

Abstract
Background
The incidence of obesity is rising, particularly among women. Microvascular dysfunction is more common with female sex, obesity, and hyperlipidemia and predicts adverse cardiovascular outcomes, but the molecular mechanisms are unclear. Because obesity is associated with mineralocorticoid receptor (MR) activation, we tested the hypothesis that MR in endothelial cells contribute to sex differences in resistance vessel dysfunction in response to cardiometabolic risk factors.
Methods and Results
Male and female endothelial cell–specific MR knockout mice and MR‐intact littermates were randomized to high‐fat‐diet–induced obesity or obesity with hyperlipidemia induced by adeno‐associated virus–based vector targeting transfer of the mutant stable form (DY mutation) of the human PCSK9 (proprotein convertase subtilisin/kexin type 9) gene and compared with control diet. Female but not male mice were sensitive to obesity‐induced endothelial dysfunction, whereas endothelial function was impaired in obese hyperlipidemic males and females. In males, obesity or hyperlipidemia decreased the nitric oxide component of vasodilation without altering superoxide production or endothelial nitric oxide synthase expression or phosphorylation. Decreased nitric oxide content in obese males was overcome by enhanced endothelium‐derived hyperpolarization–mediated relaxation along with increased SK3 expression. Conversely, in females, endothelium‐derived hyperpolarization was significantly impaired by obesity with lower IK1 expression and by hyperlipidemia with lower IK1 and SK3 expression, loss of H2O2‐mediated vasodilation, and increased superoxide production. Endothelial cell–MR deletion prevented endothelial dysfunction induced by risk factors only in females. Rather than restoring endothelium‐derived hyperpolarization in females, endothelial cell–MR deletion enhanced nitric oxide and prevented hyperlipidemia‐induced oxidative stress.
Conclusions
These data reveal distinct mechanisms driving resistance vessel dysfunction in males versus females and suggest that personalized treatments are needed to prevent the progression of vascular disease in the setting of obesity, depending on both the sex and the metabolic profile of each patient.
Keywords: aldosterone, endothelial dysfunction, microvascular dysfunction, mineralocorticoid receptor, mineralocorticoids, nitric oxide, obesity, sex differences
Subject Categories: Vascular Biology, Endothelium/Vascular Type/Nitric Oxide, Peripheral Vascular Disease
Clinical Perspective
What Is New?
This study revealed totally distinct pathophysiological mechanisms in female and male mice underlying resistance vessel endothelial dysfunction induced by obesity with or without hyperlipidemia.
The results show distinct contributions of nitric oxide and endothelium‐derived hyperpolarization in each sex and a protective role of endothelial cell–mineralocorticoid receptor deletion specifically in females.
The mechanisms of microvessel dysfunction were also different in the obesity model versus the obesity model with hyperlipidemia.
What Are the Clinical Implications?
Obesity preferentially affects women and is associated with microvessel dysfunction, and impaired resistance vessel function predicts an increased risk of cardiovascular disease; therefore, understanding mechanisms driving microvascular dysfunction in obesity is critical to prevent adverse cardiovascular consequences.
The identification of sex‐specific mechanisms of microvessel dysfunction supports the concept that personalized therapies are needed to prevent the adverse cardiovascular outcomes of obesity in males versus females and may need to be tailored to the specific associated metabolic abnormalities in each individual.
If confirmed in humans, future clinical studies could explore the efficacy of mineralocorticoid receptor antagonists to prevent microvascular endothelial dysfunction by increasing nitric oxide bioavailability specifically in obese females.
Introduction
Although cardiovascular disease (CVD) mortality has been declining because of advances in care and awareness, coronary disease mortality has recently begun to rise in women aged 35 to 54 in the United States.1 This concerning trend has been attributed to the expanding obesity epidemic, which preferentially affects women.1, 2, 3 Obesity and the associated hyperlipidemia and metabolic dysfunction are well‐known risk factors for CVD.4 One of the earliest vascular consequences of these cardiometabolic risk factors is endothelial dysfunction, characterized by impaired endothelium‐dependent relaxation.5, 6, 7 Impaired vasorelaxation decreases organ perfusion capacity and can contribute to the development of hypertension, another CVD risk factor associated with obesity. The dysfunctional vascular endothelium is also proinflammatory and prothrombotic. In these ways, endothelial dysfunction contributes to the development of atherosclerosis and progression to plaque rupture and thrombosis, the cause of cardiovascular ischemia, which is the leading cause of death in both sexes.1, 2 Although premenopausal women are protected from CVD relative to men, this protection is attenuated in obese young women, particularly those with metabolic syndrome or diabetes mellitus,8 yet the molecular mechanisms remain unknown.
Resistance microvessels are key regulators of blood flow distribution and contribute to blood pressure by modulating peripheral vascular resistance. In humans, dysfunction of resistance vessels, rather than conduit vessels, predicts 5‐year CVD risk.9 Indeed, in the large Framingham offspring cohort, female sex, high body mass index and dyslipidemia are each associated with increased incidence of microvascular endothelial dysfunction.10 Resistance vessels dilate in response to environmental changes and physiological needs to modulate blood flow to specific organs. This endothelium‐dependent vasodilation is measured experimentally by quantifying the vascular relaxation response to acetylcholine. Three components can contribute to this endothelium‐derived vasodilatory response: COX (cyclooxygenase)–derived prostanoids, endothelial nitric oxide synthase (eNOS)–derived nitric oxide (NO), and endothelium‐derived hyperpolarization (EDH). EDH is mediated by endothelial potassium channels; specifically, IK1 and SK3 channels account for EDH in resistance microvessels.11 Hydrogen peroxide (H2O2) is an additional vasodilator that activates potassium channels to contribute to EDH.12, 13 Sex differences have been described in the relative contribution of these endothelium‐derived relaxing factors to resistance vessel function in rodent models.14, 15 However, how these components are modulated in resistance vessels in response to cardiometabolic risk factors and whether there are sex differences in those responses have not been directly investigated.
Obesity in humans is associated with increased activation of the mineralocorticoid receptor (MR), a critical regulator of blood pressure by modulating renal sodium retention. The MR is a member of the steroid receptor family that is activated by mineralocorticoids such as aldosterone and by corticosteroids, in some cells under specific conditions.16, 17 Obesity is associated with increased levels of aldosterone18 and, particularly when associated with hyperglycemia, increased activity of the Rho‐family small GTPase Rac1, a ligand‐independent MR activator.19, 20, 21 In addition to the kidney, MR is expressed in vascular cells including smooth muscle cells and endothelial cells (ECs).22, 23, 24 In a mouse model of Western diet‐induced obesity, females developed higher plasma aldosterone levels compared with males.25 MR blockade in rodents improved endothelial function in females more than males in the setting of diabetes mellitus or hyperleptinemia—common complications of obesity.26, 27 These studies support a potential role for MR in endothelial dysfunction induced by cardiometabolic risk factors and suggest that the role of MR may be enhanced in females.
Studies using mice with MR specifically deleted from ECs revealed that MR in ECs (EC‐MR) does not substantially contribute to basal blood pressure or baseline endothelial function28 but rather appears to contribute to the vascular response to risk factors such as hypertension and obesity (reviewed in 29). Previous studies exploring the role of EC‐MR in endothelial function have focused on conduit vessels, predominantly the aorta. Specifically, obesity induced by a high‐fat diet (HFD) causes aortic endothelial dysfunction that was prevented in male mice lacking EC‐MR.30 In female mice, EC‐MR contributed to aortic endothelial stiffness induced by a high‐fat/high‐sucrose diet by enhancing expression and activity of the epithelial sodium channel in aortic ECs.31 However, the role of EC‐MR in resistance vessel endothelial dysfunction in response to cardiometabolic risk factors has not been well characterized, nor have sex differences been explored. Whereas conduit vessel endothelial dysfunction contributes to vascular disease by promoting vascular inflammation, stiffness, and fibrosis, conduit vessel vasodilation is not a major mechanism in regulating blood flow or blood pressure. Understanding mechanisms driving resistance vessel dysfunction is important to our understanding of organ‐specific blood flow regulation. In addition, microvessel dysfunction, rather than conduit‐vessel dysfunction, correlates with adverse CVD outcomes in humans and is more common in women, particularly those with cardiometabolic risk factors.9, 10 Indeed, MR blockade in patients with type 2 diabetes mellitus improved coronary flow reserve,32 suggesting a role for MR in human microvessel function. These data prompted the hypothesis that there are sex differences in the mechanisms by which cardiometabolic risk factors induce resistance microvessel dysfunction and that MR activation in the endothelium contributes to those mechanisms. To test this hypothesis, we examined the mechanism by which diet‐induced obesity alone or combined with hyperlipidemia produces endothelial dysfunction in small mesenteric resistance arteries from male or female mice with MR specifically deleted from EC (EC‐MR knockout [EC‐MR‐KO] mice) compared with their MR‐intact littermates and identified sex‐specific mechanisms with a role of EC‐MR only in females.
Methods
Animals
All studies were approved by the Tufts University institutional animal care and use committee in compliance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health). EC‐MR‐KO mice were generated, as described previously,28 by crossing floxed MR mice with mice containing a Cre‐recombinase gene driven by the EC‐specific VE‐cadherin promoter (EC‐MR−/−, EC‐MR‐KO). These are compared with floxed MR/VE‐cadherin‐Cre–negative littermates (EC‐MR+/+, MR‐intact). Substantial and specific deletion of MR from ECs in this model has been previously and extensively confirmed.28, 33
Cardiovascular Risk Factor Models
Four‐week‐old male or female EC‐MR‐KO and MR‐intact littermates were randomized to 3 groups: control, normal chow (0.3% NaCl; Harlan diet TD8604); obesity, HFD (Harlan Teklad TD.88137: 42% kcal from fat, 60% as saturated fat, 34% sucrose by weight) for 12 weeks; or hyperlipidemia, HFD‐induced obesity with hyperlipidemia. To induce hyperlipidemia, animals received a single injection of adeno‐associated virus (AAV)–based vector targeting transfer of the mutant stable form (DY mutation) of the human PCSK9 (proprotein convertase subtilisin/kexin type 9) gene (AAV‐hPCSK9DY) to the liver and fed with an HFD for 12 weeks, as described previously.34 An average of 5 mice per group were randomized for each myography study and 4 per group for the biochemical studies based on power calculations to achieve a power of 0.85 with an α=0.05. Body weight was evaluated at the beginning and at the end of the 12 weeks of feeding. Blood pressure was measured in a subset of mice just before euthanization by a validated tail cuff training and measurement method, as described previously.35 Fasting blood glucose (TrueBalance glucometer and strips; Trividia Health), fasting total serum cholesterol (Molecular Probes Amplex Red Cholesterol Assay; Thermo Fisher), serum aldosterone (Aldosterone RIA; Tecan), and corticosterone (Corticosterone EIA; Arbor Assays) were measured from each group, and serum estradiol levels were measured in female mice (Mouse/Rat Estradiol ELISA; Calbiochem) for all appropriate blood samples collected at the end of each study. For the estradiol measurements, 1 sample was below the level of detection of the ELISA assay and thus was assigned an estradiol value of 0.
Wire Myography
Second‐ and third‐order mesenteric resistance arteries (<300 μm) from control, obese, and hyperlipidemic EC‐MR‐KO and MR‐intact mice were isolated and mounted in a wire myograph containing warmed (37°C), aerated (95% O2, 5% CO2) physiological salt solution (in mmol/L: 130 NaCl, 4.7 KCl, 1.17 MgSO4, 0.03 EDTA, 1.6 CaCl2, 14.9 NaHCO3, 1.18 KH2PO4, and 5.5 glucose). Internal vessel circumference under (L100) was determined and the vessels were set at 0.9×L100. After 30 minutes of equilibration, 1 μmol/L phenylephrine was administered to induce a half‐maximum contraction and then relaxation responses to acetylcholine (10−9–10−5 mol/L), and the NO donor sodium nitroprusside (10−9–10−5 mol/L) were evaluated. The specific role of endothelium‐derived relaxing factors including COX‐derived prostanoids, NO, EDH, or H2O2 was evaluated by performing concentration‐response curves to acetylcholine after 30 minutes of chamber incubation with their respective inhibitors: indomethacin (10 μmol/L), L‐NAME (100 μmol/L), TRAM34 plus apamin (1 μmol/L each), or catalase (1000 U/mL).
Western Blot
Frozen mesenteric resistance arteries were ground into a fine powder on dry ice and then collected in an Eppendorf tube containing RIPA lysis buffer with PMSF (phenylmethylsulfonyl fluoride) and protease inhibitor cocktail. Samples were centrifuged (580 g; 15 minutes at 4°C), and the lysate supernatant was collected and protein concentration measured (Pierce Protein Assay BCA Kit). In addition, 15 μg of protein lysate were resolved by SDS‐PAGE, transferred to PVDF (polyvinylidene fluoride) membrane, and probed with primary antibodies for serine 1177 phosphorylated eNOS, total eNOS, β‐actin, and endothelial K+ channels IK1 and SK3, followed by incubation with appropriate antirabbit or antimouse horseradish peroxidase secondary antibody. Proteins were detected with electrochemiluminescence and quantified by densitometry by a blinded investigator.
NO Production by DAF‐2 DA (4,5‐Diaminofluorescein Diacetate) Fluorescence and Superoxide Generation by DHE (Dihydroethidium) Fluorescence
Mesenteric resistance arteries were embedded in freezing medium (Tissue‐Tek OCT), and transverse sections (10 μm) were obtained with a cryostat (−20°C). Sections were incubated for 30 minutes at 37°C in a light‐protected humidified chamber with phosphate buffer (0.1 mmol/L, pH 7.4) containing 2 mmol/L CaCl2 and 8 μmol/L DAF‐2 DA, used as NO‐sensitive dye. DHE (dihydroethidium [hydroethidine]; 5 μmol/L), a superoxide indicator dye, was topically applied to a second set of tissue sections in a light‐protected humidified chamber with phosphate buffer for 30 minutes for quantification of reactive oxygen species (ROS). Images were obtained with a fluorescence microscope (Eclipse Ti; Nikon) using a ×20 objective. The fluorescence was quantified by a blinded investigator by calculating the integrated density using ImageJ software (National Institutes of Health).
Statistical Analyses
The data, analytic methods, and study materials will be made available to other researchers by request to the corresponding author for purposes of reproducing the results or replicating the procedures.
All values are presented as mean±SEM. For Table, the normally distributed data (blood pressure, body weight, glucose) were analyzed by 3‐factor ANOVA with risk factor, genotype, and sex as the variables, followed by the Holm–Sidak post hoc test using SigmaPlot v.12.5 (Systat Software). For the nonnormally distributed data (cholesterol, aldosterone, corticosterone, estradiol) the Kruskal–Wallis test was used in R (R Foundation for Statistical Computing) to independently assess for effects of sex, genotype, and risk factor. For Figures 1, 2, 3, 4, 5, 6, 7 through 8, means were compared by 2‐factor ANOVA followed by the Bonferroni post hoc test with risk factor and genotype as the variables, using GraphPad Prism 5.0. For all data, a value of P<0.05 was considered significant.
Table 1.
Body Weight, Blood Pressure, Biochemical Parameters, and Hormones in Males and Females
| Groups | Male | Female | Statistics | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC‐MR+/+ | EC‐MR−/− | EC‐MR+/+ | EC‐MR−/− | ||||||||||
| Control | HFD | PCSK9 | Control | HFD | PCSK9 | Control | HFD | PCSK9 | Control | HFD | PCSK9 | ||
| Postdiet body weight, g | 31±1 (5) | 46±2 (4) | 39±2 (11) | 29±1 (4) | 48±1 (5) | 40±2 (11) | 21±1 (7) | 33±1 (10) | 27±1 (10) | 22±0.3 (7) | 35±1 (11) | 28±1 (11) | a , b , c , d |
| Mean arterial pressure, mm Hg | 110±4 (6) | 106±5 (5) | 98±4 (6) | 110±7 (4) | 97±6 (4) | 97±4 (7) | 102±4 (9) | 103±5 (8) | 97±4 (6) | 95±5 (8) | 90±7 (7) | 91±3 (7) | a |
| Fasting cholesterol, mg/dL | 83±14 (6) | 191±8 (3) | 1443±122 (5) | 87±6 (7) | 195±11 (3) | 1729±307 (7) | 69±10 (7) | 147±14 (10) | 1337±116 (6) | 82±11 (6) | 124±24 (6) | 1454±151 (7) | c , d |
| Fasting glucose, mg/dL | 172±18 (8) | 161±24 (8) | 146±17 (8) | 125±28 (5) | 153±21 (7) | 138±14 (8) | 111±14 (11) | 205±27 (11) | 136±18 (7) | 147±17 (10) | 166±11 (7) | 121±17 (8) | b , d |
| Estradiol, pg/mL | ··· | ··· | ··· | ··· | ··· | ··· | 5.3±0.8 (9) | 3.7±0.6 (9) | 3.4±0.4 (11) | 6.4±1.5 (9) | 5.4±1.3 (9) | 2.5±0.4 (10) | c |
| Aldosterone, pg/mL | 79±7 (9) | 123±17 (10) | 67±8 (8) | 92±12 (10) | 95±11 (10) | 48±7 (8) | 150±22 (11) | 201±25 (17) | 125±26 (6) | 183±33 (10) | 160±30 (12) | 71±21 (6) | a , d |
| Corticosterone, ng/mL | 69±7 (10) | 75±6 (10) | 134±11 (10) | 70±6 (10) | 68±4 (10) | 128±13 (10) | 148±15 (10) | 178±17 (10) | 190±28 (9) | 206±15 (10) | 110±5 (10) | 208±29 (11) | a , d , c (male) |
Normally distributed data (weight, blood pressure, glucose) were analyzed by 3‐way ANOVA; all others were analyzed by Kruskal–Wallis nonparametric testing for each variable (sex, genotype, and treatment). Data presented as mean±SEM (n). EC indicates endothelial cell; EC‐MR+/+, mice with endothelial cell mineralocorticoid receptors intact; EC‐MR−/−, mice with endothelial cell mineralocorticoid receptors knocked out; HFD, high‐fat diet; MR, mineralocorticoid receptor; PCSK9, high‐fat diet plus adenovirus expressing mutant human PCSK9 (proprotein convertase subtilisin/kexin type 9)–induced hyperlipidemia.
Significant difference between sexes.
Significant difference between control diet and HFD treatments.
Significant difference between control diet and PCSK9 treatments.
Significant difference between HFD and PCSK9 treatments.
Unless specified, significance indicators apply to both male and female groups.
Figure 1.
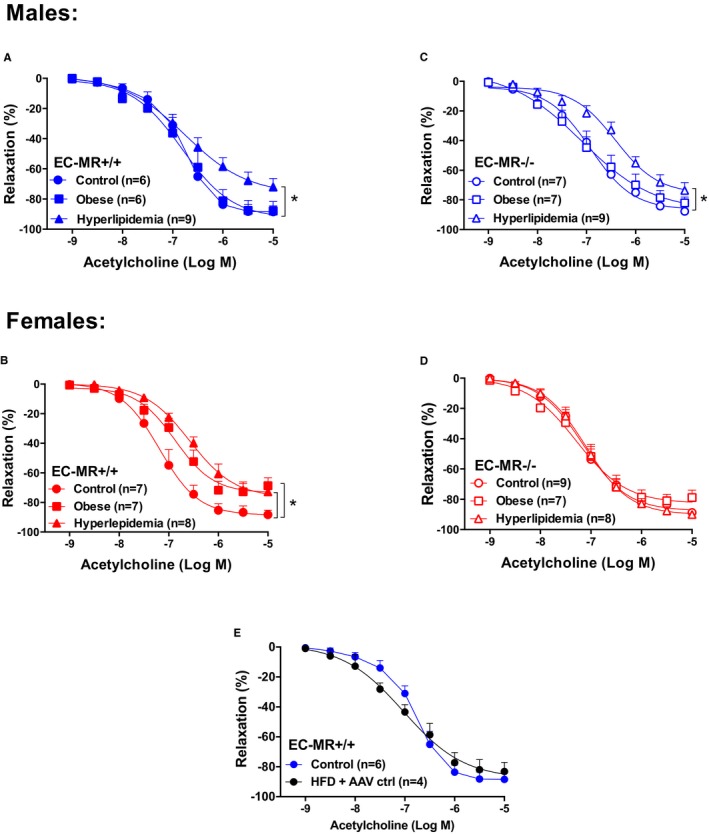
Female mesenteric resistance arteries are sensitive to obesity, and deletion of mineralocorticoid receptor (MR) in endothelial cells (EC‐MR) protects females but not males from risk factor‐induced endothelial dysfunction. Endothelium‐dependent acetylcholine‐induced relaxation in small mesenteric arteries of control, obese, and hyperlipidemic male (blue) and female (red) MR‐intact mice (EC‐MR +/+; A and B) or EC‐MR knockout mice (EC‐MR −/−; C and D). E, A single injection of control adeno‐associated virus (AAV ctrl) with a high‐fat diet (HFD) did not affect endothelium‐dependent relaxation induced by acetylcholine in obese male EC‐MR +/+ mice. Two‐factor ANOVA, *P<0.05.
Figure 2.
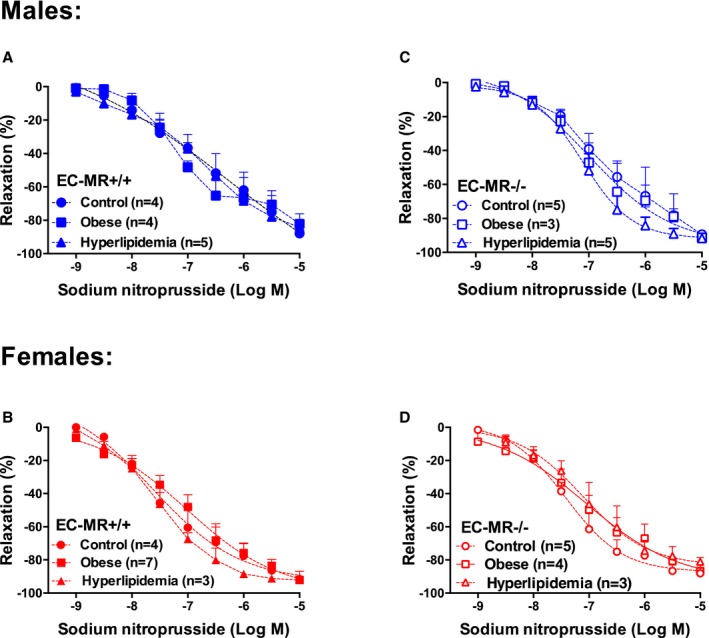
Smooth muscle cell–dependent vasodilation is not affected by risk factors or deletion of mineralocorticoid receptor (MR) in endothelial cells (EC‐MR). Sodium nitroprusside–induced relaxation in small mesenteric arteries of control, obese, and hyperlipidemic male and female MR‐intact mice (EC‐MR +/+, A and B) and EC‐MR knockout mice (EC‐MR −/−; C and D). Two‐factor ANOVA, P=not significant.
Figure 3.
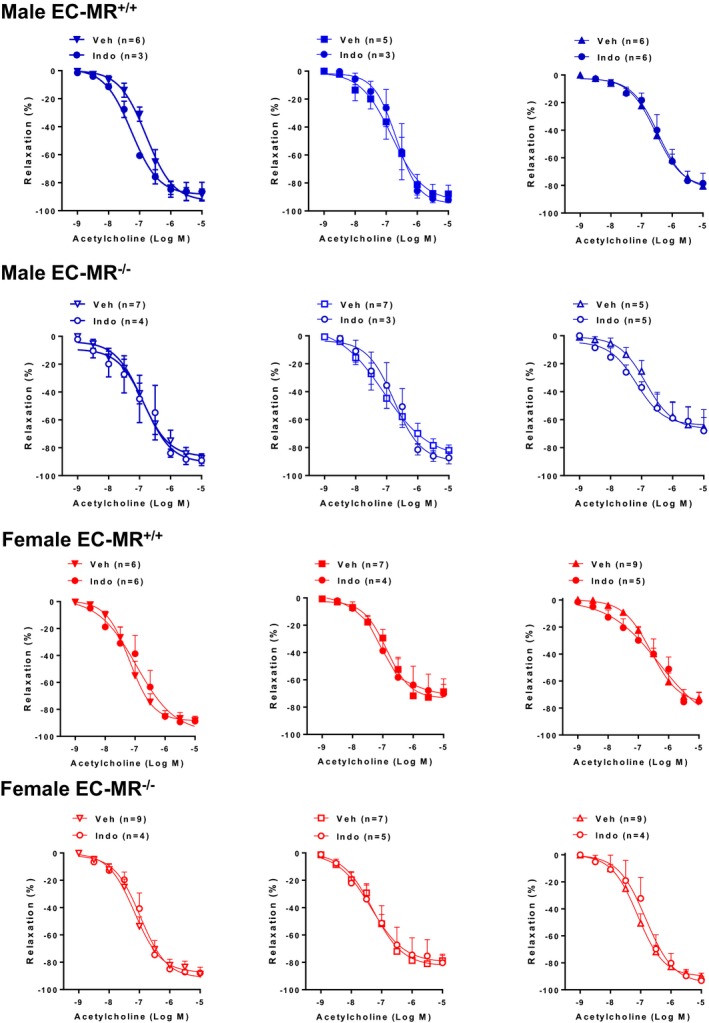
No role for COX(cyclooxygenase)–derived prostanoids in endothelium‐dependent relaxation in response to acetylcholine. Small mesenteric arteries from male and female EC‐MR–intact mice (EC‐MR+/+; closed symbols) and knockout mice (EC‐MR−/−; open symbols) were evaluated in the presence of the COX inhibitor indomethacin (Indo) or vehicle (Veh). Two‐factor ANOVA, P>0.05. EC‐MR indicates mineralocorticoid receptors in endothelial cells.
Figure 4.
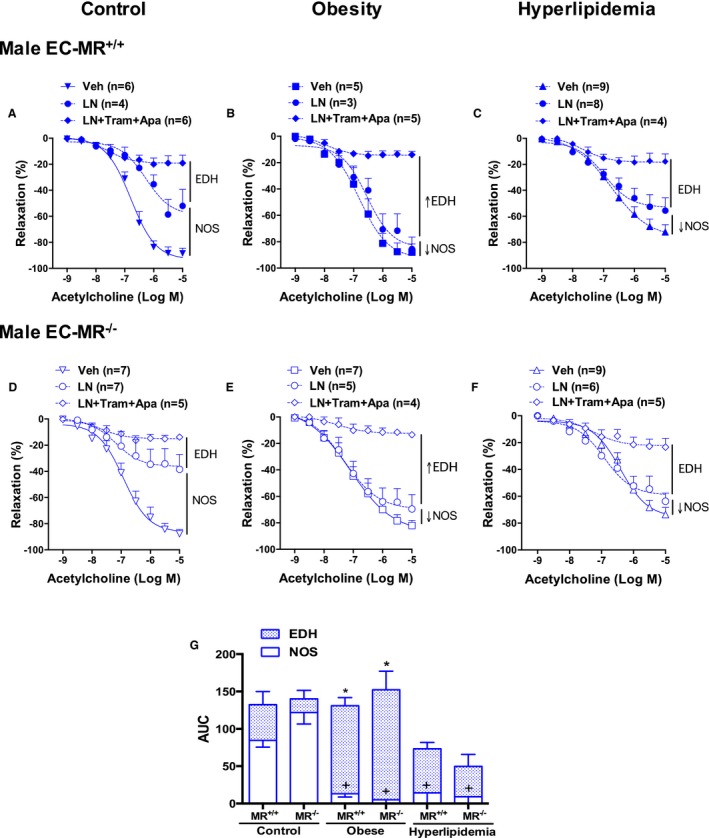
Contribution of nitric oxide synthase (NOS) and endothelium‐derived hyperpolarization (EDH) to resistance vessel endothelial function of male mice. Endothelium‐dependent acetylcholine‐induced relaxation was evaluated after NOS inhibition with L‐NAME (LN) alone, with the endothelial K+ channel blockers apamin (Apa) and TRAM34 (Tram) to inhibit EDH or with vehicle (Veh) in small mesenteric arteries of control, obese, and hyperlipidemic male EC‐MR–intact mice (EC‐MR +/+; A through C) or knockout mice (EC‐MR −/−; D through F). G, Area under the curve represents the relative contribution of NOS and EDH in males and shows that males depend more on NO and that NOS‐mediated dilation is lost with obesity and hyperlipidemia but is compensated for by increased EDH in obesity alone. Two‐factor ANOVA, *P<0.05 vs EDH in control EC‐MR +/+; + P<0.05 vs NO in control EC‐MR +/+. EC‐MR indicates mineralocorticoid receptors in endothelial cells.
Figure 5.
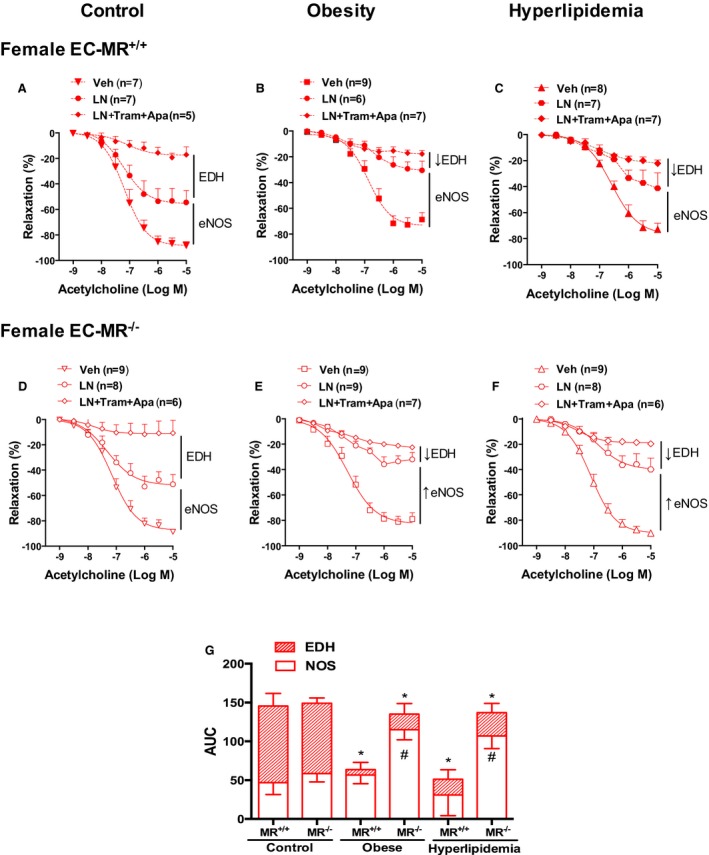
Contribution of nitric oxide synthase (NOS) and endothelium‐derived hyperpolarization (EDH) in resistance vessel endothelial function of female mice. Endothelium‐dependent acetylcholine‐induced relaxation was evaluated after NOS inhibition with L‐NAME (LN) alone, with the endothelial K+ channels blockers apamin (Apa) and TRAM34 (Tram) to inhibit EDH or with vehicle (Veh) in small mesenteric arteries of control, obese, and hyperlipidemic female EC‐MR–intact mice (EC‐MR +/+; A through C) or knockout mice (EC‐MR −/−; D through F). G, Area under the curve (AUC) represents the relative contribution of NOS and EDH in females and shows that females depend more on EDH and that the reduction in EDH in obesity and hyperlipidemia is compensated for with an increased NOS component when EC‐MR is deleted. Two‐factor ANOVA, *P<0.05 vs EDH in control EC‐MR +/+; # P<0.05 vs nitric oxide in EC‐MR +/+ under the same risk factor condition. EC‐MR indicates mineralocorticoid receptors in endothelial cells.
Figure 6.
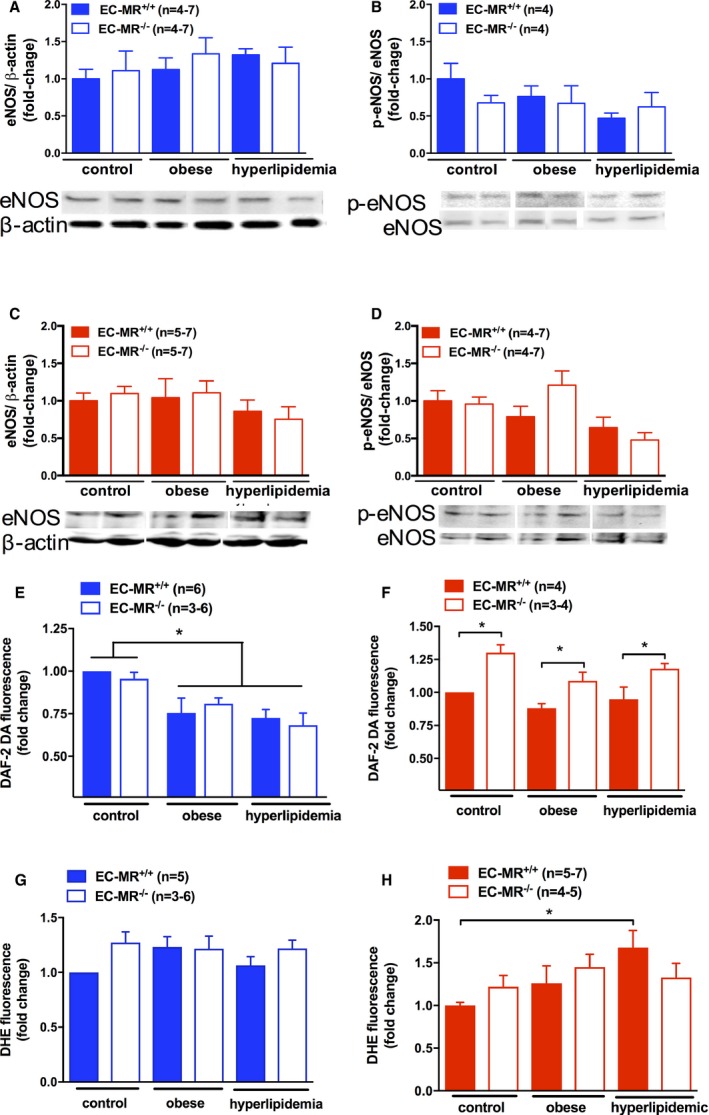
Nitric oxide (NO) and reactive oxygen species (ROS) production in male and female resistance vessels in response to risk factors. Protein expression of total endothelial nitric oxide synthase (eNOS) and serine 1177 phosphorylated eNOS (p‐eNOS) in small mesenteric arteries of control, obese, and hyperlipidemic male (A and B) and female (C and D) EC‐MR–intact mice (EC‐MR +/+) or knockout mice (EC‐MR −/−). NO levels were evaluated by quantification of DAF‐2 DA (4,5‐diaminofluorescein diacetate) fluorescence, and ROS production was evaluated by quantification of DHE (dihydroethidium [hydroethidine]) fluorescence in mesenteric artery sections from male (E and G) and female (F and H) mice. Two‐factor ANOVA: *P<0.05. EC‐MR indicates mineralocorticoid receptors in endothelial cells.
Figure 7.
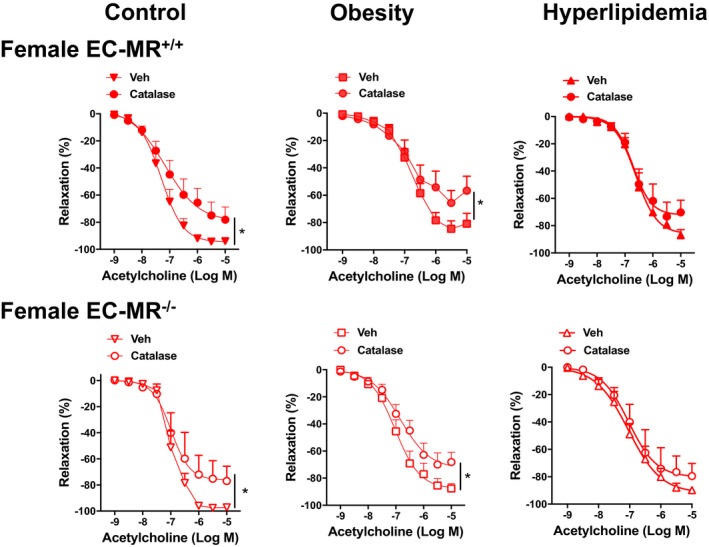
Hydrogen peroxide contributes to mesenteric microvessel relaxation in females and is lost with hyperlipidemia, with no role of mineralocorticoid receptors in endothelial cells (EC‐MR). Effect of catalase on the endothelium‐dependent relaxation to acetylcholine in mesenteric resistance arteries from control, obese, and hyperlipidemic female EC‐MR–intact mice (EC‐MR +/+; closed symbols) and knockout mice (EC‐MR −/−; open symbols). Two‐factor ANOVA, *P<0.05. EC‐MR indicates mineralocorticoid receptors in endothelial cells; Veh, vehicle.
Figure 8.
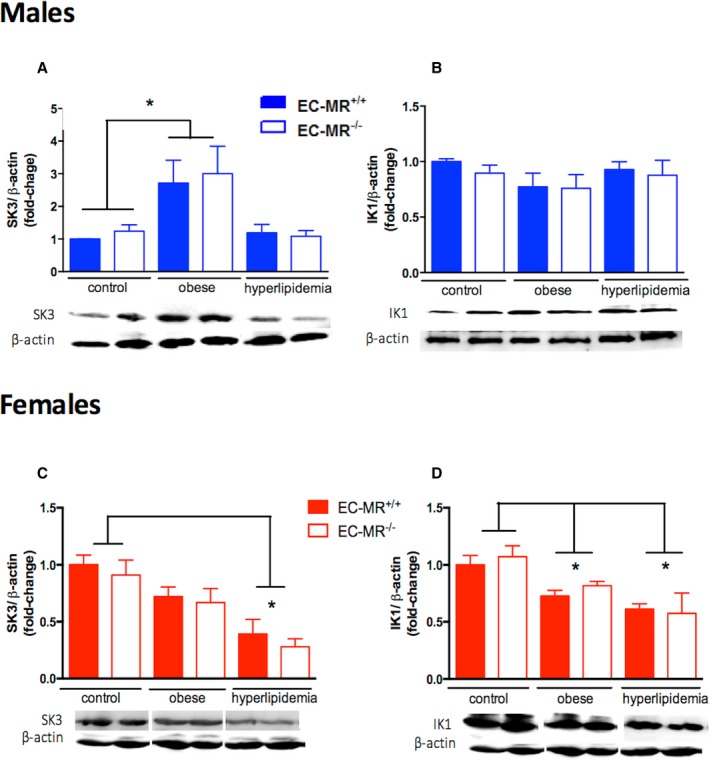
Changes in the expression of endothelial K+ channels in male and female resistance vessels in response to cardiometabolic risk factors. Protein expression of endothelial K+ channels SK3 and IK1 in small mesenteric arteries of control, obese, and hyperlipidemic male (A and B) and in female (C and D) EC‐MR–intact mice (EC‐MR +/+) or knockout mice (EC‐MR −/−; n=4–8 mice). Two‐factor ANOVA, *P<0.05. EC‐MR indicates mineralocorticoid receptors in endothelial cells.
Results
Traditional Risk Factors and Hormone Levels in Mouse Models of Obesity and Hyperlipidemia
To explore mechanisms underlying sex differences in the response of resistance vessels to cardiometabolic risk factors, male and female MR‐intact and EC‐MR‐KO littermates underwent diet‐induced obesity with or without AAV‐PCSK9DY‐induced hyperlipidemia and were compared with littermates fed normal chow (control). At the termination of the studies, traditional risk factors and hormone levels were measured in a subset of animals in each group. As expected, animals fed an HFD exhibited significantly greater body weight (obesity model), and the single injection of AAV‐hPCSK9DY induced a significant increase in serum cholesterol (hyperlipidemia model; Table). The obesity model was associated with an increase in fasting glucose consistent with metabolic dysfunction, whereas the hyperlipidemic obese animals gained slightly less weight with no significant change in serum glucose. As expected, females had lower body weight and slightly lower blood pressure than males.
Serum levels of aldosterone and corticosterone, hormones that can activate the MR, were significantly increased in females compared with males. Hyperlipidemic mice had significantly lower aldosterone and higher corticosterone levels compared with the obesity group. Serum estradiol levels were measured in females and were significantly decreased in hyperlipidemic females compared with normal‐diet controls. It is important to note that all changes in the traditional cardiovascular risk factors and hormones measured in Table, including the extent of the obesity or hyperlipidemia in both sexes, were independent of the presence or deletion of EC‐MR.
Female Mice are Susceptible to Obesity‐Associated Resistance Microvessel Dysfunction
The degree of endothelium‐dependent relaxation in response to acetylcholine was measured in small mesenteric resistance arteries. All vessels were preconstricted with phenylephrine, and there was no significant effect of genotype or diet on the contractile response to phenylephrine. As previously published regarding males,28 in control mice of either sex without risk factors, EC‐MR deletion did not affect the vasodilatory response to acetylcholine (data not shown). In EC‐MR–intact mice, 12 weeks of HFD‐induced obesity resulted in significant impairment of endothelium‐dependent relaxation only in females, with no effect in males (Figure 1A and 1B). These data suggest that in female mice, as in women,5, 36 there is increased susceptibility to microvessel endothelial dysfunction in the setting of obesity. HFD combined with hPCSK9DY‐induced hyperlipidemia was associated with endothelial dysfunction in both male and female mice (Figure 1A and 1B). Injection of control AAV combined with HFD in males did not significantly affect relaxation of mesenteric arteries (Figure 1E), suggesting that the vascular dysfunction was not induced by viral infection alone and was likely due to hyperlipidemia.
EC‐MR Deletion Protects Only Females From Endothelial Dysfunction Induced by Cardiometabolic Risk Factors
EC‐MR deletion prevented resistance vessel endothelial dysfunction associated with obesity and hyperlipidemia in females but not in males (Figure 1C and 1D), supporting a sex difference in the role of EC‐MR in the vascular response to cardiometabolic risk factors. Relaxation responses to the NO donor sodium nitroprusside were similar in all groups (Figure 2), indicating that the mechanism for the impaired vasodilatory response to acetylcholine and the sex differences resulted not from a smooth muscle cell defect but rather from an EC defect.
Sex Differences in the Contributions of NO and EDH to Microvessel Dysfunction Induced by Cardiometabolic Risk Factors
To investigate the mechanisms driving endothelial dysfunction in response to cardiometabolic risk factors, we incubated the isolated vessels with specific inhibitors of endothelium‐derived relaxation factors and again quantified endothelial‐dependent relaxation to a dose escalation of acetylcholine. Indomethacin was first used to inhibit COX‐derived prostanoid production. Prior studies showed that COX‐derived products do not contribute to vasodilation in healthy resistance vessels in rodents14 but are implicated in conduit vessel function in the setting of obesity and dyslipidemia.30, 37 We found that indomethacin does not affect acetylcholine‐induced relaxation at baseline in small mesenteric resistance arteries or in mice exposed to risk factors regardless of their sex or the presence of EC‐MR (Figure 3). These data suggest that COX‐derived products do not contribute to endothelium‐dependent relaxation in mesenteric resistance microvessels in these obesity and hyperlipidemia models.
Next, the contribution of NO and EDH to resistance vessel function was quantified. Endothelium‐dependent relaxation was measured after blockade of endothelial NO production with the eNOS inhibitor L‐NAME alone and in combination with EDH blockade with inhibitors of the endothelial potassium channels IK1 and SK3 by TRAM34 and apamin, respectively. The data reveal profound differences in the effects of cardiometabolic risk factors on the components of endothelial dilation in females versus males. In males (Figure 4), in the absence of risk factors, both eNOS and EDH contribute to vasodilation, with NOS contributing a greater proportion of mesenteric vasodilatory function than EDH (Figure 4A and 4D), consistent with prior studies.14, 15 Exposure of male mice to obesity, with or without hyperlipidemia, resulted in profound impairment in eNOS‐mediated relaxation of resistance vessels. However, in males exposed to obesity alone, decreased NO was compensated for by a significant increase in the EDH component of vasorelaxation (Figure 4B). This contrasts with hyperlipidemic obese males in which the profound decline in the NOS component is not overcome, resulting in impaired endothelium‐dependent relaxation (Figure 4C). Under all experimental conditions tested in males, EC‐MR deletion had no effect on the contributions of these endothelium‐derived relaxation factors (Figure 4D through 4F). The relative contribution of endothelium‐derived relaxation factors in small arteries of males is summarized in Figure 4G by calculating the area under each of the curves in Figure 4A through 4F.
In females, the response of the resistance vasculature to cardiometabolic risk factors was quite distinct from males (Figure 5). As published previously,14, 15 in control female mice without risk factors, EDH was a greater contributor to endothelial‐dependent relaxation than NO (Figure 5A and 5D). When exposed to obesity or hyperlipidemia, female resistance vessels exhibited a significant decline in the EDH component of acetylcholine‐induced relaxation (Figure 5B and 5C). However, in female EC‐MR‐KO mice, the impaired EDH was compensated for by a significant increase in the NOS contribution to vasodilation (Figure 5E and 5F). The relative contribution of endothelium‐derived relaxation factors in small resistance arteries of females is summarized in Figure 5G by calculating the area under each of the curves in Figure 5A through 5F. The results in Figures 4 and 5 demonstrate profound sex differences in the mechanisms underlying risk factor–induced endothelial responses of the resistance vasculature, with a protective role for EC‐MR deletion only in females.
NO and ROS Production in Male and Female Resistance Vessels in Response to Risk Factors
To investigate mechanisms for the observed changes in the NO component of resistance vessel function in response to cardiometabolic risk factors, mesenteric vessel protein lysates were used to quantify total eNOS protein expression as well as the level of eNOS phosphorylation at serine 1177, a posttranslational modification associated with enhanced eNOS activation (Figure 6A through 6D). There was no effect of obesity, hyperlipidemia, or the presence of EC‐MR on mesenteric vessel total eNOS expression (Figure 6A and 6C). The level of eNOS activation, as determined by the ratio of phosphorylated to total eNOS, was also not changed under any condition (Figure 6B and 6D). We next evaluated the overall level of bioavailable NO in mesenteric vessels in situ using the specific fluorescent probe DAF‐2 DA. In males, resistance vessel NO content was significantly reduced in mesenteric arteries from obese and hyperlipidemic mice, independent of the presence of EC‐MR (Figure 6E). In MR‐intact females, exposure to obesity with or without hyperlipidemia did not affect resistance artery NO content (Figure 6F). However, females lacking EC‐MR exhibited significantly increased NO levels compared with EC‐MR–intact female mice. These data are consistent with the functional studies in Figures 4 and 5 and support that, in males, exposure to cardiometabolic risk factors impairs resistant vessel NO bioavailability with no role for EC‐MR, whereas in females, NO levels are not affected by risk factors but are enhanced when EC‐MR is deleted. A potential explanation for changes in vascular NO levels without a change in eNOS expression or serine 1177 phosphorylation would be changes in NO bioavailability due to ROS production that inactivates NO. To test this, superoxide production was measured by DHE fluorescence in mesenteric vessel sections. DHE fluorescence was not different between male groups, regardless of risk factor exposure or the presence of EC‐MR (Figure 6G). This result suggests that changes in NO levels in small mesenteric vessels exposed to risk factors may not be related to local superoxide production in males. In females, there was a significant increase in superoxide generation in response to hyperlipidemia that was not observed in female mice lacking EC‐MR (Figure 6H).
Sex‐Specific EDH Functional Responses to Cardiometobolic Risk Factors Correlate With Resistance Vessel Potassium Channel Expression
In females, the primary response of the mesenteric microvasculature to cardiometabolic stress was decreased EDH (Figure 5). Because H2O2 can act as an EDH factor in human and mouse mesenteric vessels,12, 13 we next examined the contribution of H2O2 to endothelium‐dependent relaxation in females. As shown in Figure 7, treatment with catalase to inactivate H2O2 reduced the vasodilation in control and obese EC‐MR intact female mice, whereas catalase had no effect in hyperlipidemic mice. Thus, a small component of vasodilation in control and obese females is mediated by H2O2, and this is lost with hyperlipidemia. The role of H2O2 was unaffected by the presence of EC‐MR. Although a loss of H2O2 may contribute to endothelial dysfunction in hyperlipidemic females, this finding is insufficient to explain the near total loss of EDH seen in Figure 5, nor do these data clarify the mechanism of dysfunction in obese females without hyperlipidemia.
SK3 and IK1 are endothelial channels that contribute to EDH of smooth muscle cells resulting in vasodilation of small mesenteric resistance arteries.11 We next evaluated protein expression of these endothelial potassium channels in resistance vessels from males and females exposed to obesity with or without hyperlipidemia (Figure 8). There was a significant increase in mesenteric protein expression of SK3 (Figure 8A) in obese male mice, in which we previously showed that EDH‐mediated vasodilation was increased, thereby compensating for impaired NO (Figure 4G). There was no significant change in IK1 protein expression under any condition in males (Figure 8B). There was also no effect of EC‐MR deletion on expression of SK3 or IK1 in males under any condition, consistent with the functional studies in Figure 4. These data suggest that the compensatory increase in EDH in obese males may be due to upregulation of SK3 protein in small mesenteric arterioles, with no role for EC‐MR in regulating expression of these channels in males.
In females, the observed decline in the EDH component of resistance vessel function in the setting of obesity and hyperlipidemia (Figure 5) was associated with reduced expression of endothelial potassium channels in small mesenteric arteries (Figure 8C and 8D). SK3 expression was significantly reduced in mesenteric vessels specifically from hyperlipidemic females, whereas IK1 expression was decreased in obese female mice with or without concomitant hyperlipidemia. As with the functional data in Figure 5, in which MR deletion did not affect EDH‐mediated vasodilation, the deletion of EC‐MR did not mitigate the effects of risk factors on expression of the endothelial potassium channel proteins (Figure 8C and 8D). Combined, these data suggest that in females with obesity, endothelial dysfunction is caused by a loss of endothelium‐derived hyperpolarizing factor in association with decreased resistance vessel IK1 protein, whereas with hyperlipidemia, the mechanism may involve decreased SK3, IK1, and H2O2.
Discussion
This study compared, for the first time, the endothelial responses of mesenteric resistance microvessels to cardiometabolic risk factors in male and female mice. The results reveal sex‐specific mechanisms driving endothelial dysfunction that are further modulated depending on whether obesity is also associated with hyperglycemia or hyperlipidemia. Specifically, we showed, first, that mesenteric microvessels from males are dependent on NO for vasodilation and that when exposed to obesity with or without hyperlipidemia, males exhibit a substantial loss of NOS‐dependent relaxation in association with decreased microvessel NO content. Second, in obese males without hyperlipidemia, the deficiency of NO was compensated for by increased EDH with greater expression of the SK3 potassium channel in resistance vessels. Third, female mesenteric resistance vessels were particularly sensitive to obesity‐induced endothelial dysfunction and were more dependent on EDH for vasodilation. Fourth, in females, obesity and hyperlipidemia led to profound impairment of the EDH component of endothelium‐dependent vasodilatation. In obesity, impaired EDH was associated with decreased expression of the endothelial IK1 channel, whereas in females with hyperlipidemia, impaired EDH correlated with a decline in IK1, SK3, and H2O2 and increased oxidative stress. Fifth, in females, MR may be activated by increased serum aldosterone and corticosterone levels compared with males, and deletion of the MR from EC specifically protected females from endothelial dysfunction by increasing NO availability, thereby compensating for impaired EDH. These results support the concept that the molecular mechanisms driving microvascular endothelial dysfunction, the initial step in the development of vascular disease in response to common risk factors, may be entirely different in males and females. Based on these findings, personalized therapies that are both sex‐specific and tailored to the associated metabolic risk factor profiles need to be developed to prevent the adverse cardiovascular consequences of the growing obesity epidemic.
Previous studies have demonstrated sex differences in the mechanisms of resistance vessel vasodilation in the absence of risk factors. In small arteries, endothelium‐dependent relaxation in males was found to rely more heavily on NOS than EDH, and the opposite was found for females.14, 15, 38, 39 No role for COX‐derived prostanoids was found in either sex.12, 14, 15 We confirmed these findings in resistance vessels from healthy mice and further explored the response to common cardiometobolic risk factors. In males, we showed that obesity and hyperlipidemia profoundly impaired NO availability in mesenteric resistance microvessels. This is consistent with many prior studies demonstrating that vascular NO bioavailability is impaired by obesity and metabolic syndrome; however, much of the data supporting this concept were performed only in males, thereby missing potential sex‐specific mechanisms (reviewed in 7, 40). In addition, many such studies, regardless of sex, measured endothelial responses in the mouse aorta,27, 30 a conduit vessel that is larger and thus easier to study. Resistance vessels differ from conduit vessels, in which NO is the major endothelium‐dependent relaxation factor, and no role has yet been identified for endothelial SK and IK channels.11, 41, 42 Because aortic vasodilation does not contribute substantially to regulation of blood flow or pressure, it can be speculated that conduit vessels have fewer redundant pathways to maintain this function, but further studies are needed to test that hypothesis.
Interestingly, although the degree of mesenteric relaxation to acetylcholine in male mice exposed to 12 weeks of diet‐induced obesity appeared to be preserved, further investigation revealed that there was, in fact, profound impairment of NO availability that was overcome by a compensatory increase in the contribution of EDH. This compensatory rise in EDH was not observed when males were exposed to obesity with hyperlipidemia. Our results in male mice are consistent with a recent study in male rhesus monkeys fed a high‐fat/high‐fructose diet to induce obesity with progression to metabolic syndrome.43 In the obese male monkeys, NO bioavailability in skeletal muscle microvessels progressively declined over 2 years, but skeletal blood flow was initially preserved because of increased endothelium‐derived vasodilators. In the monkey model at 2 years, insulin resistance developed and skeletal muscle blood flow abruptly declined.43 Combining our findings in rodents with this study in primates supports the concept that, in males, early obesity‐induced impairment of microvessel NO availability is overcome by an increase in non‐NO vasodilators but that when obesity is combined with additional metabolic risk factors, this compensatory mechanism in males is lost. Further studies are needed to test this mechanism in humans; however, a recent meta‐analysis of human data demonstrated that obesity is associated with microvascular endothelial dysfunction that is further impaired when obesity is complicated by other metabolic abnormalities.44
Our study further explored potential mechanisms driving microvessel dysfunction in response to risk factors. In the endothelium, NO is produced by eNOS and mediates vasodilation by activating guanylyl cyclase in underlying smooth muscle cells. The smooth muscle cell vasodilatory response to an NO donor and the contractile response to phenylephrine were not affected by sex or risk factors, confirming that the defect is in the endothelium. Potential alterations in vascular tone or contractile responses to other agonists remain to be explored. The decline in NOS‐mediated vasodilation in obese and hyperlipidemic male mice was associated with decreased NO bioavailability measured directly in the vessels. NO bioavailability can decrease because of a decline in NO production associated with reduced eNOS expression or activity, by uncoupling of eNOS to produce ROS, or by NO inactivation through reactions with oxidative free radicals from other sources.45 We found no differences in ROS generation, total eNOS expression, or the degree of eNOS serine 1177 phosphorylation, an important posttranslational modification that enhances eNOS activity. Although increased vascular ROS generation has be found in response to obesity, dyslipidemia, and metabolic syndrome,46 in early stages of obesity, vascular oxidative stress might not be present.47, 48 We cannot exclude the possibility that risk factors modify eNOS activity and NO bioavailability through modulation of a different eNOS phosphorylation site other than serine 1177, such as the inhibitory threonine 495 phosphorylation site or others. In obese males only, the impairment in NO was associated with increased EDH‐mediated relaxation and a specific increase in mesenteric protein expression of the endothelial SK3 potassium channel. This finding is consistent with a recent study showing that EC‐specific deletion of SK3 in male mice impaired EDH‐mediated mesenteric vessel relaxation and caused hypertension, although females were not examined in this study.49 Thus, increased expression of SK3 channels leading to enhanced EDH may be an important mechanism by which males defend resistance vessel endothelial vasodilatory function from the decrease in NO induced by obesity.
By performing identical studies on male and female littermates, we found that the mechanisms driving endothelial dysfunction in response to risk factors are completely different in female mice. In response to HFD‐induced obesity, female mice developed impaired resistance vessel endothelial function. This female susceptibility to vascular dysfunction was shown in a prior study in the aorta of mice with elevated leptin, an adipokine that is increased in obesity.27 In human population studies, female sex is associated with increased risk of endothelial dysfunction specifically in microvessels but not in conduit vessels10; therefore, the focus was on resistance vessels in this study. In females, we showed that endothelial dysfunction was caused by impaired EDH‐mediated relaxation, which was distinct from the results in males. The decline in EDH in obese females was associated with a significant decrease in IK1 channel expression with a significant decline in SK3 and the hydrogen peroxide vasodilatory effect with concomitant hyperlipidemia. Interestingly, the reduction in hydrogen peroxide in vessels from hyperlipidemic females was associated with increased ROS generation evaluated by DHE fluorescence. DHE is widely used to detect superoxide generation with high sensitivity, although other oxidants can produce a 2‐electron oxidation product with similar fluorescence characteristics.50 Consequently, the results suggest increased superoxide generation concomitant with reduced hydrogen peroxide in small arteries of female hyperlipidemic mice. It is possible this is a result of an imbalance between superoxide‐generating enzymes and superoxide scavengers, but further studies are warranted to explore this possibility.
In females, obesity with hyperlipidemia was associated with significantly lower estrogen levels. Because estrogen has been shown to transcriptionally regulate SK3 and IK1 expression,15, 51 the decrease in estradiol levels in females exposed to hyperlipidemia could contribute to the reduced expression of endothelial potassium channels and impaired endothelial function. Estrogen is also known to enhance endothelial NO production by activating eNOS via phosphorylation (reviewed in 40). In this study, however, exposure to cardiometabolic risk factors did not change eNOS phosphorylation in mesenteric vessels in either sex.
Interestingly, EC‐MR deletion specifically protected females from resistance vessel endothelial dysfunction by increasing NO bioavailability. Although we could not directly measure mesenteric EC‐MR expression because of the lack of a mouse MR antibody, a prior study showed that exposure to14 weeks of HFD did not alter the level of MR mRNA expression in isolated aortic ECs.30 Rather, we showed that females had higher levels of aldosterone and corticosterone than males, supporting the potential for enhanced MR activation to play a greater role in females. In addition, the ligand‐independent activation of MR by Rac1 (Rac family small GTPase 1) has been shown to be dependent on oxidative stress52; therefore, the enhanced vascular ROS in hyperlipidemic females could also contribute to enhanced MR activation in females. This finding is consistent with previous data demonstrating that EC‐MR deletion reduced aortic oxidative stress in a Western diet–induced obesity model.31 In that model, EC‐MR deletion decreased EC stiffness by reducing endothelial sodium channel activation,31 and reduced EC stiffness is another mechanism to increase endothelial NO release.53 Furthermore, the shift from aldosterone to corticosterone in hyperlipidemic mice could contribute to the distinct mechanisms driving endothelial dysfunction in obesity compared with hyperlipidemia because MR activation in vivo has been shown to differentially modulate gene expression depending on the ligand.54 Although these clues are intriguing, additional studies are warranted to clarify the specific mechanism of vascular protection by EC‐MR deletion and to determine why this protection occurs only in females. Potential crosstalk between estrogen and MR transcriptional function is supported by a study showing an interaction between the estrogen receptor and the MR in human ECs,55 but future studies comparing intact to ovariectomized females are warranted to confirm a role for estrogen in modulating MR function in vivo.
Although additional studies are needed determine whether these sex‐specific mechanisms contribute to vascular dysfunction in obese humans, a study using vessels from intestinal surgery patients showed that human mesenteric resistance vessels also depend on a combination of NO and EDH for vasodilation and that IK1 is the predominant mediator of EDH in human mesenteric arteries.56 A study examining pig coronary arterioles, which are quite similar to those in humans, also demonstrated an enhanced role for EDH in females, with males more dependent on NO.57 Furthermore, both SK3 and IK1 were shown to be expressed in porcine coronaries of both sexes, but IK1 channels specifically contributed to EDH‐mediated relaxation in female porcine coronary vessels, with no clear role in males.57 This supports the possibility that the mechanisms identified in the current study in mouse mesenteric vessels may be similar to coronary vessels and may apply to humans. Future studies directly exploring sex differences in mechanisms of coronary microvessel dysfunction in response to obesity could have particular clinical importance. Indeed, women with cardiovascular ischemia are more likely than men to have coronary microvascular dysfunction than large vessel coronary disease,58 but the mechanisms are not clear and no treatments are available. Consequently, these women are particularly difficult to treat, so enhanced mechanistic understanding could lead to novel treatment strategies for this unmet medical need. Moreover, a common complication of obesity is heart failure with preserved ejection fraction, for which there is also no approved drug therapy. A recent large clinical trial of MR antagonism in heart failure with preserved ejection fraction was negative,59 but the trial mixed male and female patients and included patients with all causes of heart failure with preserved ejection fraction. Understanding sexual dimorphisms in the mechanisms of microvascular dysfunction in response to obesity might suggest the idea that future trials should be designed to test sex‐specific therapies for specific causes of heart failure with preserved ejection fraction, such as MR antagonism specifically in obese females.
Important limitations of this study should be acknowledged. First, all data were collected in mesenteric resistance microvessels. This choice was made because microvascular function correlates more closely with CVD outcomes than conduit vessels in humans, resistance vessels contribute to blood flow and blood pressure control, and females are more susceptible to microvessel dysfunction. Nevertheless, as described, evidence suggests distinct responses to risk factors in conduit vessels and in different microvascular beds.28 Future studies are needed to determine whether the sexual dimorphic mechanisms identified in mesenteric vessels will be relevant in other beds, particularly the coronary and cerebral vasculature. In addition, we examined the effects of obesity alone or with hyperlipidemia, a component of metabolic syndrome; however, although glucose was elevated in the obesity model, we did not quantify other measures of insulin resistance or adipose dysfunction. Importantly, EC‐MR deletion did not affect serum glucose in our study, and published data further suggest that EC‐MR does not contribute to the metabolic response or to changes in adipose inflammation in response to diet‐induced obesity. Schäfer et al put male EC‐MR‐KO mice (made with the Tie2‐Cre, so MR is deleted in leukocytes and ECs) on an HFD for 14 weeks and showed that the obesity‐induced elevation in proinflammatory cytokines in adipose tissue was not changed by EC‐MR deletion,30 nor did EC‐MR deletion affect the degree of weight gain, fat weight, fasting glucose, or glucose tolerance. Two additional studies put female EC‐MR‐KO mice (using the same Cad‐Cre as in this study) on a Western diet for 16 weeks to induce obesity with insulin resistance and found that EC‐MR deletion did not affect the change in fat mass, lean mass, body weight, or insulin resistance as measured by homeostatic model assessment of insulin resistance (HOMA‐IR).25, 31 Although tail cuff blood pressure measurement revealed no difference in response to risk factors or EC‐MR deletion in this study, the tail cuff measurement is less sensitive than telemetry. Although published studies using telemetry reveal no blood pressure difference between MR‐intact and EC‐MR‐KO male mice on a control diet28 or female mice exposed to 16 weeks of Western diet–induced obesity,25 we cannot rule out a small blood pressure difference that is below the sensitivity of this method. Finally, these studies were performed using mouse models that allow for careful control of risk factor exposure; however, the additional genetic and environmental factors that likely contribute to vascular dysfunction in humans cannot be completely modeled in rodents.
Sources of Funding
This work was supported by grants from the National Institutes of Health (HL095590 and HL119290 to Jaffe, K12HD092535 to Jaffe and DuPont and F30HL137255 to Moss), the American Heart Association (EIA18290005 to Jaffe and 17PRE32910003 to Moss), and The Sao Paulo Research Foundation (FAPESP 2014/26192‐6 to Davel).
Disclosures
None.
Acknowledgments
We wish to thank Daniel Engelbertsen in the laboratory of Andrew H. Lichtman for his assistance in establishing the AAV‐PCSK9 model.
(J Am Heart Assoc. 2018;7:e007675 DOI: 10.1161/JAHA.117.007675.)29453308
References
- 1. Mosca L, Appel LJ, Benjamin EJ, Berra K, Chandra‐Strobos N, Fabunmi RP, Grady D, Haan CK, Hayes SN, Judelson DR, Keenan NL, McBride P, Oparil S, Ouyang P, Oz MC, Mendelsohn ME, Pasternak RC, Pinn VW, Robertson RM, Schenck‐Gustafsson K, Sila CA, Smith SC Jr, Sopko G, Taylor AL, Walsh BW, Wenger NK, Williams CL; AHA . Evidence‐based guidelines for cardiovascular disease prevention in women. American Heart Association scientific statement. Arterioscler Thromb Vasc Biol. 2004;24:e29–e50. [DOI] [PubMed] [Google Scholar]
- 2. Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, Abraham JP, Abu‐Rmeileh NM, Achoki T, AlBuhairan FS, Alemu ZA, Alfonso R, Ali MK, Ali R, Guzman NA, Ammar W, Anwari P, Banerjee A, Barquera S, Basu S, Bennett DA, Bhutta Z, Blore J, Cabral N, Nonato IC, Chang JC, Chowdhury R, Courville KJ, Criqui MH, Cundiff DK, Dabhadkar KC, Dandona L, Davis A, Dayama A, Dharmaratne SD, Ding EL, Durrani AM, Esteghamati A, Farzadfar F, Fay DF, Feigin VL, Flaxman A, Forouzanfar MH, Goto A, Green MA, Gupta R, Hafezi‐Nejad N, Hankey GJ, Harewood HC, Havmoeller R, Hay S, Hernandez L, Husseini A, Idrisov BT, Ikeda N, Islami F, Jahangir E, Jassal SK, Jee SH, Jeffreys M, Jonas JB, Kabagambe EK, Khalifa SE, Kengne AP, Khader YS, Khang YH, Kim D, Kimokoti RW, Kinge JM, Kokubo Y, Kosen S, Kwan G, Lai T, Leinsalu M, Li Y, Liang X, Liu S, Logroscino G, Lotufo PA, Lu Y, Ma J, Mainoo NK, Mensah GA, Merriman TR, Mokdad AH, Moschandreas J, Naghavi M, Naheed A, Nand D, Narayan KM, Nelson EL, Neuhouser ML, Nisar MI, Ohkubo T, Oti SO, Pedroza A, Prabhakaran D, Roy N, Sampson U, Seo H, Sepanlou SG, Shibuya K, Shiri R, Shiue I, Singh GM, Singh JA, Skirbekk V, Stapelberg NJ, Sturua L, Sykes BL, Tobias M, Tran BX, Trasande L, Toyoshima H, van de Vijver S, Vasankari TJ, Veerman JL, Velasquez‐Melendez G, Vlassov VV, Vollset SE, Vos T, Wang C, Wang X, Weiderpass E, Werdecker A, Wright JL, Yang YC, Yatsuya H, Yoon J, Yoon SJ, Zhao Y, Zhou M, Zhu S, Lopez AD, Murray CJ, Gakidou E. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384:766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Writing Group Members , Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jiménez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER III, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB; American Heart Association Statistics Committee; Stroke Statistics Subcommittee . Heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation. 2016;133:e38–e360. [DOI] [PubMed] [Google Scholar]
- 4. Landmesser U, Drexler H. Endothelial function and hypertension. Curr Opin Cardiol. 2007;22:316–320. [DOI] [PubMed] [Google Scholar]
- 5. Jongh RT, Serné EH, IJzerman RG, de Vries G, Stehouwer CD. Impaired microvascular function in obesity: implications for obesity‐associated microangiopathy, hypertension, and insulin resistance. Circulation. 2004;109:2529–2535. [DOI] [PubMed] [Google Scholar]
- 6. Grizelj I, Cavka A, Bian JT, Szczurek M, Robinson A, Shinde S, Nguyen V, Braunschweig C, Wang E, Drenjancevic I, Phillips SA. Reduced flow‐and acetylcholine‐induced dilations in visceral compared to subcutaneous adipose arterioles in human morbid obesity. Microcirculation. 2015;22:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Engin A. Endothelial dysfunction in obesity. Adv Exp Med Biol. 2017;960:345–379. [DOI] [PubMed] [Google Scholar]
- 8. Pucci G, Alcidi R, Tap L, Battista F, Mattace‐Raso F, Schillaci G. Sex‐ and gender‐related prevalence, cardiovascular risk and therapeutic approach in metabolic syndrome: a review of the literature. Pharmacol Res. 2017;120:34–42. [DOI] [PubMed] [Google Scholar]
- 9. Lind L, Berglund L, Larsson A, Sundström J. Endothelial function in resistance and conduit arteries and 5‐year risk of cardiovascular disease. Circulation. 2011;123:1545–1551. [DOI] [PubMed] [Google Scholar]
- 10. Hamburg NM, Palmisano J, Larson MG, Sullivan LM, Lehman BT, Vasan RS, Levy D, Mitchell GF, Vita JA, Benjamin EJ. Relation of brachial and digital measures of vascular function in the community: the Framingham Heart Study. Hypertension. 2011;57:390–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garland CJ, Dora KA. EDH: endothelium‐dependent hyperpolarization and microvascular signalling. Acta Physiol (Oxf). 2017;219:152–161. [DOI] [PubMed] [Google Scholar]
- 12. Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H, Takeshita A. Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matoba T, Shimokawa H, Kubota H, Morikawa K, Fujiki T, Kunihiro I, Mukai Y, Hirakawa Y, Takeshita A. Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in human mesenteric arteries. Biochem Biophys Res Commun. 2002;290:909–913. [DOI] [PubMed] [Google Scholar]
- 14. Zhang R, Thor D, Han X, Anderson L, Rahimian R. Sex differences in mesenteric endothelial function of streptozotocin‐induced diabetic rats: a shift in the relative importance of EDRFs. Am J Physiol Heart Circ Physiol. 2012;303:H1183–H1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chan MV, Bubb KJ, Noyce A, Villar IC, Duchene J, Hobbs AJ, Scotland RS, Ahluwalia A. Distinct endothelial pathways underlie sexual dimorphism in vascular auto‐regulation. Br J Pharmacol. 2012;167:805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268–275. [DOI] [PubMed] [Google Scholar]
- 17. Rogerson FM, Fuller PJ. Mineralocorticoid action. Steroids. 2000;65:61–73. [DOI] [PubMed] [Google Scholar]
- 18. Bentley‐Lewis R, Adler GK, Perlstein T, Seely EW, Hopkins PN, Williams GH, Garg R. Body mass index predicts aldosterone production in normotensive adults on a high‐salt diet. J Clin Endocrinol Metab. 2007;92:4472–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoshida S, Ishizawa K, Ayuzawa N, Ueda K, Takeuchi M, Kawarazaki W, Fujita T, Nagase M. Local mineralocorticoid receptor activation and the role of Rac1 in obesity‐related diabetic kidney disease. Nephron Exp Nephrol. 2014;126:16–24. [DOI] [PubMed] [Google Scholar]
- 20. Nagase M, Fujita T. Role of Rac1‐mineralocorticoid‐receptor signalling in renal and cardiac disease. Nat Rev Nephrol. 2013;9:86–98. [DOI] [PubMed] [Google Scholar]
- 21. Shibata S, Fujita T. Mineralocorticoid receptors in the pathophysiology of chronic kidney diseases and the metabolic syndrome. Mol Cell Endocrinol. 2012;350:273–280. [DOI] [PubMed] [Google Scholar]
- 22. Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res. 2005;96:643–650. [DOI] [PubMed] [Google Scholar]
- 23. Caprio M, Newfell B, Sala Al, Baur W, Fabbri A, Rosano G, Mendelsohn ME, Jaffe IZ. Functional mineralocorticoid receptor in human vascular endothelial cells regulate inter‐cellular adhesion molecule‐1 expression and promote leukocyte adhesion. Circ Res. 2008;102:1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME, Jaffe IZ. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med. 2012;18:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jia G, Habibi J, DeMarco VG, Martinez‐Lemus LA, Ma L, Whaley‐Connell AT, Aroor AR, Domeier TL, Zhu Y, Meininger GA, Mueller KB, Jaffe IZ, Sowers JR. Endothelial mineralocorticoid receptor deletion prevents diet‐induced cardiac diastolic dysfunction in females. Hypertension. 2015;66:1159–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DeMarco VG, Habibi J, Jia G, Aroor AR, Ramirez‐Perez FI, Martinez‐Lemus LA, Bender SB, Garro M, Hayden MR, Sun Z, Meininger GA, Manrique C, Whaley‐Connell A, Sowers JR. Low‐dose mineralocorticoid receptor blockade prevents western diet‐induced arterial stiffening in female mice. Hypertension. 2015;66:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huby AC, Otvos L, Belin de Chantemèle EJ. Leptin induces hypertension and endothelial dysfunction via aldosterone‐dependent mechanisms in obese female mice. Hypertension. 2016;67:1020–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mueller KB, Bender SB, Hong K, Yang Y, Aronovitz M, Jaisser F, Hill MA, Jaffe IZ. Endothelial mineralocorticoid receptors differentially contribute to coronary and mesenteric vascular function without modulating blood pressure. Hypertension. 2015;66:988–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Davel AP, Anwar IJ, Jaffe IZ. The endothelial mineralocorticoid receptor: mediator of the switch from vascular health to disease. Curr Opin Nephrol Hypertens. 2017;26:97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schäfer N, Lohmann C, Winnik S, van Tits LJ, Miranda MX, Vergopoulos A, Ruschitzka F, Nussberger J, Berger S, Lüscher TF, Verrey F, Matter CM. Endothelial mineralocorticoid receptor activation mediates endothelial dysfunction in diet‐induced obesity. Eur Heart J. 2013;34:3515–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jia G, Habibi J, Aroor AR, Martinez‐Lemus LA, DeMarco VG, Ramirez‐Perez FI, Sun Z, Hayden MR, Meininger GA, Mueller KB, Jaffe IZ, Sowers JR. Endothelial mineralocorticoid receptor mediates diet‐induced aortic stiffness in females. Circ Res. 2016;118:935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garg R, Rao AD, Baimas‐George M, Hurwitz S, Foster C, Shah RV, Jerosch‐Herold M, Kwong RY, Di Carli MF, Adler GK. Mineralocorticoid receptor blockade improves coronary microvascular function in individuals with type 2 diabetes. Diabetes. 2015;64:236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Salvador AM, Moss ME, Aronovitz M, Mueller KB, Blanton RM, Jaffe IZ, Alcaide P. Endothelial mineralocorticoid receptor contributes to systolic dysfunction induced by pressure overload without modulating cardiac hypertrophy or inflammation. Physiol Rep. 2017;5:e13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roche‐Molina M, Sanz‐Rosa D, Cruz FM, García‐Prieto J, López S, Abia R, Muriana FJ, Fuster V, Ibáñez B, Bernal JA. Induction of sustained hypercholesterolemia by single adeno‐associated virus‐mediated gene transfer of mutant hPCSK9. Arterioscler Thromb Vasc Biol. 2015;35:50–59. [DOI] [PubMed] [Google Scholar]
- 35. McGraw AP, Bagley J, Chen WS, Galayda C, Nickerson H, Armani A, Caprio M, Carmeliet P, Jaffe IZ. Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor‐dependent mechanism. J Am Heart Assoc. 2013;2:e000018 DOI: 10.1161/JAHA.112.000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hall ME, Brinkley TE, Chughtai H, Morgan TM, Hamilton CA, Jordan JH, Stacey RB, Soots S, Hundley WG. Adiposity is associated with gender‐specific reductions in left ventricular myocardial perfusion during dobutamine stress. PLoS One. 2016;11:e0146519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li S, Liu B, Luo W, Zhang Y, Li H, Huang D, Zhou Y. Role of cyclooxygenase‐1 and ‐2 in endothelium‐dependent contraction of atherosclerotic mouse abdominal aortas. Clin Exp Pharmacol Physiol. 2016;43:67–74. [DOI] [PubMed] [Google Scholar]
- 38. Scotland RS, Madhani M, Chauhan S, Moncada S, Andresen J, Nilsson H, Hobbs AJ, Ahluwalia A. Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase‐1 double‐knockout mice: key role for endothelium‐derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation. 2005;111:796–803. [DOI] [PubMed] [Google Scholar]
- 39. Luksha L, Poston L, Gustafsson JA, Hultenby K, Kublickiene K. The oestrogen receptor beta contributes to sex related differences in endothelial function of murine small arteries via EDHF. J Physiol. 2006;577:945–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vanhoutte PM, Zhao Y, Xu A, Leung SW. Thirty years of saying NO: sources, fate, actions, and misfortunes of the endothelium‐derived vasodilator mediator. Circ Res. 2016;119:375–396. [DOI] [PubMed] [Google Scholar]
- 41. Kong BW, Vanhoutte PM, Man RY, Leung SW. 17β‐estradiol potentiates endothelium‐dependent nitric oxide‐ and hyperpolarization‐mediated relaxations in blood vessels of male but not female apolipoprotein‐E deficient mice. Vascul Pharmacol. 2015;71:166–173. [DOI] [PubMed] [Google Scholar]
- 42. Takaki A, Morikawa K, Tsutsui M, Murayama Y, Tekes E, Yamagishi H, Ohashi J, Yada T, Yanagihara N, Shimokawa H. Crucial role of nitric oxide synthases system in endothelium‐dependent hyperpolarization in mice. J Exp Med. 2008;205:2053–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chadderdon SM, Belcik JT, Bader L, Peters DM, Kievit P, Alkayed NJ, Kaul S, Grove KL, Lindner JR. Temporal changes in skeletal muscle capillary responses and endothelial‐derived vasodilators in obesity‐related insulin resistance. Diabetes. 2016;65:2249–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brant LC, Wang N, Ojeda FM, LaValley M, Barreto SM, Benjamin EJ, Mitchell GF, Vasan RS, Palmisano JN, Münzel T, Blankenberg S, Wild PS, Zeller T, Ribeiro AL, Schnabel RB, Hamburg NM. Relations of metabolically healthy and unhealthy obesity to digital vascular function in three community‐based cohorts: a meta‐analysis. J Am Heart Assoc. 2017;6:e004199 DOI: 10.1161/JAHA.116.004199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li Q, Youn JY, Cai H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J Hypertens. 2015;33:1128–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Virdis A, Neves MF, Duranti E, Bernini G, Taddei S. Microvascular endothelial dysfunction in obesity and hypertension. Curr Pharm Des. 2013;19:2382–2389. [DOI] [PubMed] [Google Scholar]
- 47. Heinonen I, Rinne P, Ruohonen ST, Ruohonen S, Ahotupa M, Savontaus E. The effects of equal caloric high fat and western diet on metabolic syndrome, oxidative stress and vascular endothelial function in mice. Acta Physiol (Oxf). 2014;211:515–527. [DOI] [PubMed] [Google Scholar]
- 48. da Silva Franco N, Lubaczeuski C, Guizoni DM, Victorio JA, Santos‐Silva JC, Brum PC, Carneiro EM, Davel AP. Propranolol treatment lowers blood pressure, reduces vascular inflammatory markers and improves endothelial function in obese mice. Pharmacol Res. 2017;122:35–45. [DOI] [PubMed] [Google Scholar]
- 49. Yap FC, Weber DS, Taylor MS, Townsley MI, Comer BS, Maylie J, Adelman JP, Lin MT. Endothelial SK3 channel‐associated Ca2+ microdomains modulate blood pressure. Am J Physiol Heart Circ Physiol. 2016;310:H1151–H1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, Harrison DG, Bhatnagar A; American Heart Association Council on Basic Cardiovascular Sciences . Measurement of reactive oxygen species, reactive nitrogen species, and redox‐dependent signaling in the cardiovascular system: a scientific statement from the American Heart Association. Circ Res. 2016;119:e39–e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jacobson D, Pribnow D, Herson PS, Maylie J, Adelman JP. Determinants contributing to estrogen‐regulated expression of SK3. Biochem Biophys Res Commun. 2003;303:660–668. [DOI] [PubMed] [Google Scholar]
- 52. Kawarazaki H, Ando K, Shibata S, Muraoka K, Fujita M, Kawarasaki C, Fujita T. Mineralocorticoid receptor‐Rac1 activation and oxidative stress play major roles in salt‐induced hypertension and kidney injury in prepubertal rats. J Hypertens. 2012;30:1977–1985. [DOI] [PubMed] [Google Scholar]
- 53. Fels J, Oberleithner H, Kusche‐Vihrog K. Ménage à trois: aldosterone, sodium and nitric oxide in vascular endothelium. Biochim Biophys Acta. 2010;1802:1193–1202. [DOI] [PubMed] [Google Scholar]
- 54. Messaoudi S, Gravez B, Tarjus A, Pelloux V, Ouvrard‐Pascaud A, Delcayre C, Samuel J, Launay JM, Sierra‐Ramos C, Alvarez de la Rosa D, Clément K, Farman N, Jaisser F. Aldosterone‐specific activation of cardiomyocyte mineralocorticoid receptor in vivo. Hypertension. 2013;61:361–367. [DOI] [PubMed] [Google Scholar]
- 55. Barrett Mueller K, Lu Q, Mohammad NN, Luu V, McCurley A, Williams GH, Adler GK, Karas RH, Jaffe IZ. Estrogen receptor inhibits mineralocorticoid receptor transcriptional regulatory function. Endocrinology. 2014;155:4461–4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chadha PS, Liu L, Rikard‐Bell M, Senadheera S, Howitt L, Bertrand RL, Grayson TH, Murphy TV, Sandow SL. Endothelium‐dependent vasodilation in human mesenteric artery is primarily mediated by myoendothelial gap junctions intermediate conductance calcium‐activated K+ channel and nitric oxide. J Pharmacol Exp Ther. 2011;336:701–708. [DOI] [PubMed] [Google Scholar]
- 57. Wong PS, Roberts RE, Randall MD. Sex differences in endothelial function in porcine coronary arteries: a role for H2O2 and gap junctions? Br J Pharmacol. 2014;171:2751–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pepine CJ, Ferdinand KC, Shaw LJ, Light‐McGroary KA, Shah RU, Gulati M, Duvernoy C, Walsh MN, Bairey Merz CN; ACC CVD in Women Committee . Emergence of nonobstructive coronary artery disease: a woman's problem and need for change in definition on angiography. J Am Coll Cardiol. 2015;66:1918–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, Clausell N, Desai AS, Diaz R, Fleg JL, Gordeev I, Harty B, Heitner JF, Kenwood CT, Lewis EF, O'Meara E, Probstfield JL, Shaburishvili T, Shah SJ, Solomon SD, Sweitzer NK, Yang S, McKinlay SM; TOPCAT Investigators . Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370:1383–1392. [DOI] [PubMed] [Google Scholar]