. 2016 doi: 10.1039/c6ob01078d. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Takenaka K, Urata S, Shima T, Kikushige Y, Tokuyama T, Iwamoto C, Nishihara M, Iwasaki H, Miyamoto T, et al. Polymorphic Sirpa is the genetic determinant for NOD-based mouse lines to achieve efficient human cell engraftment. Blood. 2013;121:1316–1325. doi: 10.1182/blood-2012-06-440354. [DOI] [PubMed] [Google Scholar]
- Zhang L, McGraw KL, Sallman DA, List AF. The role of p53 in myelodysplastic syndromes and acute myeloid leukemia: molecular aspects and clinical implications. Leuk Lymphoma. 2016;1:14. doi: 10.1080/10428194.2016.1266625. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of genome-wide screening results, Related to Figure 1.
Table S2. Summary of in vivo second screening results, Related to Figure 1.
Table S3. Summary of domain saturating mutagenesis results and a list of sgRNA sequences used, Related to Figure 1.
Table S4. Summary of mass-spectrometry results, Related to Figure 3.
Table S5. Summary of RNA-seq results, Related to Figure 4.