

Abstract
Genetic coding is generally thought to have required ribozymes whose functions were taken over by polypeptide aminoacyl-tRNA synthetases (aaRS). Two discoveries about aaRS and their interactions with tRNA substrates now furnish a unifying rationale for the opposite conclusion: that the key processes of the Central Dogma of molecular biology emerged simultaneously and naturally from simple origins in a peptide•RNA partnership, eliminating the epistemological utility of a prior RNA world. First, the two aaRS classes likely arose from opposite strands of the same ancestral gene, implying a simple genetic alphabet. The resulting inversion symmetries in aaRS structural biology would have stabilized the initial and subsequent differentiation of coding specificities, rapidly promoting diversity in the proteome. Second, amino acid physical chemistry maps onto tRNA identity elements, establishing reflexive, nanoenvironmental sensing in protein aaRS. Bootstrapping of increasingly detailed coding is thus intrinsic to polypeptide aaRS, but impossible in an RNA world. These notions underline the following concepts that contradict gradual replacement of ribozymal aaRS by polypeptide aaRS: 1) aaRS enzymes must be interdependent; 2) reflexivity intrinsic to polypeptide aaRS production dynamics promotes bootstrapping; 3) takeover of RNA-catalyzed aminoacylation by enzymes will necessarily degrade specificity; and 4) the Central Dogma’s emergence is most probable when replication and translation error rates remain comparable. These characteristics are necessary and sufficient for the essentially de novo emergence of a coupled gene–replicase–translatase system of genetic coding that would have continuously preserved the functional meaning of genetically encoded protein genes whose phylogenetic relationships match those observed today.
Keywords: aminoacyl-tRNA synthetases, bootstrapping, evolution of translation, molecular phylogeny
Introduction
I. Whence Molecular Genetics?
Gene expression consists of interpreting symbolic information stored in nucleic acid sequences. This irreversible computational process creates intrinsically novel meaning, and is thus fundamentally different from the physical chemistry underlying other natural processes, distinguishing it even from the molecular biological processes of replication and transcription. Our goal here is to integrate 1) the dual ancestry of the two aminoacyl-tRNA synthetase (aaRS) classes from opposite strands of the same primordial gene (Rodin and Ohno 1995) and 2) the mapping of amino acid physical chemistry onto tRNA base sequences and its explicit role in protein folding (Carter and Wolfenden 2015, 2016; Wolfenden et al. 2015) into a new conceptual basis for understanding how the synthesis of peptide catalysts from genetic instructions might have emerged and evolved compatibly with inheritance.
A. The Central Dogma and the Adaptor Hypothesis Imply aaRS
Crick recognized that protein synthesis must be directed by information archived in DNA sequences and that information flow proceeds unidirectionally via an intermediate RNA “message” to ribosomes. He also proposed that intervention of a third RNA component (Crick 1955) “adapted” individual amino acids to “codons” in the message (fig. 1A), placing the origin of genetics in the initially obscure relationship between collinear sequences of genes and proteins.
Fig. 1.

Information flow in molecular biology. (A) The Central Dogma is supplemented by the “adaptor” hypothesis. The dashed triangle represents the crucial elements of Crick’s original insight, which necessarily implicates both tRNA and aaRS. (B) The physico-chemical properties of the amino acids define the nano-scale “ecologies” within folded proteins, creating the intersection between genome and proteome. These ecologies drove protein folding and the selection of tRNA identity elements, analogous to a programming language. As a consequence, they also drove the selection of amino acid sequences in mRNA gene sequences (mRNA), analogous to computer programs. (C) Network analysis of the Central Dogma consists of the nodes of a tetrahedron. Embedding the triangle from (A) into the ecology in (B) reveals a uni-directional feedback cycle or self-referential element as generator of complexity in the spirit of Gödel’s incompleteness theorem (Hofstadter 1979). Genetic instructions assemble amino acids according to their physical properties in ways that, when translated according to the programming language in tRNA, yield functional proteins (enzymes, switches, regulators). AARS with cognate tRNAs furnish reflexive elements (orange arrow) connecting their gene sequences, via their folded structures, to the enzymes that enforce rules in the codon table. Physical properties of amino acids and the codon assignment table are “fixed” because they are governed by chemical equilibria. The genome and proteome are dynamically determined, driving the evolution of diversity through self-organization and natural selection of phenotypes.
Participation of the adaptor, transfer RNA (tRNA), involves creating a covalent bond between its 3′ terminus and an appropriate amino acid’s α-carboxylate group. Creating that bond, in turn, requires carboxyl group activation by ATP. In cells, activation and tRNA aminoacylation require a separate enzyme for each amino acid. These aaRS were first clearly identified by Berg and Ofengand (1958).
To execute genetic coding rules aaRSs must recognize both amino acids and tRNAs with high specificity—a process we call assignment catalysis—so that the latter can escort the former to the ribosome for protein synthesis. However, specific recognition by folded proteins depends on a complex “ecology” based on interactions between individual amino acids (fig. 1B). That chemical behavior can be accurately parameterized by two experimental phase transfer Gibbs free energies—from vapor to cyclohexane and from water to cyclohexane—related to the size and polarity, respectively, of each amino acid’s side chain (Carter and Wolfenden 2015, 2016; Wolfenden et al. 2015). Correlations between these free energies and tRNA identity elements recognized by aaRS and the distribution of amino acids between surfaces and cores after protein folding established these parameters as the axes of a kind of “periodic table” of amino acids concatenated in chains that fold to generate proteins of virtually unlimited functional diversity, in analogy with joining atoms to form molecules (Carter and Wolfenden 2016).
Implementing the irreversible attachments of amino acids to codon-specific tRNAs by aaRSs thus exploits the ecology of the amino acids within those enzymes. Proteins folded in accordance with such ecologies that, in turn, execute computationally controlled production from genes of specialized amino acid ecologies (including their own!) compose a reflexive system property known as a computational “strange loop” (fig. 1C;Hofstadter 1979). Recognizing that strange loop opens fundamentally new ways to think about what enabled the aaRS to emerge as the only proteins coded by programs written as mRNA that can, once folded, collectively interpret the programming language in tRNA. We propose that this reflexivity of functional chemistry and encoded information played a crucial role in creating genetics.
B. The RNA World Hypothesis Fails to Address Key Questions about Gene Expression
Life simultaneously requires passing genetic information from generation to generation, and catalytic synchronization of chemical reaction rates underlying the accuracy in gene replication, expression, and metabolism. Base pairing between complementary nucleic acid strands solved the former problem immediately and decisively, once the helical structure of double-stranded DNA was elucidated (Watson and Crick 1953), and pointedly highlighted the latter problem.
Unlike DNA, RNA can assume tertiary structures, consistent with proposals (Woese 1967; Crick 1968; Orgel 1968) that the earliest catalysts might have been RNAs that could “do the job of a protein” (Crick 1968). That hypothesis was sustained by the observation that, whereas proteins cannot readily store or transmit digital information, RNA has rudimentary catalytic properties (Cech 1986; Guerrier-Takada et al. 1989). The expedient conclusion that RNAs functioned as both genes and catalysts in a life form devoid of proteins was rapidly embraced as “the RNA World” (Gilbert 1986).
The clarity with which base-pairing solved the inheritance problem and the discovery of catalytic RNA short-circuited the quest to understand and answer deeper questions:
Catalytic RNA itself cannot fulfill the tasks now carried out by proteins. The term “catalytic RNA” overlooks three fundamental problems: 1) it vastly overestimates the potential catalytic proficiency of ribozymes (Wills 2016); and fails to address either 2) the computational essence of translation or 3) the requirement that catalysts not only accelerate, but more importantly, synchronize chemical reactions whose spontaneous rates at ambient temperatures differ by more than 1020-fold (Wolfenden and Snider 2001). Thus, synchronized catalysis required simultaneous evolution of genetic coding.
The nexus connecting prebiotic chemistry to biology is not replication but the translation table that maps amino acid sequences of functional proteins onto nucleotide triplet codons. The quintessential problem posed by life’s diversity (Carter and Wolfenden 2016; Wills 2016) is how that critical transformation became embedded, in parallel, into tRNA and gene sequences, together with the ribosomal read-write mechanism (Bowman et al. 2015; Petrov and Williams 2015). Spontaneous folding of RNA aptamers and the dynamics of an RNA world do not require encoding into genetic information and hence fall well short of explaining the separation of phenotype from genotype necessary for true Darwinian evolution.
Protein folding irreversibly transforms genetic information. Reversing translation by unfolding, then “reading” the sequence of a protein would require shuttling each successive amino acid through ∼20 active sites until one fitted, and then overcoming the redundancy of the genetic code. The one-way flow of genetic information enshrined in the Central Dogma (Koonin 2015) ensures that biological evolution transcends the simple population dynamics of natural selection in any RNA world.
Computational (Wills 2009, 2014; Hordijk et al. 2012) and structural (Carter and Kraut 1974) modeling argue that some mutual, interdependent process embedded information into proteins and nucleic acids. RNA research has never provided even an approximate experimental model for how a nearly random catalytic network might have progressively bootstrapped the specificity and selectivity characteristic of enzymic systems without encoded proteins (Hordijk et al. 2014). In contrast, recent biochemical and bioinformatic analysis of aaRS superfamilies provides multidimensional, deeply rooted experimental evidence for just such a process (Carter 2017). That experimental record, together with new, complementary theoretical developments (Wills and Carter 2017) motivates this communication.
C. It Is Important to Identify the Genuine Support from Experimental Data on Which the RNA World Hypothesis Rests
Selex experiments (Tuerck and Gold 1990) support a limited version of the RNA World hypothesis (Wolf and Koonin 2007; Van Noorden 2009; Yarus 2011a, 2011b; Bernhardt 2012; Breaker 2012; Robertson and Joyce 2012). RNA catalysts selected from large combinatorial libraries based originally on self-splicing introns (Wochner et al. 2011; Attwater et al. 2013; Sczepanski and Joyce 2014; Taylor et al. 2015; Horning and Joyce 2016) provide partial existence proofs for ribozymal polymerases. However, defenders (Robertson and Joyce 2012) acknowledge that no phylogenetic evidence connects these to biological ancestry. So far as is known, all biopolymers are assembled from activated monomers by catalysts from the opposite class: nucleic acids by protein polymerases, proteins by the ribosomal peptidyl transferase center (Noller et al. 1992; Noller 2004; Petrov et al. 2014; Bowman et al. 2015). The latter appears to be the principal biologically derived ribozymal catalyst in contemporary translation, although tRNA (Woese et al. 2000) and, as noted in Section IV.B, unknown ribozymal components similar to synthetic aptamers (Illangsekhare and Yarus 1999; Niwa et al. 2009; Turk et al. 2011) may once have helped catalyze tRNA aminoacylation.
Riboswitches (Breaker 2012) constitute stronger evidence for an RNA world. These sophisticated regulatory devices are widely distributed in eubacteria. Thus, it is possible to trace their ancestry. Moreover, at least one representative, the T-box riboswitch (Grundy et al. 2002) has a close molecular connection to translation because it recognizes tRNAs at both the unacylated acceptor and anticodon (Grigg et al. 2013), thereby qualifying it as a biologically relevant model for ribozymal tRNA synthetases.
Among RNAs selected for binding activities (Wilson and Szostak 1999; Fedor and Williamson 2005), the ATP-binding aptamer (Sassanfar and Szostak 1993) is a relevant example analogous to the ATP-binding function of aaRS protozymes. Unlike riboswitches, considering such synthetic aptamers evidence for an RNA world is tempered by several observations:
They have no phylogenetic connection to biological RNAs.
There is no comparable combinatorial search algorithm for identifying peptide aptamers.
An aptamer selected by Yarus with high affinity for a 50S ribosomal bi-substrate analog and an uncanny eight-nucleotide sequence identity to a sequence in the peptidyl-transferase site (Welch et al. 1995) is catalytically inactive and its apparent secondary structure is unrelated to that observed in the 50S subunit. Thus, any link between selected and biological sequence seems to be artefactual.
Finally, the ribosome itself stabilizes bi-substrate alignment, increasing the −TΔS‡ term of the activation free energy (Sievers et al. 2004; Schroeder and Wolfenden 2007). Wolfenden (2011) noted, insightfully, that a substantial challenge in understanding the catalytic power of enzymes is that as temperatures of the prebiotic earth cooled, rates of different chemical reactions slowed to differing degrees, and that this increased requirement for catalytic synchronization required catalysts that can decrease ΔH‡, which apparently excludes many, if not most ribozymes.
Koonin and colleagues (Aravind et al. 2002; Leipe et al. 2002; Koonin and Novozhilov 2009; Koonin 2011), and others (Caetano-Anolles et al. 2007; Caetano-Anollés et al. 2013; Caetano-Anollés and Caetano-Anollés 2016) argue that protein domains speciated substantially before the advent of protein-based aaRS and translation factors. Consequently, they argue, a fully developed ribozyme-based version of the contemporary universal genetic code must have first mapped RNA sequences to the amino acid sequences of peptides. We will call this fully-blown RNA World scenario the “RNA Coding World” (RCW; see also [Rodin and Rodin 2006a, 2006b; Rodin and Rodin 2008; Rodin et al. 2011]). The contemporary “Protein Coding World” (PCW), which uses aaRS enzymes to attach amino acids to cognate tRNAs, is envisaged to have evolved by a series of “takeovers,” whereby the coding functions of aaRS ribozymes were progressively replaced, without disruption, by enzymic counterparts. Notably, although Rodin and Ohno (1995) first identified bidirectional coding as a possible ancestry of the two aaRS classes, they themselves failed to recognize the logical difficulties it posed for the RCW. We articulate in Section III.B the far greater probability that such a takeover never took place and describe an alternative phylogeny in Section IV.B.
D. Contemporary aaRSs Furnish Clues about How They Became Molecular Interpreters
Understanding the evolutionary basis for the Central Dogma (fig. 1) requires asking how self-organization and selection might have produced, from nearly random origins, finely tuned ecological niches of amino acids arranged to provide the catalytic and pattern-matching capabilities necessary to operate a code using a 20-letter alphabet. We envision that this process began with a reduced alphabet administered by a small “boot block” and grew by correlated increases in alphabet size and specificity, information selected at each stage being used by the existing interpreters to ensure their own functionality, in spite of the errors that they made.
Replication and translation errors represent the most significant resistance to the emergence and gradual enhancement of biological complexity. Replicative errors increasingly limit the survival of progressively longer “genes,” risking what has been called an “Eigen catastrophe” (Eigen and Schuster 1977). Similarly, translation errors eventually limit the functional specificity required to maintain a cell’s biochemical network, leading to an “Orgel catastrophe” (Orgel 1968). Eigen (1971) and Eigen and Schuster (1977) noted that cooperation between separate, multiply interdependent molecular “information carriers” and “functional catalysts” might help an error-prone network survive that would otherwise be eliminated by competition. Connected concentric rings of components within such sets are called “hypercycles,” a concept whose advantages can be realized by other interdependent arrangements.
Early aaRS phylogenies should record the order in which enzymic aaRS appeared, either ab initio or during their takeover of ribozymal aaRSs. Section II summarizes a new interpretation of evidence, from experimental deconstruction of both aaRS classes (Chandrasekaran et al. 2013; Carter 2014, 2015, 2016, 2017; Carter et al. 2014), that all contemporary aaRS descended in modular fashion from a single bidirectional gene, whose strands coded for functional ancestors, respectively, of Class I and II synthetases. Products of that gene appear to have been optimally differentiated and crafted to establish hypercycle-like interdependence, implementing a minimal amino acid alphabet—all characteristics of the “boot block” we envision to have first enabled genetic coding. This bidirectional coding ancestry necessarily coupled contemporary Class I and II aaRS phylogenies (O’Donoghue and Luthey-Schulten 2003; Wolf and Koonin 2007; Caetano-Anollés et al. 2013) as discussed in Sections I.B and II.
Statistically significant relationships between identity elements different synthetases use to recognize tRNA and the size and polarity of amino acid sidechains supplement phylogenetic and biochemical evidence (Carter and Wolfenden 2015, 2016). Coding relationships implemented in tRNA recognition are not arbitrary, but reflect the deeply relevant inner logic of protein folding rules (Carter and Wolfenden 2015, 2016; Wolfenden et al. 2015). We consider this reflexivity and other relevant concepts in greater detail in Section III.
The aaRS pose a dilemma: either their bidirectional coding ancestry (Chandrasekaran et al. 2013; Carter et al. 2014; Carter 2015) and sequential decompositions into urzymes (Ur = primitive; Li et al. 2013; Carter 2014; Martinez et al. 2015) and protozymes (Proto = before; Martinez et al. 2015), or the previous phylogenetic analyses described in Section I.C must be wrong. Section IV.A outlines a resolution.
Results
II. AARS Class Dualities Would Have Helped to Stabilize Quasispecies Bifurcations
Three functionalities give unique status to aaRS as descendants of the earliest enzymes: 1) They accelerate by ∼1014-fold amino acid activation at the expense of two ATP phosphates, irreversibly synthesizing aminoacyl 5′AMP. Uncatalyzed rates of other reactions in protein synthesis are all orders of magnitude faster than activation, which thus limits the rate of prebiotic protein synthesis. 2) The adenosine serves as an affinity tag that increases amino acid binding 1000-fold, enhancing coding assignment specificity, especially where editing is required. 3) They acylate tRNA, covalently linking a specific amino acid to a tRNA molecule bearing a code-cognate anticodon.
Notably, two distinct sets of homologous aaRS structures, Class I and Class II (Cusack et al. 1990; Eriani et al. 1990; Ruff et al. 1991), implement these three functions in disparate ways. The two classes activate symmetrical sets of 10 amino acids. Both classes have one major (A), and two different minor subclasses (B and C) (Cusack 1994). The common origin of the two aaRS classes on opposite strands of the same ancestral gene (Rodin and Ohno 1995) remained obscure until recently (Martinez et al. 2015). Consequences of this duality at multiple structural and functional levels may have served to differentiate and stabilize early stages of genetic coding in the face of high error rates.
A. Validation of Ancestral Bidirectional Genetic Coding
Rodin and Ohno (1995) identified highly significant bidirectional coding of the class-defining active-site sequence motifs from aligned coding sequences of the two aaRS Classes aligned in opposite directions. Subsequently, it became increasingly apparent that protein-based aaRSs all descended from a single ancestral gene whose complementary strands encoded precursors to the Class I and Class II aaRS superfamilies (Carter et al. 2014; Carter 2015; Martinez et al. 2015). Bidirectional coding ancestry implies that protein aaRS gene evolution began with an early stage in which the unique information in one strand of a gene could be interpreted on the opposite strand as a different protein with a similar function. Three predictions of the Rodin-Ohno hypothesis have been confirmed:
Two successive experimental deconstructions confirmed the prediction that the most highly conserved portions of contemporary aaRSs correspond to modules capable of bidirectional alignment, and retain catalytic activity when excerpted from the full-length genes. Urzymes (Pham et al. 2007, 2010; Li et al. 2011, 2013) have ∼120–130 amino acids and retain all three translation functions of contemporary synthetases and accelerate amino acid activation by 109-fold, with significant specificity. Class I and II protozymes of ∼46 amino acids contain the ATP binding sites of the respective aaRS, bind ATP tightly, and accelerate amino acid activation 106-fold (Martinez et al. 2015).
Coding sequences retain a higher frequency of base-pairing between middle codon bases in antiparallel, in-frame alignments of Class I and II aaRS. This middle-base pairing frequency, ∼0.34, is significantly nonrandom and increases to 0.42 in comparisons between coding sequences reconstructed independently for ancestral nodes of bacterial Class I and II aaRS (Chandrasekaran et al. 2013).
By configuring Rosetta (Leaver-Fay et al. 2011) to both constrain tertiary structures and impose genetic complementarity we produced a bona fide bidirectional gene encoding both Class I and II protozymes (Martinez et al. 2015). Remarkably, all four wild-type and designed peptides from Class I and Class II have the same kcat/KM and accelerate amino acid activation by ∼106-fold. Wild-type sequences have 100-fold lower kcat and KM values than do the designed protozymes from the complementary gene, in keeping with the possibility that their wild-type sequences may include amino acid binding determinants lost in the designed protozymes. The protozymes extend a linear relationship between transition state stabilization free energy and the number of residues of the constructs. Notably, the same slopes and intercepts relating rate acceleration to number of residues (Martinez et al. 2015) are found separately for Class I and II constructs.
Bidirectional, in-frame coding is a strange idea. Base-pairing is part of an inversion symmetry operator that generates the sequence and (using helical symmetry operators) the structure of the opposite strand. Because the opposite strand sequence can be retrieved using this inversion operator, a gene’s unique information is contained in one strand. That unique information, however, has two different functional interpretations. Validating (1)–(3) of the Rodin-Ohno hypothesis revealed higher-order symmetries relating Class I and II gene products (Carter et al. 2014; Carter 2015), as discussed in Sections II.C–II.E.
B. Bidirectional Coding Implies Quasispecies Bifurcation
The simplest imaginable code to encode useful information would have required discriminating between at least two kinds of amino acids. The interesting scenarios (Wills 2004) thus entail generating the full code from simple 2- or 4-letter alphabets via transitions that increased the effective size neff of the amino acid and codon alphabets. Nested instabilities (Wills 2004) allow code-expanding transitions to attractor states with progressively larger values of neff. These transitions connect dynamic states with significant error rates and thus entail broad distributions of functional protein sequences whose encoding genes are called “quasispecies,” so we call the corresponding transitions “quasispecies bifurcations” (fig. 2).
Fig. 2.
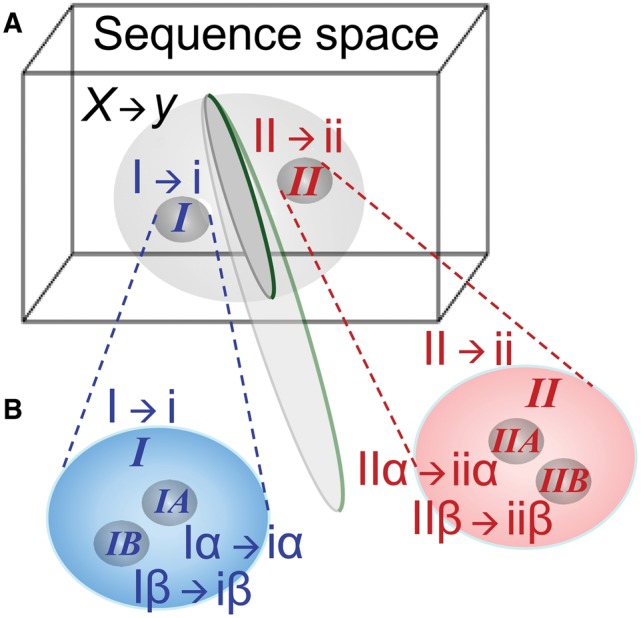
Quasispecies bifurcations in aaRS gene or protein sequence space. (A) A single undifferentiated quasispecies making random assignments X → y of codons (X) to amino acids (y) cannot transmit genetic information. Nor can it easily bifurcate to a pair of narrower quasispecies. Bidirectional coding ancestry of the contemporary aaRS created suitable quasispecies de novo {I, II; red and blue; bold italics explicitly indicating Class I, II aaRS} each separately supporting binary coding assignments I → i and II → ii of specific subsets of codons {I, II} to corresponding subsets of amino acids {i, ii}. That double-helical gene with dual single-strand interpretations overcame the initial and most substantial barrier to the emergence of genetic coding by partitioning protein sequence space decisively into two functionally distinct populations. The plane between the I and II quasispecies is a local representation of the inversion operator that transforms a sequence into its complement read in the reverse direction. (B) Daughter population distributions derived from nearly simultaneous bifurcation of the two ancestral binary coding quasispecies into smaller separate sub-populations of genes and assignment catalysts operating a 4-letter code {Iα → iα, Iβ → iβ, IIα → iiα, IIβ → iiβ,}. Genetic coding bidirectionality is preserved through complementary gene pairs Iα⇔IIα and Iβ⇔IIβ. Recapitulating the bifurcation would further specialize related species, each step being progressively easier, owing to the increased coding specificity, but eventually losing the ability to use information in both strands of genes.
The TrpRS, LeuRS, and HisRS urzymes (Li et al. 2013) and the designed Class I/II protozyme gene (Martinez et al. 2015) furnish substantive experimental representations of the ancestral assignment catalysts envisioned by Wills (2004). All four protozymes exhibit high ATP affinity and the Class I protozymes possess a consensus phosphate binding site composed entirely of oriented backbone NH groups (Hol et al. 1978). Thus, it seems plausible and of obvious interest that protozymes coded using fewer than the canonical 20 amino acids might retain substantial catalytic activity.
A coding system assigning dual classes of functionally differentiated amino acids {α,β} in a crude binary fashion to tRNAs with anticodons complementary to codons {Α,Β} could bifurcate into two versions to produce four-member amino acid and codon alphabets, {α, β, χ, δ} and {Α, Β, Χ, Δ}, increasing the coding capacity from 2 to 4 letters, and expanding the 2 × 2 translation table into a 4 × 4 table. In simulations (Wills 2004, 2009), the hierarchically nested embedding of assignment activities in the protein sequence space geometrically mirrored the decomposition of the alphabets. The system showed stepwise coding self-organization, first from a non-coding state to the execution of a binary code {A→a, B→b} and then from the binary code to the expanded four-dimensional code {Α→α, B→β, Χ→χ, Δ→δ} (fig. 2), anticipating experimental studies of the two synthetase Classes (Martinez et al. 2015).
A puzzling hierarchy of inversion symmetries in the structural, functional, and evolutionary biology of contemporary aaRSs makes sense if the aaRSs were created by such bifurcations. Ancestral bidirectional coding would have decisively partitioned sequence space, dividing it between sequences related most closely to each of the two strands. Translated products of each strand would then have differentiated the functional specificities retained by sequences surrounding the centroids of the two populations. The bidirectional coding complementarity constraint increases selection pressure for coding by steepening the fitness landscape, decisively enforcing more robust coding cooperation than for independent genes for the Class I and II urzymes. Finally, the reduced volumes of sequence space and enhanced functional specialization of the two bidirectionally coded quasispecies suggest that fewer mutations were necessary for neofunctionalization of subsequent duplications, successively easing subsequent bifurcations as neff increased during the bidirectional coding regime.
C. Experimental Deconstructions of Class I and II aaRS Reveal Parallel Structural Hierarchies
Superimposing Class I and II aaRS catalytic domains reveals small invariant cores, distinct from idiosyncratic elements unique to each amino acid. Like Russian Matryoshka dolls, parallel deconstruction of both Class I and II aaRS families reveals nested, increasingly conserved modular catalysts of nearly equal molecular mass (Carter 2014): catalytic domains (200–350 residues), urzymes (120–130 residues; Pham et al. 2007, 2010; Li et al. 2011, 2013), and protozymes (46 residues; Martinez et al. 2015), each retaining conserved portions from its preceding construct.
Urzymes retain all necessary functions of full-length aaRS, albeit with lower proficiency and specificity, and are analogous to using “molecule” to define the smallest unit of matter that retains all properties of a chemical substance. Protozymes, on the other hand, approach the smallest polypeptide catalysts, but have not yet been shown either to acylate tRNA or to discriminate significantly between different amino acids, hence are perhaps more analogous to “atoms.”
Published evidence that experimental urzyme catalytic activities arise neither from tiny amounts of wild-type enzyme nor from unrelated, but highly active contaminants includes the following (Pham et al. 2010; Li et al. 2011): 1) empty vector controls have no activity; 2) protease cleavage of tagged fusion proteins releases cryptic activity; 3) mutations alter activity; 4) amino acid KM values differ from WT values; and, most importantly; 5) single turnover active-site titration experiments show presteady-state burst sizes demonstrating that 35–75% of molecules transiently form tight transition-state complexes. Experimental assays of protozymes were validated by showing that active-site mutants H18A (Class I) and R113A (Class II) eliminated activity of the respective catalyst (Martinez et al. 2015).
Modular accretions in the structurally unrelated Class I and II protein superfamilies exhibit parallel accelerations of the rate-limiting step of protein synthesis over a 108-fold range. Experimental transition-state stabilization free energies track linearly with number of residues in deconstructed constructs from both classes, justifying the identification of these constructs as snapshots in the parallel evolution of both synthetase classes (Martinez et al. 2015). Urzymes retain ∼60% of the full-length transition state stabilization free energy observed in modern synthetases. Protozymes from both Class I and II aaRS retain only the ATP binding sites, but exhibit ∼40% of the full-length transition-state stabilization.
These accelerations document that multiple protein families can synchronize chemical reactions over a very broad range, from the uncatalyzed rate to that observed in contemporary organisms. RNA has not been shown capable of parallel rate accelerations over such a dynamic range either in parallel families or with similar increases in mass, underscoring the superior ability of polypeptide catalysts to adaptively synchronize cellular chemistry.
D. Folded Class I and II AARS Tertiary Structures Are “Inside Out”
Binary patterns coding for protein secondary structures (Kamtekar et al. 1993; Patel et al. 2009) are reflected across complementary coding strands. They are determined by positions of hydrophobic residues (Muñoz and Serrano 1994). The heptapeptide repeat, a–g, with hydrophobic amino acids in positions a, e, f, is diagnostic for alpha helix. Alternation of hydrophobic side chains, especially when they include side chains with branched β-carbon atoms, is a predictor of β-structure.
Soluble globular proteins have hydrophobic cores and water-soluble surfaces. The distribution of amino acids in folded proteins between these two extreme environments is spanned by a two-dimensional “basis set” furnished by the experimental free energies of transfer between vapor and cyclohexane and between water and cyclohexane (Carter and Wolfenden 2015; Wolfenden et al. 2015). The contemporary genetic code respects this dichotomy to an extraordinary degree, as codons for virtually all core side chains are anticodons for surface side chains (Zull and Smith 1990). Complementary codons for proline and glycine, most often associated with turns, mean that such sequence-directed turn formation also reflects across codes from antiparallel strands. Thus, the folded products from a bidirectional gene will tend to have comparable secondary structures, with opposite polarities. By these criteria, Class I and II aaRS urzymes are both antiparallel and “inside out.”
E. Ancestral Bidirectional Genetic Coding Underlies the aaRS Class Distinction
tRNA acceptor stem identity elements represent a code for amino acid side-chain size and other descriptors including side chain carboxylation and β-branching. Evidence that the much smaller aaRS urzymic cores accelerate tRNA aminoacylation rates (Li et al. 2013) now makes it increasingly likely that an early “operational genetic code” (Schimmel et al. 1993; Schimmel 1996) used acceptor stem bases to specify the most significant difference between Class I and Class II amino acids.
Ancestral tRNAs may have been only about half the size and consisted of only the acceptor and TΨC loops of modern tRNAs. Doubling of this ancestral structure has been proposed to have created the anticodon and dihydrouridine loops with the anticodon initially serving as a proxy for the identity elements in the acceptor stem (Di Giulio 1992, 2004, 2008; Rodin et al. 1996; Rodin and Rodin 2008). Any successful model for the emergence of genetic coding from an RNA-based system of molecular information processing should thus be consistent with these two observations as well as with the phylogenies of the two aaRS Classes.
Class I and II aaRS amino acid substrate specificities, especially those from ancestral codes, are related by inversion with respect to side chain size (Carter et al. 2014; Carter 2015). Modern aaRSs prefer their cognate amino acids by ∼5.5 kcal/mol, ∼80% of which comes from allosteric influences of more recently acquired modules on the urzyme activities. Lacking insertion- and anticodon-binding domains, Class I LeuRS and Class II HisRS urzymes are relatively nonspecific (Carter et al. 2014; Carter 2015). Experimental values show that they have similar and complementary specificities. LeuRS urzyme prefers Class I substrates; HisRS urzyme prefers Class II substrates, both by ∼1 kcal/mol. They are therefore capable of making the correct choice between Class I and Class II amino acids roughly four times in five. That fidelity is too promiscuous to support more than “statistical ensembles” of peptides, as hypothesized by Woese (1965a, 1965b) and Woese et al. (1966). Thus, urzymes would have been the predominant assignment catalysts within a much broader population of molecular types, with the properties of a “quasispecies-like” cloud as defined by Eigen and Schuster (1977) that would have included many species with lower specificity and/or catalytic proficiency.
The only statistically significant distinction between amino acids activated by Class I and Class II aaRS is their sizes (Carter and Wolfenden 2015; Wolfenden et al. 2015): Class I amino acids are significantly larger than those from Class II. Accounting for the solvent exposure of amino acids in folded proteins entails both size and polarity and is therefore two-dimensional (Carter and Wolfenden 2015, 2016). Class II amino acids migrate significantly toward water interfaces during protein folding, whereas Class I amino acids migrate toward cores. Thus, the ancestral bidirectional gene likely enforced the difference between large and small side chains, and prefigured the requirements for encoding surface and core amino acids in folded tertiary structures.
F. An Ancient Hypercycle-like Interdependence Relates Catalytic Residues in Each Class
Active-site amino acids in aaRS occur in three sets of signature sequences (Eriani et al. 1990; Carter 1993). Class I HIGH and KMSKS sequences and Class II Motifs 1 and 2 are present in the respective urzymes. The HIGH/Motif 2 signature is present in the protozymes. As these motifs provided the original evidence for bidirectional coding (Rodin and Ohno 1995), and contain active-site residues, it comes as no surprise that the respective active-sites utilize different catalytic residues. In fact, all residues contributing to catalysis by Class I active sites must be activated by Class II aaRS, and conversely, residues needed for Class II activity must be activated by Class I aaRS (Carter et al. 2014; Carter 2015, 2017). This functional “anti-homology” dates from the earliest Class I and II catalysts. Interdependence induces a coupling between the two bidirectional gene products similar to that proposed by Eigen (Eigen 1971; Eigen and Schuster 1977) to induce cooperation and mitigate competition, thereby increasing the overall semirandom genetic content that could survive deterioration at given copy-error rates.
G. Class I and II Genes, Gene Products, Mechanisms, and Specificities Are Maximally Differentiated
An important barrier to the emergence of diversity from quasi-random reproductive processes is the strong tendency of mutant daughter species to regress to the centroid of the distributions from which they originate (Eigen et al. 1988). The centroids behave as “strong attractors.” Inversion symmetries relating Class I and II aaRS, described in Sections II.B–II.E suggest that their genes, gene products, functions, and substrates are inherently differentiated to survive successive quasispecies bifurcations necessary for enhanced genetic coding to emerge from populations of low sequence identity and modest specificity:
Bidirectional coding complementarity means that individual ancestral Class I and II gene sequences are as difficult as possible to interconvert from one to the other by serial mutation.
Descent of the Class I and II aaRS from a bidirectional gene stabilizes two quasispecies that can presumably begin to interpret binary sequence patterns, decisively overcoming the barrier posed by the strong attraction of a single quasispecies.
Reduced population size and enhanced functional specialization of the two bidirectionally coded quasispecies suggest that fewer mutations are necessary for neofunctionalization, successively easing subsequent bifurcations during the bidirectional coding regime.
Distinct properties of protozymes and urzymes point to successive emergence during the bidirectional coding era of their ATP-, amino-acid-, and pyrophosphate-binding sites, consistent with modular construction of aaRS functions.
Inverted folding instructions give rise to “inside out” Class I and II tertiary structures that are as different as possible from one another, and thus minimally vulnerable to mutations that might fuse the two quasispecies by regression to the common centroid.
Catalytic residues in Class I and II aaRS are entirely segregated. Thus, throughout their early evolution, the two Classes formed a hypercycle-like network (fig. 3). By arguments from Eigen and Schuster (Eigen et al. 1988) and Wills (Wills 2009), their interdependence defended them against corruption by molecular parasites during growth of catalytic networks.
Class I and II amino acids are themselves optimally separated on the basis of 1) size, 2) polarity, and hence 3) their ultimate destination in folded proteins.
Fig. 3.
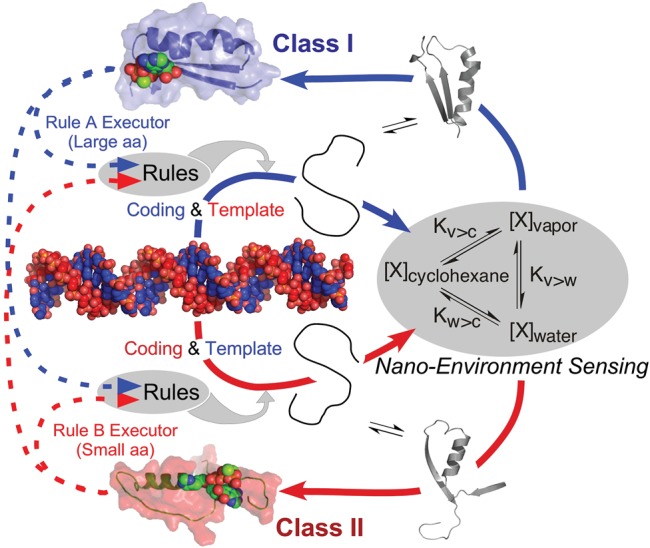
Reflexivity is an exclusive property of protein aaRS. The putative ancestral amino acid activating protozyme gene, substantiated experimentally in Martinez et al. (2015) furnishes two assignment catalysts, each executing a complementary assignment rule, one for large, the other for small amino acid sidechains. Each also contributes to the translation of the other. As the assignment catalysts are proteins, their folding reactions are governed by the phase transfer equilibria of the amino acids, sensing the nano-environment in a necessary prelude to function. The fundamental circularity and interdependence of this feedback loop enable the protozyme gene to bootstrap the evolution of increasingly specific genetic coding.
III. Bidirectionality Furnishes Four Properties Indispensable for Self-Organization of Coding
Avoiding multiple stop codons on both strands of a bidirectionally coded ancestral gene would mandate that each of the four bases have a functionally coded meaning when it occurs as an (internal) codon middle base (see, e.g., Delarue 2007). This would imply a (possibly redundant) alphabet of four letters. Such a reduced repertoire is consistent with that expected for an ancestral tRNA acceptor stem, in keeping with the fact that the contemporary acceptor stem code distinguishes best between 1) large and small, 2) β-branched versus unbranched, and 3) carboxylate versus noncarboxylate side chains (Carter and Wolfenden 2015). Presumably, selection subsequently drove both the code and primordial coding sequences to capture and employ additional symbolic information for precisely those chemical properties—size and polarity—that determine how the 20 amino acids direct proteins into unique tertiary structures (Carter and Wolfenden 2016).
Bidirectional coding of enzymic aaRS impacts four properties that favor much more rapid and efficient evolution of gene expression than would have been possible for ribozymal aaRS. These properties are developed with greater mathematical rigor in a separate paper (Wills and Carter 2017).
A. Any Set of aaRSs Forms an Interdependent Catalytic Network
Structural variants in any functional aaRS population must respond coordinately to two different chemical signals—amino acid and tRNA. Because contemporary aaRS are proteins, their functional structures all depend on all aaRS functionalities and so still form hypercycle-like networks. Interdependence implies that both their mRNA sequences and the tRNA programming language coevolved from simpler ancestors with fewer distinctions between them, whose discrete ancestries lead to successively simpler levels of interdependence as the root is approached. As Class I and II aaRS active-site catalytic residues must be activated by the opposite class (Carter et al. 2014; Carter 2015, 2017), bidirectional coding ancestry anchors interdependence in the earliest ancestral quasispecies.
B. Reflexivity of Protein-Based Assignment Catalysis Offers Superior Paths to Code Bootstrapping and Optimal Gene Sequences
The aaRS molecular biological interpreters are the first and, probably the only products of mRNA blueprints that can implement the translation table embodied in tRNA. Accumulating reflexive genetic information—genes whose expression by rules can, in turn, execute those expression rules—is an intrinsic architectural feature of the PCW that is absent from any RCW. Rapid self-organization of coding in the PCW is driven by reflexive, in-parallel sensing (fig. 3) of the amino acid phase transfer equilibria that drive folding and thus enable aaRS to recognize both the symbolic information in tRNA (i.e., the syntax) and the chemistry of enzymes (i.e., the semantics) embedded in the coding language (fig. 1C).
Coding rules follow from folding rules that generate functional assignment catalysts from sequence. Ribozymal and enzymatic functions are coupled to very different nano-environmental effects. RNA folding depends largely on base pairing because the four nucleotide bases are otherwise almost undifferentiated, having only two sizes and solvent phase transfer equilibria that differ by at most –3.7 kcal/mol in their transfer free energies from chloroform to water (Cullis and Wolfenden 1981; see supplementary fig. S2, Supplementary Material online). The corresponding phase transfer equilibria of the 20 canonical amino acids (Radzicka and Wolfenden 1988) exhibit approximately 5-fold greater variations in polarity and 26-fold greater variation in size. These differences and the dominance of backbone-backbone hydrogen bonding result in profoundly different protein folding rules.
The universal genetic code is a nearly unique selection from an inconceivably large number of possible codes and must have been discovered by bootstrapping. It efficiently maps the chemical properties of amino acids onto the sequence space of triplet codons (Carter and Wolfenden 2016) and is almost ideally robust to mutation (Freeland and Hurst 1998; Koonin and Novozhilov 2009). Bidirectional ancestry restricted the tiny fraction of the possible codes that share that optimality to an even smaller subset by requiring anticorrelated coding of amino acid physical properties (Zull and Smith 1990; Chandrasekaran et al. 2013). Discovery of such a rare, highly optimized code through random-sampling natural selection has a vanishingly small probability, reminiscent of Levinthal’s protein folding paradox (Dill and Chan 1997).
Far more likely to produce such a result is a series of feedback-accelerated symmetry-breaking phase transitions that could bootstrap the earliest translation system into existence from less well-organized chemistry. The initial binary coding need not have made high accuracy distinctions between codons and amino acids. Rather, it need only have had a kinetically self-sustaining bias in assignment probabilities, consistent with distinguishable aaRS quasi-species. For clarity, we henceforth refer to executors of assignment catalysis as RNA or protein “translatases,” to distinguish them from contemporary aaRS.
The bootstrapping metaphor integrates local environmental sensing directly into the generation of function. We envision a minimal, low fidelity instruction set or “boot block” whose realization has been substantially demonstrated (Martinez et al. 2015); and whose feedback-sensitivity (fig. 3) enabled self-improvement by elaborating its own resources, much like installing an operating system in a computer at startup. Increasingly specific coding assignments during successive transition steps could take hold only by conferring new selective advantage(s) to the evolving genes, that is mRNA sequences, that encode them. In this way, such a system could express new meaning in a snowball effect beyond the specific level of fidelity and complexity already achieved. The mechanistic implementation of reflexivity (fig. 3) makes it clear that the requisites for accelerating a bootstrapped discovery of coding are built into the PCW, but absent in the RCW (fig. 4).
Fig. 4.
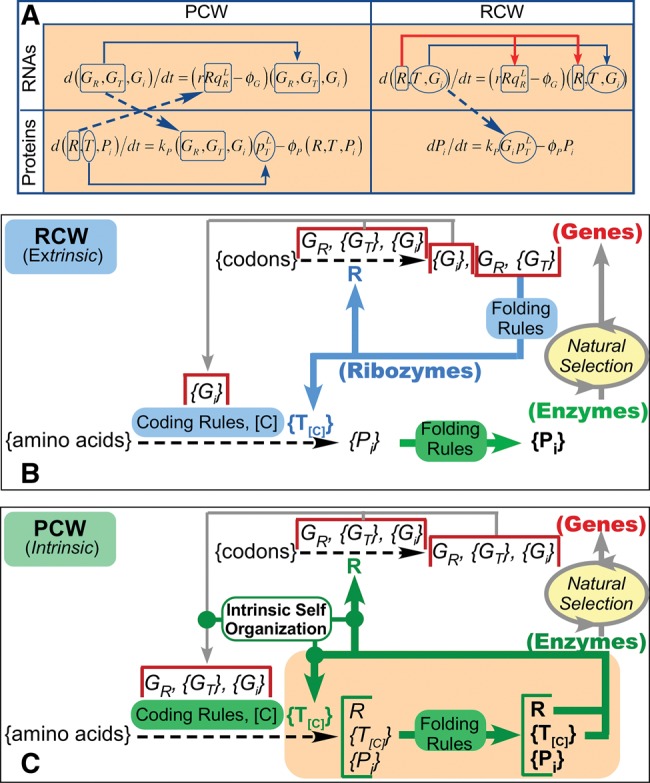
Feedback in ribozymal (RCW) and protein (PCW) GRT networks. (A) Coupled Replicase and Translatase production. Differential equations for gene expression in PCW and RCW are compared for RNAs and Proteins (eqs. 30 and 31 of Wills and Carter [2017]). Solid lines indicate autocatalytic acceleration. Dashed arrows form a (hyper)cycle coupling production dynamics of replicase and translatase in the PCW, but not in any RCW. (B) In an RCW, coding rules [C] are implemented by ribozymal assignment catalysts {T[C]} that cannot sense the phase transfer equilibria accessible to protein assignment catalysts. Thus, natural selection is the only feedback cycle. Non-aaRS functional proteins {Pi} furnish the only source of selective advantage, and have no direct influence on the coding rules. (C) In the PCW, coding rules are executed by proteins that must first fold. A tighter feedback loop (green arrows) is a structural feature of the reaction network (see also fig. 3). Protein folding rules determine the function of the assignment catalysts and therefore also the eventual choice of codon assignments, substantially accelerating self-organization.
Differential equations governing expression dynamics (fig. 4A;Wills and Carter 2017) reinforce the transcendent difference between coding rules derived in an RCW and in the PCW. Synthesis of protein translatases (aaRS) is autocatalytic (horizontal arrows) in the PCW, but not in an RCW. Coding rules executed by ribozymes (fig. 4B) are based on RNA folding rules and intrinsically insensitive to protein folding rules and/or functionality. Thus, reflexive feedback cannot trigger bootstrapping of higher-functioning encoded proteins in an RCW because its assignment catalysts contain no proteins. Variant ribozymal aaRSs capable of improved assignments would have to progressively prove their advantages for the relevant unit of selection, presumably a protocell. The resulting slow, indirect Darwinian evolution could discover protein folding rules and robustness against mutation only from non-aaRS protein performance. The extrinsic self-organization resulting from mutation and higher-level selection in an RCW provides no direct feedback procedure for discovering a translation table that embodies an ordered symbolic encoding of amino acid sidechain chemistry in folded proteins.
Reflexivity in the PCW (fig. 4C) accelerates self-organization in genetic coding, essentially as dynamic phase transitions, because nano-environmental sensing couples coding rules directly to protein folding rules. AARS tertiary structures—positioning amino acids distant in primary structure close to one another in space—as determined by amino acid phase transfer equilibria (fig. 3), furnish the aaRS specificity required to determine the coding rules. Sensitivity of the code to the phase transfer equilibria of amino acid side chains allows those equilibria to feed directly back onto protein aaRS folding and function, naturally producing a refined map of the phase equilibria that govern protein folding and function in the existing code, via the tRNA identity elements (Wolfenden et al. 1979, 2015; Radzicka and Wolfenden 1988; Carter and Wolfenden 2016). Thus, in the PCW nanoscale control of chemistry, in this case coding, is determined directly by its outcome.
A PCW also coordinates and optimizes discovery of gene sequences by placing amino acids with different properties in different positions in accordance with their effects on a folded protein. For aminoacylation functionalities to serve as “assignment catalysis” relevant to coding, their specificity for the relevant amino acid must also have gained parallel specificities choosing primitive “codons” in precursor mRNA. A PCW automatically pressures an evolving code to discover and refine partitions between amino acids that give the genetic representation of functional properties best adapted for survival: an error-minimized code in which amino acids with similar chemical properties are assigned to similar codons. This argument extends to every stage of code expansion. Enhancements that incorporated new amino acids into the programming language had to coevolve with messages able to exploit them. Thus, code evolution in a PCW will inevitably target both near-optimal folded protein functionality and an encoding that represents survival fitness as precisely as possible (fig. 4C).
For these reasons, de novo emergence of genetic coding into a peptide•RNA world appears to have introduced such overwhelming influence on a choice of codons optimally able to represent the effect of an amino acid entering the developing ecology inside a folding protein that it must be seen as enormously more rapid and probable than coding emerging in an RNA World.
C. Fidelity: Any Simple PCW Taking over a More Sophisticated Ribozymal Coding Will Increase the Overall Error Rate, Degrade Fitness, and Hence Be Eliminated by Purifying Selection
The evident simplicity of the earliest coding apparatus in the PCW poses an insuperable barrier to its taking over a more sophisticated coding apparatus in an RCW. The PCW is rooted in phylogenetically based ancestors capable only of the simplest coding assignments—one or at most two bits—and consequently also in a coding system necessarily operating at high error rates. Reducing error rates in both replication and translation must certainly have required larger alphabets. To be selected, the functionality of such primordial coding must already have exceeded that of whatever preceded it. Its low specificity appears to rule out scenarios involving proteins “taking over” catalytic functions from any sophisticated preexisting RNA catalysts.
A separate paper (Wills and Carter 2017) treats this problem in an extension of earlier mathematical models of coding self-organization (Bedian 1982, 2001; Wills 1993, 1994, 2004) by comparing the dynamic stability of coexisting ribozyme- and protein-operated assignment catalysts. We confirm analytically the intuitive conclusion that translation errors would inevitably be higher for any hybrid coding situation driven simultaneously by separate ribozymal and protein translatases than they would be for an optimized system with only one type of aaRS. If both types of translatases effect codon-to-amino acid assignments at different characteristic rates and accuracies the hybrid system will necessarily operate at intermediate error rates. As Equations (23–27) of Wills and Carter (2017) make abundantly clear, introducing any significant population of intrinsically less accurate protein translatases to an extant ribozymal coding apparatus will undermine the role of the ribozymal translatases, possibly threatening the protein domain with extinction by indirectly undermining the selective advantage of ribozymal translatases.
Newly emerging protein-based assignment catalysts must, therefore, have been far less specific than the preexisting ribozymal assignment catalysts envisioned, for example, by Wolf and Koonin (2007), and cannot have been selected within an advanced RCW because their very rudimentary functionality would corrupt any preexisting ribozymal translation system of higher specificity and diversity. The problem will be extreme for rudimentary ancestral protein aaRSs that operate a low-dimensional translation table, substantially reducing the accuracy of the extant ribozymal population, making survival of proteins dependent on the elimination of the protein translatases by an analog of purifying selection.
The following considerations reinforce the conclusion that no hybrid set of protein and ribozymal aaRS and/or replicases can have superior fitness to those of a preexisting RCW:
The more sophisticated the preexisting RCW, the harder it would have been for early stages of PCW code development to compete. Conversely, the detailed inversion symmetries arising from bidirectionally coded genes (Section I) all point to the key role of these asymmetries in enforcing differentiation early in the evolution of the genetic code, when it was most vulnerable to parasites with incorrect specificities.
The dramatic rate acceleration by aaRS protozymes on the other hand represents a decisive selective advantage in a pepide•RNA world, first by harnessing the chemical free energy transfer of NTP utilization and then by providing a flow of activated amino acids.
RNA sequences destined to evolve into genes once an accurate translation system had evolved would have had no obvious selective advantage unless the emergent PCW code was practically identical to that operating in the RCW.
Thus, even were an RCW to have existed, it would be irrelevant to contemporary biology if the PCW had to recapitulate the entire genesis of the code. Nor, of course, does any evidence remain of such ribozymal amino acid activating catalysts, or, indeed of ribozymal polymerases. Finally, if the branching phylogenies of protein aaRS provided opportunity for self-organizing quasispecies bifurcations, and their evident reflexivity greatly accelerated the search for an optimal code, then, an extensive phase of ribozymal protein synthesis no longer fills any theoretical deficiency in accounting for the genetic code. Thus, it is our view that nature did not reinvent its “operating system” (Bowman et al. 2015).
Any coding system must maintain templates to specify either the sequences of ribozymal aaRSs or encode the sequences of protein aaRSs. In an RCW all such templates must somehow survive essentially as parasites, in a world of RNA replicators. A ribozymal coding system consisting only of ribozymal translatase species could be functionally autonomous. However, the attractor state of a hybrid ribozymal/protein aaRS system is one in which the protein population also contributes to the overall rate of translation of any genetic template, and more importantly, to its overall error rate. Either way, the only path to current molecular biology appears to require protein aaRS genes to emerge in concert with other essential encoded protein genes. That requirement highlights the problems arising from coordinating inheritance with gene expression. We therefore turn our attention to the dynamics of template replication and its effect on the evolution of translation.
Mixed ribozymal and enzymatic protein replicases pose an analogous problem. Copying of genetic information lies at the heart of Darwinian evolution. Introducing a protein replicase into an RCW with sophisticated and accurate information copying generates a problem similar to that for the advent of protein translatases. Any protein replicase less accurate than the ribozymal replicase—as expected for the first such proteins to emerge into an RCW—would diminish the probability of correctly copying all genes, including that coding for the ribozymal replicase. Since the system evolution has been optimized under the constraint of the ribozymal replicase’s performance, the system will risk an error catastrophe unless selection purges it of the emergent protein replicase. By these arguments, gene expression and replication by functional protein replicases could not have emerged efficiently from a world in which either function was already performed at a higher level by ribozymes.
D. Efficiency: Minimizing Dissipative Losses
Progressive mutational loss of reflexivity progressively increases the coding error rate (Wills 1994), resulting in the dissipation of free energy flows and ultimately in what have been called “error catastrophes.” Error rates impede self-organization at multiple levels. The bootstrapping requirement (Section III.B) and the instability of hybrid coding assignment systems with substantially different error rates (Section III.C) may reflect inherently complementary arguments for efficient coupling, both thermodynamic and computational, between self-organization of information storage (replication) and readout (translation). We examine here the possible coupling between error generation during replication and translation.
Studies of gene–replicase–translatase (GRT) systems reveal that gene replication and coded expression are interdependent. Living systems now produce proteins from information encoded in genes using protein translatases whose genes are copied using protein polymerases. Could self-organization of both processes be so strongly coupled that they emerged simultaneously? Such coupling is not only possible (Smith et al. 2014) but it occurs spontaneously (Füchslin and McCaskill 2001; Markowitz et al. 2006; Wills et al. 2015). GRT systems are intrinsically spatially self-organizing, and unlike the hypothetical RCW no extrinsic, higher level units of selection—that is compartmentation—are required to assure their survival. The dynamics of the RNA domains of the PCW and RCW (fig. 4A;Wills and Carter 2017) make it evident that gene and protein production in the PCW are tightly coupled through the population variables representing the genes and replicase enzyme. Furthermore, translatases in the protein domain are cooperatively autocatalytic.
Events in the RCW protein domain (fig. 4A), however, have no effect on the value of any RNA domain variable, so replication and catalyzed coding assignment dynamics are completely autonomous in the RNA domain of the RCW. Moreover, the protein domain is utterly dependent on the RNA domain through the variables that represent the populations of encoding genes and the accuracy of the ribozymal translatase population.
Impedance matching argues for coevolution of replication and translation. Errors quite literally (Gladstone 2016) slow the accumulation of information and hence the growth of complexity in many situations. Just as power transfer in dissipative electronic structures is optimal if input and output impedances match, so molecular biological organization observed in life’s informational systems may have evolved most efficiently by matching improvements in the accuracy of information transfer for nucleic acid replication and protein synthesis at successive development stages. Paraphrasing a recent definition of “information impedance matching” of information sources to receivers in a different context (Martin 2005), reading out genetic information with as little dissipation as possible requires readout machinery (translation) with approximately the level of noise present in the information sources (replication). Thus, it appears that natural selection and self-organization provide efficient coupling between replication and translation, as if the two processes were impedance matched.
If errors in either process are either too high or too low, the system will dissipate energy unnecessarily, reducing the readout efficiency. In other words, at any evolutionary stage of developments in molecular biology, the selective effect of the “replicases” and the fidelity of the “translatases” (and any associated accessories) need to limit noise to comparable levels in order to optimize the efficiency of information transfer at that stage.
Viewed another way, overcoming the dual risks of Eigen- and Orgel-like error catastrophes in information storage and readout implicit in highly coupled molecular biological systems seems equally unlikely, until one takes account of the fact that natural selection is a self-organizing force that staves off the potential error catastrophe that threatens information storage (Eigen 1971). Likewise, coding self-organization (Wills 1993) staves off the potential error catastrophe in translation (Orgel 1963). Neither system can be expected to operate unless each limits deleterious effects of the error rate of the other.
We conjecture in figure 5 that progressive increases in the dimension of the codon table, neff, enhance coding evolution efficiency by matching noise in genetic information maintenance (replication errors and quasi-neutral drift in sequence space) to that from the translation error rate, thereby coupling biological information storage and readout as indicated by dashed lines in figure 4A (Wills and Carter 2017). Our heuristic use of impedance-matching—well-established in physics—is supported by the following observations: 1) Error rates appear to be a valid metric for emerging biological complexity over quite large timescales (Lewis et al. 2016). 2) Michaelis Menten parameters for the LeuRS and HisRS2 urzymes (Carter et al. 2014; Carter 2015) suggest that, whereas they are quite impressive catalysts, their specificities for cognate amino acids are well below those necessary to stabilize populations of full-length aaRS, which have much higher fidelities. 3) Structural studies of the TrpRS urzyme show that its high rate acceleration arises from what appears to be a molten globular ensemble (Sapienza et al. 2016). In other words, it is a less complex molecule—in a higher entropy state—than a properly folded protein. 4) The million-fold rate accelerations of both wild type and designed Class I and II protozymes (Martinez et al. 2015) suggest that the manifold of catalytically competent polypeptides is far larger than previously thought possible. 5) Presumptive error rates for the aaRS constructs therefore exhibit a monotonic decline with increasing mass, and by implication, increasing complexity.
Fig. 5.
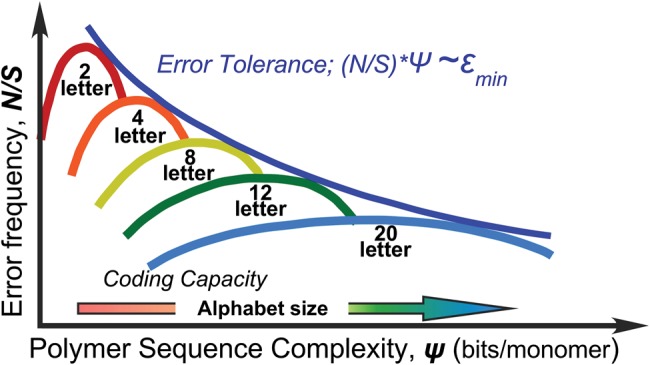
Impedance-matching eases elaboration of coding from a 2-letter amino acid alphabet to a full 20 letter alphabet. Noise, N, in the genetic signal, S, on the y-axis, serves as the primary obstacle opposing information transfer in translation. Increased polymer sequence complexity, Ψ (bits of information transmitted per codon or amino acid incorporated into a protein sequence), on the x-axis, must be accompanied by reduced error rates. The error tolerance curve is a hyperbola in which the product of error frequency by complexity, (N/S)*Ψ, is proportional to the minimum energetic cost of an error, ɛmin, as estimated by Schneider (2010). By analogy to the gears on a bicycle’s derailleur, enlarging the alphabet size increases coding capacity, providing a series of matches with the hyperbolic bounding error tolerance curve (dark blue), easing the path to increased fidelity by enabling stepped increases in coding capacity and polymer sequence complexity.
Direct bootstrapping of genetic information and encoded functional proteins from a PCW is thus far more plausible than any scenario in which there was an initial RNA World by three criteria—reflexive feedback (Section III.B), degraded specificity in hybrid systems (Section III.C), and the need to match the complexity of coding to that of protein function (Section III.D).
IV. Scenarios for Early aaRS Speciation and Coevolution of Replication and Readout
Phylogenetic ancestries of contemporary Class I and II aaRS project convincingly back to a single gene. The simplicity of such a gene and the mapping of amino acid chemistry to tRNA identity elements furnish a conceptually consistent “boot block” (fig. 3) substantially reducing the challenge of understanding how genetic coding might have emerged from a peptide/RNA partnership. Moreover, the detailed inversion symmetries help to explain how such a gene would enforce the initial differentiation necessary to break the powerful forces that make quasispecies centroids strong attractors, substantially strengthening arguments that no genetic code could have preceded the earliest coded protein aaRS. Dual-coding genetic quasispecies exemplified experimentally by the protozyme gene described by Martinez et al. (2015) and the urzyme gene proposed by Pham et al. (2007) are thus presumptive ancestors to both Class I and II aaRS superfamilies and the universal genetic code itself.
A. Why Do Established Protein Phylogenies Suggest Late aaRS Speciation?
Takeover of ribozyme-based computational translation must lead in a plausible way to the observed phylogeny of contemporary aaRS superfamilies. We believe the conclusion that aaRSs developed after the advent of fully functional proteins based on a 12–20 amino acid alphabet (Aravind et al. 2002; Leipe et al. 2002; Koonin and Novozhilov 2009; Koonin 2011) rests on two questionable phylogenic assumptions: 1) that domains (∼250 amino acids) are the basic unit of remote protein ancestry and 2) that Class I and II aaRS arose independently. The former assumption fails to account appropriately for the highly mosaic nature of contemporary proteins (Pham et al. 2010; Li et al. 2011). The latter ignores the bidirectional coding ancestry of Class I and II aaRS urzymes and protozymes, for which experimental evidence is now exceptionally strong (Pham et al. 2010; Li et al. 2011, 2013; Carter 2014, 2015, 2016, 2017; Carter et al. 2014; Martinez et al. 2015).
The low fidelity of aaRS urzymes implies that they represent an important, but early stage in the evolution of complexity and hence that deep phylogenies based on aligning intact contemporary aaRS sequences (Aravind et al. 1998; Wolf et al. 1999; Leipe et al. 2002; Wolf and Koonin 2007) are probably misleading, especially in the case of the pre-LUCA heritage of the aaRSs themselves (Wolf et al. 1999; Wolf and Koonin 2007). Notably, neither domain database (SCOP [Murzin et al. 1995; Andreeva et al. 2008]; CATH [Pearl et al. 2003]) has been compiled at sufficiently high resolution to identify the Class I and II urzymes as ancestral forms. Large insertions within aaRS catalytic domains likely accumulated segmentally, from exogenous genetic modules with their own previous ancestry (Pham et al. 2007), subsequent to their initial evolutionary speciation. Mosaicity in the multiple sequence alignments, akin to horizontal gene transfer (Leipe et al. 2004; Soucy et al. 2015) albeit in shorter segments than those considered by Wolf et al. (1999), could obscure deeper ancestral evolution of the urzymes.
The alternative phylogeny of Class I aaRS in figure 6 traces ancestries from a single gene by two distinct processes—speciation of the bidirectional gene (I) and strand specialization to transcend its limitations (II). It accounts for the increase in structural multiplexing and independent parallel evolution of insertion elements and anticodon-binding domains during a period in which protein synthesis operated with a gradually increasing alphabet size that ultimately required editing domains (III) to achieve the requisite fidelity of the contemporary proteome in the era relevant to previous phylogenies.
Fig. 6.

Alternative evolution of Class I aaRS catalytic domains. This scenario redefines the Class I CP1 insertion between the N- and C-terminal modules, Ncore (blue) and Ccore (red), of the Class I Urzymes, both of which are portrayed as ancestral to all Rossmannoid superfamilies (adapted from fig. 4 of Aravind et al. [2002]). The initial CP1 insertion (white) is the origin of most subsequent elaborations of the Class I catalytic domains that appear to have provided the requisite increases in specific amino acid recognition (Carter 2015). Idiosyncratic Class I anticodon-binding domains are not considered here. We distinguish three phases of aaRS evolution: (I) bidirectionally coded with Class II; limited diversity; (II) CP1 enforces strand specialization; and (III) hydrolytic editing enhances specificity.
B. A Plausible Scenario for Coevolution of Inheritance and Gene Expression
To highlight how conclusions from Section III change how we think translation might have emerged, we outline a plausible scenario for the coemergence of information storage and readout. High noise initially permits co-option of unrefined functionalities grouping related effects averaged over large but separate regions of sequence space, and is gradually brought under control by refining distinguishable specificities and selecting their genes. Structural diversity and complexity can then develop simultaneously with increases in the dimension of the codon table (fig. 5), consistent with impedance matching. Although aspects of this scenario resemble previously outlined marginal scenarios (Martin 2005), its scope, continuity, and its logical, experimental, and phylogenetic support are assembled here for the first time.
The origin of contemporary translation was most likely an intimate coevolution of both polymer classes (Carter and Kraut 1974; Carter 1975). Arguments developed in Section III.D imply that replication and translation are necessarily more tightly coupled by the need for informational impedance-matching than is envisioned in the RNA World hypothesis. The overriding challenge associated with the emergence of the genetic code is to develop a scenario in which prebiotic chemistry produces biology reflexively, through cooperation between nucleic acids and proteins (or their precursors), in improving both inheritance and function from a bidirectional coding ancestry.
The following arguments must remain hypotheses until experimental investigation, perhaps guided by ideas expressed here, convincingly establish or rule them out. Our recent use of protein design and modular engineering in the experimental colonization of the void that previously existed between prebiotic organic chemistry (Patel et al. 2015; Sutherland 2016) and the Last Universal Common Ancestor (Forterre et al. 2005; Wong 2005; Xue et al. 2005; Fournier et al. 2011; Fournier and Alm 2015; Wong et al. 2016) argues that such experimentation can now be fruitful on a larger scale.
Structural complementarities were identified between extended polypeptide secondary structures and nucleic acids before the discovery of catalytic RNA (Carter and Kraut 1974; Carter 1975; Church et al. 1977; Warrant and Kim 1978). Stability as complexes appeared to depend largely on their complementary van der Waals surfaces, arising from opposite chiralities of amino acids and ribose in biological polymers. The short polymers required—six to eight amino acids and less than half a turn of RNA double helix—suggested they might have been more stable if their polypeptide and polynucleotide components formed hairpins (Berezovsky et al. 2000).
Stereochemically templated cross-catalysis plausibly accounted for the simultaneous appearance of bidirectional coding and catalysis. Helix radii of RNA and double-stranded extended peptides formed optimal van der Waals contacts between the two components at precisely the integral, indefinitely repeating stoichiometry of two amino acids per base (Carter and Kraut 1974; Carter 1975). Integral stoichiometry enabled a putative rudimentary stereochemical coding. Moreover, specific hydrogen bonding between peptide carboxyl groups and RNA 2′ OH groups oriented the 3′ OH group as a likely nucleophile, consistent with the observed 5′–3′ linkages in biological nucleic acids. These coincidences also suggested possible templated cross catalysis, each polymer accelerating the elongation of the other.
Successive recombinational inverted repeats of complementary polypeptide•polynucleotide complexes increased their lengths from ∼12 to ∼23 to ∼46 amino acids and from ∼3 to ∼6 to ∼12 base pairs. Partial complementarity of the 5′ and 3′-terminal halves of the Class I protozyme gene (Carter 2015) suggests coding by an ancestral RNA hairpin. Peptides of at least 46 amino acids produced by stereochemical coding based on complementary van der Waals surfaces plausibly then began to exhibit ATP dependent carboxyl group activation (Martinez et al. 2015), potentiating peptide synthesis. Polypeptide catalytic activities may thus have preceded indirect, symbolic coding (Kamtekar et al. 1993; Moffet et al. 2003; Patel et al. 2009). Ligation might then have assembled protogenes and a proto-ribosome.
Bidirectional coding and the wobble effect (Crick 1966) would have required a triplet code, enabling more than 4 codons. We encounter here a substantive broken symmetry. The protozyme gene (138 bases) is 6-fold longer than whatever putative RNA hairpin might have been associated with the earliest ∼46-residue peptides arising via stereochemical coding. Assuming that such a system could have sustained reproduction nevertheless leaves us with a 6-fold gap between the relative stoichiometries of templated cross catalysis and the first true gene expression. Transitions from an initial state in which protein synthesis is initiated without information-bearing genetic templates is envisioned in the theory of coding self-organization (Bedian 1982) by GRT systems (Eigen et al. 1988; Wills 1993; Füchslin and McCaskill 2001).
Symbolic coding emulated the direct stereochemical coding, preserving complementary van der Waals surfaces of peptide and RNA backbones. What continuity might have connected direct, stereochemical coding to indirect, symbolic coding by introducing messenger RNA and the use of adaptors to give the messages meaning? The tRNA acceptor stem “operational RNA code” (Schimmel et al. 1993; Schimmel 1996)—β-branched side chains favoring extended β-structure and alternating small/large side chains (Carter and Wolfenden 2015, 2016)—is necessary and sufficient to encode peptides allowing van der Waals access on one face to assume structures complementary to the RNA minor groove (Carter and Kraut 1974; Carter 1975). That symbolic coding could therefore have reimplemented precisely those features necessary to preserve molecular mechanisms that sustained the earlier, direct stereochemical coding, smoothing the transition between different stoichiometries.
The earliest genetic coding substantially enhanced nature’s ability to engineer nanoscale chemistry. Wills’s (2016) description of the substantial 106- to 109-fold intrinsic advantage that proteins have over ribozymes (supplementary fig. S1, Supplementary Material online) depends on the expanded amino acid alphabet. How much better catalysts could peptides have been as catalysts specified by a four-letter alphabet comparable to that of ribozymes? Realization that tertiary structures in water result from a two-dimensional basis set of phase transfer free energies (supplementary fig. S2, Supplementary Material online; Carter and Wolfenden 2015, 2016; Wolfenden et al. 2015) suggests that the average alphabet consisting of two amino acids each from Classes I and II would exhibit an ∼50-fold enhancement in nanoscale chemical engineering over ribozymes. The supplement discusses this point more fully.
The ancestral bidirectional gene produced two amino acid activating enzymes, Class I with a modest specificity for larger amino acids, Class II with a similar specificity for smaller amino acids, in keeping with the contemporary specificities of Class I and II aaRS and urzymes. An obvious question is: how limited an amino acid alphabet is consistent with catalytic activity of such protozyme genes? Extant experimental results, however, show only that by utilizing the full genetic code the two gene products created from opposite strands can both accelerate amino acid activation ∼106-fold. The Class I protozyme possesses a consensus phosphate binding site (Hol et al. 1978), suggesting that its catalytic activity may arise from backbone configurations, and not depend entirely on “catalytic residues.”
The earliest catalysts of aminoacylation may have combined ancestral aaRS with ribozymes (Turk et al. 2010, 2011). It is unknown whether aminoacylation by aaRS protozymes required assistance from such ribozymes. tRNA acceptor stem ID elements likely composed the earliest connection between aminoacylated RNAs and a gene sequence (Schimmel et al. 1993; Rodin et al. 1996, 2009, 2011; Henderson and Schimmel 1997; Rodin and Rodin 2008). Dependence of aaRS tRNA affinity on acquiring an additional, anticodon-binding domain suggests that, in contrast to amino acid activation, aminoacylation may have originated in polypeptide•RNA collaboration and was later taken over by urzymes with the rudimentary capability to recognize tRNA acceptor stems (Li et al. 2013).
Emerging control of intermediary metabolism may have exploited catalytic phosphoryl-transfer mechanisms from amino acid activation. Recent studies of prebiotic reaction networks (Powner et al. 2009, 2010; Powner and Sutherland 2010; Patel et al. 2015; Sutherland 2016) reveal a previously unsuspected coherence and interdependence in generating precursors for building nucleic acids, polypeptides, and lipids. That “protometabolism” resembles much of intermediary metabolism in biology and simulations of Sousa et al. (2015) and Gánti (2003). A crucial difference between the chemistry of prebiotic and intermediary metabolism is that the primary energy source of the former is light, whereas nucleotide triphosphate hydrolysis drives biological chemistry.
Most biological ribozymes catalyze phosphoryl transfer, either hydrolyzing or ligating other RNAs, suggesting possible previous roles in intermediary metabolism. However, aaRS protozymes catalyze biosynthetic use of ATP (Martinez et al. 2015) and the Class I protozyme is fundamentally a P-loop module whose secondary structure occurs in >120 protein superfamilies (Cammer and Carter 2010), including many enzymes from intermediary metabolism, suggesting much closer relationships. Indeed, Smith (1995) has noted a persistent theme in the construction of the enzymes of nucleotide metabolism, with emphasis on phosphoribosyltransferases: they exploit “… construction of larger structures from modules with a catalytic function.” It might be fruitful to correlate reaction rates in Sutherland’s prebiotic network with the phylogenies of such enzymes.
Discussion
The continuing search for ever better RNA replicases (Wochner et al. 2011; Attwater et al. 2013; Sczepanski and Joyce 2014; Taylor et al. 2015; Horning and Joyce 2016) has achieved notable success. However, we argue here for a more holistic and ambitious set of goals than those fueling that search, anticipating that data (Section II) and theory (Section III and Wills and Carter 2017) will stimulate discussion and further research on questions relevant to the origins of biology’s genetic readout mechanism. A high degree of coherence connects theories of self-organization to the experimental, structural, and phylogenetic aspects of the evolution of the aaRS enzymes that implement gene expression today.
Pronounced inversion symmetries in the amino acid substrates, catalytic residues, tertiary, and secondary structures are evident in phylogenetic, structural, and biochemical data for contemporary Class I and II aaRS and arise from their bidirectional coding ancestry, which maximizes functional use of sequence space.
Inversion symmetries assure maximal structural and functional differentiation between the two classes, a necessary precondition for their survival in competition with parasitic molecular forms.
tRNA identity elements that implement coding efficiently capture the amino acid phase equilibria that drive protein folding and are optimal for bidirectional coding (Zull and Smith 1990).
Bidirectional coding combines with nano-environment sensing to create a reflexive feed-back cycle to guide rapid evolutionary emergence of protein aaRS genes by bootstrapping rapidly to an optimal coding table and mRNA sequences. Ribozymal assignment catalysts lack such reflexivity.
Hybrid system expression dynamics show that any emerging PCW with any coding table of dimension lower than that of a preexisting RCW would necessarily have been eliminated by purifying selection before it had sufficient time to expand the dimension of its coding table.
Coupling of dynamic equations for GRT systems suggest that matching of error rates maximized the probability of launching replication and translation.
(1)–(6) imply that replication and readout emerged simultaneously from a peptide•RNA partnership. We outline a more probable scenario than an RNA world for the origin of biology.
Molecular constructs (Section I) enhance the ability to test specific elements of proposed scenarios.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by The National Institute of General Medical Sciences (grant numbers R01-78227 and R01-90406 to C.W.C. Jr). This publication was made possible also through the support of a grant from the John Templeton Foundation. The opinions expressed in this publication are those of the author(s) and do not necessarily reflect the views of the John Templeton Foundation. H. Fried (Cursor Scientific Editing and Writing, LLC) and anonymous referees made many useful suggestions on earlier drafts.
References
- Andreeva A, Howorth D, Chandonia J-M, Brenner SE, Hubbard TJP, Chothia C, Murzin AG.. 2008. Data growth and its impact on the SCOP database: new developments. Nucl Acids Res. 36(Database issue): D419–D425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Anantharaman V, Koonin EV.. 2002. Monophyly of class I aminoacyl tRNASynthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide-binding domains: implication for protein evolution in the RNAWorld. PROTEINS: Struct Funct Genet. 481: 1–14. [DOI] [PubMed] [Google Scholar]
- Aravind L, Leipe DD, Koonin EV.. 1998. Toprim—a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res. 2618: 4205–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwater J, Wochner A, Holliger P.. 2013. In-ice evolution of RNA polymerase ribozyme activity. Nat Chem. 512: 1101–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedian V. 1982. The possible role of assignment catalysts in the origin of the genetic code. Orig Life 122: 181–204. [DOI] [PubMed] [Google Scholar]
- Bedian V. 2001. Self-description and the origin of the genetic code. Biosystems 60(1–3): 39–47. [DOI] [PubMed] [Google Scholar]
- Berezovsky IN, Grosberg AY, Trifonov EN.. 2000. Closed loops of nearly standard size: common basic element of protein structure. FEBS Lett. 466(2–3): 283–286. [DOI] [PubMed] [Google Scholar]
- Berg P, Offengand EJ.. 1958. An enzymatic mechanism for linking amino acids to RNA. Proc Nat Acad Sci U S A. 442: 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt HS. 2012. The RNA world hypothesis: the worst theory of the early evolution of life (except for all the others). Biol Direct. 7:23.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman JC, Hud NV, Williams LD.. 2015. The ribosome challenge to the RNA world. J Mol Evol. 80(3–4): 143–161. [DOI] [PubMed] [Google Scholar]
- Breaker RR. 2012. Riboswitches and the RNAWorld. Cold Spring Harb Perspect Biol. 4:a003566.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano-Anollés D, Caetano-Anollés G.. 2016. Piecemeal buildup of the genetic code, ribosomes, and genomes from primordial tRNA building blocks. Life 64: 43.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano-Anolles G, Kim HS, Mittenthal JE.. 2007. The origin of modern metabolic networks inferred from phylogenomic analysis of protein architecture. Proc Natl Acad Sci U S A. 10422: 9358–9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano-Anollés G, Wang M, Caetano-Anollés D.. 2013. Structural phylogenomics retrodicts the origin of the genetic code and uncovers the evolutionary impact of protein flexibility. PLoS ONE 88: e72225.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammer S, Carter CW Jr.. 2010. Six rossmannoid folds, including the Class I aminoacyl-tRNA synthetases, share a partial core with the anticodon-binding domain of a Class II aminoacyl-tRNA synthetase. Bioinformatics 266: 709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CW., Jr. 2015. What RNA world? Why a peptide/RNA partnership merits renewed experimental attention. Life 51: 294–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CW Jr, Kraut J.. 1974. A proposed model for interaction of polypeptides with RNA. Proc Natl Acad Sci U S A. 712: 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CW., Jr. 2016. An alternative to the RNA world. Nat Hist. 1251: 28–33. [PMC free article] [PubMed] [Google Scholar]
- Carter CW. 2017. Coding of Class I and II Aminoacyl-tRNA Synthetases. In: Advances in Experimental Medicine and Biology. Boston, MA: Springer, DOI https://doi.org/10.1007/5584_2017_93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CW., Jr. 1993. Cognition mechanism and evolutionary relationships in aminoacyl-tRNA synthetases. Annu Rev Biochem. 621: 715–748. [DOI] [PubMed] [Google Scholar]
- Carter CW., Jr. 1975. Cradles for molecular evolution. New Sci. 784–787. [Google Scholar]
- Carter CW., Jr. 2014. Urzymology: experimental access to a key transition in the appearance of enzymes. J Biol Chem. 28944: 30213–30220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CW, Li L, Weinreb V, Collier M, Gonzalez-Rivera K, Jimenez-Rodriguez M, Erdogan O, Kuhlman B, Ambroggio X, Williams T, et al. 2014. The Rodin-Ohno hypothesis that two enzyme superfamilies descended from one ancestral gene: an unlikely scenario for the origins of translation that will not be dismissed. Biol Direct. 91: 11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CW Jr, Wolfenden R.. 2016. Acceptor-stem and anticodon bases embed amino acid chemistry into tRNA. RNA Biol. 132: 145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CW Jr, Wolfenden R.. 2015. tRNA acceptor-stem and anticodon bases form independent codes related to protein folding. Proc Natl Acad Sci U S A. 11224: 7489–7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cech TR. 1986. The intervening sequence RNA of tetrahymena is an enzyme. Sci Am. 2555: 64–75.2430331 [Google Scholar]
- Chandrasekaran SN, Yardimci G, Erdogan O, Roach JM, Carter CW Jr.. 2013. Statistical evaluation of the Rodin-Ohno hypothesis: sense/antisense coding of ancestral Class I and II aminoacyl-tRNA synthetases. Mol Biol Evol. 307: 1588–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church GM, Sussman JL, Kim SH.. 1977. Secondary structural complementarity between DNA and proteins. Proc Natl Acad Sci U S A. 744: 1458–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick FHC. 1966. Codon-anticodon pairing: the wobble hypothesis. J Mol Biol. 192: 548–555. [DOI] [PubMed] [Google Scholar]
- Crick FHC. 1955. On degenerate templates and the adaptor hypothesis. Unpublished; https://profiles.nlm.nih.gov/ps/retrieve/Narrative/SC/p-nid/153, last accessed October 15, 2017.
- Crick FHC. 1968. The origin of the genetic code. J Mol Biol. 383: 367–379. [DOI] [PubMed] [Google Scholar]
- Cullis PM, Wolfenden R.. 1981. Affinities of nucleic acid bases for solvent water? Biochemistry 2011: 3024–3028. [DOI] [PubMed] [Google Scholar]
- Cusack S. 1994. Evolutionary implications. Nat Struct Mol Biol. 111: 760. [Google Scholar]
- Cusack S, Berthet-Colominas C, Härtlein M, Nassar N, Leberman R.. 1990. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 Å. Nature 3476290: 249–255. [DOI] [PubMed] [Google Scholar]
- Delarue M. 2007. An asymmetric underlying rule in the assignment of codons: possible clue to a quick early evolution of the genetic code via successive binary choices. RNA 13:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giulio M. 1992. On the origin of the transfer RNA molecule. J Theor Biol. 1592: 199–214. [DOI] [PubMed] [Google Scholar]
- Di Giulio M. 2004. The origin of the tRNA molecule: implications for the origin of protein synthesis. J Theor Biol. 2261: 89–93. [DOI] [PubMed] [Google Scholar]
- Di Giulio M. 2008. Transfer RNA genes in pieces are an ancestral character. EMBO Rep. 99: 820.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill K, Chan HS.. 1997. From Levinthal to pathways to funnels. Nat Struct Biol. 41: 10–19. [DOI] [PubMed] [Google Scholar]
- Eigen M. 1971. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 5810: 465–523. [DOI] [PubMed] [Google Scholar]
- Eigen M, McCaskill JS, Schuster P.. 1988. Molecular quasi-species. J Phys Chem. 9224: 6881–6891. [Google Scholar]
- Eigen M, Schuster P.. 1977. The hypercyde: a principle of natural self-organization Part A: emergence of the hypercycle. Naturwissenschaften 6411: 541–565. [DOI] [PubMed] [Google Scholar]
- Eriani G, Delarue M, Poch O, Gangloff J, Moras D.. 1990. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 3476289: 203–206. [DOI] [PubMed] [Google Scholar]
- Fedor MJ, Williamson JR.. 2005. The catalytic diversity of RNAs. Nat Rev Mol Cell Biol. 65: 399–412. [DOI] [PubMed] [Google Scholar]
- Forterre P, Gribaldo S, Brochier C.. 2005. Luca: à la recherche du plus proche ancêtre commun universel. Med Sci. 21:860–865. [DOI] [PubMed] [Google Scholar]
- Fournier GP, Alm EJ.. 2015. Ancestral reconstruction of a pre-LUCA aminoacyl-tRNA synthetase ancestor supports the late addition of trp to the genetic code. J Mol Evol. 80(3–4): 171–185. [DOI] [PubMed] [Google Scholar]
- Fournier GP, Andam CP, Alm EJ, Gogarten JP.. 2011. Molecular evolution of aminoacyl tRNA synthetase proteins in the early history of life. Orig Life Evol Biosph. 416: 621–632. [DOI] [PubMed] [Google Scholar]
- Freeland SJ, Hurst LD.. 1998. The genetic code is one in a million. J Mol Evol. 473: 238–248. [DOI] [PubMed] [Google Scholar]
- Füchslin RM, McCaskill JS.. 2001. Evolutionary self-organization of cell-free genetic coding. Proc Natl Acad Sci U S A. 9816: 9185–9190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gánti T. 2003. The principles of life. Oxford: Oxford University Press. [Google Scholar]
- Gilbert W. 1986. The RNA world. Nature 3196055: 618. [Google Scholar]
- Gladstone E. 2016. Error in information diffusion processes. Ithaca (NY: ): Cornell University. [Google Scholar]
- Grigg JC, Chen Y, Grundy FJ, Henkin TM, Pollack L, Ke A.. 2013. T box RNA decodes both the information content and geometry of tRNA to affect gene expression. Proc Natl Acad Sci U S A. 11018: 7240–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy FJ, Winkler WC, Henkin TM.. 2002. tRNA-mediated transcription antitermination in vitro: codon-anticodon pairing independent of the ribosome. Proc Natl Acad Sci U S A. 9917: 11121–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier-Takada C, Lumelsky N, Altman S.. 1989. Specific interactions in RNA enzymes-substrate complexes. Science 2464937: 1578–1584. [DOI] [PubMed] [Google Scholar]
- Henderson BS, Schimmel P.. 1997. RNA-RNA interactions between oligonucleotide substrates for aminoacylation. Bioorg Med Chem. 56: 1071–1079. [DOI] [PubMed] [Google Scholar]
- Hofstadter DR. 1979. Gödel, Escher, Bach: an eternal golden braid. New York: Basic Books, Inc. [Google Scholar]
- Hol WG, van Duijnen PT, Berendsen HJ.. 1978. The α-helix dipole and the properties of proteins. Nature 2735662: 443–446. [DOI] [PubMed] [Google Scholar]
- Hordijk W, Steel M, Kauffman S.. 2012. The structure of autocatalytic sets: evolvability, enablement, and emergence. Acta Biotheor. 604: 379–392. [DOI] [PubMed] [Google Scholar]
- Hordijk W, Wills PR, Steel M.. 2014. Autocatalytic sets and biological specificity. Bull Math Biol. 761: 201–224. [DOI] [PubMed] [Google Scholar]
- Horning DP, Joyce GF.. 2016. Amplification of RNA by an RNA polymerase ribozyme. Proc Natl Acad Sci U S A. 11335: 9786–9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illangasekare M, Yarus M.. 1999. A tiny RNA that catalyzes both aminoacyl-tRNA and peptidyl-RNA. RNA 511: 1482–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamtekar S, Schiffer JM, Xiong H, Babik JM, Hecht MH.. 1993. Protein design by binary patterning of polar and non-polar amino acids. Science 2625140: 1680–1685. [DOI] [PubMed] [Google Scholar]
- Koonin EV. 2011. The logic of chance: the nature and origin of biological evolution. Upper Saddle River (NJ: ): Pearson Education; FT Press Science. [Google Scholar]
- Koonin EV. 2015. Why the Central Dogma: on the nature of the great biological exclusion principl. Biol Direct 10:52.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Novozhilov AS.. 2009. Origin and evolution of the genetic code: the universal enigma. IUBMB Life 612: 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A, Jacak R, Stranges PB, Kuhlman B.. 2011. A generic program for multistate protein design. PLoS ONE 67: e20937.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipe DD, Koonin EV, Aravind L.. 2004. STAND, a class of P-Loop NTPases including animal and plant regulators of programmed cell death: multiple, complex domain architectures, unusual phyletic patterns, and evolution by horizontal gene transfer. J Mol Biol. 3431: 1–28. [DOI] [PubMed] [Google Scholar]
- Leipe DD, Wolf YI, Koonin EV, Aravind L.. 2002. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol. 3171: 41–72. [DOI] [PubMed] [Google Scholar]
- Lewis CA Jr, Crayle J, Zhou S, Swanstrom R, Wolfenden R.. 2016. Cytosine deamination and the precipitous decline of spontaneous mutation during Earth’s history. Proc Natl Acad Sci U S A. 11329: 8194–8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Francklyn C, Carter CW Jr.. 2013. Aminoacylating urzymes challenge the RNA world hypothesis. J Biol Chem. 28837: 26856–26863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Weinreb V, Francklyn C, Carter CW Jr.. 2011. Histidyl-tRNA synthetase urzymes: Class I and II aminoacyl-tRNA synthetase urzymes have comparable catalytic activities for cognate amino acid activation. J Biol Chem. 28612: 10387–10395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz S, Drummond A, Nieselt K, Wills PR.. 2006. Simulation model of prebiotic evolution of genetic coding In: Rocha LM, Yaeger LS, Bedau MA, Floreano D, Goldstone RL, Vespignani A, editors. Artificial life. Cambridge (MA: ): MIT Press; p. 152–157. [Google Scholar]
- Martin P. 2005. Spatial interpolation in other dimensions. Corvallis, OR: Oregon State University. [Google Scholar]
- Martinez-Rodriguez L, Erdogan O, Jimenez-Rodriguez M, Gonzalez-Rivera K, Williams T, Li L, Weinreb V, Collier M, Chandrasekaran SN, Ambroggio X, et al. 2015. Functional Class I and II amino acid activating enzymes can be coded by opposite strands of the same gene. J Biol Chem. 29032: 19710–19725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffet DA, Foley J, Hecht MH.. 2003. Midpoint reduction potentials and heme binding stoichiometries of de novo proteins from designed combinatorial libraries. Biophys Chem. 105(2–3): 231–239. [DOI] [PubMed] [Google Scholar]
- Muñoz V, Serrano L.. 1994. Intrinsic secondary structure propensities of the amino acids, using statistical f-y matrices: comparison with experimental scales. PROTEINS: Struct Funct Genet. 204: 301–311. [DOI] [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard TJP, Chothia C.. 1995. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 2474: 536–540. [DOI] [PubMed] [Google Scholar]
- Niwa N, Yamagishi Y, Murakami H, Suga H.. 2009. A flexizyme that selectively charges amino acids activated by a water-friendly leaving group. Bioorg Med Chem Lett. 1914: 3892–3894. [DOI] [PubMed] [Google Scholar]
- Noller H. 2004. The driving force for molecular evolution of translation. RNA 1012: 1833–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noller HF, Hoffarth V, Zimniak L.. 1992. Unusual resistance of peptidyl transferase to protein extraction procedures. Science 2565062: 1416–1419. [DOI] [PubMed] [Google Scholar]
- O’Donoghue P, Luthey-Schulten Z.. 2003. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol Mol Biol Rev. 67:550–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel LE. 1968. Evolution of the genetic apparatus. J Mol Biol. 383: 381–393. [DOI] [PubMed] [Google Scholar]
- Orgel LE. 1963. The maintenance of the accuracy of protein synthesis and its relevance to ageing. Proc Natl Acad Sci USA. 49:517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel BH, Percivalle C, Ritson DJ, Duffy CD, Sutherland JD.. 2015. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nat Chem. 7:301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SC, Bradley LH, Jinadasa SP, Hecht MH.. 2009. Cofactor binding and enzymatic activity in an unevolved superfamily of de novo designed 4-helix bundle proteins. Prot Sci. 187: 1388–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl FM, Bennett CF, Bray JE, Harrison AP, Martin N, Shepherd A, Sillitoe I, Thornton J, Orengo CA.. 2003. The CATH database: an extended protein family resource for structural and functional genomics. Nucleic Acids Res. 311: 452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AS, Bernier CR, Hsiao C, Norris AM, Kovacs NA, Waterbury CC, Stepanov VG, Harvey SC, Fox GE, Wartell RM, et al. 2014. Evolution of the ribosome at atomic resolution. Proc Natl Acad Sci U S A. 11128: 10251–10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AS, Williams LD.. 2015. The ancient heart of the ribosomal large subunit: a response to caetano-anolles. J Mol Evol. 80(3–4): 166–170. [DOI] [PubMed] [Google Scholar]
- Pham Y, Kuhlman B, Butterfoss GL, Hu H, Weinreb V, Carter CW Jr.. 2010. Tryptophanyl-tRNA synthetase Urzyme: a model to recapitulate molecular evolution and investigate intramolecular complementation. J Biol Chem. 28549: 38590–38601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham Y, Li L, Kim A, Erdogan O, Weinreb V, Butterfoss G, Kuhlman B, Carter CW Jr.. 2007. A minimal TrpRS catalytic domain supports sense/antisense ancestry of Class I and II aminoacyl-tRNA synthetases. Mol Cell. 256: 851–862. [DOI] [PubMed] [Google Scholar]
- Powner MW, Gerland B, Sutherland JD.. 2009. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 4597244: 239–242. [DOI] [PubMed] [Google Scholar]
- Powner MW, Sutherland JD.. 2010. Phosphate-mediated interconversion of ribo- and arabino-configured prebiotic nucleotide intermediates. Angew Chem Int Ed. 4927: 4641–4643. [DOI] [PubMed] [Google Scholar]
- Powner MW, Sutherland JD, Szostak JW.. 2010. Chemoselective multicomponent one-pot assembly of purine precursors in water. J Am Chem Soc. 13246: 16677–16688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzicka A, Wolfenden R.. 1988. Comparing the polarities of the amino acids: side-chain distribution coefficients between the vapor phase, cyclohexane, 1-ctanol, and neutral aqueous solution. Biochemistry 275: 1664–1670. [Google Scholar]
- Robertson MP, Joyce GF.. 2012. The origins of the RNA world. Cold Spring Harb Perspect Biol. 45: a003608.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodin A, Rodin SN, Carter CW Jr.. 2009. On primordial sense-antisense coding. J Mol Evol. 695: 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodin AS, Szathmáry E, Rodin SN.. 2011. On origin of genetic code and tRNA before translation. Biol Direct 6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodin SN, Ohno S.. 1995. Two types of aminoacyl-tRNA synthetases could be originally encoded by complementary strands of the same nucleic acid. Orig Life Evol Biosph. 256: 565–589. [DOI] [PubMed] [Google Scholar]
- Rodin SN, Rodin A.. 2006a. Origin of the genetic code: first aminoacyl-tRNA synthetases could replace isofunctional ribozymes when only the second base of codons was established. DNA Cell Biol 25:365–375. [DOI] [PubMed] [Google Scholar]
- Rodin SN, Rodin A.. 2006b. Partitioning of aminoacyl-tRNA synthetases in two classes could have been encoded in a strand-symmetric RNA world. DNA Cell Biol. 2511: 617–626. [DOI] [PubMed] [Google Scholar]
- Rodin SN, Rodin A, Ohno S.. 1996. The presence of codon-anticodon pairs in the acceptor stem of tRNAs. Proc Natl Acad Sci U S A. 9310: 4537–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodin SN, Rodin AS.. 2008. On the origin of the genetic code: signatures of its primordial complementarity in tRNAs and aminoacyl-tRNA synthetases. Heredity 1004: 341–355. [DOI] [PubMed] [Google Scholar]
- Ruff M, Krishnaswamy S, Boeglin M, Poterszman A, Mitschler A, Podjarny A, Rees B, Thierry JC, Moras D.. 1991. Class II aminoacyl transfer RNA synthetases: crystal structure of yeast aspartyl-tRNA synthetase complexed with tRNAAsp. Science 2525013: 1682–1689. [DOI] [PubMed] [Google Scholar]
- Sapienza PJ, Li L, Williams T, Lee AL, Carter CW Jr.. 2016. An ancestral tryptophanyl-tRNA synthetase precursor achieves high catalytic rate enhancement without ordered ground-state tertiary structures. ACS Chem Biol. 116: 1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassanfar M, Szostak JW.. 1993. An RNA motif that binds ATP. Nature 3646437: 550–553. [DOI] [PubMed] [Google Scholar]
- Schimmel P. 1996. Origin of genetic code: a needle in the haystack of tRNA sequences. Proc Natl Acad Sci U S A. 9310: 4521–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmel P, Giegé R, Moras D, Yokoyama S.. 1993. An operational RNA code for amino acids and possible relationship to genetic code. Proc Natl Acad Sci U S A. 9019: 8763–8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD. 2010. A brief review of molecular information theory. Nano Commun Netw. 13: 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder GK, Wolfenden R.. 2007. The rate enhancement produced by the ribosome: an improved model. Biochemistry 4613: 4037–4044. [DOI] [PubMed] [Google Scholar]
- Sczepanski JT, Joyce GF.. 2014. A cross-chiral RNA polymerase ribozyme. Nature 5157527: 440–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers A, Beringer M, Rodnina MV, Wolfenden R.. 2004. The ribosome as an entropy trap. Proc Natl Acad Sci U S A. 10121: 7897–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JI, Steel M, Hordijk W.. 2014. Autocatalytic sets in a partitioned biochemical network. J Syst Chem. 5:2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JL. 1995. Enzymes of nucleotide synthesis. Curr Opin Struct Biol. 56: 752–757. [DOI] [PubMed] [Google Scholar]
- Soucy SM, Huang J, Gogarten JP.. 2015. Horizontal gene transfer: building the web of life. Nat Rev Genet. 168: 472.. [DOI] [PubMed] [Google Scholar]
- Sousa FL, Hordijk W, Steel M, Martin WF.. 2015. Autocatalytic sets in E. coli metabolism. J Syst Chem. 61: 4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland John D. 2016. ‘The Origin of Life—Out of the Blue’, Angew. Chem. Int. Ed., 55: 104–121. [DOI] [PubMed] [Google Scholar]
- Taylor AI, Pinheiro VB, Smola MJ, Morgunov AS, Peak-Chew S, Cozens C, Weeks KM, Herdewijn P, Holliger P.. 2015. Catalysts from synthetic genetic polymers. Nature 5187539: 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuerk C, Gold L.. 1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T7 DNA polymerase. Science 2494968: 505–510. [DOI] [PubMed] [Google Scholar]
- Turk RM, Chumachenko NV, Yarus M.. 2010. Multiple translational products from a five-nucleotide ribozyme. Proc Natl Acad Sci USA. 10710: 4585–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk RM, Illangasekare M, Yarus M.. 2011. Catalyzed and spontaneous reactions on ribozyme ribose. J. Am. Chem. Soc. 13315: 6044–6050. [DOI] [PubMed] [Google Scholar]
- Van Noorden R. 2009. RNA world easier to make. Nature 459, published online. [Google Scholar]
- Warrant RW, Kim S-H.. 1978. a-Helix-double helix interaction shown in the structure of a protamine-transfer RNA complex and a nucleoprotamine model. Nature 2715641: 130–135. [DOI] [PubMed] [Google Scholar]
- Watson JD, Crick FHC.. 1953. A structure for deoxyribose nucleic acid. Nature 1714356: 737–738. [DOI] [PubMed] [Google Scholar]
- Welch M, Chastang J, Yarus M.. 1995. An inhibitor of ribosomal peptidyl transferase using transition-state analogy. Biochemistry 342: 385–390. [DOI] [PubMed] [Google Scholar]
- Wills PR. 1993. Self-organization of genetic coding. J Theor Biol. 1623: 267–287. [DOI] [PubMed] [Google Scholar]
- Wills PR. 1994. Does information acquire meaning naturally? Ber Bunsengesellschaft Phys Chem. 989: 1129–1134. [Google Scholar]
- Wills PR. 2004. Stepwise evolution of molecular biological coding In: Pollack J, Bedau M, Husbands P, Ikegami T, Watson RA, editors. Artificial life IX. Cambridge: MIT Press; p. 51–56. [Google Scholar]
- Wills PR. 2009. Informed generation: physical origin and biological evolution of genetic code script interpreters. J Theor Biol. 2573: 345–358. [DOI] [PubMed] [Google Scholar]
- Wills PR. 2014. Spontaneous mutual ordering of nucleic acids and proteins. Orig Life Evol Biosph. 444: 293–298. [DOI] [PubMed] [Google Scholar]
- Wills PR. 2016. The generation of meaningful information in molecular systems. Phil Trans R Soc A. 3742063: 20150016.. [DOI] [PubMed] [Google Scholar]
- Wills PR, Carter CW Jr. 2017. Insuperable problems of an initial genetic code emerging from an RNA world. BioSystems, http://dx.doi.org/10.1016/j.biosystems.2017.09.006. [DOI] [PMC free article] [PubMed]
- Wills PR, Nieselt K, McCaskill JS.. 2015. Emergence of coding and its specificity as a physico-informatic problem. Orig Life Evol Biosph. 45: 249–255. [DOI] [PubMed] [Google Scholar]
- Wilson DS, Szostak JW.. 1999. In vitro selection of functional nucleic acids. Annu Rev Biochem. 68:611–647. [DOI] [PubMed] [Google Scholar]
- Wochner A, Attwater J, Coulson A, Holliger P.. 2011. Ribozyme-catalyzed transcription of an active ribozyme. Science 3326026: 209–212. [DOI] [PubMed] [Google Scholar]
- Woese C. 1967. The genetic code. New York: Harper & Row. [Google Scholar]
- Woese CR. 1965a. On the origin of the genetic code. Proc Natl Acad Sci U S A. 546: 1546–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR. 1965b. Order in the origin of the genetic code. Proc Natl Acad Sci U S A. 54:71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR, Dugre DH, Saxinger WC, Dugre SA.. 1966. The molecular basis for the genetic code. Proc Natl Acad Sci U S A. 554: 966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR, Olsen GJ, Ibba M, Soll D.. 2000. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev. 641: 202–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Aravind L, Grishin NV, Koonin EV.. 1999. Evolution of aminoacyl-tRNA synthetases—analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 98: 689–710. [PubMed] [Google Scholar]
- Wolf YI, Koonin EV.. 2007. On the origin of the translation system and the genetic code in the RNA world by means of natural selection, exaptation, and subfunctionalization. Biol Direct. 2:14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfenden R. 2011. Benchmark reaction rates, the stability of biological molecules in water, and the evolution of catalytic power in enzymes. Annu Rev Biochem. 80:645–667. [DOI] [PubMed] [Google Scholar]
- Wolfenden R, Cullis PM, Southgate CCF.. 1979. Water, protein folding, and the genetic code. Science 2064418: 575–577. [DOI] [PubMed] [Google Scholar]
- Wolfenden R, Lewis CA, Yuan Y, Carter CW Jr.. 2015. Temperature dependence of amino acid hydrophobicities. Proc Natl Acad Sci U S A. 11224: 7484–7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfenden R, Snider MJ.. 2001. The depth of chemical time and the power of enzymes as catalysts. Acc Chem Res. 3412: 938–945. [DOI] [PubMed] [Google Scholar]
- Wong JT-F. 2005. Coevolution theory of the genetic code at age thirty. BioEssays 274: 416–425. [DOI] [PubMed] [Google Scholar]
- Wong JT-F, Ng S-K, Mat W-K, Hu T, Xue H.. 2016. Coevolution theory of the genetic code at age forty: pathway to translation and synthetic life. Life 6:12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue H, Ng S-K, Tong K-L, Wong JT-F.. 2005. Congruence of evidence for a Methanopyrus-proximal root of life based on transfer RNA and aminoacyl-tRNA synthetase genes. Gene 3602: 120–130. [DOI] [PubMed] [Google Scholar]
- Yarus M. 2011a. Getting past the RNA world: the initial Darwinian Ancestor. Cold Spring Harb Perspect Biol. 3:a003590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarus M. 2011b. Life from an RNA world: the ancestor within. Cambridge (MA: ): Harvard University Press. [Google Scholar]
- Zull JE, Smith SK.. 1990. Is genetic code redundancy related to retention of structural information in both DNA strands? Trends Biochem. Sci. 157: 257–261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.