

ABSTRACT
The human oral microbiota encompasses representatives of many bacterial lineages that have not yet been cultured. Here we describe the isolation and characterization of previously uncultured Desulfobulbus oralis, the first human-associated representative of its genus. As mammalian-associated microbes rarely have free-living close relatives, D. oralis provides opportunities to study how bacteria adapt and evolve within a host. This sulfate-reducing deltaproteobacterium has adapted to the human oral subgingival niche by curtailing its physiological repertoire, losing some biosynthetic abilities and metabolic independence, and by dramatically reducing environmental sensing and signaling capabilities. The genes that enable free-living Desulfobulbus to synthesize the potent neurotoxin methylmercury were also lost by D. oralis, a notably positive outcome of host association. However, horizontal gene acquisitions from other members of the microbiota provided novel mechanisms of interaction with the human host, including toxins like leukotoxin and hemolysins. Proteomic and transcriptomic analysis revealed that most of those factors are actively expressed, including in the subgingival environment, and some are secreted. Similar to other known oral pathobionts, D. oralis can trigger a proinflammatory response in oral epithelial cells, suggesting a direct role in the development of periodontal disease.
KEYWORDS: evolution, genome analysis, oral microbiology, periodontitis, proteomics
IMPORTANCE
Animal-associated microbiota likely assembled as a result of numerous independent colonization events by free-living microbes followed by coevolution with their host and other microbes. Through specific adaptation to various body sites and physiological niches, microbes have a wide range of contributions, from beneficial to disease causing. Desulfobulbus oralis provides insights into genomic and physiological transformations associated with transition from an open environment to a host-dependent lifestyle and the emergence of pathogenicity. Through a multifaceted mechanism triggering a proinflammatory response, D. oralis is a novel periodontal pathobiont. Even though culture-independent approaches can provide insights into the potential role of the human microbiome “dark matter,” cultivation and experimental characterization remain important to studying the roles of individual organisms in health and disease.
INTRODUCTION
Periodontitis is one of the most prevalent human diseases around the world. Moderate stages affect more than half of adults, while the severe form, involving tooth loss, occurs in 10 to 20% of the population (1, 2). Periodontitis has a complex polymicrobial etiology, with current models implicating synergistic and dysbiotic interactions between members of the gingival microbial community (including species present at low abundance) as the disease onset (3–6). Plaque biomass accumulation followed by changes in the composition of the microbiota lead to and advance gum inflammation (gingivitis). Further increase in microbial biomass, exacerbated by poor hygiene and genetic predisposition, sustain additional shifts in the microbial community composition, which becomes increasingly anaerobic in deepening periodontal pockets as periodontitis develops (6). At that stage, microbial signaling, with some species playing keystone disease roles, interferes with the normal host immune defense and orchestrates a persistent inflammatory response, resulting in tissue and bone loss (7–9).
Among the first bacteria implicated in the etiology of periodontal disease were Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia (10). More recent microbiome sequencing and disease association studies suggest that a much larger number of species (from over 700 that constitute the human oral microbiome) likely play a role in the development of oral disease, highlighting that periodontitis has a complex polymicrobial etiology (11–14). Some species are potentially beneficial, contributing to a healthy oral homeostasis, while others are associated with various stages of gingivitis and periodontitis, potentially acting as keystone species and/or virulent components of the disease-inducing microbiota (6). Studies with pure cultures, defined communities in vitro, and animal models have revealed proinflammatory characteristics and interspecies signaling that result in community shifts and a host response leading to tissue destruction (15, 16). However, approximately one-third of the oral microbial species-level taxa are still uncultured, including dozens of disease-associated lineages (17). To better understand the complex interactions between oral bacteria and their role in the transition from health to disease, cultivation remains a critical initial step (17).
Based on rRNA relative sequence abundance, Desulfobulbus sp. strain HOT041 (Deltaproteobacteria) has been one of the uncultured bacteria strongly associated with advanced periodontitis (12, 14). Deltaproteobacteria have low taxonomic diversity in the human microbiome, and other species have been linked, based on relative abundance, to either oral disease (Desulfomicrobium orale and Desulfovibrio fairfieldensis) (18, 19) or intestinal and gynecologic disorders (Desulfovibrio piger and Desulfovibrio intestinalis) (20–22). As with other oral species, it has remained unclear if those organisms play a role in the etiology of periodontitis or their increased abundance is a consequence of the disease state, with deep subgingival pockets favoring the proliferation of strict anaerobes. No host-associated species of Desulfobulbus has been cultivated so far, although multiple free-living relatives have been isolated and characterized from aquatic and sediment samples (23–29). All free-living Desulfobulbus species are sulfate reducers, strict anaerobes, largely prototrophic, and use a variety of electron donors and acceptors. Adaptation to a host-associated lifestyle provides relative environmental stability and close interaction with other host-recruited species. These factors often lead to genome reduction, metabolic codependence/specialization, and sometimes emergence of pathogenicity through lateral gene transfer (29–31). Previous single-cell genomic data suggested Desulfobulbus sp. strain HOT041 is capable of sulfate reduction (32), which we used here to selectively enrich for, isolate, and characterize that organism as the first host-associated member of the Desulfobulbus genus. Its physiological, genomic, and immunomodulatory characteristics provide new insights into adaptation to the human host-associated lifestyle and the emergence of pathogenicity.
RESULTS AND DISCUSSION
Enrichment cultures and isolation of a human oral Desulfobulbus strain in pure culture.
We established culture enrichments of sulfate-reducing bacteria using human oral subgingival samples collected from an individual with chronic periodontitis. Upon repeated passage in sulfate-lactate-based medium, we obtained a stable, simple community composed of Fusobacterium nucleatum, a common human oral bacterium, and Desulfobulbus sp. strain HOT041. The composition of this coculture was characterized using small subunit (SSU) rRNA amplicon sequencing (Illumina MiSeq), fluorescence in situ hybridization (FISH), and quantitative PCR (qPCR), revealing that F. nucleatum outnumbered Desulfobulbus sp. strain HOT041 by 5- to 10-fold in stationary phase (Fig. 1A and B). During time course cocultivation experiments, the growth of Desulfobulbus sp. strain HOT041 always exhibited a lag phase, but growth continued for days after F. nucleatum reached stationary phase. As initial experiments to isolate Desulfobulbus sp. strain HOT041 in pure culture by serial dilutions were unsuccessful, we hypothesized that the organism required metabolites produced by F. nucleatum in the coculture and, because microscopic analysis did not reveal a stable physical interaction between the two organisms, that Desulfobulbus sp. strain HOT041 could acquire them directly from the coculture medium. Such dependence on metabolic cofactors produced by other community members, including Fusobacterium, has been described for other oral bacteria as well (33–35). We therefore supplemented fresh culture medium with filter-sterilized medium from stationary-phase cocultures (cell-free spent medium [CFS]), and by applying three successive rounds of dilution to extinction, we obtained pure cultures of Desulfobulbus sp. strain HOT041. Purity was determined microscopically by FISH and by deep rRNA amplicon and genome sequencing. The isolate was named Desulfobulbus oralis, its SSU rRNA being 99.6% identical to that of Desulfobulbus sp. strain HOT041 clone R004 (36).
FIG 1 .

Desulfobulbus sp. strain HOT041 (D. oralis) in coculture with Fusobacterium nucleatum and in pure culture. (A) FISH using fluorescent oligonucleotide probes specific for Deltaproteobacteria (green) and universal Bacteria (red). (B). Growth of Desulfobulbus sp. strain HOT041 and F. nucleatum in coculture monitored by species-specific qPCR (with error bars based on three replicates). (C and D) Scanning electron micrographs of the D. oralis isolate. The arrowheads point to membrane vesicles.
Physiological characterization of D. oralis.
D. oralis is Gram negative, nonmotile, and nonsporulating, with rod-shaped cells 1 to 2 μm in length and 0.3 μm in diameter. Cells usually occur singly or in pairs, although they sometimes form longer chains. In scanning electron micrographs, we observed the presence of outer membrane vesicles (25 to 50 nm in diameter) on some cells (Fig. 1C and D). Such vesicles, common in Gram-negative organisms, including known oral pathobionts, have been shown to play important roles in interspecies interaction and to contain factors that trigger an inflammatory response (37, 38).
For routine cultivation, we grew D. oralis anaerobically at 37°C in a minimal defined liquid medium with lactate as the sole carbon and electron source and sulfate as the electron acceptor, supplemented with 5% (vol/vol) F. nucleatum CFS. No growth occurred in the presence of trace levels of oxygen or in the absence of F. nucleatum CFS. We found that the soluble, growth-promoting component(s) of CFS could not be inactivated by heating or protease treatment and could not be substituted for by defined supplements such as vitamins, amino acids, volatile fatty acids, siderophores, or complex nutrients (yeast extract, peptone, or brain heart infusion [BHI]), individually or in combinations. Therefore, the nature of the critical component(s) provided to D. oralis by F. nucleatum remains unknown, but such a requirement has been documented for other human oral bacteria as well, including Chloroflexi sp. strain HOT-439 (34) and Fretibacterium fastidiosum (35). Within the oral cavity, this type of interspecies dependence has become evident and is likely the main reason for recalcitrance to cultivation of many yet-uncultured species that likewise have lost the ability to synthesize a number of compounds and therefore must rely on other members of the microbiota (17, 39). CFS from a type strain of F. nucleatum (ATCC 23726) also supported the growth of D. oralis, indicating its physiological dependence is not restricted to the strain it coenriched with, although we have not extended that analysis to additional strains or species.
D. oralis is quite limited in the types of electron donors and acceptors it can utilize as an energy source for respiratory growth. Lactate and pyruvate are the only electron donors capable of being respired, while only sulfate or thiosulfate served as electron acceptors, out of all combinations tested (see Table S1 in the supplemental material). Lactate was utilized with stoichiometric (1:1) production of acetate as the metabolic by-product (Fig. 2A) and the release of sulfide. Respiratory growth on pyruvate was very weak. We also tested D. oralis for its ability to ferment organic substrates in the absence of an electron acceptor (sulfate respiration). Growth was supported by pyruvate and weakly by amino acids but not by lactate, glucose, mucin, or peptone. Pyruvate was fermented to acetate and propionate in a 2:1 ratio (Fig. 2B). In the subgingival space, sulfate could originate from both blood and serum, where its concentration is relatively low (0.15 mM) (40), but also could result from the connective tissue and bone breakdown during periodontal disease progression (40, 41). Lactate and pyruvate are released by other members of the oral microbiota and are important nutrient sources within the microbial oral biofilm (e.g., see reference 42). Therefore, we expect that D. oralis grows in vivo both by lactate-sulfate respiration and by pyruvate fermentation, depending on the local microenvironment and disease state. D. oralis expands therefore the list of confirmed sulfate reducers in the oral cavity, which includes species of Desulfovibrio and Desulfomicrobium. Sulfate reducers have long been associated with periodontal disease and their abundance correlated with disease severity (43). The availability of nutrients and especially sulfate may be limiting factors in their proliferation, as these organisms are specialized users of that terminal electron acceptor (44).
FIG 2 .
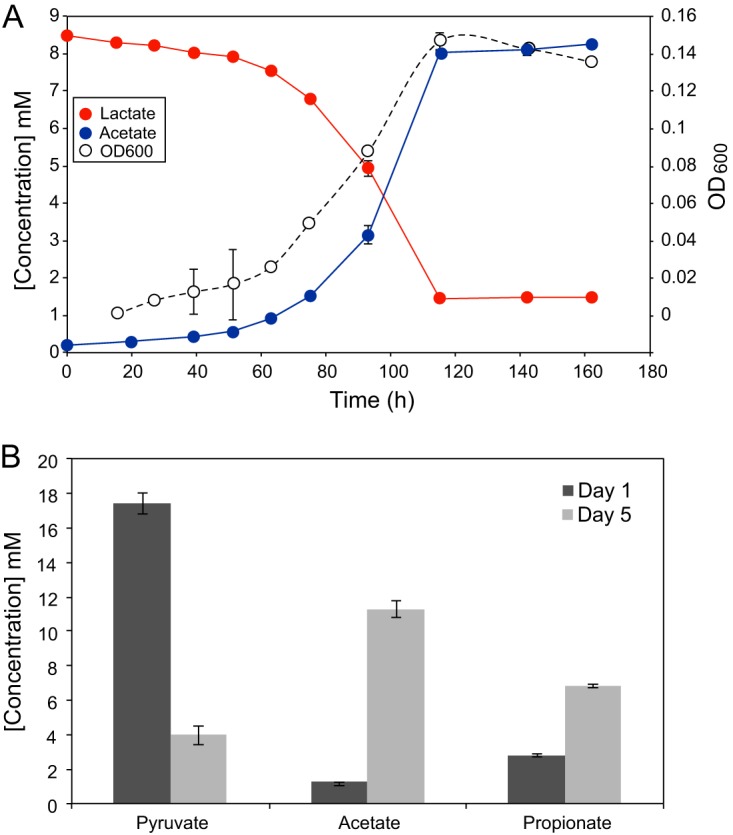
Growth of D. oralis by respiration and by fermentation. (A) Time course cultivation using sulfate and lactate as the electron donor-acceptor pair, with the release of acetate. (B) Substrate utilization and metabolic by-products of pyruvate fermentation. The error bars are based on three culture replicates.
D. oralis physiological characteristics compared to related species of the Desulfobulbus genus. Download TABLE S1, PDF file, 0.1 MB (100.5KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Comparative physiology and emergence of host association within the Desulfobulbus genus.
Host association leads to a reduced metabolic versatility, as the chemical and biological environment is relatively stable. This was evident as we compared the ability of several Desulfobulbus species to utilize various growth substrates (Table S1). All of the free-living isolates are able to utilize a wider range of substrates as electron donors and acceptors and for fermentation than D. oralis. Notably, propionate respiration and lactate fermentation, common traits among all free-living Desulfobulbus species, have been lost in D. oralis. Within the Desulfobulbus genus, host association is not restricted to humans, as closely related uncultured species have been detected in the oral microbiota of domesticated dogs (45) and cats (46). We performed a phylogenetic analysis using full-length SSU rRNA gene sequences, which suggested that mammalian host association was likely the result of an ancestral oral colonization by a free-living Desulfobulbus species, most closely related to Desulfobulbus oligotrophicus (Fig. 3). Additional sequences from other mammalian lineages as well as cultivation of other host-associated Desulfobulbus would be required to better understand the coevolution of these bacteria with their hosts.
FIG 3 .
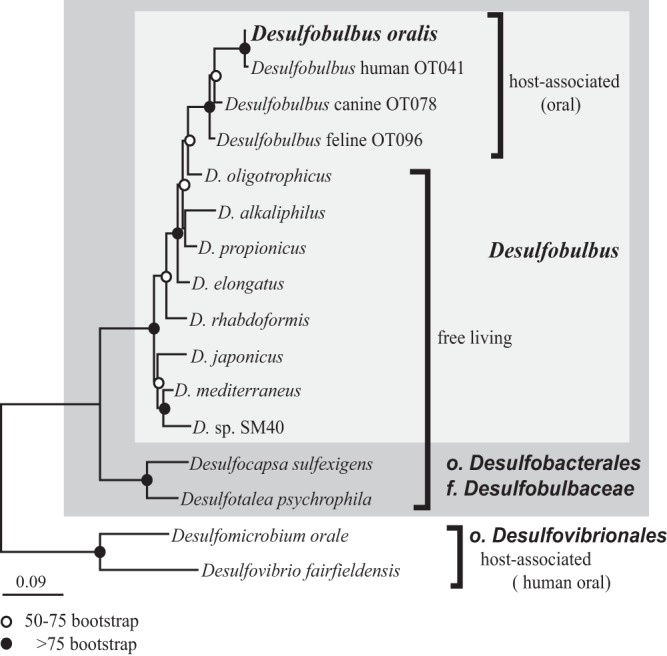
Maximum likelihood phylogeny of Desulfobulbus and related Deltaproteobacteria. The tree is based on full-length SSU rRNA gene sequences. Node support codes are based on 100 bootstrap replicates.
Genome sequencing, annotation, and metabolic reconstruction.
We obtained the complete genome sequence of D. oralis using long-read sequencing (PacBio). The assembly resulted in a single contig (>600-fold coverage), which was circularized as a single chromosome of 2,774,417 bp, with a G+C content of 60%. We further used mapping of Illumina short reads to correct any potential sequence and assembly errors. Gene prediction and annotation were performed in both GenBank and IMG and supplemented with annotations based on Pfam, KEGG, and eggNOG (47) (see Data Set S1 in the supplemental material). The D. oralis genome encodes 2,395 proteins (GenBank), contains 51 tRNAs, and has 2 rRNA operons. A slightly larger number of proteins were predicted with IMG (2,442), and we identified the differences as small hypothetical proteins and mobile genetic elements (transposases).
Functional gene annotations for D. oralis. Download DATA SET S1, XLSX file, 0.7 MB (695.2KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Compared to genomes of environmental Desulfobulbus species (Table S1), the genome of D. oralis is 30 to 50% smaller. There is also limited gene synteny based on comparison with the only closed genome of an environmental Desulfobulbus species (D. propionicus) (Fig. 4), suggesting numerous chromosomal rearrangements associated with genomic fragmentation. For metabolic reconstruction, we combined assignments from IMG, KEGG’s BlastKOALA, and MetaCyc, with similarity searches, domain architecture analyses, sequence alignments, and phylogenetic reconstruction for comparisons of enzymes and pathways with other sulfate reducers and oral bacteria.
FIG 4 .

Genomic comparison of D. oralis and D. propionicus. Matching orthologs are connected by internal arc lines, the color scale indicating the degree of pairwise sequence identity at the protein level. External tick lines indicate genes in each genome that contain protein domains enriched (blue) or depleted (red) in D. oralis (based on data in Data Set S2). The outer histograms (light blue) indicate the relative abundance of each detected protein in D. oralis cells (bottom) or culture medium (top). Secreted proteins significantly enriched in the medium are identified and indicated by purple asterisks and lines.
Enriched and depleted functional domains (Pfam) and gene families (COG) in D. oralis compared to free-living Desulfobulbus. Download DATA SET S2, XLSX file, 0.1 MB (37.4KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
As physiological characterization of D. oralis revealed streamlining and loss of some of the free-living Desulfobulbus respiratory and fermentative capabilities, the first objective in metabolic reconstruction was to predict the bioenergetics and carbon flow associated with respiratory versus fermentative growth in the oral environment. The dissimilatory sulfate reduction pathway (48), common to all Desulfobulbus species, could be reconstructed based on the D. oralis genome and is proposed to follow transformations represented schematically in Fig. 5. Sulfate enters the cell via a SulP permease and is activated by adenylation via a sulfate adenylyl transferase (Sat). The resulting pyrophosphate is hydrolyzed by a manganese-dependent inorganic pyrophosphatase (ppaC), which drives the otherwise endergonic activation step. The adenosine 5′-phosphosulfate is converted to sulfite by APS reductase (Apr), a heterodimeric iron-sulfur flavoprotein complex, which receives electrons from a membrane-bound oxidoreductase complex (QmoABC) that interacts with the quinone pool as part of the energy conservation cycle (49). Sulfite is further reduced to sulfide by the dissimilatory sulfite reductase (Dsr), a large enzyme complex that includes cytoplasmic, membrane-anchored and periplasmic subunits, including cytochromes b and c, quinone-interacting domains, and iron-sulfur centers that mediate electron transfer and proton translocation (Fig. 5). Another energy-conserving membrane complex present in D. oralis shares homology with the membrane-bound NADH dehydrogenase complex Nuo and is encoded by an 11-gene cluster. However, the NuoEFG subunits, responsible for oxidizing NADH, are missing, so the complex may oxidize a different electron donor, such as reduced ferredoxin, similar to other sulfate reducers (50). The Nuo complex is complete in D. propionicus.
FIG 5 .

Metabolic reconstruction of D. oralis, with emphasis on membrane processes, energy conservation, and pathogenicity potential. Selected genes encoding enzymes, complexes, and other proteins proposed to function in such cellular processes are indicated by numbers in red, corresponding to GenBank genes in Data Set S1. The putative EtfAB-Ldh complex is depicted in gray.
A cytoplasmic Hdr-Flx complex that can perform an alternative mode of energy conservation, flavin-based electron bifurcation (FBEB) (51), is present in D. oralis but absent in free-living Desulfobulbus. The complex appears to have originated from Desulfovibrio by horizontal gene transfer (HGT), and is most closely related to that of the human oral pathogen D. fairfieldensis. The FlxABC complex has flavin adenine dinucleotide (FAD), NAD(P)-binding sites, and an iron-sulfur center, representing a distinct type of NAD(P)H dehydrogenase, which was proposed to oxidize NAD(P)H using both ferredoxin and a high-redox disulfide center (DsrC) as electron acceptors (52). The reverse reaction, NAD+ recycling to NADH, can be coupled in environmental Desulfovibrio with aldehyde conversion to ethanol during pyruvate fermentation, using electrons coming from reduced ferredoxin through HdrABC (52). While we did not detect alcohol production by D. oralis, a related aldehyde detoxification process could occur through an NADPH-dependent alcohol dehydrogenase (CAY53_10125), most closely related to genes from human- and animal-associated actinobacteria. The interconversion between reduced and oxidized forms of NAD(H) and NADP(H) is performed by NfnAB, also an electron-bifurcating, ferredoxin-dependent flavoprotein. Another complex with a role in detoxification is the cytochrome c-containing nitrite reductase, NrfAH. This membrane complex, present in pathogenic bacteria, detoxifies nitric oxide produced by host cells as a defense mechanism against bacterial invasion (53).
Oxidation of lactate to pyruvate during sulfate respiration is catalyzed by a peripheral membrane-associated multisubunit lactate dehydrogenase (LutABC) that uses the menaquinone pool as the electron acceptor. The complex is encoded in the same operon with a lactate permease (LutP) and appears to have been acquired from host-associated Desulfovibrio, lacking close homologues in environmental Desulfobulbus. Lactate is an important shared substrate among many oral bacteria, and the acquisition of the Lut system by D. oralis likely reflects an important adaptation to this environment following colonization. Pyruvate is further decarboxylated to acetyl coenzyme A (acetyl-CoA) by a pyruvate:ferredoxin oxidoreductase, generating reduced ferredoxin. D. oralis has lost both phosphate acetyltransferase and acetate kinase, and we hypothesize that acetate release occurs via CoA transfer by a succinate:acetate-CoA transferase that was likely acquired from a host-associated Campylobacter strain (see Fig. S1 in the supplemental material), followed by transphosphorylation by succinate thiokinase and nucleoside diphosphate kinase (Fig. 5). This pathway is consistent with the observed 1:1 stoichiometry of lactate conversion to acetate (Fig. 2A). It would result in the production of one ATP per lactate via substrate-level phosphorylation and an additional amount of ATP via sulfate respiration that depends on the number of protons translocated across the cytoplasmic membrane and the coupling number of the FoF1 ATPase.
Phylogenetic analysis (FastTree) of selected horizontally acquired proteins in D. oralis. Download FIG S1, PDF file, 0.2 MB (201KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
For free-living Desulfobulbus, lactate can also be fermented. An electron-bifurcating, cytoplasmic lactate dehydrogenase-flavoprotein complex (Ldh-Etf) oxidizes lactate to pyruvate by coupling the endergonic NAD+-dependent oxidation of lactate to the exergonic electron transfer from reduced ferredoxin to NAD+ resulting in the oxidation of one ferredoxin molecule and the production of 2 NADH molecules (54). For every three pyruvates produced from lactate, one is converted to acetate, generating reduced ferredoxin and an ATP by substrate-level phosphorylation, while two more pyruvates are reduced to propionate. This would result in an expected product ratio of 2:1 propionate to acetate (55). Even though we did not detect fermentative growth on lactate, D. oralis has a potential Ldh-Etf complex encoded in its genome. It is therefore possible that it may ferment lactate in vivo and that the inability to do so under pure culture conditions is due to insufficient protein expression. In fact, based on whole-cell proteomic analysis (detailed below) we found that the Ldh complex subunit was present at extremely low levels (~0.001% of the proteome), 2 orders of magnitude below that of the Etf subunits. In the absence of lactate fermentation, D. oralis can nevertheless ferment pyruvate through the propanoate cycle. In this case, for every three pyruvate molecules, two can be converted to two acetate molecules with the generation of two ATPs and two reduced ferredoxins, while one additional pyruvate serves as an electron acceptor and is reduced to propionate. This results in the expected ratio of 1:2 propionate to acetate during pyruvate fermentation, consistent with our observed results (Fig. 2B).
D. oralis has incomplete pathways for the biosynthesis of several amino acids (serine, threonine, cysteine, tyrosine, and phenylalanine) as well as for biotin, molybdenum cofactor, and cobalamin. Amino acid and vitamin auxotrophies are common among bacteria that live within complex communities such as the oral microbiota. However, D. oralis has additional, yet unidentified dependencies; simple supplementation with amino acids, peptides, and/or vitamins did not relieve the requirement for the Fusobacterium culture filtrate. Nucleotide biosynthesis and conversion appear fully functional, and D. oralis can also synthesize a variety of fatty acids and sugars for membrane, peptidoglycan, and lipid A assembly. Future elucidation of this nutritional requirement may enable more rapid isolation of other fastidious microorganisms from human microbiomes.
Genomic fingerprints for adaptation to a host-associated environment and potential pathogenicity.
Desulfobulbus oralis has a reduced genome that displays limited conserved gene synteny compared to its closest sequenced free-living relative (Fig. 4), as well as evidence for both gene loss and gene gain. Aside from becoming dependent on exogenous amino acids and vitamins, gene loss has affected hydrogenases, ferredoxin oxidoreductases, cytochromes, the dissimilatory nitrate reductase, and pyruvate kinase. This restricts D. oralis from utilizing a wider range of electron donors and acceptors and fermentation substrates that are not present at high concentrations in the oral environment (nitrate, short-chain fatty acids, and alcohols). As very few organisms from the oral microbiota are capable of sulfate respiration, the restricted energy metabolism in D. oralis may reflect its adaptation to a metabolic niche in which there are few competitor species, with no selection to maintain metabolic versatility typical of free-living Desulfobulbus species.
Following host colonization, cohabitation with bacteria not present in open environments presents the opportunity for horizontal gene transfer and acquisition of physiological traits that are absent in free-living species. Horizontal gene transfer is an important factor influencing the human oral microbiota and its individual species’ positive and negative interactions with the host (56, 57). Based on sequence similarity and phylogenetic analyses, we found evidence for multiple horizontal gene transfer events. Sixty percent of the D. oralis genes have their closest homologues in the genomes of free-living Desulfobulbus and potentially can be regarded as inherited from the ancestral lineage that colonized the mammalian oral environment (Data Set S1). Approximately 200 predicted protein-encoding genes (~8% of the genome) have no distinguishable homologues, most of them being quite short and without recognizable protein domains. Out of the remaining genes, >250 (10%) are most closely related to homologues from other Deltaproteobacteria and have the oral Desulfovibrio fairfieldensis and Desulfomicrobium orale species as the top hits, suggesting they might have been acquired from such physiologically related lineages. Among those genes are clusters encoding a variety of transporters and energy conservation complexes (lactate dehydrogenase, glycerol-3-phosphate dehydrogenase, and heterodisulfide reductase). Other potential horizontally acquired genes include those coding for transporters, drug and antibiotic efflux pumps, hydrolases, glycosyl transferases involved in cell membrane biosynthesis, clustered regularly interspaced short palindromic repeat (CRISPR) system proteins, and other enzymes and regulators from a wide range of oral bacteria, including Actinomyces, Streptococcus, Porphyromonas, Tanerella, Prevotella, Neisseria, and Campylobacter (Fig. S1). As far as gene absence, it was notable to find that the hgcA and hgcB genes, present in all environmental Desulfobulbus species identified so far (58), have been lost in D. oralis. Those genes encode a corrinoid iron-sulfur protein (HgcA) and a ferredoxin (HgcB) that are responsible for the microbial synthesis of methylmercury, a potent neurotoxin (59). While the biological role of HgcA-B is still unknown, the finding that D. oralis has lost that biochemical potential represents a highly significant positive outcome of adaptation to the oral microbiota, especially considering mercury amalgam tooth fillings continue to be used and are present in many individuals.
To get a more quantitative estimate of gene family loss and gain in the D. oralis genome relative to its free-living relatives, we performed functional abundance profile analyses in IMG (see Data Set S2 in the supplemental material). At the protein domain (Pfam) level, several categories of transposases and other mobile elements (integrases and endonucleases) are highly enriched in D. oralis compared to other Desulfobulbus species. Phylogenetically, many of those genes appear to be related to Desulfomicrobium and may reflect multiple genetic exchanges between these two related sulfate reducers in the oral microbiota. At the gene family level (COG), aside from mobile elements, COG families involved in membrane lipid biosynthesis and transporters are enriched. Considering that D. oralis relies on exogenous amino acids, it is not unexpected to see that amino acid and peptide ABC transporters are at higher abundance than in other Desulfobulbus species, and some of the genes were likely acquired from other oral bacteria by HGT. Four genes (9825 and 9840 to 9855) that encode members of an ankyrin repeat protein family that may be involved in the interaction with human cells (60), absent in free-living Desulfobulbus, were also apparently acquired from oral Synergistetes (Fig. S1).
With respect to gene loss, the most pronounced differences between D. oralis and its free-living relatives were observed in the category of signal transduction and gene regulation (Data Set S2). Indeed, this functional category shows the steepest dependence on the total number of genes in Bacteria and Archaea (61); however, D. oralis presents significant deviation from the general trend. While the number of genes in D. oralis is only 30% smaller than that in a free-living D. propionicus, the number of signal transduction genes in D. oralis is 4.5 times smaller: 72 genes (see Data Set S3 in the supplemental material) versus 320 genes in D. propionicus (data from the MiST database [62]). Such deviations were proposed to reflect particular biological phenomena as well as environmental conditions in a microbial habitat (63). Thus, we explain a disproportionate loss of signal transduction capabilities in D. oralis as a direct result of adaptation to a much more stable niche, where a constant temperature, pH, and set of nutrients eliminate the need to monitor and respond to various environmental factors. The absence of swimming motility in D. oralis is yet another conspicuous example of adaptation to a new environment via gene loss. While free-living representatives of Desulfobulbus contain a full complement of flagellar genes and chemotaxis machinery (more than 50 genes), D. oralis lost all of these genes. However, it retained type IVa pili (TFP), which are essential for active translocation across solid surfaces, including PilT, coded for by a marker gene for TFP-based motility, as well as two regulatory chemosensory systems that belong to the TFP class (see Data Set S4 in the supplemental material). Thus, despite the loss of flagella and chemotaxis, D. oralis is capable of actively navigating toward beneficial microenvironments within the oral cavity.
Complete list of signal transduction proteins. Download DATA SET S3, XLSX file, 0.1 MB (29.1KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Genes encoding components of type IV pili and chemosensory systems controlling type IV pilus-based motility. Download DATA SET S4, XLSX file, 0.1 MB (59.5KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
As the relatively streamlined metabolism of D. oralis does not suggest it could provide the human cells with vitamins or other beneficial factors, we searched for potential genes that could mediate evasion of the host defense system and a potential role in pathogenesis. One such candidate gene family encodes eight proteins containing the Sel1 domain, characteristic of solenoid proteins involved in a variety of signal transduction processes (64). Three of the D. oralis Sel1 domain proteins are related to proteins that are involved in the bacterial interaction with human cells, including evasion of lysosomal host defense mechanisms, such as Legionella LpnE and LidL (65) and yet uncharacterized proteins from opportunistic pathogens like Neisseria, Haemophilus, and Kingella (66–69) (Fig. S1). Another relative of those proteins is EsiB (Escherichia coli secretory immunoglobulin A-binding protein), a pathogenic E. coli marker (70) confirmed to have host immunomodulatory activity by interacting with secretory IgA, thus leading to inhibition of neutrophil activation and evasion of host defense mechanisms (71, 72). This ultimately allows E. coli to avoid clearance by the host’s immune system and permits further infection. A similar role could be employed by D. oralis to avoid negative effects of host immune responses in periodontal disease progression, allowing for its proliferation and increase in abundance.
D. oralis also encodes genes for leukotoxin/hemolysin production and export through type I secretion systems. Leukotoxins belong to the RTX toxin family, broadly distributed among Gram-negative bacteria and with diverse biological functions. Leukotoxins produced by the periodontal pathobiont Aggregatibacter actinomycetemcomitans are lethal to human monocytes and human T lymphocytes in vitro (73, 74) and are a main virulence factor within deep periodontal pockets in aggressive periodontitis (75–77). Genes encoding other RTX family homologues include the gene coding for adenylate cyclase/hemolysin, which is the major virulence factor of Bordetella (78–80), as well as genes involved in an outer membrane translocation complex involved in RTX protein processing and export (76). Based on these findings, we propose that leukotoxin/hemolysin systems of D. oralis, acquired as three distinct operons (see Data Set S5 in the supplemental material), could play roles in the putative pathogenicity of D. oralis by contributing to host immune evasion, manipulation, and neutralization of defense mechanisms.
Select genes and gene clusters (operons) encoding potential toxins, pathogenicity factors, and their secretion machinery. Download DATA SET S5, XLSX file, 0.1 MB (18.1KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Proteomic analysis of D. oralis.
While genome analysis revealed both native and acquired physiological potential, including the possibility that D. oralis may play an active role in the etiology of periodontal disease, simple gene presence is not sufficient evidence to assume that every potential gene product is actually expressed. We therefore applied a proteomic approach to determine the relative amounts of every expressed protein within the cell and whether specific proteins are released into the extracellular space. Because the main focus was on proteins that may contribute to pathogenesis, some of which were predicted to be membrane anchored while others were secreted into the extracellular space, we collected both cells and filtered culture supernatant. Three separate culture replicates were performed, and the samples for analyses were collected in late log phase.
One of the primary metabolic by-products of D. oralis sulfate respiration, hydrogen sulfide, is well-known to trigger an inflammatory response at the leukocyte-endothelium interface, acting as a primary signaling molecule and synergizing with the proinflammatory action of virulence factors (81, 82). As we wanted to test the synthesis and potential proinflammatory action of potential virulence factors encoded by D. oralis, we grew the cells fermentatively, in the absence of sulfate. The whole-cell proteome and the soluble proteins released into the medium were fragmented and analyzed by bottom-up, two-dimensional liquid chromatography-tandem mass spectrometry (2D-LC-MS/MS). A total of 1,628 proteins were detected, representing 70% of the predicted proteome (see Data Sets S1 and S6 in the supplemental material), which is similar to coverage that has been previously obtained on other organisms with reduced genomes (83, 84). Considering that proteins containing multiple transmembrane domains are difficult to detect in tryptic digests (here we detected only 40% of the proteins predicted to contain more than 1 transmembrane domain) we conclude that a very large fraction of the D. oralis genetic potential (>80%) was expressed under the tested condition. We also detected most of the putative virulence factors (hemolysins, leukotoxin, and Sel1 proteins) and the proteins involved in their processing and export. In terms of dynamic range, we detected proteins spanning 5 orders of magnitude in relative abundance, from 2% of the cell total cellular proteome (sulfite reductase) to 2e−5% (a type IV transporter subunit). The most abundant proteins were determined to be enzymes involved in energy conversion and central metabolism, protein translation, chaperones, regulators, and several transporters. While the high level of proteins involved in respiratory growth may appear surprising, it has been shown in other sulfate reducers grown under fermentative conditions that the sulfate reduction pathway is constitutively expressed (85). This indicates that the sulfate reduction capacity of D. oralis is actively maintained, and when periodontal sulfate levels are adequate, it may be the primary energy source.
Normalized proteome data. Download DATA SET S6, XLSX file, 0.4 MB (399.5KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
To identify if any of the putatively expressed virulence factors were secreted, we compared the proteome of whole cells against their secretome. We identified 12 proteins that were significantly enriched in the culture medium relative to their abundance in whole cells (Fig. 4 and 6; Data Set S6). With the exception of two, a DNA binding factor and an unknown protein, all others were predicted hemolysin family proteins and were enriched between 40- and 100-fold in the culture medium, suggesting that the cells actively secreted them. In vivo, such secreted hemolysins could diffuse through the epithelial layer, causing cytotoxicity, lysis, and modulation of the host immune response.
FIG 6 .

Proteomics and cytokine stimulation implicating D. oralis in proinflammatory responses. (a) D. oralis proteins that are secreted from the cell (indicated in red) based on enrichment analysis. Only proteins that had at least a 4-fold enrichment and P < 0.01 (based on three biological replicates) were considered. The table on the right identifies those secreted proteins and compares them to their closest homologues in the human oral microbiota or the free-living Desulfobulbus. (b) Cytokine stimulation in oral epithelial keratinocytes by D. oralis cells. Keratinocytes were incubated with D. oralis cells or with the medium control for 48 h, and cytokine stimulation was measured using a multiplex bead assay. Each bar represents the mean ± standard deviation of the mean from triplicate measurements. Asterisks indicate results significantly different from the keratinocyte medium-only control: *, P < 0.05; **, P < 0.01.
D. oralis cytokine stimulation in oral epithelial keratinocytes.
During the initial stages of periodontal disease, there is an acute inflammatory response to some of the oral microbiota resulting in the recruitment of polymorphonuclear leukocytes stimulated by host chemokines and/or cytokines, such as interleukin-8 (IL-8) (86). As inflammation progresses from acute to chronic, an increase in the production of proinflammatory mediators such as IL-1β, IL-6, IL-8, and tumor necrosis factor alpha (TNF-α) by various host cells, including oral epithelial keratinocytes, results in the recruitment of additional immune cells such as macrophages, T cells, and B cells (86). There is still insufficient data to distinguish the relative roles of oral bacterial lipopolysaccharide (LPS) and surface-associated and secreted proteins in cytokine induction. Various recognized periodontal pathobionts trigger distinct types of inflammatory responses (87–89), and some (e.g., Porphyromonas gingivalis) can modulate that by interfering with chemokine production (90).
Based on the finding that D. oralis secretes and has surface-associated proteins (e.g., leukotoxin, hemolysins, and EsiB-Sel1) that have been shown in other oral bacteria to be linked to an inflammatory response in periodontitis, we analyzed its ability to induce proinflammatory cytokine production in vitro. We used a human immortalized oral keratinocyte cell line (OKF6/TERT-2), which has been previously used as a model to study the interaction of oral bacteria with human epithelial cells (88, 91). Cells and filtered medium from the same D. oralis cultures used for proteomic analyses were applied to epithelial cells in culture, and the production of 13 human inflammatory cytokines/chemokines was measured after 48 h using bead-based immunoassays. We detected a significant stimulation of IL-1β release by the keratinocytes exposed to D. oralis culture filtrate, while IL-1β, alpha interferon (IFN-α), IFN-γ, monocyte chemoattractant protein 1 (MCP-1), IL-6, IL-8, and IL-18 were stimulated when keratinocytes were exposed to D. oralis cells (Fig. 6; see Fig. S2 and S3 in the supplemental material). The differences in proinflammatory responses between culture medium and whole cells are due to protein concentration differences, as the enriched proteins in the medium (secreted leukotoxins/hemolysins) and the bacterial cell surface agonists (e.g., Sel1 proteins, surface pili, and LPS) may activate different pathways. The strongest stimulation was observed for IL-8 (P < 0.01). IL-8 is one of the most important chemokines associated with periodontitis, being elevated at the disease site and attracting neutrophil polymorphs that further increase proinflammatory mediators (86). The observed release of the other cytokines/chemokines is expected to stimulate B- and T-cell differentiation as well as attracting macrophages and natural killer cells to the site of infection. The absence of stimulation of IL-10 and IL-12—both anti-inflammatory cytokines—does not support a potential beneficial role of D. oralis. On the other hand, the complex response triggered by D. oralis in epithelial cells indicates that this bacterium is not merely a colonizer in the low-oxygen deep periodontal pockets but actively participates in inflammation. During periodontal disease progression, the gingival extracellular matrix breaks down and glycosaminoglycans (GAGs) are released (41, 92). D. oralis encodes several sulfatases that could liberate inorganic sulfate from GAGs, potentially playing an active role in matrix destruction (93). In addition, the hydrogen sulfide produced by D. oralis during sulfate respiration can lead to apoptosis in keratinocytes (82, 94) and further elicits a proinflammatory response in oral cells (95, 96). These features illustrate the capacity of D. oralis to create a dysbiotic oral environment and actively contribute to the pathogenesis of periodontal disease in a novel and compounded way not described for other oral pathogens so far. These results emphasize that cultivation of novel bacteria, only associated with disease based on sequence data, is important to begin to fully understand their role in the oral cavity.
Cytokine/chemokine stimulation in oral epithelial keratinocytes by Desulfobulbus oralis supernatant. Download FIG S2, PDF file, 0.1 MB (109.5KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Cytokine/chemokine stimulation in oral epithelial keratinocytes by Desulfobulbus oralis cells. Download FIG S3, PDF file, 0.1 MB (115.3KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Expression of D. oralis pathogenicity genes in periodontitis.
A recent clinical metatranscriptomic study identified Deltaproteobacteria as one of the bacterial lineages that increase in prevalence and transcriptional activity in periodontitis (97). We therefore aimed at determining the functional profile of D. oralis and specifically the relative abundance of its pathogenicity gene transcripts, based on the transcriptome sequencing (RNA-seq) data generated from oral clinical samples. We determined that in the data sets from three individuals with periodontitis, the sequences that mapped to D. oralis protein-encoding genes (~3,000 to 25,000 reads) represent 0.1 to 0.5% of the total number of microbial reads. Even though the overall coverage of the D. oralis transcriptome by the RNA-seq data sets was shallow (<2-fold), across the three data sets >80% of the predicted gene transcripts were detected (Data Set S1), with ~65% of the genes represented by at least two reads. This correlates with the overall number of proteins we detected in D. oralis grown in pure laboratory cultures and supports the hypothesis that most genes in this bacterium are expressed both in vitro and in the natural subgingival environment. While the coverage level was too low to enable a robust global comparison of the transcriptional versus proteome profile, we analyzed the relative levels of mRNAs encoding putative proinflammatory response-inducing proteins. Several of the hemolysin-type genes were among the top 50 most abundant transcripts (0.2 to 1% of the mapped reads), with four of them being part of a highly expressed operon (CAY53_915-940) (Data Set S5). This indicates that such factors, which have been linked to inflammatory response in other oral bacteria, are being actively produced by D. oralis in the subgingival space and likely play a role in the development and progression of periodontal disease.
Description of Desulfobulbus oralis sp. nov.
Desulfobulbus oralis sp. nov. (o.ra’lis. NL masc. adj. oralis, of the mouth). Cells are nonmotile, non-spore-forming Gram-negative rods 1 to 2 μm in length occurring singly, in pairs, and sometimes chains. Growth occurred at 37°C with optimum pH 7 to 7.1 under strictly anaerobic conditions, with cysteine as a reducing agent. D. oralis incompletely oxidizes lactate and pyruvate to acetate, using sulfate or thiosulfate as electron acceptors and releasing sulfide. The organism does not utilize citrate, propionate, acetate, hydrogen, formate, succinate, glucose, fructose, ethanol, propanol, fumarate, or malate as electron donors or sulfite and nitrate as acceptors. In the absence of electron acceptors, pyruvate and amino acids are fermented, while lactate, peptone, mucin, and dextrose are not. Growth requires the addition of 5% Fusobacterium nucleatum cell-free spent medium (CFS).
D. oralis type strain HOT041/ORNL was isolated from a human subgingival sample from a patient with periodontitis at Ohio State University College of Dentistry, Columbus, OH. The genome is represented by a circular chromosome of 2.77 Mbp and with a G+C content of 59.76 mol%.
MATERIALS AND METHODS
Clinical sample collection.
Subgingival biofilm and crevicular fluid were obtained from a patient with periodontitis at the Ohio State University College of Dentistry. Informed consent was obtained under a protocol approved by the Ohio State University Institutional Review Board (IRB; no. 2007H0064). Following removal of supergingival plaque with gauze, sterile endodontic paper points were inserted into the gingival sulcus and left for 1 min. Afterward they were placed into liquid dental transport medium (Anaerobe Systems, Morgan Hill, CA) and used for inoculation into primary enrichment medium.
Microbial cultivation.
Primary oral enrichments were passaged (2% vol/vol) in an ATCC 1249 modified Baar’s medium containing (per liter) 2 g MgSO4⋅7H2O, 1 g (NH4)2SO4, 0.5 g K2HPO4, 1 g CaCl2⋅2H2O, 5 g sodium citrate, 1.17 g sodium propionate, 1.17 g sodium acetate, 1.5 ml l(+)-lactic acid (85% wt/vol [Sigma]), 1 g yeast extract, and 0.5 mg resazurin. The medium was degassed with N2-CO2 (80/20 vol/vol) and sterilized for 15 min at 121°C, followed by addition of sterile degassed cysteine (1 mM final concentration) and 10 ml Fe(NH4)2(SO4)2–5% (wt/vol) and pH adjustment to 7.2 with 8 M NaOH. Cultures were maintained in 15-ml glass tubes fitted with butyl rubber stoppers under N2-CO2 (80/20 vol/vol) at 37°C for 1 to 2 weeks. Cell growth resulted in the release of hydrogen sulfide, which formed a black iron sulfide precipitate and was also detected using lead acetate indicator paper. Stocks were preserved by freezing at −80°C, after addition of dimethyl sulfoxide (DMSO) to 10% (vol/vol). Cell-free spent medium (CFS) supernatant was prepared by filtering cultures through two 0.22-μm-pore Nalgene Rapid-Flow sterile filters and degassing with N2 (100%). Pure cultures were obtained by three successive dilutions to extinction transfers in liquid medium. For culture enrichments of D. oralis, the medium was supplemented with 25% (vol/vol) coculture CFS. Pure cultures of D. oralis were grown in a modified lactate-Baar basal medium containing (per liter of distilled water): 2 g MgSO4⋅7H2O, 1 g (NH4)2SO4, 10 mM MOPS (morpholinepropanesulfonic acid), 10 mM HEPES, 10 mM sodium lactate, 0.1% yeast extract (stimulatory for growth but not strictly required), 2.5 mM cysteine, and 0.5 mg resazurin (0.5%). The pH of the medium was adjusted to 7 to 7.1 with 8 M NaOH, degassed with N2 (100%), and autoclaved for 30 min at 121°C. After cooling, sterile K2HPO4 (0.5 M, pH 7) was added to a 3 mM final concentration, and CFS of F. nucleatum grown in brain heart infusion broth (BHI) supplemented with 0.2% glucose was added to 5% (vol/vol).
Cellular and molecular analyses.
Fluorescence in situ hybridization (FISH) with the oligonucleotide probes EUB338 (Bacteria) and Delta495a (Deltaproteobacteria) was performed as previously described (32). For DNA isolation, we used proteinase K digestion followed by phenol-chloroform extraction (83). PCR amplification of the 16S rRNA gene was carried out using the universal primer set 27F (5′-AGAGTTTGATYMTGGCTCAG-3′) and 1492R (5′-TACGGYTACCTTGTTACGACTT-3′) or a specific D. oralis set, DsbF (5′-AGTTAGCCGGTGCTTCCT-3′) and 1492R. Amplification with Lucigen 98HotStart supermix used the following thermal profile: 94°C for 3 min, followed by 32 cycles of 94°C for 20 s, 60°C for 30 s, and 72°C for 90 s, with a final extension of 72°C for 5 min. Following purification, the PCR products were sequenced directly using Sanger chemistry (ABI 3730) or were first cloned into pCR2.1-TOPO (Invitrogen, Carlsbad, CA). Sequence data were analyzed using Geneious v.10 (98) and confirmed the in silico-inferred primer specificity. To quantitatively determine the coculture composition, we developed a qPCR assay using 16S rRNA gene primers specific for F. nucleatum (5′-CAGCGTTTGACATCTTAGGA-3′ and 5′-ATCGTAGGCAGTATCGCAT-3′) and D. oralis (5′-GGTGGTGCCTTCTTTGAA-3′ and 5′-TTCCTCCGGTTTGACACC-3′). Amplification was with BioRad iQ SYBR green supermix in a CFX96 real-time C1000 thermal cycler (Bio-Rad, Hercules, CA), using a plasmid-cloned 16S rRNA gene insert and the following thermal profile: 95°C for 3 min, followed by 40 cycles of 95°C for 20 s and 66°C for 30 s. Sanger sequencing confirmed primer specificity for each species and primer pair.
For scanning electron microscopy, cells from 20 ml of culture were collected onto a 0.1-μm-pore polyvinylidene difluoride (PVDF) syringe filter, washed with phosphate-buffered saline (PBS; pH 7), and fixed with 3% glutaraldehyde in PBS for 1 h at room temperature, followed by three 10-min wash steps with PBS. Fixed cells were resuspended in 2% osmium tetraoxide in PBS for 1 h, collected by centrifugation at 10,000 × g for 5 min, and washed three times in water (10 min each). On the final wash, cells were deposited onto a 3- by 4-mm silicon chip (Ted Palla, Inc., CA) for 15 min followed by dehydration in an ethanol series (50, 70, 95, and 100%) for 10 min each. The cells were dried in a Ladd critical-point dryer and then gold coated using an SPI sputter coater and examined on a Zeiss Auriga focused-ion-beam scanning electron microscope.
Physiological characterization.
Optimum pH was determined in lactate-Baar medium containing 5% F. nucleatum CFS and 15 mM MES (morpholineethanesulfonic acid), MOPS, and/or HEPES, with the pH adjusted between 5 and 8 at 0.5-pH-unit intervals. Growth was monitored for 1 week at 37°C by daily measurements of optical density at 600 nm (OD600). Utilization of electron donors was determined in lactate-Baar medium containing 5% F. nucleatum CFS and each substrate at 10 mM. The substrates tested include citrate, propionate, acetate, lactate, formate, fumarate, succinate, malate, pyruvate (all as sodium salts), glucose, fructose, ethanol, and propanol. Hydrogen utilization was determined in the presence of sodium acetate and H2-CO2 (80/20 vol/vol). Utilization of electron acceptors was determined with sodium lactate (10 mM) in minimal sulfate-free medium containing (per liter of distilled water) 2 g MgCl2, 1 g NH4Cl, 1 g yeast extract, 10 mM MOPS, 10 mM HEPES, 3 mM K2HPO4, 2.5 mM cysteine, 0.5 mg resazurin, and 5% F. nucleatum CFS and containing 20 mM sodium sulfate, sulfite, thiosulfate, or nitrate. Fermentative capabilities were determined in the same basal medium used for the electron acceptor test, however, lacking addition of electron donor-acceptor pairs. Substrates tested for fermentation include sodium lactate (20 mM), sodium pyruvate (20 mM), peptone (0.1%), mucin (0.25%), synthetic amino acid supplements (Y1501; Sigma) (0.2%), or dextrose (0.2%).
F. nucleatum CFS susceptibility to inactivation was tested by treatment (2 h at 37°C) with proteinase K (0.2 mg/ml) or incubation at 80°C for 14 h prior to supplementation into lactate-Baar medium at 5% vol/vol. For testing potential replacement of CFS with defined amino acids, vitamins, and siderophores, yeast synthetic dropout medium (amino acids) was added to 0.2% and ATCC vitamins to 1% in lactate-Baar medium prior to inoculation. The siderophore mixture contained a 1:1:1 mix of pyoverdines, 2,3-dihydroxybenzoic acid, and ferrichrome and was added to a final concentration of 1 μg/ml. The cultures were monitored by optical density and microscopic examination for 2 weeks. Very weak growers were transferred to fresh media and monitored for growth up to 1 month. Growth was considered positive if an increase in OD600 occurred after three consecutive passages. All experiments were performed in triplicate.
To determine metabolic by-products of lactate respiration and pyruvate fermentation, soluble analytes were measured via high-performance liquid chromatography (HPLC) using a Waters Breeze 2 system (Waters Corp., Milford, MA) equipped with a refractive index detector (model 2414) and an Aminex HPX-87H column (Bio-Rad Laboratories). Sulfuric acid (5 mM) was used as the mobile phase at a flow rate of 0.6 ml/min. Peak areas and retention times were compared against known standards.
Genomic sequencing and analysis.
D. oralis genomic DNA isolated using the phenol-chloroform method described previously (83) was used to construct a large-insert (20-kb) library for sequencing on a PacBio RS II system (Institute for Genome Sciences, University of Maryland School of Medicine). We generated 154,149 reads, with an N50 length of 20.2 kbp and median of 14 kbp, which were assembled into one polished contig 2,796,768 bp in length, with 668 mean coverage, using PacBio SMRT Portal. We also sequenced in-house a small insert D. oralis genomic library on an Illumina MiSeq platform. Approximately 4 million reads (2 × 250 nucleotides [nt]) were mapped on the PacBio contig using Geneious v.10. (98). Individual indel discrepancies were resolved based on both read coverage and inspection of their effect of disruption/truncation of predicted ORFs in that region. Contig end overlaps were identified, which resulted in circularization of the initial contig into one chromosome of 2,774,417 bp. In addition to the annotations provided by the NCBI and IMG platforms, which included Pfam, COG, TIGRFam, knockout (KO), EC, secretion signal, and transmembrane domains, we also analyzed the predicted proteins and their functions through MetaCyc, eggNOG, and TransportDB. For metabolic reconstruction, we combined the use of the IMG networks, KEGG-BlastKOALA, and MetaCyc-Pathway Tools with individual gene searches (blast and hmmsearch), sequence alignments, and phylogenetic reconstruction using homologues from related sulfate reducers, acetogens, or human microbiome bacteria. Sequence alignments were performed using Muscle, and maximum likelihood phylogenetic reconstructions were based on PhyML or FastTree, all implemented in Geneious. Genomic alignments were displayed using Circos (99).
Proinflammatory cytokine secretion assays.
Late-log cultures of D. oralis grown under pyruvate fermentation condition (50 ml, three replicates) were chilled on ice and spun by centrifugation (4°C, 10,000 × g for 10 min). The cell pellets were flash-frozen at −80°C, and the spent medium was passed through a 0.22-μm-pore Millipore filter and frozen at −20°C.
Immortalized human oral keratinocytes (OKF6/TERT-2) were grown on keratinocyte-SFM 1× medium (Invitrogen, Carlsbad, CA) supplemented with epidermal growth factor (0.3 ng/ml), bovine pituitary extract (50 μg/ml), 1 IU/ml penicillin, and 1 μg/ml streptomycin to reach ~50% confluence, in 12-well tissue culture plates (Corning, Inc., Corning, NY) precoated with 3 mg/ml collagen (StemCell Technologies, Vancouver, Canada). To each well, we added 100 μl of (i) D. oralis filtered spent medium (100 μl), (ii) D. oralis cells resuspended in keratinocyte medium, (iii) fresh D. oralis medium, or (iv) fresh keratinocyte medium (negative controls), each in triplicate for every triplicate D. oralis biological sample. Keratinocytes were incubated for 48 h at 37°C, after which medium samples were collected, clarified by centrifugation, and frozen in aliquots at −20°C. Cytokine concentrations were measured with the LEGENDplex human inflammation panel (13-plex) (BioLegend, San Diego, CA) following the manufacturer’s instructions, using a Cytopeia influx flow cytometer (BD, Franklin Lakes, NJ). Statistical analysis was performed using GraphPad Prism version 7. One-way analysis of variance (ANOVA; nonparametric) with Tukey correction was used to investigate significant differences between independent groups of data. Statistical differences were considered significant at P < 0.05.
Whole-cell and secreted proteome data acquisition.
Samples from the same three replicate D. oralis cultures used for the proinflammatory assay were also used for proteomic analysis, conducted as detailed in reference 100. Cell pellets were resuspended in 4% sodium deoxycholate (SDC) in 100 mM ammonium bicarbonate (ABC) and ultrasonically disrupted. The crude protein extract was clarified by centrifugation and reduced with 10 mM dithiothreitol (DTT), and cysteine residues were blocked with 30 mM iodoacetamide (IAA). The proteins were then collected on top of a 10-kDa cutoff spin column filter. D. oralis spent culture medium samples (~12 ml) were concentrated on a 5-kDa filter. Collected proteins were washed with ABC and digested with sequencing-grade trypsin (Sigma). Peptides were collected by centrifugation, acidified to 1% formic acid (FA) followed by extraction with ethyl acetate, and concentrated. The peptide mixture was analyzed by an automated, two-dimensional liquid chromatography-tandem mass spectrometry (2D-LC-MS/MS) system (100) using the Ultimate 3000 RS system in-line with a Q Exactive Plus (QE+) mass spectrometer (Thermo, Fisher Scientific).
Proteomic analyses.
All MS/MS spectra were searched against a constructed FASTA database containing all proteins predicted from the sequenced genome of Desulfobulbus oralis, concatenated with common contaminants and reversed-decoy sequences using Myrimatch version 2.2 (101). Peptides were identified and proteins inferred using IDPicker version 3.1 (102) with an experimental false-discovery rate (FDR) of <1% at the peptide level. Peptide abundances were derived in IDPicker by extracting precursor intensities/area under the curve from chromatograms with the following parameters: retention time of ±90 s and mass tolerance of ±5 ppm. Protein abundances were calculated by summing together the intensities of all identified peptides and normalized by their respective protein lengths. Protein abundances were log2 transformed and median centered across all samples in InfernoRDN version 1.1 (103).
To investigate enriched secreted proteins, we performed Student’s t tests between the cell pellet and the spent medium (supernatant) samples using the Perseus software platform (104). We limited the comparison to those proteins observed in all supernatant experiments to better focus on proteins that could be secreted and/or induce inflammatory response. Proteins with missing values were imputed from a normal distribution (width of 0.3 and shift of 1.8). A protein was considered significantly enriched in the supernatant if it passed the threshold of a Benjamini-Hochberg FDR of <0.01 and log2 fold change of >2 in the comparison of supernatant versus cell pellet.
Metatranscriptomic data analysis.
MiSeq data for 10 healthy subjects and 6 with periodontitis (97) were downloaded from MG-RAST (http://metagenomics.anl.gov/linkin.cgi?project=5148). The reads were mapped to the D. oralis genome with bowtie2 v.2.2.6 (with parameters --very-sensitive-local—no-unal -X 1000—score-min G,20,28—no-mixed—rg-id). The resulting sam files were converted and sorted with samtools 0.1.19, alignments were inspected with IGV 2.3.55, and mapping was quantitated with htseq-count v.0.6.1p1. The code used is available at https://github.com/cliffbeall/Doralis-metatrans.
Accession number(s).
The final assembled and annotated D. oralis genome has been deposited in GenBank under GenBank accession no. CP021255 and in JGI IMG under genome ID no. 2711768589.
Availability of data.
The proteomic raw and searched data files are available via the MassIVE (MSV000081569) and ProteomeXchange (PXD007870) proteome databases. The D. oralis HOT041/ORNL type strain has been deposited at ATCC and is also available from the corresponding author upon request.
ACKNOWLEDGMENTS
We acknowledge the University of Tennessee Advanced Microscopy and Imaging Center for instrument use and for scientific and technical assistance with scanning electron microscopy. We acknowledge the Genomics Resource Center of the University of Maryland Institute for Genome Science for PacBio sequencing and the Genomics Core at the University of Tennessee Knoxville for Sanger sequencing. We thank Steve Allman, Zamin Yang, Dawn Klingeman, and Ann Wymore for assistance with flow cytometry, sequencing, and tissue culture. We also thank Jim Rheinwald, Anna Mandinova, and Kristina Todorova for providing the OKF6/TERT-2 cell line and advice on its cultivation.
This research was fully funded by grant R01DE024463 from the National Institute of Dental and Craniofacial Research of the U.S. National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article has been authored by UT-Battelle, LLC, under contract no. DE-AC05-00OR22725 with the U.S. Department of Energy. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
K.L.C., E.M.M., and R.K.M. performed the laboratory microbial cultivation, physiological analyses, and molecular work. K.L.C. conducted the scanning electron microscopy imaging. K.L.C., C.J.B., J.G.E., A.M.G., S.S.J., I.B.J., and M.P. performed genomic and metabolic data analyses. K.L.C. and P.C. conducted the proinflammatory assays and data analysis. W.X. and R.J.G. conducted the proteomic data acquisition and analysis. J.G.E., A.L.G., R.L.H., I.B.Z., E.J.L., and M.P. oversaw the project and participated in data interpretation. K.L.C. and M.P. wrote the manuscript, with input from the other authors.
Footnotes
Citation Cross KL, Chirania P, Xiong W, Beall CJ, Elkins JG, Giannone RJ, Griffen AL, Guss AM, Hettich RL, Joshi SS, Mokrzan EM, Martin RK, Zhulin IB, Leys EJ, Podar M. 2018. Insights into the evolution of host association through the isolation and characterization of a novel human periodontal pathobiont, Desulfobulbus oralis. mBio 9:e02061-17. https://doi.org/10.1128/mBio.02061-17.
Contributor Information
Floyd E. Dewhirst, The Forsyth Institute.
Maria Gloria Dominguez Bello, New York University School of Medicine.
REFERENCES
- 1.Dye BA. 2012. Global periodontal disease epidemiology. Periodontol 2000 58:10–25. doi: 10.1111/j.1600-0757.2011.00413.x. [DOI] [PubMed] [Google Scholar]
- 2.Kassebaum NJ, Bernabé E, Dahiya M, Bhandari B, Murray CJ, Marcenes W. 2014. Global burden of severe periodontitis in 1990–2010: a systematic review and meta-regression. J Dent Res 93:1045–1053. doi: 10.1177/0022034514552491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Darveau RP. 2010. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol 8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 4.Hajishengallis G, Lamont RJ. 2012. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol 27:409–419. doi: 10.1111/j.2041-1014.2012.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diaz PI, Hoare A, Hong BY. 2016. Subgingival microbiome shifts and community dynamics in periodontal diseases. Calif Dent Assoc J 44:421–435. [PubMed] [Google Scholar]
- 7.Khan SA, Kong EF, Meiller TF, Jabra-Rizk MA. 2015. Periodontal diseases: bug induced, host promoted. PLoS Pathog 11:e1004952. doi: 10.1371/journal.ppat.1004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohlrich EJ, Cullinan MP, Seymour GJ. 2009. The immunopathogenesis of periodontal disease. Aust Dent J 54(Suppl 1):S2–S10. doi: 10.1111/j.1834-7819.2009.01139.x. [DOI] [PubMed] [Google Scholar]
- 9.Hajishengallis G. 2014. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol 35:3–11. doi: 10.1016/j.it.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL. 1998. Microbial complexes in subgingival plaque. J Clin Periodontol 25:134–144. doi: 10.1111/j.1600-051X.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 11.Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, Gamonal J, Diaz PI. 2013. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J 7:1016–1025. doi: 10.1038/ismej.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, Podar M, Leys EJ. 2012. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J 6:1176–1185. doi: 10.1038/ismej.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu B, Faller LL, Klitgord N, Mazumdar V, Ghodsi M, Sommer DD, Gibbons TR, Treangen TJ, Chang YC, Li S, Stine OC, Hasturk H, Kasif S, Segrè D, Pop M, Amar S. 2012. Deep sequencing of the oral microbiome reveals signatures of periodontal disease. PLoS One 7:e37919. doi: 10.1371/journal.pone.0037919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oliveira RR, Fermiano D, Feres M, Figueiredo LC, Teles FR, Soares GM, Faveri M. 2016. Levels of candidate periodontal pathogens in subgingival biofilm. J Dent Res 95:711–718. doi: 10.1177/0022034516634619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biyikoğlu B, Ricker A, Diaz PI. 2012. Strain-specific colonization patterns and serum modulation of multi-species oral biofilm development. Anaerobe 18:459–470. doi: 10.1016/j.anaerobe.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Millhouse E, Jose A, Sherry L, Lappin DF, Patel N, Middleton AM, Pratten J, Culshaw S, Ramage G. 2014. Development of an in vitro periodontal biofilm model for assessing antimicrobial host modulatory effects of bioactive molecules. BMC Oral Health 14:80. doi: 10.1186/1472-6831-14-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wade WG, Thompson H, Rybalka A, Vartoukian SR. 2016. Uncultured members of the oral microbiome. Calif Dent Assoc J 44:447–456. [PubMed] [Google Scholar]
- 18.Langendijk PS, Kulik EM, Sandmeier H, Meyer J, van der Hoeven JS. 2001. Isolation of Desulfomicrobium orale sp. nov. and Desulfovibrio strain NY682, oral sulfate-reducing bacteria involved in human periodontal disease. Int J Syst Evol Microbiol 51:1035–1044. doi: 10.1099/00207713-51-3-1035. [DOI] [PubMed] [Google Scholar]
- 19.Loubinoux J, Bisson-Boutelliez C, Miller N, Le Faou AE. 2002. Isolation of the provisionally named Desulfovibrio fairfieldensis from human periodontal pockets. Oral Microbiol Immunol 17:321–323. doi: 10.1034/j.1399-302X.2002.170510.x. [DOI] [PubMed] [Google Scholar]
- 20.Ichiishi S, Tanaka K, Nakao K, Izumi K, Mikamo H, Watanabe K. 2010. First isolation of Desulfovibrio from the human vaginal flora. Anaerobe 16:229–233. doi: 10.1016/j.anaerobe.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Loubinoux J, Valente FM, Pereira IA, Costa A, Grimont PA, Le Faou AE. 2002. Reclassification of the only species of the genus Desulfomonas, Desulfomonas pigra, as Desulfovibrio piger comb. nov. Int J Syst Evol Microbiol 52:1305–1308. doi: 10.1099/00207713-52-4-1305. [DOI] [PubMed] [Google Scholar]
- 22.Rowan F, Docherty NG, Murphy M, Murphy B, Calvin Coffey J, O’Connell PR. 2010. Desulfovibrio bacterial species are increased in ulcerative colitis. Dis Colon Rectum 53:1530–1536. doi: 10.1007/DCR.0b013e3181f1e620. [DOI] [PubMed] [Google Scholar]
- 23.El Houari A, Ranchou-Peyruse M, Ranchou-Peyruse A, Dakdaki A, Guignard M, Idouhammou L, Bennisse R, Bouterfass R, Guyoneaud R, Qatibi AI. 2017. Desulfobulbus oligotrophicus sp. nov., a sulfate-reducing and propionate-oxidizing bacterium isolated from a municipal anaerobic sewage sludge digester. Int J Syst Evol Microbiol 67:275–281. doi: 10.1099/ijsem.0.001615. [DOI] [PubMed] [Google Scholar]
- 24.Kharrat H, Karray F, Bartoli M, Ben Hnia W, Mhiri N, Fardeau ML, Bennour F, Kamoun L, Alazard D, Sayadi S. 2017. Desulfobulbus aggregans sp. nov., a novel sulfate reducing bacterium isolated from marine sediment from the Gulf of Gabes. Curr Microbiol 74:449–454. doi: 10.1007/s00284-017-1211-4. [DOI] [PubMed] [Google Scholar]
- 25.Lien T, Madsen M, Steen IH, Gjerdevik K. 1998. Desulfobulbus rhabdoformis sp. nov., a sulfate reducer from a water-oil separation system. Int J Syst Bacteriol 48:469–474. doi: 10.1099/00207713-48-2-469. [DOI] [PubMed] [Google Scholar]
- 26.Sass A, Rütters H, Cypionka H, Sass H. 2002. Desulfobulbus mediterraneus sp. nov., a sulfate-reducing bacterium growing on mono- and disaccharides. Arch Microbiol 177:468–474. doi: 10.1007/s00203-002-0415-5. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki D, Ueki A, Amaishi A, Ueki K. 2007. Desulfobulbus japonicus sp. nov., a novel Gram-negative propionate-oxidizing, sulfate-reducing bacterium isolated from an estuarine sediment in Japan. Int J Syst Evol Microbiol 57:849–855. doi: 10.1099/ijs.0.64855-0. [DOI] [PubMed] [Google Scholar]
- 28.Sorokin DY, Tourova TP, Panteleeva AN, Muyzer G. 2012. Desulfonatronobacter acidivorans gen. nov., sp. nov. and Desulfobulbus alkaliphilus sp. nov., haloalkaliphilic heterotrophic sulfate-reducing bacteria from soda lakes. Int J Syst Evol Microbiol 62:2107–2113. doi: 10.1099/ijs.0.029777-0. [DOI] [PubMed] [Google Scholar]
- 29.Widdel F, Pfennig N. 1982. Studies on dissimilatory sulfate-reducing bacteria that decompose fatty-acids II. Incomplete oxidation of propionate by Desulfobulbus propionicus gen. nov., sp. nov. Arch Microbiol 131:360–365. doi: 10.1007/BF00411187. [DOI] [PubMed] [Google Scholar]
- 30.Bentley SD, Parkhill J. 2015. Genomic perspectives on the evolution and spread of bacterial pathogens. Proc Biol Sci 282:20150488. doi: 10.1098/rspb.2015.0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bliven KA, Maurelli AT. 2016. Evolution of bacterial pathogens within the human host. Microbiol Spectr 4:1–8. doi: 10.1128/microbiolspec.VMBF-0017-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campbell AG, Campbell JH, Schwientek P, Woyke T, Sczyrba A, Allman S, Beall CJ, Griffen A, Leys E, Podar M. 2013. Multiple single-cell genomes provide insight into functions of uncultured deltaproteobacteria in the human oral cavity. PLoS One 8:e59361. doi: 10.1371/journal.pone.0059361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson H, Rybalka A, Moazzez R, Dewhirst FE, Wade WG. 2015. In vitro culture of previously uncultured oral bacterial phylotypes. Appl Environ Microbiol 81:8307–8314. doi: 10.1128/AEM.02156-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vartoukian SR, Adamowska A, Lawlor M, Moazzez R, Dewhirst FE, Wade WG. 2016. In vitro cultivation of “unculturable” oral bacteria, facilitated by community culture and media supplementation with siderophores. PLoS One 11:e0146926. doi: 10.1371/journal.pone.0146926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vartoukian SR, Downes J, Palmer RM, Wade WG. 2013. Fretibacterium fastidiosum gen. nov., sp. nov., isolated from the human oral cavity. Int J Syst Evol Microbiol 63:458–463. doi: 10.1099/ijs.0.041038-0. [DOI] [PubMed] [Google Scholar]
- 36.Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, Sahasrabudhe A, Dewhirst FE. 2001. Bacterial diversity in human subgingival plaque. J Bacteriol 183:3770–3783. doi: 10.1128/JB.183.12.3770-3783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwechheimer C, Kuehn MJ. 2015. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat Rev Microbiol 13:605–619. doi: 10.1038/nrmicro3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie H. 2015. Biogenesis and function of Porphyromonas gingivalis outer membrane vesicles. Future Microbiol 10:1517–1527. doi: 10.2217/fmb.15.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vartoukian SR, Palmer RM, Wade WG. 2010. Strategies for culture of “unculturable” bacteria. FEMS Microbiol Lett 309:1–7. doi: 10.1111/j.1574-6968.2010.02000.x. [DOI] [PubMed] [Google Scholar]
- 40.Krebs HA. 1950. Chemical composition of blood plasma and serum. Annu Rev Biochem 19:409–430. doi: 10.1146/annurev.bi.19.070150.002205. [DOI] [PubMed] [Google Scholar]
- 41.Armitage GC. 2004. Analysis of gingival crevice fluid and risk of progression of periodontitis. Periodontol 2000 34:109–119. doi: 10.1046/j.0906-6713.2002.003427.x. [DOI] [PubMed] [Google Scholar]
- 42.Brown SA, Whiteley M. 2007. A novel exclusion mechanism for carbon resource partitioning in Aggregatibacter actinomycetemcomitans. J Bacteriol 189:6407–6414. doi: 10.1128/JB.00554-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langendijk PS, Hanssen JTJ, Van der Hoeven JS. 2000. Sulfate-reducing bacteria in association with human periodontitis. J Clin Periodontol 27:943–950. doi: 10.1034/j.1600-051x.2000.027012943.x. [DOI] [PubMed] [Google Scholar]
- 44.Boopathy R, Robichaux M, LaFont D, Howell M. 2002. Activity of sulfate-reducing bacteria in human periodontal pocket. Can J Microbiol 48:1099–1103. doi: 10.1139/w02-104. [DOI] [PubMed] [Google Scholar]
- 45.Dewhirst FE, Klein EA, Thompson EC, Blanton JM, Chen T, Milella L, Buckley CMF, Davis IJ, Bennett ML, Marshall-Jones ZV. 2012. The canine oral microbiome. PLoS One 7:e36067. doi: 10.1371/journal.pone.0036067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dewhirst FE, Klein EA, Bennett ML, Croft JM, Harris SJ, Marshall-Jones ZV. 2015. The feline oral microbiome: a provisional 16S rRNA gene based taxonomy with full-length reference sequences. Vet Microbiol 175:294–303. doi: 10.1016/j.vetmic.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, Rattei T, Mende DR, Sunagawa S, Kuhn M, Jensen LJ, von Mering C, Bork P. 2016. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44:D286–D293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rabus R, Venceslau SS, Wöhlbrand L, Voordouw G, Wall JD, Pereira IA. 2015. A post-genomic view of the ecophysiology, catabolism and biotechnological relevance of sulphate-reducing prokaryotes. Adv Microb Physiol 66:55–321. doi: 10.1016/bs.ampbs.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 49.Duarte AG, Santos AA, Pereira IA. 2016. Electron transfer between the QmoABC membrane complex and adenosine 5′-phosphosulfate reductase. Biochim Biophys Acta 1857:380–386. doi: 10.1016/j.bbabio.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 50.Schut GJ, Boyd ES, Peters JW, Adams MW. 2013. The modular respiratory complexes involved in hydrogen and sulfur metabolism by heterotrophic hyperthermophilic archaea and their evolutionary implications. FEMS Microbiol Rev 37:182–203. doi: 10.1111/j.1574-6976.2012.00346.x. [DOI] [PubMed] [Google Scholar]
- 51.Buckel W, Thauer RK. 2013. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na(+) translocating ferredoxin oxidation. Biochim Biophys Acta 1827:94–113. doi: 10.1016/j.bbabio.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 52.Ramos AR, Grein F, Oliveira GP, Venceslau SS, Keller KL, Wall JD, Pereira IA. 2015. The FlxABCD-HdrABC proteins correspond to a novel NADH dehydrogenase/heterodisulfide reductase widespread in anaerobic bacteria and involved in ethanol metabolism in Desulfovibrio vulgaris Hildenborough. Environ Microbiol 17:2288–2305. doi: 10.1111/1462-2920.12689. [DOI] [PubMed] [Google Scholar]
- 53.Poock SR, Leach ER, Moir JW, Cole JA, Richardson DJ. 2002. Respiratory detoxification of nitric oxide by the cytochrome c nitrite reductase of Escherichia coli. J Biol Chem 277:23664–23669. doi: 10.1074/jbc.M200731200. [DOI] [PubMed] [Google Scholar]
- 54.Weghoff MC, Bertsch J, Müller V. 2015. A novel mode of lactate metabolism in strictly anaerobic bacteria. Environ Microbiol 17:670–677. doi: 10.1111/1462-2920.12493. [DOI] [PubMed] [Google Scholar]
- 55.Seeliger S, Janssen PH, Schink B. 2002. Energetics and kinetics of lactate fermentation to acetate and propionate via methylmalonyl-CoA or acrylyl-CoA. FEMS Microbiol Lett 211:65–70. doi: 10.1111/j.1574-6968.2002.tb11204.x. [DOI] [PubMed] [Google Scholar]
- 56.Roberts AP, Kreth J. 2014. The impact of horizontal gene transfer on the adaptive ability of the human oral microbiome. Front Cell Infect Microbiol 4:124. doi: 10.3389/fcimb.2014.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ. 2011. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- 58.Podar M, Gilmour CC, Brandt CC, Soren A, Brown SD, Crable BR, Palumbo AV, Somenahally AC, Elias DA. 2015. Global prevalence and distribution of genes and microorganisms involved in mercury methylation. Sci Adv 1:e1500675. doi: 10.1126/sciadv.1500675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parks JM, Johs A, Podar M, Bridou R, Hurt RA Jr, Smith SD, Tomanicek SJ, Qian Y, Brown SD, Brandt CC, Palumbo AV, Smith JC, Wall JD, Elias DA, Liang L. 2013. The genetic basis for bacterial mercury methylation. Science 339:1332–1335. doi: 10.1126/science.1230667. [DOI] [PubMed] [Google Scholar]
- 60.Al-Khodor S, Price CT, Kalia A, Abu Kwaik Y. 2010. Functional diversity of ankyrin repeats in microbial proteins. Trends Microbiol 18:132–139. doi: 10.1016/j.tim.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Konstantinidis KT, Tiedje JM. 2004. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc Natl Acad Sci U S A 101:3160–3165. doi: 10.1073/pnas.0308653100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ulrich LE, Zhulin IB. 2010. The MiST2 database: a comprehensive genomics resource on microbial signal transduction. Nucleic Acids Res 38:D401–D407. doi: 10.1093/nar/gkp940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ulrich LE, Koonin EV, Zhulin IB. 2005. One-component systems dominate signal transduction in prokaryotes. Trends Microbiol 13:52–56. doi: 10.1016/j.tim.2004.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mittl PR, Schneider-Brachert W. 2007. Sel1-like repeat proteins in signal transduction. Cell Signal 19:20–31. doi: 10.1016/j.cellsig.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 65.Hubber A, Roy CR. 2010. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu Rev Cell Dev Biol 26:261–283. doi: 10.1146/annurev-cellbio-100109-104034. [DOI] [PubMed] [Google Scholar]
- 66.Swann RA, Holmes B. 1984. Infective endocarditis caused by Kingella denitrificans. J Clin Pathol 37:1384–1387. doi: 10.1136/jcp.37.12.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Minamoto GY, Sordillo EM. 1992. Kingella denitrificans as a cause of granulomatous disease in a patient with AIDS. Clin Infect Dis 15:1052–1053. doi: 10.1093/clind/15.6.1052. [DOI] [PubMed] [Google Scholar]
- 68.Rajanna DM, Manickavasagam J, Jewes L, Capper R. 2011. Retropharyngeal abscess from an unusual organism—Kingella denitrificans—in a patient on low-dose methotrexate. Ear Nose Throat J 90:15–17. [DOI] [PubMed] [Google Scholar]
- 69.Chiang WF, Cheng CJ, Chau T, Hsiao PJ, Lin SH. 2014. A tale of two patients: refractory peritonitis with umbilical hernias. Perit Dial Int 34:817–819. doi: 10.3747/pdi.2013.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moriel DG, Bertoldi I, Spagnuolo A, Marchi S, Rosini R, Nesta B, Pastorello I, Corea VA, Torricelli G, Cartocci E, Savino S, Scarselli M, Dobrindt U, Hacker J, Tettelin H, Tallon LJ, Sullivan S, Wieler LH, Ewers C, Pickard D, Dougan G, Fontana MR, Rappuoli R, Pizza M, Serino L. 2010. Identification of protective and broadly conserved vaccine antigens from the genome of extraintestinal pathogenic Escherichia coli. Proc Natl Acad Sci U S A 107:9072–9077. doi: 10.1073/pnas.0915077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Egmond M, Damen CA, van Spriel AB, Vidarsson G, Van Garderen E, van de Winkel JGJ. 2001. IgA and the IgA Fc receptor. Trends Immunol 22:205–211. doi: 10.1016/S1471-4906(01)01873-7. [DOI] [PubMed] [Google Scholar]
- 72.Pastorello I, Rossi Paccani S, Rosini R, Mattera R, Ferrer Navarro M, Urosev D, Nesta B, Lo Surdo P, Del Vecchio M, Rippa V, Bertoldi I, Gomes Moriel D, Laarman AJ, van Strijp JA, Daura X, Pizza M, Serino L, Soriani M. 2013. EsiB, a novel pathogenic Escherichia coli secretory immunoglobulin A-binding protein impairing neutrophil activation. mBio 4:e00206-13. doi: 10.1128/mBio.00206-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Taichman NS, Dean RT, Sanderson CJ. 1980. Biochemical and morphological characterization of the killing of human monocytes by a leukotoxin derived from Actinobacillus actinomycetemcomitans. Infect Immun 28:258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mangan DF, Taichman NS, Lally ET, Wahl SM. 1991. Lethal effects of Actinobacillus actinomycetemcomitans leukotoxin on human T lymphocyes. Infect Immun 59:3267–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Slots J, Reynolds HS, Genco RJ. 1980. Actinobacillus actinomycetemcomitans in human periodontal disease: a cross-sectional microbiological investigation. Infect Immun 29:1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Linhartová I, Bumba L, Mašín J, Basler M, Osička R, Kamanová J, Procházková K, Adkins I, Hejnová-Holubová J, Sadílková L, Morová J, Sebo P. 2010. RTX proteins: a highly diverse family secreted by a common mechanism. FEMS Microbiol Rev 34:1076–1112. doi: 10.1111/j.1574-6976.2010.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Åberg CH, Kelk P, Johansson A. 2015. Aggregatibacter actinomycetemcomitans: virulence of its leukotoxin and association with aggressive periodontitis. Virulence 6:188–195. doi: 10.4161/21505594.2014.982428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Confer DL, Eaton JW. 1982. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 217:948–950. doi: 10.1126/science.6287574. [DOI] [PubMed] [Google Scholar]
- 79.Hanski E. 1989. Invasive adenylate cyclase toxin of Bordetella pertussis. Trends Biochem Sci 14:459–463. doi: 10.1016/0968-0004(89)90106-0. [DOI] [PubMed] [Google Scholar]
- 80.Mouallem M, Farfel Z, Hanski E. 1990. Bordetella pertussis adenylate cyclase toxin: intoxication of host cells by bacterial invasion. Infect Immun 58:3759–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. 2006. Hydrogen-sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J 20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- 82.Greabu M, Totan A, Miricescu D, Radulescu R, Virlan J, Calenic B. 2016. Hydrogen-sulfide, oxidative stress and periodontal diseases: a concise review. Antioxidants (Basel) 5:1–13. doi: 10.3390/antiox5010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wurch L, Giannone RJ, Belisle BS, Swift C, Utturkar S, Hettich RL, Reysenbach AL, Podar M. 2016. Genomics-informed isolation and characterization of a symbiotic Nanoarchaeota system from a terrestrial geothermal environment. Nat Commun 7:12115. doi: 10.1038/ncomms12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Giannone RJ, Huber H, Karpinets T, Heimerl T, Küper U, Rachel R, Keller M, Hettich RL, Podar M. 2011. Proteomic characterization of cellular and molecular processes that enable the Nanoarchaeum equitans-Ignicoccus hospitalis relationship. PLoS One 6:e22942. doi: 10.1371/journal.pone.0022942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Otwell AE, Callister SJ, Zink EM, Smith RD, Richardson RE. 2016. Comparative proteomic analysis of Desulfotomaculum reducens MI-1: insights into the metabolic versatility of a Gram-positive sulfate- and metal-reducing bacterium. Front Microbiol 7:191. doi: 10.3389/fmicb.2016.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sahingur SE, Yeudall WA. 2015. Chemokine function in periodontal disease and oral cavity cancer. Front Immunol 6:214. doi: 10.3389/fimmu.2015.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Henderson B, Nair SP, Ward JM, Wilson M. 2003. Molecular pathogenicity of the oral opportunistic pathogen Actinobacillus actinomycetemcomitans. Annu Rev Microbiol 57:29–55. doi: 10.1146/annurev.micro.57.030502.090908. [DOI] [PubMed] [Google Scholar]
- 88.Ramage G, Lappin DF, Millhouse E, Malcolm J, Jose A, Yang J, Bradshaw DJ, Pratten JR, Culshaw S. 2017. The epithelial cell response to health and disease associated oral biofilm models. J Periodont Res 52:325–333. doi: 10.1111/jre.12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stathopoulou PG, Benakanakere MR, Galicia JC, Kinane DF. 2010. Epithelial cell pro-inflammatory cytokine response differs across dental plaque bacterial species. J Clin Periodontol 37:24–29. doi: 10.1111/j.1600-051X.2009.01505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Darveau RP, Belton CM, Reife RA, Lamont RJ. 1998. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun 66:1660–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wöllert T, Rollenhagen C, Langford GM, Sundstrom P. 2012. Human oral keratinocytes: a model system to analyze host-pathogen interactions. Methods Mol Biol 845:289–302. doi: 10.1007/978-1-61779-539-8_19. [DOI] [PubMed] [Google Scholar]
- 92.Li Y, He J, He Z, Zhou Y, Yuan M, Xu X, Sun F, Liu C, Li J, Xie W, Deng Y, Qin Y, VanNostrand JD, Xiao L, Wu L, Zhou J, Shi W, Zhou X. 2014. Phylogenetic and functional gene structure shifts of the oral microbiomes in periodontitis patients. ISME J 8:1879–1891. doi: 10.1038/ismej.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Langendijk PS, Hagemann J, van der Hoeven JS. 1999. Sulfate-reducing bacteria in periodontal pockets and in healthy oral sites. J Clin Periodontol 26:596–599. doi: 10.1034/j.1600-051X.1999.260906.x. [DOI] [PubMed] [Google Scholar]
- 94.Calenic B, Yaegaki K, Murata T, Imai T, Aoyama I, Sato T, Ii H. 2010. Oral malodorous compound triggers mitochondrial-dependent apoptosis and causes genomic DNA damage in human gingival epithelial cells. J Periodont Res 45:31–37. doi: 10.1111/j.1600-0765.2008.01199.x. [DOI] [PubMed] [Google Scholar]
- 95.Dufton N, Natividad J, Verdu EF, Wallace JL. 2012. Hydrogen-sulfide and resolution of acute inflammation: a comparative study utilizing a novel fluorescent probe. Sci Rep 2:499. doi: 10.1038/srep00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chi XP, Ouyang XY, Wang YX. 2014. Hydrogen sulfide synergistically upregulates Porphyromonas gingivalis lipopolysaccharide-induced expression of IL-6 and IL-8 via NF-κB signalling in periodontal fibroblasts. Arch Oral Biol 59:954–961. doi: 10.1016/j.archoralbio.2014.05.022. [DOI] [PubMed] [Google Scholar]
- 97.Szafrański SP, Deng ZL, Tomasch J, Jarek M, Bhuju S, Meisinger C, Kühnisch J, Sztajer H, Wagner-Döbler I. 2015. Functional biomarkers for chronic periodontitis and insights into the roles of Prevotella nigrescens and Fusobacterium nucleatum; a metatranscriptome analysis. NPJ Biofilms Microbiomes 1:15017. doi: 10.1038/npjbiofilms.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Krzywinski MI, Schein JE, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Clarkson SM, Giannone RJ, Kridelbaugh DM, Elkins JG, Guss AM, Michener JK. 2017. Construction and optimization of a heterologous pathway for protocatechuate catabolism in Escherichia coli enables bioconversion of model aromatic compounds. Appl Environ Microbiol 83:e01313-17. doi: 10.1128/AEM.01313-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tabb DL, Fernando CG, Chambers MC. 2007. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J Proteome Res 6:654–661. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ma ZQ, Dasari S, Chambers MC, Litton MD, Sobecki SM, Zimmerman LJ, Halvey PJ, Schilling B, Drake PM, Gibson BW, Tabb DL. 2009. IDPicker 2.0: improved protein assembly with high discrimination peptide identification filtering. J Proteome Res 8:3872–3881. doi: 10.1021/pr900360j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Taverner T, Karpievitch YV, Polpitiya AD, Brown JN, Dabney AR, Anderson GA, Smith RD. 2012. DanteR: an extensible R-based tool for quantitative analysis of -omics data. Bioinformatics 28:2404–2406. doi: 10.1093/bioinformatics/bts449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. 2016. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
D. oralis physiological characteristics compared to related species of the Desulfobulbus genus. Download TABLE S1, PDF file, 0.1 MB (100.5KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Functional gene annotations for D. oralis. Download DATA SET S1, XLSX file, 0.7 MB (695.2KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Enriched and depleted functional domains (Pfam) and gene families (COG) in D. oralis compared to free-living Desulfobulbus. Download DATA SET S2, XLSX file, 0.1 MB (37.4KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Phylogenetic analysis (FastTree) of selected horizontally acquired proteins in D. oralis. Download FIG S1, PDF file, 0.2 MB (201KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Complete list of signal transduction proteins. Download DATA SET S3, XLSX file, 0.1 MB (29.1KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Genes encoding components of type IV pili and chemosensory systems controlling type IV pilus-based motility. Download DATA SET S4, XLSX file, 0.1 MB (59.5KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Select genes and gene clusters (operons) encoding potential toxins, pathogenicity factors, and their secretion machinery. Download DATA SET S5, XLSX file, 0.1 MB (18.1KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Normalized proteome data. Download DATA SET S6, XLSX file, 0.4 MB (399.5KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Cytokine/chemokine stimulation in oral epithelial keratinocytes by Desulfobulbus oralis supernatant. Download FIG S2, PDF file, 0.1 MB (109.5KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Cytokine/chemokine stimulation in oral epithelial keratinocytes by Desulfobulbus oralis cells. Download FIG S3, PDF file, 0.1 MB (115.3KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.