

Abstract
The anti-apoptotic protein survivin is highly expressed in cancer cells but has a very low expression in fully differentiated adult cells. Overexpression of survivin is positively correlated with cancer cell resistance to chemotherapy and radiotherapy, cancer cell metastasis, and poor patient prognosis. Therefore, selective targeting survivin represents an attractive strategy for the development of anticancer therapeutics. Herein, we reported the extensive structural modification of our recently discovered selective survivin inhibitor UC-112 and the synthesis of thirty-three new analogs. The structure-activity relationship (SAR) study indicated that replacement of the benzyloxy moeity in UC-112 with an indole moiety was preferred to other moieties. Among these UC-112 analogs, 10f, 10h, 10k, 10n showed the most potent antiproliferative activities. Interestingly, they were more potent against the P-glycoprotein overexpressing cancer cell lines compared with the parental cancer cell lines. Mechanistic studies confirmed that new analogs maintained their unique selectivity against survivin among the IAP family members. In vivo study using 10f in a human A375 melanoma xenograft model revealed that it effectively inhibited melanoma tumor growth without observable acute toxicity. Collectively, this study strongly supports the further preclinical development of selective survivin inhibitors based on the UC-112 scaffold.
Keywords: selective survivin inhibitors, structure-activity relationships, antiproliferative activities, P-glycoprotein overexpression
Graphical Abstract
1. Introduction
Survivin is the smallest member in the family of the inhibitor of apoptosis proteins (IAP). Its expression is very low in healthy tissues and is highly expressed in tumors [1]. The overexpression of survivin in tumor cells has been positively correlated with their development of multidrug resistance and radiation resistance [2, 3]. For example, it is well established that cancer cells with high expression of survivin (e.g., prostate, ovarian and colorectal cancer cells) develop resistance to cisplatin and vincristine much easier than tumors cells with low expression of survivin [4]. Survivin is therefore considered as a cancer specific biomarker and an attractive therapeutic target for the development of anticancer therapy [5] [6].
Several reported strategies have been applied to block the anti-apoptotic ability of survivin. These strategies include but are not limited to introducing recombinant cell-permeable dominant-negative survivin protein [7, 8], obstructing protein translation using antisense oligonucleotides [9], and developing small-molecule based survivin antagonists [10, 11]. The crystal structure of human survivin was obtained in the early of this century, revealing its unusual bow tie-shape dimer structure [12]. However, targeting survivin using small-molecule survivin inhibitors has been proven to be challenging because no experimentally validated binding pocket in survivin has been identified yet. The difficulty in developing a survivin specific small-molecule inhibitor also lies in the fact that survivin interacts with many other proteins to mediate apoptosis and mitosis, such as p53, STAT3, caspases and elements in the notch signaling pathway [13–16]. A number of scaffolds have been reported in the literature to significantly suppress the expression of survivin and some have entered clinical trials for cancer treatment [17–26]. Notable examples are listed in Figure 1. However, none of these molecules has gained FDA approval currently. The most widely studied survivin inhibitor is YM155 which was initially discovered by Astellas Pharma in 2007. YM155 has an IC50 in the sub-nanomolar range against several types of cancer cell lines [27]. It inhibits survivin expression by inhibiting survivin promotor activity, instead of directly degrading survivin protein. In clinical trials, YM155 was evaluated as monotherapy or combinational forms for patient with different cancer types, including blood cancer and solid tumors [28]. Unfortunately, YM155 was withdrawn in phase II clinical trials due to its low ability to degrade survivin and systemic toxicity.
Figure 1.

Examples of reported survivin inhibitors.
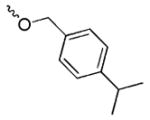
We have recently discovered UC-112 (structure shown in Figure 1), 5-((benzyloxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol, through virtual screening followed by biology validation [25]. UC-112 is a potent and selective survivin inhibitor. UC-112 selectively degrades survivin protein via the proteasome pathway, activating caspases 3/7 and 9, and thus leads to cancer cell apopotosis. Unlike YM155 which is highly susceptible to P-glycoprotein (P-gp) mediated drug efflux pump mediated drug resistance, UC-112 has demonstrated to be effective for the P-gp overexpressing multidrug-resistant cancer cell lines. Subsequent structural modifications of UC-112 results in MX-106 (Figure 1), which shows improvements in suppressing survivin expression both in vitro and in vivo [29]. According to the structure-activity relationship (SAR) analyses, the 8-hydroxyquinoline and the pyrrolidine in UC-112 are essential for maximum activity; hydrophobic substituent on the benzene is beneficial to activity. With this observation, we hypothesize that (1): the benzyloxy moiety in UC-112 is amicable to modifications; (2) conformational restricted analogs formed by reducing the flexibility of the benzyloxy moiety in this scaffold can improve the activity. With these hypotheses in mind, we herein show our extensive effort on modifying the benzyloxy moiety in the UC-112 scaffold. Thirty-three UC-112 analogs were synthesized and evaluated for activities.
2. Experimental
2.1. General methods
All solvents and chemical reagents were obtained from commercial sources and directly used without further purification. Glassware was oven-dried before use. All reactions were performed under an argon atmosphere. TLC was performed on silica gel 60 GF254 and monitored under UV light or visualized using phosphomolybdic acid reagent. Flash chromatography was performed on 230–400 mesh silica gel (Fisher Scientific, Pittsburgh, PA). NMR spectra were obtained on a Bruker Ascend 400 (Billerica, MA) spectrometer or a Varian Inova-500 spectrometer (Agilent Technologies, Santa Clara, CA). Chemical shifts are given in ppm with tetramethylsilane (TMS) as an internal reference. All coupling constants (J) are given in Hertz (Hz). HR-MS were obtained on Waters Acquity UPLC linked to Waters Acquity Photodiode Array Detector and Waters Acquity Single Quadrupole Mass Detector. The purity (≥95%) of the compounds was verified by the HPLC study performed on BEH C18 (2.1 x 50mm, 1.7um) column using a mixture of solvent acetonitrile/water (with 0.1% formic acid) at a flow rate of 0.3 mL/min and monitoring by UV absorption at 254 nm.
2.2. Chemistry
General procedure for synthesis of 1a–1d, 9a–9v
To a stirred suspension of 5-chloromethyl-8-quinolinol hydrochloride (1.0 mmol, 230 mg), sodium carbonate (6.0 mmol, 636 mg) and amine or indole (1.1 mmol) in acetonitrile (10 mL) was added potassium iodide (0.3 mmol, 50 mg). The mixture was refluxed for 6 h and then filtered through celite; the residue was washed with acetonitrile. The combined solution was evaporated under vacuum to give the crude material which was directly used for next step without purification.
General procedure for synthesis of 2a–2f, 6a–6f, 8 and 10a–10v
To a stirred suspension of 1a–1g or 5a–5g or 7 or 9a–9v (0.5 mmol) and paraformaldehyde (0.5 mmol, 15 mg) in ethanol (6 mL) was added pyrrolidine (0.45 mmol, 32 mg). The mixture was refluxed for 1–5 h and solvent was evaporated under reduced pressure to give the crude which was purified with flash chromatography on silica. Elution with dichloromethane-methanol (10:0–10:1) gave desired compounds 2a–2f, 6a–6f, 8 and 10a–10v (34–81% yield).
5-((5-fluoro-1H-indazol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (2a)
1H NMR (400 MHz, Chloroform-d) δ 8.84 (dd, J = 4.1, 1.6 Hz, 1H), 8.74 (dd, J = 4.2, 1.7 Hz, 1H), 8.19 (dd, J = 8.6, 1.6 Hz, 1H), 7.68 (ddt, J = 9.2, 4.6, 1.0 Hz, 1H), 7.64 (d, J = 0.9 Hz, 1H), 7.38 – 7.29 (m, 2H), 7.10 – 6.99 (m, 2H), 5.87 (s, 2H), 4.02 (s, 2H), 2.84 – 2.62 (m, 4H), 1.88 (p, J = 3.3 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 154.42, 148.97, 146.09, 139.73, 131.76, 129.97, 126.91, 122.72, 122.64, 122.04, 120.14, 119.55, 119.45, 118.11, 117.64, 117.35, 102.88, 102.64, 57.23, 55.15, 53.86, 23.73. HRMS: calculated for C22H22FN4O [M+H]+ 377.1778, found 377.1790. Purity: 95.8% by HPLC.
5-((1H-benzo[d][1,2,3]triazol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (2b)
1H NMR (400 MHz, Chloroform-d) δ 10.42 (s, 1H), 8.78 (dd, J = 4.2, 1.6 Hz, 1H), 8.44 (dd, J = 8.6, 1.6 Hz, 1H), 7.96 (dt, J = 7.9, 1.2 Hz, 1H), 7.44 – 7.17 (m, 5H), 6.11 (s, 2H), 3.95 (s, 2H), 2.64 (td, J = 5.6, 4.5, 2.3 Hz, 4H), 1.81 (p, J = 3.4 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 153.99, 148.74, 146.31, 139.56, 132.80, 131.60, 129.24, 127.27, 126.51, 123.86, 121.78, 119.95, 119.23, 118.18, 109.99, 57.00, 53.69, 50.30, 23.61. HRMS: calculated for C21H22N5O [M+H]+ 360.1824, found 360.1838. Purity: 95.3% by HPLC.
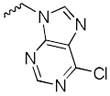
5-((6-chloro-9H-purin-9-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (2c)
1H NMR (400 MHz, Chloroform-d) δ 8.83 (s, 1H), 8.79 (dd, J = 4.2, 1.5 Hz, 1H), 8.69 (s, 1H), 8.60 (dd, J = 8.6, 1.5 Hz, 1H), 8.46 (s, 1H), 7.49 (dd, J = 8.6, 4.2 Hz, 1H), 5.80 (s, 2H), 4.45 (s, 2H), 3.30 (s, 4H), 2.16 – 2.11 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 152.66, 151.86, 151.44, 151.06, 148.77, 146.38, 138.38, 132.97, 132.61, 131.42, 127.15, 123.55, 121.89, 111.79, 53.29, 52.30, 44.19, 23.40. HRMS: calculated for C20H20ClN6O [M+H]+ 395.1387, found 395.1396. Purity: 95.5% by HPLC.
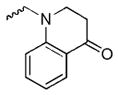
1-((8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)methyl)-2,3-dihydroquinolin-4(1H)-one (2d)
1H NMR (400 MHz, Chloroform-d) δ 8.95 (d, J = 4.1 Hz, 1H), 8.31 (dd, J = 8.5, 2.2 Hz, 1H), 8.05 – 7.96 (m, 1H), 7.50 – 7.37 (m, 2H), 7.23 (s, 1H), 6.93 – 6.78 (m, 2H), 4.82 (s, 2H), 4.01 (d, J = 1.9 Hz, 2H), 3.46 (t, J = 6.7 Hz, 2H), 2.73 (d, J = 6.1 Hz, 4H), 2.65 (t, J = 6.9 Hz, 2H), 1.90 (p, J = 3.3, 2.6 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 193.59, 153.06, 151.91, 148.78, 135.58, 131.40, 128.43, 127.46, 126.45, 121.38, 121.19, 120.37, 117.93, 117.47, 113.20, 57.30, 53.71, 52.18, 47.92, 38.28, 23.64. HRMS: calculated for C24H26N3O2 [M+H]+ 388.2025, found 388.2040. Purity: 95.0% by HPLC.
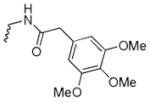
N-((8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)methyl)-2-(3,4,5-trimethoxyphenyl)acetamide (6a)
1H NMR (400 MHz, Methanol-d4) δ 8.78 (d, J = 4.1 Hz, 1H), 8.33 (d, J = 8.3 Hz, 1H), 7.40 (dd, J = 8.5, 4.2 Hz, 1H), 7.31 (s, 1H), 6.55 (s, 2H), 4.74 (s, 1H), 3.97 (s, 2H), 3.75 (s, 3H), 3.70 (s, 6H), 3.45 (s, 2H), 3.37 (d, J = 1.7 Hz, 2H), 2.72 (s, 4H), 1.89 (s, 4H). 13C NMR (101 MHz, Chloroform-d) δ 170.31, 153.57, 153.23, 148.70, 139.74, 132.09, 130.25, 128.56, 126.40, 123.13, 121.48, 106.39, 60.84, 56.10, 53.88, 44.15, 41.04, 29.72, 23.67. HRMS: calculated for C26H32N3O5 [M+H]+ 466.2342, found 466.2332. Purity: 95.4% by HPLC.
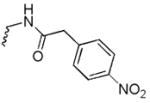
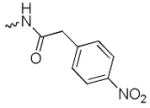
N-((8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)methyl)-2-(4-nitrophenyl)acetamide (6b)
1H NMR (400 MHz, DMSO-d6) δ 8.85 (dd, J = 4.1, 1.5 Hz, 1H), 8.66 (t, J = 5.6 Hz, 1H), 8.37 (dd, J = 8.5, 1.6 Hz, 1H), 8.23 – 8.12 (m, 2H), 7.60 – 7.47 (m, 3H), 7.39 (s, 1H), 4.64 (d, J = 5.5 Hz, 2H), 3.78 (s, 2H), 3.64 (s, 2H), 2.51 (dt, J = 3.7, 1.8 Hz, 4H), 1.73 (p, J = 3.0 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 168.60, 150.52, 147.83, 146.27, 144.49, 138.53, 132.32, 130.27, 128.84, 125.56, 123.79, 123.27, 121.25, 119.86, 54.06, 53.48, 41.98, 23.21. HRMS: calculated for C23H25N4O4 [M+H]+ 421.1876, found 421.1879. Purity: 95.5% by HPLC.
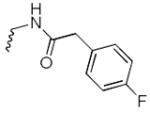
2-(4-fluorophenyl)-N-((8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)methyl)acetamide (6c)
1H NMR (400 MHz, DMSO-d6) δ 8.92 (dd, J = 4.1, 1.5 Hz, 1H), 8.60 (t, J = 5.5 Hz, 1H), 8.43 (dd, J = 8.6, 1.6 Hz, 1H), 7.70 – 7.49 (m, 2H), 7.36 – 7.23 (m, 2H), 7.18 – 7.04 (m, 2H), 4.63 (d, J = 5.5 Hz, 2H), 4.25 (s, 2H), 3.47 (s, 2H), 3.04 (s, 4H), 1.89 (p, J = 3.4 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 169.78, 151.66, 148.21, 138.45, 132.69, 132.54, 130.80, 130.72, 129.54, 126.66, 124.53, 122.12, 114.92, 114.71, 54.87, 53.08, 41.30, 22.74. HRMS: calculated for C23H25FN3O2 [M+H]+ 394.1931, found 394.1942. Purity: 95.2% by HPLC.
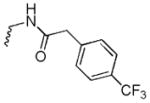
N-((8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)methyl)-2-(4-(trifluoromethyl)phenyl)acetamide (6d)
1H NMR (400 MHz, Chloroform-d) δ 8.86 (dd, J = 4.1, 1.6 Hz, 1H), 8.20 (dd, J = 8.6, 1.6 Hz, 1H), 7.61 – 7.51 (m, 2H), 7.44 – 7.30 (m, 3H), 7.15 (s, 1H), 5.76 (s, 1H), 4.75 (d, J = 5.4 Hz, 2H), 3.95 (s, 2H), 3.61 (s, 2H), 2.71 (td, J = 5.4, 4.4, 2.3 Hz, 4H), 1.88 (p, J = 3.3 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 169.35, 153.19, 148.75, 139.66, 138.87, 131.95, 129.60, 128.54, 126.38, 125.80, 125.76, 122.91, 121.56, 118.00, 57.19, 53.85, 43.48, 41.06, 23.67. HRMS: calculated for C24H25F3N3O2 [M+H]+ 444.1899, found 444.1918. Purity: 98.9% by HPLC.
N-(8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)-2-(4-nitrophenyl)acetamide (6e)
1H NMR (400 MHz, Methanol-d4) δ 8.93 (dd, J = 4.2, 1.6 Hz, 1H), 8.34 (dd, J = 8.6, 1.6 Hz, 1H), 8.32 – 8.22 (m, 2H), 7.80 – 7.67 (m, 5H), 7.64 (dd, J = 8.6, 4.2 Hz, 1H), 7.60 (s, 1H), 7.40 – 7.29 (m, 3H), 4.58 (s, 2H), 4.02 (s, 2H), 3.49 – 3.40 (m, 4H), 3.37 (s, 1H), 2.11 (p, J = 4.0 Hz, 4H). 13C NMR (101 MHz, Methanol-d4) δ 172.52, 150.46, 144.50, 144.08, 132.70, 131.57, 129.11, 127.64, 125.91, 125.68, 124.65, 124.13, 118.68, 112.18, 55.15, 54.10, 43.53, 23.87. HRMS: calculated for C22H23N4O4 [M+H]+ 407.1719, found 407.1719. Purity: 95.0% by HPLC.
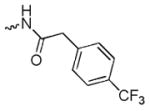
N-(8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)-2-(4-(trifluoromethyl)phenyl)acetamide (6f)
1H NMR (400 MHz, Methanol-d4) δ 8.88 (dd, J = 4.2, 1.6 Hz, 1H), 8.27 (dd, J = 8.5, 1.6 Hz, 1H), 7.68 (q, J = 8.4 Hz, 4H), 7.62 – 7.52 (m, 2H), 7.49 (d, J = 3.3 Hz, 2H), 4.33 (s, 2H), 3.95 (s, 2H), 3.57 (s, 1H), 3.19 – 3.05 (m, 4H), 2.07 – 1.94 (m, 4H). 13C NMR (101 MHz, Methanol-d4) δ 173.04, 153.35, 149.98, 141.48, 139.98, 132.77, 131.03, 130.88, 127.53, 126.53, 126.49, 124.52, 123.41, 116.23, 55.54, 54.87, 45.89, 43.67, 24.15. HRMS: calculated for C23H23F3N3O2 [M+H]+ 430.1742, found 430.1747. Purity: 98.5% by HPLC.
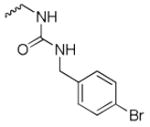
1-(4-bromobenzyl)-3-((8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)methyl)urea (8)
1H NMR (400 MHz, Methanol-d4) δ 8.83 (dd, J = 4.2, 1.6 Hz, 1H), 8.50 (dd, J = 8.6, 1.6 Hz, 1H), 7.53 (dd, J = 8.6, 4.2 Hz, 1H), 7.50 – 7.41 (m, 2H), 7.35 (s, 1H), 7.29 – 7.14 (m, 2H), 4.71 (s, 2H), 4.32 (s, 2H), 4.06 (s, 2H), 3.00 – 2.64 (m, 4H), 1.92 (p, J = 3.3 Hz, 4H). 13C NMR (101 MHz, Methanol-d4) δ 160.61, 153.65, 149.21, 140.87, 140.46, 133.91, 132.51, 130.13, 129.60, 126.10, 122.61, 121.58, 57.17, 54.69, 44.13, 42.06, 24.45. HRMS: calculated for C23H26BrN4O2 [M+H]+ 469.1239, found 469.1261. Purity: 95.1% by HPLC.
5-((1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10a)
1H NMR (400 MHz, Acetone-d6) δ 9.88 (s, 1H), 8.63 (dd, J = 4.1, 1.5 Hz, 1H), 8.37 (dd, J = 8.6, 1.6 Hz, 1H), 7.43 (dt, J = 7.9, 1.0 Hz, 1H), 7.39 (s, 1H), 7.30 (dd, J = 8.5, 4.1 Hz, 1H), 7.23 (dt, J = 8.1, 0.9 Hz, 1H), 6.94 (ddd, J = 8.2, 7.1, 1.2 Hz, 1H), 6.84 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.79 (p, J = 1.1 Hz, 1H), 4.33 (s, 2H), 3.73 (s, 2H), 2.48 – 2.35 (m, 4H), 1.61 (p, J = 3.2 Hz, 4H). 13C NMR (101 MHz, Acetone-d6) δ 150.30, 148.40, 139.97, 133.87, 129.95, 127.65, 127.35, 123.94, 123.78, 122.17, 121.71, 120.79, 119.64, 119.42, 115.57, 112.18, 112.13, 55.12, 54.51, 28.69, 24.28. HRMS: calculated for C23H24N3O [M+H]+ 358.1919, found 358.1924. Purity: 95.0% by HPLC.
5-((2-methyl-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10b)
1H NMR (400 MHz, Chloroform-d) δ 10.51 (s, 1H), 9.06 – 8.84 (m, 1H), 8.70 – 8.31 (m, 2H), 7.55 – 7.25 (m, 3H), 7.21 – 7.08 (m, 1H), 7.01 (t, J = 7.5 Hz, 1H), 6.96 (s, 1H), 4.39 (s, 2H), 3.86 (s, 2H), 2.63 (d, J = 6.1 Hz, 4H), 1.99 – 1.64 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 151.58, 148.21, 139.74, 135.50, 132.17, 132.09, 129.01, 127.09, 127.02, 126.27, 120.83, 120.76, 119.07, 118.44, 118.28, 110.39, 109.07, 57.81, 53.62, 26.51, 23.65, 11.89. HRMS: calculated for C24H26N3O [M+H]+ 372.2076, found 372.2075. Purity: 97.1% by HPLC.
5-((2-ethyl-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10c)
1H NMR (400 MHz, Chloroform-d) δ 8.90 (dd, J = 4.2, 1.5 Hz, 1H), 8.46 (dd, J = 8.6, 1.5 Hz, 1H), 8.15 (s, 1H), 7.39 (dd, J = 8.5, 4.1 Hz, 1H), 7.34 (dt, J = 8.0, 1.3 Hz, 2H), 7.17 – 7.10 (m, 1H), 7.05 – 6.99 (m, 1H), 6.95 (s, 1H), 4.42 (s, 2H), 3.88 (s, 2H), 2.73 – 2.61 (m, 6H), 1.84 (h, J = 3.2 Hz, 4H), 1.19 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 151.56, 148.23, 137.86, 135.42, 131.98, 129.01, 127.04, 126.94, 126.33, 120.99, 120.74, 119.21, 118.48, 118.31, 110.39, 108.36, 57.73, 53.58, 26.27, 23.63, 19.60, 13.99. HRMS: calculated for C25H28N3O [M+H]+ 386.2232, found 386.2214. Purity: 97.4% by HPLC.
5-((4-fluoro-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10d)
1H NMR (400 MHz, Chloroform-d) δ 8.75 (dd, J = 4.1, 1.6 Hz, 1H), 8.35 – 8.24 (m, 1H), 8.22 (d, J = 16.3 Hz, 1H), 7.26 (dd, J = 8.5, 4.1 Hz, 1H), 7.19 (d, J = 4.0 Hz, 2H), 7.14 – 6.96 (m, 2H), 6.69 (ddd, J = 11.2, 7.4, 1.3 Hz, 1H), 6.43 (s, 1H), 4.48 (s, 2H), 3.98 (s, 2H), 2.75 (t, J = 5.3 Hz, 4H), 1.83 (p, J = 3.3 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 158.80, 156.31, 151.66, 148.24, 139.66, 139.24, 139.12, 133.02, 128.09, 127.00, 126.67, 122.89, 122.54, 122.46, 120.86, 118.08, 114.67, 107.34, 107.30, 104.62, 104.43, 57.33, 53.68, 29.04, 29.01, 23.63. HRMS: calculated for C23H23FN3O [M+H]+ 376.1825, found 376.1818. Purity: 98.0% by HPLC.
5-((4-nitro-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10e)
1H NMR (400 MHz, Chloroform-d) δ 9.42 (s, 1H), 8.71 (dd, J = 4.1, 1.6 Hz, 1H), 8.15 (dd, J = 8.5, 1.6 Hz, 1H), 7.76 (dd, J = 7.9, 0.9 Hz, 1H), 7.56 (dd, J = 8.1, 0.9 Hz, 1H), 7.23 – 7.16 (m, 2H), 7.13 (t, J = 8.0 Hz, 1H), 6.98 (s, 1H), 6.50 (s, 1H), 4.43 (s, 2H), 3.86 (s, 2H), 2.60 (q, J = 5.0, 4.0 Hz, 4H), 1.75 (p, J = 3.2 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 151.75, 148.20, 143.28, 139.64, 139.29, 133.11, 128.31, 128.06, 127.02, 126.56, 120.92, 120.57, 118.92, 118.25, 117.41, 115.04, 57.43, 53.63, 30.04, 23.61. HRMS: calculated for C23H23N4O3 [M+H]+ 403.1770, found 403.1776. Purity: 96.0% by HPLC.
5-((5-fluoro-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10f)
1H NMR (400 MHz, Methanol-d4) δ 8.76 (dd, J = 4.2, 1.6 Hz, 1H), 8.47 (dd, J = 8.6, 1.6 Hz, 1H), 7.43 (dd, J = 8.6, 4.2 Hz, 1H), 7.33 – 7.24 (m, 2H), 7.09 (dd, J = 9.9, 2.5 Hz, 1H), 6.88 (s, 1H), 6.83 (td, J = 9.1, 2.5 Hz, 1H), 4.41 (s, 2H), 3.98 (s, 2H), 2.79 – 2.62 (m, 4H), 1.86 (p, J = 3.3 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 149.30, 147.52, 138.66, 133.02, 132.87, 128.79, 127.09, 126.14, 125.98, 125.34, 120.91, 119.97, 113.96, 112.33, 109.07, 108.82, 103.41, 103.18, 53.73, 53.40, 27.44, 23.10. HRMS: calculated for C23H23FN3O [M+H]+ 376.1825, found 376.1819. Purity: 96.2% by HPLC.
1-((8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)methyl)-1H-indole-5-carbonitrile (10g)
1H NMR (400 MHz, Chloroform-d) δ 8.90 (dd, J = 4.2, 1.6 Hz, 1H), 8.05 – 7.98 (m, 2H), 7.46 (d, J = 1.1 Hz, 2H), 7.37 (dd, J = 8.6, 4.1 Hz, 1H), 7.05 (d, J = 3.3 Hz, 1H), 6.97 (s, 1H), 6.57 (d, J = 3.3 Hz, 1H), 5.63 (d, J = 0.8 Hz, 2H), 3.95 (s, 2H), 2.78 – 2.59 (m, 4H), 1.88 (p, J = 3.3 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 148.92, 139.77, 137.78, 130.85, 129.83, 128.56, 128.16, 126.75, 126.29, 124.79, 121.79, 120.67, 120.19, 118.25, 110.31, 102.96, 102.87, 57.42, 53.75, 47.51, 23.66. HRMS: calculated for C24H23N4O [M+H]+ 383.1872, found 383.1889. Purity: 95.0% by HPLC.
5-((5-nitro-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10h)
1H NMR (400 MHz, Chloroform-d) δ 10.95 (s, 1H), 8.90 (dd, J = 4.1, 1.6 Hz, 1H), 8.59 (d, J = 2.2 Hz, 1H), 8.11 (dd, J = 9.1, 2.2 Hz, 1H), 8.03 (dd, J = 8.6, 1.6 Hz, 1H), 7.43 (d, J = 9.1 Hz, 1H), 7.36 (dd, J = 8.6, 4.1 Hz, 1H), 7.08 (d, J = 3.3 Hz, 1H), 6.97 (s, 1H), 6.66 (dd, J = 3.3, 0.8 Hz, 1H), 5.71 – 5.52 (m, 2H), 3.94 (s, 2H), 2.68 (td, J = 5.8, 4.9, 2.5 Hz, 4H), 1.94 – 1.77 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 154.00, 148.93, 141.88, 139.77, 139.02, 130.74, 130.72, 128.05, 128.01, 126.20, 121.72, 120.03, 118.53, 118.33, 117.47, 109.34, 104.44, 57.54, 53.74, 47.71, 23.66. HRMS: calculated for C23H23N4O3 [M+H]+ 403.1770, found 403.1773. Purity: 96.9% by HPLC.
Ethyl 1-((8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)methyl)-1H-indole-5-carboxylate (10i)
1H NMR (400 MHz, Chloroform-d) δ 10.22 (s, 1H), 9.53 (s, 1H), 8.82 (dd, J = 4.1, 1.5 Hz, 1H), 8.43 (d, J = 1.5 Hz, 1H), 8.25 (dd, J = 8.6, 1.6 Hz, 1H), 7.91 (dd, J = 8.6, 1.6 Hz, 1H), 7.35 (d, J = 8.6 Hz, 1H), 7.33 – 7.25 (m, 1H), 7.15 (s, 1H), 6.64 (s, 1H), 4.86 – 4.35 (m, 4H), 3.94 (s, 2H), 2.68 (d, J = 6.1 Hz, 4H), 2.15 – 1.72 (m, 4H), 1.42 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 167.92, 151.88, 148.20, 139.21, 132.87, 127.93, 127.02, 126.91, 125.98, 124.36, 123.23, 121.70, 121.51, 120.82, 118.48, 116.71, 111.02, 60.58, 57.72, 53.69, 27.89, 23.63, 14.50. HRMS: calculated for C26H28N3O3 [M+H]+ 430.2131, found 430.2146. Purity: 97.4% by HPLC.
5-((6-fluoro-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10j)
1H NMR (400 MHz, Chloroform-d) δ 8.89 (dd, J = 4.1, 1.6 Hz, 1H), 8.06 (dd, J = 8.6, 1.6 Hz, 1H), 7.55 (dd, J = 8.7, 5.4 Hz, 1H), 7.35 (dd, J = 8.6, 4.1 Hz, 1H), 7.07 (dd, J = 10.0, 2.3 Hz, 1H), 6.98 (s, 1H), 6.94 – 6.84 (m, 2H), 6.46 (dd, J = 3.3, 0.9 Hz, 1H), 5.52 (s, 2H), 3.94 (s, 2H), 2.84 – 2.54 (m, 4H), 1.87 (p, J = 3.3 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 153.67, 148.82, 131.04, 127.98, 127.94, 127.90, 126.34, 125.19, 121.84, 121.74, 121.59, 120.87, 118.40, 108.56, 108.31, 101.92, 96.07, 95.80, 57.58, 53.74, 47.44, 23.66. HRMS: calculated for C23H23FN3O [M+H]+ 376.1825, found 376.1820. Purity: 97.2% by HPLC.
5-((6-chloro-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10k)
1H NMR (400 MHz, Chloroform-d) δ 8.89 (dd, J = 4.2, 1.6 Hz, 1H), 8.06 (dd, J = 8.6, 1.6 Hz, 1H), 7.55 (dd, J = 8.5, 0.6 Hz, 1H), 7.42 – 7.35 (m, 2H), 7.16 – 7.04 (m, 2H), 6.93 (d, J = 3.2 Hz, 1H), 6.46 (dd, J = 3.3, 0.9 Hz, 1H), 5.56 (s, 2H), 4.03 (s, 2H), 2.79 (s, 4H), 1.91 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 151.11, 148.11, 138.50, 136.38, 132.16, 129.92, 128.93, 127.05, 126.04, 125.73, 122.16, 121.81, 121.72, 119.44, 118.55, 110.18, 101.40, 53.22, 53.10, 46.64, 22.98. HRMS: calculated for C23H23ClN3O [M+H]+ 392.1530, found 392.1532. Purity: 95.2% by HPLC.
5-((6-bromo-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10l)
1H NMR (400 MHz, Chloroform-d) δ 10.37 (s, 1H), 8.88 (dd, J = 4.1, 1.6 Hz, 1H), 8.02 (dd, J = 8.5, 1.6 Hz, 1H), 7.60 – 7.53 (m, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.34 (dd, J = 8.5, 4.1 Hz, 1H), 7.23 (dd, J = 8.4, 1.7 Hz, 1H), 6.95 (s, 1H), 6.87 (d, J = 3.2 Hz, 1H), 6.44 (dd, J = 3.3, 0.9 Hz, 1H), 5.51 (s, 2H), 3.93 (s, 2H), 2.68 (p, J = 3.9, 3.5 Hz, 4H), 1.86 (p, J = 3.3 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 153.71, 148.82, 139.75, 137.18, 131.02, 128.14, 128.04, 127.64, 126.29, 122.99, 122.32, 121.60, 120.71, 118.44, 115.40, 112.50, 102.02, 57.55, 53.74, 47.35, 23.67. HRMS: calculated for C23H23BrN3O [M+H]+ 436.1024, found 436.1041. Purity: 98.8% by HPLC.
5-((6-nitro-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10m)
1H NMR (400 MHz, Chloroform-d) δ 9.54 (s, 1H), 8.91 (dd, J = 4.1, 1.6 Hz, 1H), 8.44 (d, J = 1.9 Hz, 1H), 8.03 (ddd, J = 10.6, 8.7, 1.8 Hz, 2H), 7.68 (d, J = 8.8 Hz, 1H), 7.36 (dd, J = 8.5, 4.1 Hz, 1H), 7.18 (d, J = 3.1 Hz, 1H), 7.07 (s, 1H), 6.57 (dd, J = 3.2, 0.9 Hz, 1H), 5.68 (s, 2H), 3.97 (s, 2H), 2.80 – 2.58 (m, 4H), 1.88 (p, J = 3.3 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 154.18, 148.98, 143.15, 139.82, 134.90, 133.59, 133.14, 130.85, 128.54, 126.33, 121.78, 121.01, 119.85, 118.49, 115.28, 106.50, 102.79, 57.57, 53.77, 47.81, 23.68. HRMS: calculated for C23H23N4O3 [M+H]+ 403.1770, found 403.1760. Purity: 96.6% by HPLC.
5-((6-methoxy-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10n)
1H NMR (400 MHz, Chloroform-d) δ 8.84 (dd, J = 4.1, 1.6 Hz, 1H), 8.27 (dd, J = 8.5, 1.6 Hz, 1H), 7.90 (s, 1H), 7.46 (d, J = 8.7 Hz, 1H), 7.28 (dd, J = 8.6, 4.1 Hz, 1H), 7.15 (s, 1H), 6.84 (d, J = 2.2 Hz, 1H), 6.78 (dd, J = 8.6, 2.3 Hz, 1H), 6.48 (dt, J = 2.4, 1.2 Hz, 1H), 4.37 (s, 2H), 3.94 (s, 2H), 3.84 (s, 3H), 2.78 – 2.57 (m, 4H), 1.85 (p, J = 3.2 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 156.64, 151.75, 148.30, 139.78, 137.20, 132.77, 127.84, 127.02, 126.12, 121.79, 121.39, 120.68, 119.51, 118.59, 115.79, 109.35, 94.71, 57.79, 55.71, 53.78, 28.13, 23.67. HRMS: calculated for C24H26N3O2 [M+H]+ 388.2025, found 388.2027. Purity: 96.3% by HPLC.
5-((7-fluoro-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10o)
1H NMR (400 MHz, DMSO-d6) δ 11.34 (s, 1H), 8.81 (dd, J = 4.2, 1.5 Hz, 1H), 8.53 (dd, J = 8.6, 1.6 Hz, 1H), 7.65 – 7.40 (m, 2H), 7.40 – 7.24 (m, 1H), 7.12 (s, 1H), 7.02 – 6.68 (m, 2H), 4.41 (s, 2H), 3.80 (s, 2H), 2.50 (d, J = 6.3 Hz, 4H), 1.68 (h, J = 3.2 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 149.39, 147.52, 138.67, 132.80, 128.81, 126.06, 125.99, 124.43, 124.21, 124.08, 120.93, 119.94, 118.50, 118.44, 114.96, 114.94, 114.92, 105.83, 105.67, 53.80, 53.37, 27.42, 23.13. HRMS: calculated for C23H23FN3O [M+H]+ 376.1825, found 376.1826. Purity: 96.8% by HPLC.
5-((7-methyl-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10p)
1H NMR (400 MHz, Chloroform-d) δ 9.74 (s, 1H), 8.84 (dd, J = 4.2, 1.6 Hz, 1H), 8.28 (dd, J = 8.6, 1.6 Hz, 1H), 8.18 (s, 1H), 7.50 (d, J = 7.7 Hz, 1H), 7.30 – 7.24 (m, 1H), 7.19 (s, 1H), 7.11 – 6.99 (m, 2H), 6.57 (d, J = 2.0 Hz, 1H), 4.41 (s, 2H), 3.95 (s, 2H), 2.82 – 2.62 (m, 4H), 2.47 (s, 3H), 1.97 – 1.78 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 151.66, 148.24, 139.72, 136.04, 132.86, 127.96, 127.07, 126.84, 126.36, 122.60, 122.55, 120.77, 120.43, 119.56, 118.27, 116.56, 116.05, 57.47, 53.70, 28.15, 23.64, 16.58. HRMS: calculated for C24H26N3O [M+H]+ 372.2076, found 372.2063. Purity: 95.1% by HPLC.
5-((2,3-dimethyl-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10q)
1H NMR (400 MHz, Chloroform-d) δ 10.57 (s, 1H), 8.94 (dd, J = 4.2, 1.6 Hz, 1H), 8.28 (dd, J = 8.6, 1.6 Hz, 1H), 7.57 (dd, J = 8.2, 1.7 Hz, 1H), 7.45 (dd, J = 8.5, 4.2 Hz, 1H), 7.20 – 6.97 (m, 3H), 6.27 (s, 1H), 5.54 (d, J = 1.2 Hz, 2H), 3.75 (s, 2H), 2.72 – 2.46 (m, 4H), 2.33 (s, 3H), 2.24 (s, 3H), 1.79 (p, J = 3.2 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 152.79, 148.63, 136.57, 132.66, 130.54, 128.85, 125.44, 124.48, 122.49, 121.21, 120.86, 118.93, 118.14, 118.08, 108.86, 107.25, 57.51, 53.38, 43.50, 23.59, 10.16, 8.98. HRMS: calculated for C25H28N3O [M+H]+ 386.2232, found 386.2240. Purity: 97.7% by HPLC.
5-((5-methoxy-2-methyl-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10r)
1H NMR (400 MHz, Chloroform-d) δ 10.18 (s, 1H), 8.87 (dd, J = 4.2, 1.5 Hz, 1H), 8.41 (dd, J = 8.5, 1.6 Hz, 1H), 8.20 (s, 1H), 7.36 (dd, J = 8.5, 4.1 Hz, 1H), 7.23 – 7.07 (m, 1H), 6.93 (s, 1H), 6.74 (dt, J = 3.9, 1.9 Hz, 2H), 4.32 (s, 2H), 3.85 (s, 2H), 3.69 (s, 3H), 2.84 – 2.41 (m, 4H), 2.22 (s, 3H), 1.93 – 1.70 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 153.84, 151.55, 148.23, 133.02, 132.05, 130.56, 129.45, 127.11, 126.99, 126.11, 120.78, 118.19, 110.94, 110.51, 108.94, 100.76, 60.40, 57.64, 55.84, 53.58, 26.55, 23.61, 14.21, 11.99. HRMS: calculated for C25H28N3O2 [M+H]+ 402.2182, found 402.2185. Purity: 97.6% by HPLC.
ethyl 5-fluoro-1-((8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)methyl)-1H-indole-2-carboxylate (10s)
1H NMR (400 MHz, Chloroform-d) δ 9.11 (s, 1H), 8.89 (dd, J = 4.1, 1.6 Hz, 1H), 8.50 (dd, J = 8.6, 1.7 Hz, 1H), 7.50 – 7.40 (m, 1H), 7.39 – 7.31 (m, 1H), 7.10 – 6.92 (m, 3H), 4.83 (s, 2H), 4.39 (q, J = 7.1 Hz, 2H), 3.92 (s, 2H), 2.81 – 2.57 (m, 4H), 1.85 (p, J = 3.2 Hz, 4H), 1.28 (t, J = 7.1 Hz, H). 13C NMR (101 MHz, Chloroform-d) δ 162.08, 151.67, 148.29, 139.62, 132.67, 131.99, 127.19, 127.03, 125.53, 125.36, 120.92, 118.13, 114.82, 114.56, 112.84, 112.75, 106.00, 105.77, 61.03, 57.34, 53.58, 27.27, 23.60, 14.30. HRMS: calculated for C26H27FN3O3 [M+H]+ 448.2036, found 448.2050. Purity: 97.2% by HPLC.
2-(1-((8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)methyl)-6-methoxy-1H-indol-3-yl)acetonitrile (10t)
1H NMR (400 MHz, DMSO-d6) δ 10.73 (s, 1H), 8.83 (dd, J = 4.1, 1.5 Hz, 1H), 8.46 (dd, J = 8.5, 1.6 Hz, 1H), 7.51 (dd, J = 8.5, 4.1 Hz, 1H), 7.45 (d, J = 8.5 Hz, 1H), 7.27 (s, 1H), 6.82 (d, J = 2.3 Hz, 1H), 6.71 (dd, J = 8.6, 2.3 Hz, 1H), 4.48 (s, 2H), 4.01 (s, 2H), 3.79 (s, 2H), 3.73 (s, 3H), 2.65 – 2.36 (m, 4H), 1.66 (h, J = 3.1 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 155.56, 150.07, 147.70, 136.18, 134.12, 132.38, 128.89, 126.01, 123.74, 121.44, 121.14, 120.12, 119.12, 118.06, 108.84, 99.92, 94.63, 55.17, 54.86, 54.25, 53.41, 27.93, 23.16, 12.18. HRMS: calculated for C26H27N4O2 [M+H]+ 427.2134, found 427.2135. Purity: 95.6% by HPLC.
5-((6-chloro-5-fluoro-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10u)
1H NMR (400 MHz, DMSO-d6) δ 11.04 (s, 1H), 8.80 (dd, J = 4.2, 1.5 Hz, 1H), 8.50 (dd, J = 8.6, 1.6 Hz, 1H), 7.64 – 7.44 (m, 3H), 7.40 (d, J = 10.3 Hz, 1H), 7.18 (d, J = 1.7 Hz, 1H), 4.38 (s, 2H), 3.80 (s, 2H), 2.60 – 2.40 (m, 4H), 1.69 (q, J = 3.7, 3.3 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 149.38, 147.54, 138.68, 132.74, 128.79, 126.18, 125.89, 120.94, 120.04, 114.37, 112.36, 104.96, 104.73, 53.75, 53.39, 27.23, 23.11. HRMS: calculated for C23H22ClFN3O [M+H]+ 410.1435, found 410.1440. Purity: 95.2% by HPLC.
5-((5,6-dichloro-1H-indol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (10v)
1H NMR (400 MHz, DMSO-d6) δ 11.15 (s, 1H), 8.82 (d, J = 4.2 Hz, 1H), 8.52 (d, J = 8.5 Hz, 1H), 7.67 (s, 1H), 7.59 (s, 1H), 7.55 – 7.38 (m, 2H), 7.22 (s, 1H), 4.41 (s, 2H), 3.82 (s, 2H), 2.51 (d, J = 6.7 Hz, 4H), 1.76 – 1.45 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 149.67, 147.74, 138.71, 135.34, 132.96, 128.96, 127.06, 126.24, 126.00, 123.27, 121.20, 120.92, 119.93, 113.89, 113.00, 53.61, 53.47, 27.26, 23.15. HRMS: calculated for C23H22Cl2N3O [M+H]+ 426.1140, found 426.1156. Purity: 96.9% by HPLC.
General procedure for synthesis of 5a–5f
To a stirred solution of compound 4 (1.0 mmol, 140 mg) or 5-amino-8-hydroxyquinoline dihydrochloride and carboxylic acid (1.2 mmol) in DMF-H2O (1:5) were added oxyma (2.0 mmol, 284 mg), EDCI (2.0 mmol, 310 mg), and NaHCO3 (6.0 mmol, 636 mg). After stirring at room temperature for 12 h, the solution was extracted with EtOAc. The combined organic layers were washed with brine (3 mL), and then dried over Na2SO4. Concentration of organic phase gave crude product which was directly used for next step without purification.
Synthesis of 5-(azidomethyl)quinolin-8-ol (3)
To a stirred solution of 5-chloromethyl-8-quinolinol hydrochloride (10 mmol, 2.3 g) in acetone (20 mL) were added sodium azide (30 mmol, 1.95 g) at room temperature. After refluxed for 20 h, the mixture was filtered and the residue was washed with acetone. Combined organic solvent was removed under reduced pressure to give crude product which was purified with flash chromatography on silica. Elution with hexanes/ethylacetate (10:1–1:1) gave desired compound 3 (1.54 g, 77%). 1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.92 (dd, J = 4.1, 1.6 Hz, 1H), 8.50 (dd, J = 8.6, 1.6 Hz, 1H), 7.67 (dd, J = 8.6, 4.1 Hz, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.08 (d, J = 7.8 Hz, 1H), 4.83 (s, 2H).
Synthesis of 5-(aminomethyl)quinolin-8-ol (4)
A suspension of azide 3 (2.00 g, 10 mol) and 10% Pd/C (0.15 g) in ethylacetate (15 mL) was hydrogenated overnight, the reaction mixture was filtered off and washed with dichloromethane-methanol (1:1). The combined filtration was evaporated under vacuum to give the oily crude which was purified with flash chromatography on silica. Compound 4 was eluted out with dichloromethane/methanol (15:0–10:1) (1.18 g, 68%). 1H NMR (400 MHz, DMSO-d6) δ 8.86 (dd, J = 4.1, 1.6 Hz, 1H), 8.57 (dd, J = 8.6, 1.6 Hz, 1H), 7.58 (dd, J = 8.5, 4.1 Hz, 1H), 7.44 (dd, J = 7.8, 0.9 Hz, 1H), 7.02 (d, J = 7.8 Hz, 1H), 4.10 (d, J = 0.8 Hz, 2H).
Synthesis of 1-(4-bromobenzyl)-3-((8-hydroxyquinolin-5-yl)methyl)urea (7)
To a stirred solution of compound 4 (1.0 mmol, 140 mg) and 4-bromobenzyl isocyanate (1.0 mmol, 212 mg) in anhydrous dichloromethane (5 mL) were added catalytic amount of trimethylamine (0.1 mmol, 10.1 mg). After stirring at room temperature for 5 h, solvent was removed under reduced pressure to give crude product which was directly used for next step without purification.
2.3. Cell culture and reagents
Human melanoma A375, M14, WM164, RPMI7951, and M14/MDR1 cell lines were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA), and cultured in DMEM media (Mediatech, Inc., Manassas, VA) at 37 °C in a humidified atmosphere containing 5% CO2. The culture media were supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 1% antibiotic-antimycotic mixture (Sigma-Aldrich, St. Louis, MO). Compounds were dissolved in dimethylsulfoxide (DMSO; Sigma-Aldrich) to make a stock solution of 10 mM. Compound solutions were freshly prepared by diluting stocks with cell culture medium before use (final solution contained less than 0.5% DMSO). 5000 cells in logarithm growing phase were seeded overnight into each well of a 96-well plate. Then the cells were continuously incubated for 48 h with sequential diluted compound solution (100 μM to 3 nM, 100 μL per well) in cell culture medium. The cell viability was determined in MTS assay and IC50 was calculated (n = 4), following similar procedures as described previously [25, 29–31]. Dulbecco’s modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin and trypsin 0.25% were purchased from Hyclone (GE Healthcare Life Science, Pittsburgh, PA). Phosphate buffered saline (PBS) was purchased from Invitrogen GIBCO (Grand Island, NY). Dimethyl sulfoxide (DMSO) and 3-(4,5-dimethylthiazole-2-yl)-2,5-biphenyl tetrazolium bromide (MTT) were purchased from Sigma Chemical Co (St. Louis, MO). The P-glycoprotein (P-gp) overexpressing KB-C2 cell line was established from a parental human epidermoid carcinoma cell line KB-3-1, by a step-wise selection of KB-3-1 in increasing concentrations of colchicine up to 2 μg/mL [32]. SW620/Ad300, which is also a P-gp overexpressing drug resistant cell line, was established by stepwise exposure of the parental human colon cancer cell line SW620 to increasing concentrations of doxorubicin up to 300 ng/mL[33]. The KB-3-1 and KB-C2 cell lines were generously provided by Dr. Shin-Ichi Akiyama (Kagoshima University, Japan), and the SW620 and SW620/Ad300 cell lines were kindly provided by Dr. Susan E. Bates (Columbia University, NY, USA) and Dr. Robert W. Robey (NIH, MD, USA). All the cell lines were grown in DMEM supplemented with 10% FBS and 100 unit/mL penicillin/streptomycin in a humidified incubator containing 5% CO2 at 37 °C.
2.4. Cytotoxicity assay
A375, M14, WM164, RPMI7951, and M14/MDR1 were seeded in 96-well plates at a concentration of 1,000–5,000 cells per well, depending on growth rate of the cell line. After overnight incubation, the media was replaced and cells were treated with the test compounds at 10 concentrations ranging from 0.03 nM to 1 μM plus a vehicle control for 72 h in four replicates. Following treatment, the MTS reagent (Promega, Madison, WI) was added to the cells and incubated in dark at 37°C for at least 1 h. Absorbance at 490 nm was measured using a plate reader (DYNEX Technologies, Chantilly VA). Percentages of cell survival versus drug concentrations were plotted, and the IC50 (concentration that inhibited cell growth by 50% of untreated control) values were obtained by nonlinear regression analysis using GraphPad Prism (GraphPad Software, San Diego, CA).
2.5. Cytotoxicity against P-gp overexpressing cell lines by MTT assay
The MTT colorimetric assay was used to measure the sensitivity of the cells against the synthesized compounds. The assay detects the formazan product formed from the reduction of MTT in active cells thus assesses the cell viability[34]. Cells were seeded in 96-well plates at 5000 cells/well (KB-3-1 or KB-C2 cells) or at 7,000 cells/well (SW620 or SW620/Ad300 cells) in 180 μL completed medium and cultured overnight. Then various concentrations of the compounds (20 μL) were added to the designated wells. After 72 h continuous drug incubation, 20 μL of MTT reagent (4 mg/mL) was added to each well and the plates were incubated at 37 °C for 4 h. Subsequently, the medium was removed and 100 μL of DMSO were added to dissolve the formazan crystals in each well. The absorbance was determined at 570 nm by the accuSkan™ GO UV/Vis Microplate Spectrophotometer (Fisher Sci., Fair Lawn, NJ). The IC50 values of each compound on each cell line were calculated from the survival curves to represent the cytotoxicity of the compounds. The fold of drug resistance was calculated by dividing the IC50 of the P-gp overexpressing cells by that of the parental cells. Two known P-gp substrates, YM155 and paclitaxel, were used as positive controls for P-gp overexpressing cell lines. On the other hand, cisplatin, which is not a substrate of P-gp, was used as negative control.
2.6. Liver microsomes stability assay
NADPH regenerating agent solutions A (catalog#: 451220) and B (catalog#: 451200) and mouse liver microsomes (CD-1, mixture of male, catalog#:452701, and female, catalog#: 452702) were obtained from BD Gentest (Woburn, MA). Liver microsomes stability assay was conducted following literature reports [35, 36]. For each test compound, the mouse liver microsomal solution was prepared by adding 0.058 mL of concentrated mouse liver microsomes (20 mg/mL protein concentration) to 1.756 mL of 0.1 M potassium phosphate buffer (pH 7.4) containing 5 μL of 0.5 M EDTA to make a 0.6381 mg/mL (protein) microsomal solution. Each test compound (2.2 μL of 10 mM DMSO solution) was added directly to 1.79 mL of mouse liver microsomal solution and 90 μL was transferred to wells in 96-well plates (0, 0.25, 0.5, 1, 2, and 4 h time points each in triplicate). The NADPH regenerating agent was prepared by mixing 0.113 mL of NADPH regenerating agent Solutions A, 0.023 mL of solution B and 0.315 mL of 0.1 M potassium phosphate buffer (pH 7.4) for each tested compound. To each well of the 96-well plate, 22.5 μL of the NADPH regenerating agent was added to initiate the reaction, and the plate was incubated at 37 °C for each time point (0, 0.25, 0.5, 1, 2, and 4 h time points each in triplicate). The reaction was quenched by adding 225 μL of cold acetonitrile containing warfarin (4 mg/mL) as internal control to each well. All of the plates were centrifuged at 4,000 rpm for 20 min and the supernatants (100 μL) were transferred to another 96-well plates for analysis on UPLC–MS (Waters Acquity UPLC linked to Waters Acquity Photodiode Array Detector and Waters Acquity Single Quadrupole Mass Detector) on Acquity UPLC BEH C18 1.7 mm (2.1x50 mm) column by running 90–5% gradient for water (+0.1% formic acid) and acetonitrile (+0.1% formic acid) in 2 min. The area under the single ion recording (SIR) channel for the test compound divided by the area under the SIR for internal control at 0 time concentration was considered as 100% to calculate remaining concentration at each time point. The terminal phase rate constant (ke) was estimated by linear regression of logarithmic transformed concentration versus the data, where ke = slope × (−ln10). The half life t1/2 was calculated as ln2/ke. The intrinsic clearance (CLint,app)= (0.693/in vitro t1/2) x (1 mL incubation volume/0.5 mg of microsomal protein) × (45 mg microsomal protein/gram of liver) x (55 g of liver/kg body weight) [37, 38].
2.7. Western blotting
To determine the change of protein levels of survivin and closely related IAPs, lysates of A375 or M14 melanoma cells treated by the compound solution for 24 h were used for western blotting analysis. Primary rabbit antibodies against IAP proteins including survivin (#2808), XIAP (#2045), cIAP1 (#7065), Livin (#5471), Cleaved PARP (#9185) and the loading control protein GAPDH (HRP Conjugate) (#3683) were purchased from Cell Signaling Technology, Inc. (Danvers, MA) and used according to manufacture instructions as reported previously.
2.8. Molecular modeling
The molecular docking studies were conducted in Schrodinger Molecular Modeling Suite 2014 (Schrodinger Inc., Portland, OR) following previously described procedures [25, 29]. Ligand was prepared to generate various conformation before being docked into the SMAC AVPI binding site of a human survivin crystal structure (Protein Data Bank entry: 3UIH). Prior to molecular dynamic calculation, the docking to minimize the energy of potential ligand binding poses was performed. Results were visualized using the Maestro interface of the Schrodinger software.
2.9. In vivo xenograft model
All animal experiments were performed in accordance with the NIH animal use guidelines and protocol approved by the Institutional Animal Care and Use Committee at the University of Tennessee Health Science Center. Nude mice, 6–8 weeks old, were purchased from Envigo (Indianapolis, IN).
Logarithmic growth phase A375 cells (5 × 107 cells per mL) were prepared in phenyl red-free, FBS-free media and mixed with Matrigel immediately before injecting into mice. Tumors were established by injecting 100 μL of this mixture subcutaneously in the dorsal flank of each mouse (2.5 × 106 cells). After tumor volumes reached approximately 150 mm3 mice were randomized into control or treatment groups (n=7~8). 10f or paclitaxel was dissolved in a 1:1 ratio of PEG300: PBS solution to produce desired concentrations. The vehicle control solution was formulated with equal parts PEG300 and PBS only. 100 μl of the drug treatment or vehicle control was administered via i.p. injection every other day two weeks.
Tumor volume was measured three times a week with a caliper and calculated by using the formula a×b2×0.5, where a and b represented the larger and smaller diameters, respectively. Tumor growth inhibition (TGI) at the conclusion of the experiments was calculated as 100 – 100 ×((T − T0)/(C − C0)), where T, T0, C and C0 are the mean tumor volume for the specific group on the last day of treatment, mean tumor volume of the same group on the first day of treatment, mean tumor volume for the vehicle control group on the last day of treatment and mean tumor volume for the vehicle control group on the first day of treatment, respectively. Animal activity and body weights were monitored during the entire experiment period to assess potential acute toxicity. At the end of the experiment, mice were sacrificed and the tumors were weighed. Tumors and tissues were dissected out and preserved in 10% buffered formalin phosphate solution.
3. Results and Discussion
3.1. Chemistry
Scheme 1 showed the syntheses of UC-112 analogs 2a–2d. Starting material 5-chloromethyl-8-quinolinol hydrochloride was synthesized by following a reported procedure [29] and was treated with commercially available amines in the presence of sodium hydride or sodium carbonate and potassium iodide to give 1a–1d. 1a–1d was then refluxed with paraformaldehyde and pyrrolidine in ethanol to provide analogs 2a–2d.
Scheme 1. Synthesis of 2a–2d.

Reagents and conditions: (a) NaH, DMF or KI, Na2CO3, MeCN, reflux; (b) parafomaldehyde, pyrrolidine, ethanol, reflux.
Scheme 2 depicted the synthesis to obtain analogs 6a–6f and 8. 5-chloromethyl-8-quinolinol hydrochloride was refluxed with sodium azide in acetone to yield azide 3. 3 was subsequently hydrogenated in the presence of a catalytic equivalent of 10% Pd/C to provide amine 4. Treatment of 4 with carboxylic acids, EDCI and ethyl cyanohydroxyiminoacetate (oxyma) was able to generate amide 5a–5d. Refluxing the mixture of amide, paraformaldehyde and pyrrolidine in ethanol successfully gave the amide analogs 6a–6d. Analogs 6e and 6f were synthesized by following the same method starting from commercially available 5-amino-8-hydroxyquinoline dihydrochloride. Treatment of amine 4 using a catalytic equivalent of triethylamine generated the urea intermediate 7, which was subjected to Mannich reaction to give urea analog 8.
Scheme 2. Synthesis of 6a–6f and 8.

Reagents and conditions:(a) NaN3, acetone, reflux; (b) H2, Pd/C(10%), ethyl acetate; (c) Et3N, DCM; (d): parafomaldehyde, pyrrolidine, ethanol, reflux; (e) Oxyma, EDCI, NaHCO3, DMF-H2O.
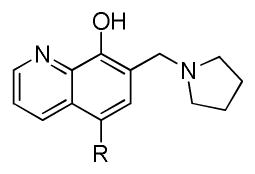
Indole analogs 10a–10v with either mono-substituent or di-substituent were prepared by following Scheme 3. Briefly, 5-chloromethyl-8-quinolinol hydrochloride was refluxed together with indoles in the presence of sodium carbonate and a catalytic amount of potassium iodide in acetonitrile to generate intermediate 9a–9v. Treatment of 9a–9v with paraformaldehyde and pyrrolidine subsequently furnished analogs 10a–10v.
Scheme 3. Synthesis of 10a–10v.

Reagents and conditions: (a) NaH, DMF or KI, Na2CO3, MeCN, reflux; (b) parafomaldehyde, pyrrolidine, ethanol, reflux.
3.2. In vitro anti-proliferative assay
To reduce the flexibility of benzyloxy in UC-112, we replaced it with different substructures and have synthesized thirty-three new UC-112 analogs. All analogs were evaluated using MTS assays for their cytotoxicity in human melanoma cell lines including A375, WM1641, M14, RPMI7951, and M14/MDR1. IC50 values were reported in μM and calculated from at least three independent experiments, each performed in duplicates.
3.2.1. In vitro growth inhibitory effects of UC-112 analogs with modification of benzyloxy moiety
Our preliminary SAR investigation of UC-112 emphasized the replacement of the benzyloxy with other substructures and has led to the syntheses of analogs 2a–2d, 6a–6f, 8 and 10a. Their in vitro assay result was shown in Table 1. Compared to the reference compound MX-106, 2a (5-fluoroindazole analog), 2b (benzotriazole analog) and 2c (4-chloropurine analog) exhibited 3~5-fold reduced activities. Replacement of the benzyloxy with the other moiety as shown in compounds 2d resulted in significant reduction of inhibitory effects (> 3-fold). Replacing the benzyloxy with phenyl acetamides (analogs 6a–6d) was detrimental to the antiproliferative activities. Shortening the linkage by one methylene in 6b and 6d resulted in analogs 6e–6f and was revealed to not affect the activities. The introduction of a benzylurea (analog 8) dramatically attenuated the antiproliferative activity. Among all analogs in this series, non-substituted indole analog 10a, exhibited the most potent activity and was comparable to that of MX-106, having an average IC50 of 0.9 μM.
Table 1.
In vitro growth inhibitory effects (μM) of UC-112 analogs with modification of benzyloxy moiety.
| |||||||
|---|---|---|---|---|---|---|---|
| compound | R | A375 | WM164 | RPMI7951 | M14 | M14/MDR1 | RIb |
| 2a |
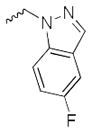
|
3.6±0.2 | 8.6±1.1 | 11.1±0.7 | 5.4±0.3 | 7.0±0.1 | 1.3 |
| 2b |
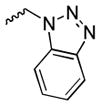
|
1.9±0.1 | >30.0 | 6.5±0.2 | 2.3±0.1 | 3.1±0.1 | 1.3 |
| 2c |
|
3.9±0.1 | 56.4±1.1 | 17.4±1.2 | 4.1±0.2 | 7.9±0.1 | 1.9 |
| 2d |
|
14.1±0.7 | 33.9±1.1 | 21.0±0.4 | 11.5±0.4 | 14.3±0.2 | 1.2 |
| 6a |
|
>30.0 | 13.0 ±0.5 | >30.0 | >30.0 | >30.0 | NDa |
| 6b |
|
4.7 ±0.2 | 3.7 ±0.3 | 26.2±6.2 | 5.1±0.1 | 10.7±1.2 | 2.1 |
| 6c |
|
3.5 ±0.3 | 2.8 ±0.2 | 18.4±0.3 | 8.6±0.2 | 7.4±1.2 | 0.9 |
| 6d |
|
1.5 ±0.1 | 1.6 ±0.1 | 12.9±0.2 | >30.0 | 12.2±0.2 | ND |
| 6e |
|
6.0±1.3 | 24.9 ± 1.0 | 25.3±2.4 | 6.7±0.3 | 12.2±0.5 | 1.8 |
| 6f |
|
2.9±0.15 | ND | 6.8±0.6 | 2.4±0.1 | 3.3±0.2 | 1.4 |
| 8 |
|
>30.0 | 20.18±0.49 | 17.3±0.3 | >30.0 | 8.7±0.2 | ND |
| 10a |
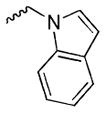
|
0.8 ±0.1 | 0.9 ±0.1 | 4.4±0.3 | 0.8 ±0.1 | 2.1±0.1 | 2.6 |
| UC-112 |
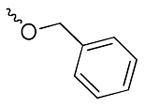
|
1.9 ±0.6 | ND | ND | 2.1 ±0.4 | 3.2 ±0.5 | 1.5 |
| MX-106 |
|
0.9 ±0.1 | 1.1 ±0.2 | ND | 0.8 ±0.2 | 1.8 ±0.4 | 2.3 |
ND: Not Determined.
Resistance index (RI) is calculated by dividing IC50 values on multidrug-resistant cell line M14/MDR1 by IC50 values on the matching sensitive parental cell line M14.
3.2.2. In vitro growth inhibitory effects of UC-112 analogs with mono- or di-substitution on the indole moiety
Because the indole analog 10a had an equipotency to that of MX-106, we subsequently focused our effort to investigate the substitutional effect on the indole ring. Analogs 10b–10p with mono-substituent and analogs 10q–10v with di-substituents were then synthesized.
The in vitro assay result for analogs 10a–10p was shown in Table 2. For the 2-position substitutued analogs, 10b with a methyl group and 10c with an ethyl group showed slightly reduced activities compared to 10a. Introducing substituents to the 4-position on the indole diminished the antiproliferative activity, which was demonstrated by analogs 10d and 10e. For analogs that had substituents on the 5-position of indole, comparable activities to that of 10a were observed, for example, compounds 10f, 10g and 10h had IC50 values ranging from 0.7 to 1.1 μM; analog 10i that had bulky ester functional group showed ~2-fold reduced activity in comparison with MX-106. In the series of analogs with substituents on the 6-position of the indole (10j–10n), they were generally equipotent to MX-106 and 10a, having IC50 values as low as 0.7 μM. Analogs having substituents on the 7-position of indole were slightly less potent than corresponding 5- or 6-position substituted counterparts.
Table 2.
In vitro growth inhibitory effects (μM) of UC-112 analogs with mono-substituent on the indole moiety.
| structure | compound | R | A375 | WM164 | RPMI7951 | M14 | M14/MDR1 | RIb |
|---|---|---|---|---|---|---|---|---|
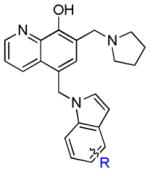
|
10a | H | 0.8 ±0.1 | 0.9 ±0.1 | 4.4±0.3 | 0.8 ±0.1 | 2.1±0.1 | 2.6 |
| 10b | 2-Me | 2.2 ±0.2 | 7.5±1.2 | 2.2±0.2 | 1.4 ±0.2 | 0.7±0.1 | 0.5 | |
| 10c | 2-Et | 2.0 ±0.1 | 8.9±1.4 | 3.2±0.4 | 1.5 ±0.2 | 1.1±0.1 | 0.7 | |
| 10d | 4-F | 1.6 ±0.3 | 6.5±0.6 | 2.2±0.2 | 1.2 ±0.2 | 0.8±0.1 | 0.7 | |
| 10e | 4-NO2 | 1.8 ±0.1 | 7.0±0.3 | 0.6±0.1 | 2.1 ±0.1 | 0.6±0.1 | 0.3 | |
| 10f | 5-F | 0.7±0.2 | 0.7±0.2 | NDa | 1.9±0.4 | ND | ND | |
| 10g | 5-CN | 1.0 ±0.1 | 1.1 ±0.1 | ND | 5.1±0.3 | ND | ND | |
| 10h | 5-NO2 | 0.7 ±0.1 | 0.8 ±0.1 | 3.7±0.3 | ND | 2.2±0.1 | ND | |
| 10i | 5-COOEt | 1.9 ±0.2 | 4.7±0.6 | 1.5±0.1 | 2.4±0.2 | 0.8±0.1 | 0.3 | |
| 10j | 6-F | 0.9±0.1 | 0.7±0.1 | 2.7±0.5 | 1.3±0.1 | 1.3±0.1 | 1.0 | |
| 10k | 6-Cl | 0.9±0.4 | 0.9±0.4 | 7.7±0.5 | 0.8±0.4 | 3.8±0.1 | 4.8 | |
| 10l | 6-Br | 0.9±0.1 | 1.4 ±0.2 | 5.3±0.6 | 3.5±0.3 | 0.7±0.1 | 0.2 | |
| 10m | 6-NO2 | 0.9±0.1 | 0.7±0.1 | 4.3±0.7 | 2.8±0.2 | 2.8±0.2 | 1.0 | |
| 10n | 6-OMe | 0.8±0.2 | 0.7±0.1 | 6.2±0.4 | 0.9±0.4 | 2.8±0.1 | 3.1 | |
| 10o | 7-F | 1.7±0.2 | 1.9±0.2 | ND | 2.6±0.4 | ND | ND | |
| 10p | 7-Me | 2.0±0.2 | 11.5±0.8 | 2.6±0.2 | 1.4±0.1 | 0.9±0.1 | 0.6 |
ND: Not Determined.
Resistance index (RI) is calculated by dividing IC50 values on multidrug-resistant cell line M14/MDR1 by IC50 values on the matching sensitive parental cell line M14.
Six di-substituted indole analogs were synthesized in this series and their in vitro assay result was shown in Table 3. All the six analogs did not show any improvement of activity compared to their mono-substituted counterparts. 10s–10v were designed based on the most potent mono-substituted analogs 10f, 10k and 10n. Incorporation of a secondary substituent to 5-fluoroindole, 6-chloro, or 6-methoxyindole generally was not beneficial to activity; for example, 10s and 10u showed reduced cytotoxicities compared to 10f and had IC50 values of >1.5 μM; the 3-cyano-6-methoxy indole analog 10t was less potent than the mono- substituted counterpart 10n; 5,6-dichloroindole analog 10v showed cytotoxicity comparable to that of 5-chloroindole analog 10k.
Table 3.
In vitro growth inhibitory effects (μM) of UC-112 analogs with di-substituents on the indole moiety.
| structure | compound | R | A375 | WM164 | RPMI7951 | M14 | M14/MDR1 | RIa |
|---|---|---|---|---|---|---|---|---|
|
|
10q | 2,3-(Me)2 | 1.9 ±0.2 | 7.1±0.8 | 8.2±0.6 | 1.5±0.2 | 1.0±0.1 | 0.7 |
| 10r | 2-Me-5-OMe | 1.9±0.2 | 4.6±0.1 | 2.1±0.1 | 2.0±0.1 | 1.0±0.1 | 0.5 | |
| 10s | 2-COOEt-5-F | 2.0 ±0.2 | 8.5±0.8 | 3.6±0.4 | 1.3 ±0.1 | 1.0±0.1 | 0.7 | |
| 10t | 3-CN-6-OMe | 1.7±0.1 | 16.7±0.5 | 3.7±0.1 | 1.6 ±0.1 | 1.9±0.1 | 1.2 | |
| 10u | 5-F-6-Cl | 2.0±0.2 | 4.5±0.2 | 1.7±0.1 | 2.0 ±0.2 | 1.0±0.1 | 0.5 | |
| 10v | 5,6-(Cl)2 | 0.7±0.1 | 0.9±0.1 | 9.3±1.1 | 8.5±0.2 | 2.2±0.1 | 0.3 |
Resistance index (RI) is calculated by dividing IC50 values on multidrug-resistant cell line M14/MDR1 by IC50 values on the matching sensitive parental cell line M14.
3.3. Inhibitory effect against P-gp overexpressing cell lines
P-gp belongs to the family of ATP-binding cassette (ABC) transporters and is encoded by the ABCB1 gene. P-gp is responsible for the decline of concentrations of extensive anticancer drugs in multidrug resistant cells. Therefore, the ability to overcome P-gp mediated drug-resistance is a favorable property for drug candidates.
In addition to the M14 melanoma cell line, its P-gp overexpressed daughter line M14/MDR1 was also tested to evaluate the abilities of new analogs to overcome P-gp mediated drug-resistance. The result was shown in Table 1–3. According to assay result, more than twelve of the twenty-two indole analogs exhibited more potent inhibitory effects against P-gp overexpressing M14/MDR1 cell line than the parental drug sensitive M14 cell line and had resistance index (RI) less than 1, indicating that new indole analogs had significant ability to circumvent drug-resistance mediated by P-gp overexpression. It is worth noting that five out of the six di-substituted indole analogs have RIs less than 1, suggesting that a secondary substituent on the indole is beneficial for overcoming drug-resistance.
In addition to melanoma cell lines, cervical and colorectal carcinoma cell lines were also tested to confirm the capabilities of the new analogs to circumvent P-gp mediated drug resistance. The result is summarized in Table 4. The small-molecular survivin inhibitor YM-155 showed remarkable cytotoxicity against KB-3-1 and SW620 cell lines with IC50 values of 4.9 nM and 3.9 nM, respectively. YM155, however, displayed significantly reduced activities against the corresponding P-gp overexpressing cell lines (KB-C2 and SW620/Ad300) and had IC50 values of more than 20 μM. Similarly, paclitaxel, a known substrate of P-gp, was not effective against P-gp overexpressing cell lines. In contrast, our indole analogs 10f, 10h, 10k, 10n and the previously reported MX-106 showed stronger inhibitory effects against P-gp overexpressing cell lines (KB-C2 and SW620/Ad300) than their corresponding parent cell lines (KB-3-1 and SW620). While the IC50 values range from 1.28 to 1.71 μM in the non-resistant KB-3-1 cell line for 10f, 10h, 10k and 10n, their potency against the P-gp overexpressing KB-C2 cell line significantly inrease (IC50 values range from 0.17 to 0.44 μM). Similarly, 10f, 10h, 10k and 10n had IC50 values ranging from 0.16 to 0.31 μM against non-resistant SW620 cell line while their activities in KB-C2 cell line were 3–6 folds more potent with IC50 values ranging from 0.027 to 0.1 μM. Collectively, these new indole analogs of UC-112 can show potential to reverse drug-resistance mediated by P-gp overexpression.
Table 4.
In vitro cytotoxicity of 10f, 10h, 10k and 10n in P-gp overexpressed cell lines.
| Compound | IC50 ± SD (μM) | RIa | IC50 ± SD (μM) | RI | ||
|---|---|---|---|---|---|---|
|
|
|
|||||
| KB-3-1 | KB-C2 | SW620 | SW620/Ad300 | |||
| 10f | 1.36 ± 0.19 | 0.17 ± 0.09 | 0.13 | 0.16 ± 0.04 | 0.027 ± 0.006 | 0.17 |
|
| ||||||
| 10h | 1.39 ± 0.41 | 0.18 ± 0.05 | 0.13 | 0.19 ± 0.09 | 0.06 ± 0.02 | 0.32 |
|
| ||||||
| 10k | 1.71 ± 0.22 | 0.25 ± 0.05 | 0.15 | 0.24 ± 0.09 | 0.05 ± 0.03 | 0.21 |
|
| ||||||
| 10n | 1.28 ± 0.25 | 0.44 ± 0.28 | 0.34 | 0.31 ± 0.05 | 0.10 ± 0.04 | 0.32 |
|
| ||||||
| MX-106 | 1.25 ± 0.10 | 0.33 ± 0.12 | 0.26 | 0.15 ± 0.01 | 0.03± 0.01 | 0.20 |
|
| ||||||
| YM-155 | 0.0049 ± 0.0005 | 37.30 ± 3.97 | 7612.24 | 0.0039 ± 0.0056 | 23.37 ± 2.37 | 5992.31 |
|
| ||||||
| Paclitaxel | 0.0005 ± 0.0001 | 0.25 ± 0.01 | 500.00 | 0.030 ± 0.001 | 1.97 ± 0.36 | 65.67 |
|
| ||||||
| Doxorubicin | 0.37 ± 0.07 | 1.69 ± 0.44 | 4.57 | 0.21 ± 0.03 | 15.24 ± 2.89 | 72.57 |
|
| ||||||
| Cisplatin | 1.15 ± 0.13 | 1.73 ± 0.11 | 1.50 | 1.79 ± 0.06 | 5.12 ± 0.65 | 2.86 |
Resistance index (RI) is calculated by dividing IC50 values on multidrug-resistant cell line M14/MDR1 by IC50 values on the matching sensitive parental cell line M14.
3.4. In vitro metabolic stability study
Prior to in vivo study, the in vitro metabolic stabilities of analogs 10f, 10h, 10k and 10n were examined by measuring their half-life upon incubation with mouse, rat, and human liver microsomes in the presence of an NADPH regeneration system. The result was summarized in Table 5. All compounds possessed acceptable stability profile in three microsome species. They were more stable in human microsome than in mouse and rat microsomes. Among the four analogs, 10f with a 5-fluoro was the most stable against mouse and human microsomes.
Table 5.
In vitro microsomal stabilities of compounds 10f, 10h, 10k and 10n.
| compound | Mouse | Rat | Human | |||
|---|---|---|---|---|---|---|
|
|
|
|
||||
| t1/2 (h) | Clint(ml/min/kg) | t1/2 (h) | Clint(ml/min/kg) | t1/2 (h) | Clint(ml/min/kg) | |
|
|
|
|
||||
| 10f | 1.36±0.06 | 42.1 | 1.29±0.03 | 36.1 | 4.59±0.39 | 4.5 |
|
| ||||||
| 10h | 1.17±0.06 | 48.7 | 1.20±0.05 | 39.0 | 2.16±0.19 | 9.6 |
|
| ||||||
| 10k | 1.20±0.07 | 47.8 | 1.62±0.10 | 29.0 | 3.50±0.36 | 5.9 |
|
| ||||||
| 10n | 0.88±0.06 | 65.1 | 1.26±0.06 | 37.0 | 2.80±0.18 | 7.4 |
3.5. Indole based UC-112 analogs maintain their selective inhibition for survivin among IAPs
To determine whether our new UC-112 analogs maintains their selective inhibition of survivin as the prototype UC-112 does, we performed the western blotting analyses using 10f, 10h, 10k and 10n in A375 cell line at different concentrations and the result was shown in Figure 2. All four compounds strongly inhibited survivin expression in A375 cell line in a concentration dependent manner. In contrast, the levels of other IAP family proteins such as C-IAP1, XIAP, and Livin, were not affected, indicating high selectivity of survivin inhibition among the IAP proteins. Consitent with survivin inhibition, all four analogs effectively induced cancer cell apoptosis, as indicated by the elevated level of cleaved PARP in Figure 2.
Figure 2.

Western blot assay of A375 cells treated with different doses of 10f, 10h, 10k and 10n for 24 h.
3.6. Molecular modeling study
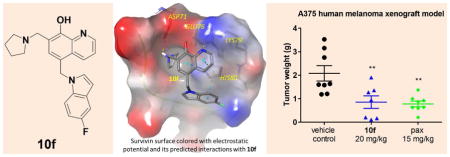
To explain the observed strong potency of 10f, a molecular modeling study was developed using the complex of human survivin-SMAC AVPI (PDB entry: 3UIH) and the result was shown in Figure 3A and 3B. 10f formed appealing hydrogen bonding and π-π stacking interactions with the survivin protein BIR domain: (1) two hydrogen bonding interactions between the 8-hydroxyquinoline of 10f and residues Glu76 and Lys79; (2) two hydrogen bonding interactions between the pyrrolidine of 10f and residue Asp71; (3) π-π stacking interaction between the 8-hydroxyquinoline of 10f and residue His80; (4) π-π stacking interaction between the 5-F indole of 10f and residue His80; (5) π-cation interaction between the pyrrolidine of 10f and residue Trp67 (red line in Figure 3B). The molecular modeling result suggested that substitute on the 7-position of the indole ring will introduce the repulse between the 8-hydroxyquinoline and indole ring and further lead to attenuated π-π stacking interactions mentioned above, for example, 10p and 10q exhibited significantly decreased potency in comparison with 10a.
Figure 3. Potential binding mode of 10f to survivin.

(A) the best docking pose of 10f binding in survivin (survivin PDB: 3UIH), shown with electron potential color-coded survivin surface (red: electron negative potential; blue: electron positive potential). Interactions between 10f and nearby residues in survivin are shown. (B) Types of interactions are shown with color-coded lines between 10f with residues in survivin protein in this 2D interaction map.
3.7. In vivo anti-tumor efficacy assessment
As one of the most potent indole analog, 10f not only exhibited the most favorable stability against mouse and human liver microsomes but also significantly inhibited the survivin expression at a low concentration; therefore 10f was selected for further evaluating antitumor efficacy in vivo. An A375 melanoma xenograft model in nude mice was used. Tumors were implanted by inoculating mice with A375 human melanoma cancer cells subcutaneously in their hindflank. After the development of viable tumors, mice were treated every other day for two weeks by i.p. injection with 20 mg/kg 10f, 15 mg/kg paclitaxel, or vehicle solution only. After 15 days of treatment, the groups receiving 20 mg/kg 10f had significantly smaller tumor volumes than the vehicle control group with a calculated TGI of 68.6% (Figure 4A). This was very similar to the paclitaxel treated group which averaged a TGI of 70.6% compared to the vehicle control group. One-way ANOVA analysis followed by Dunnett’s multiple comparison test also revealed that there was a significant reduction in final tumor volume (P < 0.0001) for all treatment groups compared to the vehicle control group. These results are in accord with the tumor final weights, where the average tumor weight for the control group was 2.08 ± 0.32 grams, and 10f and paclitaxel groups averaged 0.85 ± 0.26 grams and 0.78 ± 0.12 grams, respectively (Figure 4B). Statistically analysis was performed the same and yielded an overall P value of 0.0012, and a significant difference (P < 0.01) for each treated group compared to the control group. Mouse body weight was measured and animal activity was monitored throughout the experiment, and no significant deviations in animal weight or behavior was observed (Figure 4C).
Figure 4. 10f inhibits tumor growth in vivo.

(A) Average tumor volumes ± SEM in an A375 xenograft model in nude mice (n=7~8). (B) Individual tumor weights. Long line represents mean and error bars represent SEM. Statistical significance was determined by one-way ANOVA (P= 0.0012) analysis followed by Dunnett’s multiple comparison test. P < 0.01 for each treatment group compared to the control group. (C) Average mouse body weights ± SEM. Graph represents percent change in body weight compared to the starting weight.
4. Conclusion
In this report, thirty-three new analogs of UC-112 were synthesized and their SAR was investigated. The result showed that most indole analogs exhibited potencies stronger than or comparable to that of UC-112 and MX-106. The most potent activities were observed in 10f, 10h, 10k and 10n. As compared to YM-155, a widely studied survivin promoter inhibitor, our new indole analogs of UC-112 showed more potent activities against P-gp expressing cancer cell lines than parental cancer cell lines. Mechanistic study suggested that new indole analogs of UC-112 selectively suppressed the expression of the survivin protein in a dose-dependent manner without affecting other members of the IAP family. In an in vivo xenograft model in nude mice, 10f exhibited significant inhibitory effect of tumor growth. These results indicated in-depth investigation of these novel scaffolds is warranted in the future.
Supplementary Material
Research highlights.
A series of novel UC-112 analogs was prepared and evaluated as potential antitumor agents.
Most of these analogs show similar or increased potency against P-gp overexpressed cancer cell lines compared with their drug sensitive parental cancer cell lines.
Compound 10f was found to be the most potent and metabolically stable analog.
Compound 10f selectively inhibited survivin expression among the IAP protein family, induced apoptosis, and significantly suppress melanoma tumor growth in vivo.
Acknowledgments
This work was supported by NIH grants 1R01CA193609, 1S10OD010678-01, and 1S10RR026377-01 to W.L. Additional support from the University of Tennessee College of Pharmacy Drug Discovery Center is acknowledged. We are thankful to Dr. Shin-Ichi Akiyama (Kagoshima University, Japan) for providing the KB-3-1 and KB-C2 cell lines. We thank Dr. Susan E Bates (Columbia University, NY, USA) and Dr. Robert W. Robey (NIH, MD, USA) for providing the SW620 and SW620/Ad300 cell lines. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Dr. Lei Yang at St. Jude Children’s Research Hospital for the metabolic stability evaluation for compounds 10f, 10h, 10k and 10n.
Abbreviations
- ABC
ATP-binding cassette
- IAP
inhibitor of apoptosis proteins
- P-gp
P-glycoprotein
- RI
resistance index
- SAR
structure-activity relationships
- TGI
Tumor growth inhibition
- TMS
tetramethylsilane
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 2.Chakravarti A, Zhai GG, Zhang M, Malhotra R, Latham DE, Delaney MA, Robe P, Nestler U, Song Q, Loeffler J. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene. 2004;23:7494–7506. doi: 10.1038/sj.onc.1208049. [DOI] [PubMed] [Google Scholar]
- 3.Cheung CH, Chen HH, Kuo CC, Chang CY, Coumar MS, Hsieh HP, Chang JY. Survivin counteracts the therapeutic effect of microtubule de-stabilizers by stabilizing tubulin polymers. Mol Cancer. 2009;8:43. doi: 10.1186/1476-4598-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pennati M, Folini M, Zaffaroni N. Targeting survivin in cancer therapy: fulfilled promises and open questions. Carcinogenesis. 2007;28:1133–1139. doi: 10.1093/carcin/bgm047. [DOI] [PubMed] [Google Scholar]
- 5.Ryan BM, O’Donovan N, Duffy MJ. Survivin: a new target for anti-cancer therapy. Cancer Treat Rev. 2009;35:553–562. doi: 10.1016/j.ctrv.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Duffy MJ, O’Donovan N, Brennan DJ, Gallagher WM, Ryan BM. Survivin: a promising tumor biomarker. Cancer Lett. 2007;249:49–60. doi: 10.1016/j.canlet.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 7.O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio PC, Altieri DC. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A. 2000;97:13103–13107. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan H, Thomas J, Liu T, Raj D, London N, Tandeski T, Leachman SA, Lee RM, Grossman D. Induction of melanoma cell apoptosis and inhibition of tumor growth using a cell-permeable Survivin antagonist. Oncogene. 2006;25:6968–6974. doi: 10.1038/sj.onc.1209676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Talbot DC, Davies J, Callies S, Andre V, Lahn M, Ang J, De Bono JS, Ranson M. First human dose study evaluating safety and pharmacokinetics of LY2181308, an antisense oligonucleotide designed to inhibit survivin. J Clin Oncol. 2008;26 [Google Scholar]
- 10.Xiao M, Li W. Recent Advances on Small-Molecule Survivin Inhibitors. Curr Med Chem. 2015;22:1136–1146. doi: 10.2174/0929867322666150114102146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roy K, Singh N, Kanwar RK, Kanwar JR. Survivin Modulators: An Updated Patent Review (2011–2015) Recent Pat Anticancer Drug Discov. 2016;11:152–169. doi: 10.2174/1574892811666160229121815. [DOI] [PubMed] [Google Scholar]
- 12.Chantalat L, Skoufias DA, Kleman JP, Jung B, Dideberg O, Margolis RL. Crystal structure of human survivin reveals a bow tie-shaped dimer with two unusual alpha-helical extensions. Mol Cell. 2000;6:183–189. [PubMed] [Google Scholar]
- 13.Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–3257. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- 14.Chandele A, Prasad V, Jagtap JC, Shukla R, Shastry PR. Upregulation of survivin in G2/M cells and inhibition of caspase 9 activity enhances resistance in staurosporine-induced apoptosis. Neoplasia. 2004;6:29–40. doi: 10.1016/s1476-5586(04)80051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Gambosova K, Cooper ZA, Holloway MP, Kassai A, Izquierdo D, Cleveland K, Boney CM, Altura RA. EGF regulates survivin stability through the Raf-1/ERK pathway in insulin-secreting pancreatic beta-cells. Bmc Mol Biol. 2010;11:66. doi: 10.1186/1471-2199-11-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Li D, Liu H, Xu H, Zheng H, Qian F, Li W, Zhao C, Wang Z, Wang X. Notch-1 signaling facilitates survivin expression in human non-small cell lung cancer cells. Cancer Biol Ther. 2011;11:14–21. doi: 10.4161/cbt.11.1.13730. [DOI] [PubMed] [Google Scholar]
- 17.Coumar MS, Tsai FY, Kanwar JR, Sarvagalla S, Cheung CH. Treat cancers by targeting survivin: just a dream or future reality? Cancer Treat Rev. 2013;39:802–811. doi: 10.1016/j.ctrv.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Xia W, Bisi J, Strum J, Liu L, Carrick K, Graham KM, Treece AL, Hardwicke MA, Dush M, Liao Q, Westlund RE, Zhao S, Bacus S, Spector NL. Regulation of survivin by ErbB2 signaling: therapeutic implications for ErbB2-overexpressing breast cancers. Cancer Res. 2006;66:1640–1647. doi: 10.1158/0008-5472.CAN-05-2000. [DOI] [PubMed] [Google Scholar]
- 19.Wang HQ, Jin JJ, Wang J. Arctigenin enhances chemosensitivity to cisplatin in human nonsmall lung cancer H460 cells through downregulation of survivin expression. J Biochem Mol Toxicol. 2014;28:39–45. doi: 10.1002/jbt.21533. [DOI] [PubMed] [Google Scholar]
- 20.Ling X, Cao S, Cheng Q, Keefe JT, Rustum YM, Li F. A novel small molecule FL118 that selectively inhibits survivin, Mcl-1, XIAP and cIAP2 in a p53-independent manner, shows superior antitumor activity. Plos One. 2012;7:e45571. doi: 10.1371/journal.pone.0045571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wall NR, O’Connor DS, Plescia J, Pommier Y, Altieri DC. Suppression of survivin phosphorylation on Thr(34) by flavopiridol enhances tumor cell apoptosis. Cancer Res. 2003;63:230–235. [PubMed] [Google Scholar]
- 22.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Oh SW, Kim HY, Moon SH, Ha JR, Kahn M. A small molecule inhibitor of beta-catenin/cyclic AMP response element-binding protein transcription (Vol 101, pg 12682, 2004) P Natl Acad Sci USA. 2004;101:16707–16707. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang CC, Heller JD, Kuo J, Huang RC. Tetra-O-methyl nordihydroguaiaretic acid induces growth arrest and cellular apoptosis by inhibiting Cdc2 and survivin expression. Proc Natl Acad Sci U S A. 2004;101:13239–13244. doi: 10.1073/pnas.0405407101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, Tominaga F, Hatakeyama S, Kinoyama I, Matsuhisa A, Kudoh M, Sasamata M. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–8021. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Li W. Discovery of novel second mitochondria-derived activator of caspase mimetics as selective inhibitor of apoptosis protein inhibitors. J Pharmacol Exp Ther. 2014;349:319–329. doi: 10.1124/jpet.113.212019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi J, Dong Z, Liu J, Peery RC, Zhang S, Liu JY, Zhang JT. Effective Targeting of the Survivin Dimerization Interface with Small-Molecule Inhibitors. Cancer Res. 2016;76:453–462. doi: 10.1158/0008-5472.CAN-15-1874. [DOI] [PubMed] [Google Scholar]
- 27.Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, Tominaga F, Hatakeyama S, Kinoyama I, Matsuhisa A, Kudou M, Sasamata M. YM155, a Novel Small-Molecule Survivin Suppressant, Induces Regression of Established Human Hormone-Refractory Prostate Tumor Xenografts (vol 67, pg 8014, 2007) Cancer Res. 2012;72:3886–3886. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- 28.Rauch A, Hennig D, Schafer C, Wirth M, Marx C, Heinzel T, Schneider G, Kramer OH. Survivin and YM155: how faithful is the liaison? Biochim Biophys Acta. 2014;1845:202–220. doi: 10.1016/j.bbcan.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Xiao M, Wang J, Lin Z, Lu Y, Li Z, White SW, Miller DD, Li W. Design Synthesis and Structure-Activity Relationship Studies of Novel Survivin Inhibitors with Potent Anti-Proliferative Properties. Plos One. 2015;10:e0129807. doi: 10.1371/journal.pone.0129807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Chen J, Miller DD, Li W. Synergistic combination of novel tubulin inhibitor ABI-274 and vemurafenib overcome vemurafenib acquired resistance in BRAFV600E melanoma. Molecular cancer therapeutics. 2014;13:16–26. doi: 10.1158/1535-7163.MCT-13-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang DJ, Wang J, Li W, Miller DD. Structural Optimization of Indole Derivatives Acting at Colchicine Binding Site as Potential Anticancer Agents. ACS medicinal chemistry letters. 2015;6:993–997. doi: 10.1021/acsmedchemlett.5b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akiyama S, Fojo A, Hanover JA, Pastan I, Gottesman MM. Isolation and genetic characterization of human KB cell lines resistant to multiple drugs. Somatic cell and molecular genetics. 1985;11:117–126. doi: 10.1007/BF01534700. [DOI] [PubMed] [Google Scholar]
- 33.Robey RW, Shukla S, Finley EM, Oldham RK, Barnett D, Ambudkar SV, Fojo T, Bates SE. Inhibition of P-glycoprotein (ABCB1)- and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor, CBT-1((R)) Biochemical pharmacology. 2008;75:1302–1312. doi: 10.1016/j.bcp.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47:936–942. [PubMed] [Google Scholar]
- 35.Mahindroo N, Connelly MC, Punchihewa C, Yang L, Yan B, Fujii N. Amide conjugates of ketoprofen and indole as inhibitors of Gli1-mediated transcription in the Hedgehog pathway. Bioorg Med Chem. 2010;18:4801–4811. doi: 10.1016/j.bmc.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di L, Kerns EH, Li SQ, Petusky SL. High throughput microsomal stability assay for insoluble compounds. Int J Pharm. 2006;317:54–60. doi: 10.1016/j.ijpharm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 37.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 38.Lu C, Li P, Gallegos R, Uttamsingh V, Xia CQ, Miwa GT, Balani SK, Gan LS. Comparison of intrinsic clearance in liver microsomes and hepatocytes from rats and humans: evaluation of free fraction and uptake in hepatocytes. Drug Metab Dispos. 2006;34:1600–1605. doi: 10.1124/dmd.106.010793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.