

Abstract
The mutational process in bacteria is biased toward A and T, and most species are GC-rich relative to the mutational input to their genome. It has been proposed that the shift in base composition is an adaptive process—that natural selection operates to increase GC-contents—and there is experimental evidence that bacterial strains with GC-rich versions of genes have higher growth rates than those strains with AT-rich versions expressing identical proteins. Alternatively, a nonadaptive process, GC-biased gene conversion (gBGC), could also increase the GC-content of DNA due to the mechanistic bias of gene conversion events during recombination. To determine what role recombination plays in the base composition of bacterial genomes, we compared the spectrum of nucleotide polymorphisms introduced by recombination in all microbial species represented by large numbers of sequenced strains. We found that recombinant alleles are consistently biased toward A and T, and that the magnitude of AT-bias introduced by recombination is similar to that of mutations. These results indicate that recombination alone, without the intervention of selection, is unlikely to counteract the AT-enrichment of bacterial genomes.
Keywords: bacterial genomes, recombination, biased gene conversion, G+C contents, sequence evolution
Introduction
Bacteria and Archaea display wide variation in their genomic base compositions, which range among sequenced genomes from 13% to 75% GC (McCutcheon and Moran 2010; Thomas etal. 2008). This compositional variation has long been considered to result from genomic differences in the underlying patterns of mutations, such that organisms with higher GC-contents incurred more mutations toward G and C nucleotides (Freese and Strack 1962; Sueoka 1962). Because the most extreme differences in GC-contents occur at nucleotide sites under low selective constraints (Muto and Osawa 1987), and because repeated attempts to link genomic base composition to selective agents have proven unsuccessful, the prevalent view was that the differences in base composition among species were selectively neutral.
Comparative sequence analyses performed on multiple taxa show that, even in bacterial groups with high genomic GC-contents, mutation is universally biased toward A and T, implying a role of natural selection in shaping base composition (Hershberg and Petrov 2010; Hildebrand etal. 2010; Long etal. 2015). Moreover, E. coli strains expressing GC-rich versions of genes exhibit higher growth rates than those expressing the identical protein from AT-rich versions (Raghavan etal. 2012), indicating that increased GC-contents can improve cellular fitness. Various selective pressures have been proposed to account for the differences in base composition among bacterial species, including metabolic costs (Rocha and Danchin 2002) and environmental factors, such as temperature, oxygen, UV radiation, or nitrogen fixation (McEwan etal. 1998; Musto etal. 2004; Naya etal. 2002; Reichenberger etal. 2015; Singer and Ames 1970), that may shift genomes toward a particular GC-content. Additionally, adaptive codon usage bias can generate nucleotide compositions that differ from the mutational input at purportedly neutral sites (Hershberg and Petrov 2009, 2008). But despite continued efforts to elucidate the target of selection, the key driver of genomic GC-content remains enigmatic (Rocha and Feil 2010).
Recombination has been invoked as an alternative to selection as a process for increasing GC-content of genomes. Because mismatches induced by recombination are repaired by gene conversion events that are mechanistically biased toward G and C, recombining regions become enriched in G and C (Duret and Galtier 2009). GC-biased gene conversion (gBGC) was originally cited as a nonselective mechanism responsible for the compositional variation within mammalian genomes (Galtier etal. 2001), and it has recently been extended to account for differences among bacteria (Lassalle etal. 2015). Recombination can also increase the efficiency of selection by disrupting the linkage between sites under different selective constraints (Hill and Robertson 1966). As a consequence, recombination would promote the removal of any detrimental AT-biased mutations, thereby increasing GC-content. Thus, there are two routes by which recombination will promote shifts in base composition—one adaptive and one nonadaptive—and although they operate in distinct manners, both can have similar effects on genomic GC-content.
Recombination rates are highly variable across bacteria (Vos and Didelot 2009) making it possible that recombination has been a major contributor to the differences in base compositions of bacterial genomes. Several studies have reported a positive association between recombination and GC-content within and across bacterial species (Lassalle etal. 2015; Touchon etal. 2009), whereas others have claimed a less pronounced effect (Hildebrand etal. 2010; Yahara etal. 2015). Part of the difficulty in establishing a relationship between recombination and base composition is due to the imprecision with which the rates and scale of recombination events are estimated. But more importantly, a correlation between recombination and GC-content does not identify the process responsible for increasing GC-contents: if most recombinant alleles, like mutations, are AT-biased, then selection might well be the source of GC-enrichment.
In this study, we establish the direct impact of recombination on the nucleotide composition in 93 species of Bacteria and Archaea by focusing on individual alleles that display an unambiguous signal of recent recombination. We show that in a majority of species recombinant alleles are biased toward A and T, and that recombination is rarely sufficient to counteract the AT-bias introduced by mutations. Moreover, in every species, there is stronger purifying selection acting on new alleles introduced by recombination than by mutations, even at synonymous codon positions.
Results
Neither Recombination Nor Mutation is Biased Toward GC
We inferred the status of each polymorphic allele as originating by mutation or by recombination in the core genomes of 93 microbial species (91 Bacteria and 2 Archaea, see supplementary table S1, Supplementary Material online). We assigned homoplasies as arising unambiguously from recombination (as opposed to convergent mutations) using two methods (fig. 1), and applied parsimony to establish the original allelic state of each polymorphic site.
Fig. 1.
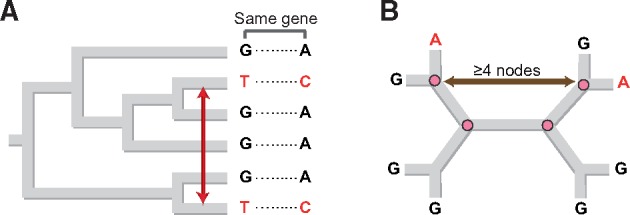
Identification of recombinant alleles. (A) Homoplasies were identified as those single nucleotide polymorphisms (SNPs) whose distributions are incongruent with the strain phylogeny as determined by both distance-based and topology-based methods. To distinguish homoplasies attributable to recombination from those generated by convergent mutations, analyses were confined to cases where two or more homoplasies within the same gene contained identical SNPs at the identical locations exhibiting the identical distribution among strains, a circumstance unlikely to occur by independent mutations. (B) The ancestral state of each homoplasy was determined from the number of nodes separating the recombinant SNP in two or more strains. We considered a polymorphic allele to represent the acquired state (i.e., as having been introduced by recombination) if present in strains separated by at least four nodes in the unrooted tree of each species.
We tested the accuracy of these procedures against 120 data sets simulated under an array of diverse parameters (supplementary table S2, Supplementary Material online). Our method presents a very low rate of false positives: only two of 170,587 recombinant alleles were inferred incorrectly. These two alleles were inferred in simulated genomes where population size, mutation rate and recombination rate were set at the lowest values (supplementary table S2, Supplementary Material online), resulting in sequences with very few polymorphisms and poorly resolved tree topologies. Note that the vast majority of the real data sets contain much higher levels of polymorphisms (supplementary table S1, Supplementary Material online) and that even in simulated sequences with very low polymorphism, false positives represented <2% of the inferred recombinant alleles. Therefore, the stringency of this method causes a negligible rate of false positives.
Looking first at the changes occurring at 4-fold degenerate sites (GC4), the vast majority of species displayed an excess of transitions over transversions; and in particular, G/C to A/T transitions (fig. 2) represent the most common changes incurred both by recombination and by mutations. (Note that for all species, transition and transversion patterns are corrected for GC-contents at 4-fold degenerate sites). Limiting the analysis to include only recently acquired recombinant alleles and recent mutations (fig. 2) revealed that the input of new alleles by recombination and by mutations at GC4 were similar to one another, and to the patterns observed when all polymorphisms at these sites are considered and for alleles at each other codon positions (supplementary fig. S1, Supplementary Material online).
Fig. 2.

Comparison of nucleotide changes introduced by recombination and mutation. Cumulative proportions of SNPs at 4-fold degenerate sites (GC4) for each of the six types of nucleotide changes, as calculated for all alleles introduced by recombination new alleles introduced by recombination all alleles introduced by mutations new alleles introduced by mutations. Values were normalized by the GC-contents at 4-fold degenerate sites for each species prior to calculating overall proportions, and species with fewer than 50 polymorphic sites for a given category of alleles were excluded.
Prior studies have shown that the underlying pattern of mutations would lead to an enrichment of A and T in most bacterial species (Hershberg and Petrov 2010; Hildebrand etal. 2010), so we separately compared the impact of mutation and of recombination on base composition. We calculated GCeq, the GC-content expected based solely on the patterns of recombination or mutation for each species. In agreement with previous results (Hershberg and Petrov 2010; Hildebrand etal. 2010), mutations lead to a lower GC-content than the current GC-content (i.e., GCeq < GC) (fig. 3 and supplementary fig. S2, Supplementary Material online) in the majority of species. Only a few of the more AT-rich species (e.g., Fusobacterium nucleatum, Bacillus cereus, Bacillus thuringiensis, Streptococcus mitis) would evolve toward a higher GC-content based solely on the mutational input to the genome.
Fig. 3.

Equilibrium GC content inferred from new polymorphisms relative to actual GC content. GC4eq is the expected GC-content at 4-fold degenerate sites for a given species when based on new alleles introduced by recombination (left panel) and new alleles introduced by mutations (right panel). GC4eq values were normalized by the GC-contents at 4-fold degenerate sites for each species, and species with fewer than 50 polymorphic sites for any given category of allele were excluded. Points in shaded area below the diagonal denote species that are GC-rich relative to the input of polymorphisms by recombination or mutation.
Focusing solely on alleles originating by gene exchange, we observe that recombination, like mutation, leads to the enrichment of A and T in most bacterial species (fig. 3 and supplementary fig. S2, Supplementary Material online)—a trend that is even more pronounced when considering only the most recently acquired recombinant alleles. Again, only a few species with AT-rich genomes (e.g., Bacillus thuringiensis, Streptococcus mitis) undergo an increase in GC-content based on their recombinational patterns. Consistent results were obtained when analyzing the other codon positions (supplementary fig. S3, Supplementary Material online). In summary, the spectrum of changes introduced by recombination resembles that of mutations, and in the vast majority of bacterial species, both are biased toward A and T.
Recombination can change the frequency and distribution of polymorphic sites in a species, but all variant sites are ultimately produced by mutations. Although we focus on recent recombination events to mitigate the effects of selection, these sites might display any bias that is already present in the mutational events that produced these variants; and therefore, recent recombinant alleles would exhibit the AT-bias imprinted by mutation before being transferred by recombination. To filter out any effect that the original mutations have on the AT-bias observed in recombination, we directly compared the degree of nucleotide bias of recombinant alleles relative to that of recent mutations.
To quantify and compare the degree of AT-bias caused by recombination and mutations in each species, we calculated the ratio (B) of AT-converting changes relative to GC-converting changes (after correcting for the GC-content at each codon position). A value of B > 1 is indicative of an enrichment of A and T, and B < 1 is indicative of an enrichment of G and C. Changes introduced by recent mutations and by recent recombination events display similar B values (Bnew-mut = 2.0; Bnew-recomb = 1.8) (fig. 4A), denoting a slight preference toward AT-converting changes. The AT-bias is more pronounced for changes introduced by mutations (P < 0.05, paired Student’s t-test); however, recombination alone is AT-biased and unlikely to counteract the effects of mutation on GC-content, since both processes present biases of comparable intensities.
Fig. 4.

Impact of recombination and mutations on genomic nucleotide composition and codon usage. (A) The metric B represents the number of changes from G or C to A or T relative to the number of changes from A or T to G or C at 4-fold degenerate sites. B > 1 indicates an enrichment toward A and T, and B < 1 indicates an enrichment toward G and C. (B) The metric δ denotes the shift in codon usage caused by synonymous changes in the coding sequences of genes constituting the core genome of each species. δ > 0 indicates that nucleotide changes led toward more commonly used codons, and δ < 0 indicates that nucleotide changes led toward less frequently used codons. Values shown are for all alleles introduced by recombination new alleles introduced by recombination; all alleles introduced by mutations; new alleles introduced by mutations. Values were normalized by the GC-contents at 4-fold degenerate sites for each species prior to calculating overall proportions, and species with fewer than 50 polymorphic sites for a given category of alleles were excluded.
When considering the entire set of changes introduced by recombination at 4-fold degenerate sites—not just those caused by recent events—the numbers of AT-converting and GC-converting changes approach parity (B = 1), meaning that they reflect the current GC-content of the genome at these sites. This shift toward higher GC when including older alleles is likely to reflect the cumulative effects of selection in that the base composition of recombinant alleles at first and second codon positions (where there are stronger selective constraints) lies closer to the equilibrium value for a genome (supplementary fig. S4, Supplementary Material online).
Recombinant Polymorphisms are Exposed to Higher Levels of Purifying Selection
Because recombination is expected to increase the efficiency of selection by unlinking genomic sites (Hill and Robertson 1966), we tested for signals of enhanced selection at recombinant sites by comparing the dN/dS ratios of alleles introduced by recombination to those introduced by mutations (fig. 5). Cumulatively, the dN/dS ratios of recombinant polymorphisms are >2-fold lower than those attributable to mutations (P < 0.00001, paired Student’s t-test) indicative of stronger selection acting on sites introduced by recombination. The same ratio is observed for the subset of recent recombinant alleles relative to recent mutations (P < 0.00001, paired Student’s t-test). Because recombinant alleles are, on average, older than recent mutations—since variants must first arise by mutation before they are transferred—this implies that selection either is more effective or has been acting longer on recombinant sites.
Fig. 5.
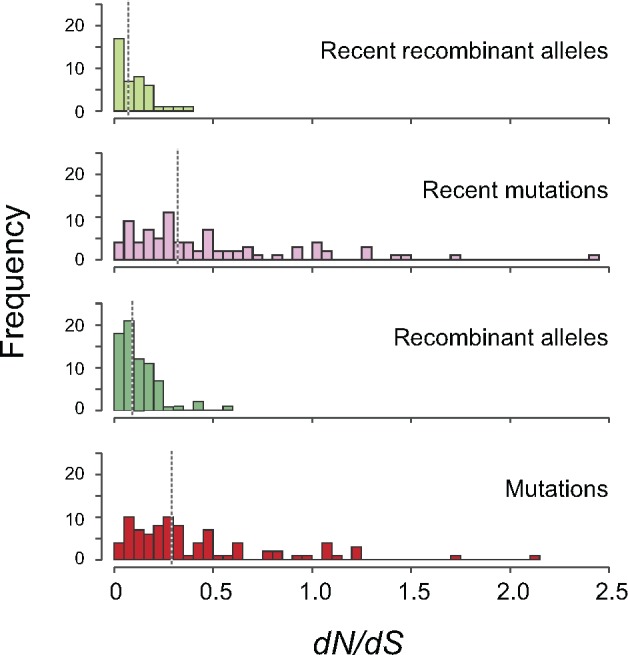
Testing for selection on nucleotide changes introduced by recombination and mutation. dN/dS ratios were calculated from the concatenate of the core genome of each species. Values shown are for all alleles introduced by recombination; new alleles introduced by recombination; all alleles introduced by mutations; new alleles introduced by mutations.
We hypothesize that increased selection on recombinant alleles, not biased gene conversion, is responsible for the GC enrichment of recombinant alleles relative to mutations. Although our analyses focused on recent events of recombination and mutation in order to reduce the effects of selection, several species still displayed signs of strong purifying selection—even at 4-fold degenerate sites (fig. 5). Recent recombinants are defined as homoplasic alleles present in two terminal tips of a tree, whereas recent mutations are defined as those alleles present at a single tip of a tree. Since recent recombinant alleles involve an additional event (i.e., a mutational event that produces the polymorphism must precede the event of recombination), then, recent recombination events are, on average, slightly older than recent mutations, and selection should have had more time to act upon these sites. Although recent recombinant sites might be expected to have a slightly stronger bias toward GC relative to recent mutations; in the majority of species (20/25), both mutation and recombination were similarly biased towards AT (fig. 6). Only in five species (Pseudomonas putida, P. stutzeri, Rhizobium leguminosarum, Rhodococcus fasciens, and Serratia marcescens) is there less AT-enrichment of recombinant alleles than mutations. In general, those species in which recombinant alleles are less AT-enriched (lower right quadrant, fig. 6) display the strongest purifying selection on recent recombinant alleles (dN/dS ≤ 0.05), consistent with the hypothesis that any GC-bias associated with recombination is introduced by selection. If gBGC were driving this bias, one expects a stronger GC-enrichment in the most highly recombining species; however, these five GC-biased species did not present higher recombination rates or biased sample sizes relative to other species (supplementary fig. S6, Supplementary Material online), again indicating that the GC-bias in this species is unlikely driven by gBGC.
Fig. 6.
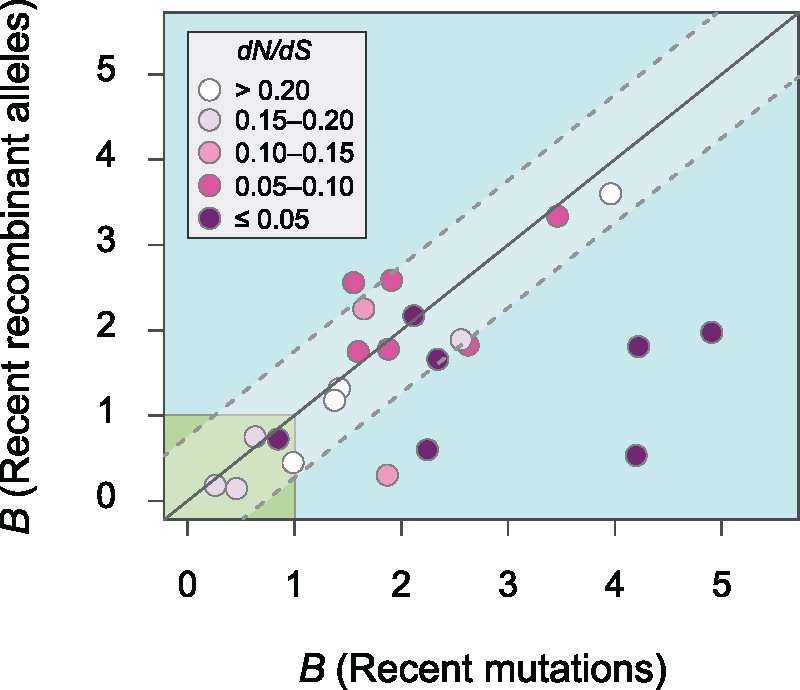
Nucleotide bias of recent recombinant alleles and recent mutations. The metric B represents the number of changes from G or C to A or T relative to the number of changes from A or T to G or C at 4-fold degenerate sites. On each axis, a value of B < 1 indicates an enrichment toward G and C (lower left corner), and value of B > 1 indicates an enrichment toward A and T. The solid diagonal line indicates identical nucleotide bias for recent recombinant alleles and recent mutations; the dotted lines represent half of the standard deviation. Values were normalized by the GC-contents at 4-fold degenerate sites for each species prior to calculating overall proportions, and species with fewer than 50 polymorphic sites for a given category of alleles were excluded. As indicated in the key, the color shading of dots denotes the dN/dS ratio of recombinant alleles for a species. All phylogenetic trees of species included in this analysis display average bootstrap values >70, and the list of included species and the corresponding average bootstrap values are indicated in bold in supplementary table S1, Supplementary Material online.
Although our analyses focused on 4-fold-degenerate sites to diminish the contribution of selection, adaptive codon usage might still impose constraints at these sites (Hershberg and Petrov 2009, 2008). To determine if selection for codon usage is acting on these sites, we examined how synonymous changes introduced by recombination and by mutation alter codon usage frequencies for the entire set of core genes of each species. Both mutations and recombinant alleles show evidence of an overall negative impact on codon usage: changes caused by each of the processes tended to shift codons toward those less commonly used in the core genome (fig. 4B). Recent recombinant alleles have a less detrimental effect on codon usage than do recent mutations (P < 0.01, paired Student’s t-test), suggesting that selection is more efficient at purging recombinant alleles causing shifts toward less favorable codons.
Discussion
Recombination, through the action of GC-biased gene conversion (gBGC), has been invoked as a nonadaptive process responsible for increasing the GC-content of genomic regions. Rates of recombination are positively associated with GC-content in multiple animals, and gBGC has been invoked to explain the variation in GC-contents within the human genome and the formation of isochores, by which recombination has mechanistically increased the genomic GC-content of certain regions (Galtier etal. 2001). Therefore, gBGC also offers a particularly attractive hypothesis to explain the variation in base composition observed among bacterial genomes (Lassalle etal. 2015). There is convincing evidence that bacterial base composition does not result from a strictly neutral process (Hildebrand etal. 2010), but despite repeated attempts over the past 50 years, the mechanism by which selection operates on base composition of bacterial genomes has yet to be identified. Therefore, recombination, specifically, gBGC, could serve as alternative to selection that operates across bacteria, such that those species with more recombination have higher base compositions.
To determine the impact of recombination on the evolution of bacterial base composition, we inferred recombination on a site-by-site basis, instead of the more common approach of correlating recombination rates to the GC-content of genes or even larger genomic regions (Lassalle etal. 2015; Touchon etal. 2009; Yahara etal. 2015). This allowed us to directly infer nucleotide replacements and revealed that recombination is not typically biased towards G and C as predicted by biased gene conversion.
When considering all analyzed species together, recent recombination events are slightly, but significantly, less biased toward AT than are recent mutations. The increased GC content of recombined alleles might, at first glance, be viewed as evidence of gBGC; however, there are several factors indicating that this pattern is not caused by gene conversion. First, when evaluating species individually, recent recombination events are, in fact, less GC-biased than recent mutations in 28% of the analyzed species, such that recombination causes many genomes to become more AT-rich. Second, recent recombination events display clear signs of enhanced or more prolonged selection than do recent mutations. Thus, selection on base composition appears to be acting more effectively on recombinant sites than on mutations, as previously suggested (Hildebrand etal. 2010; Touchon etal. 2009). Because bacteria reproduce clonally, all of the alleles in a strain are linked to a common fate. Therefore, the success of an allele depends strongly on its genetic background: neutral or slightly detrimental alleles can hitchhike to fixation due to the presence of an allele(s) under strong positive selection in the same genotype or can be lost due to the counter-selection of strongly detrimental allele(s). By reducing linkage among sites—and concomitantly reducing background selection and hitchhiking—recombination helps purge deleterious alleles and increases the frequency of beneficial alleles. Third, in most species, the compositional biases introduced by recombinant alleles would not be sufficient to counteract the AT-bias introduced by mutations (fig. 3), as has been previously reported for Mycobacterium tuberculosis (Namouchi etal. 2012).
The overall effects of recombination on genomic base composition depend on the relative frequencies of mutations relative to recombination and the biases introduced by each. The gBGC hypothesis predicts a positive association between GC-content and recombination rate. Among the 93 species analyzed, 15 exhibit a significant positive correlation, and seven exhibit a significant negative correlation, between GC-content and the relative frequency of recombination (Spearman’s Rho, P < 0.05, supplementary table S1, Supplementary Material online).
Previous evidence of associations between GC-contents and recombination rates has been contradictory (Hildebrand etal. 2010; Lassalle etal. 2015; Yahara etal. 2015), and the differences among studies can often be traced to the methodologies used to infer recombination. For example, Lassalle etal. (2015) used PHI (Bruen etal. 2006) to classify genes into two categories—those that have recombined and those that have not—and reported an association between GC-content and the proportion of recombining genes in a genome. However, a dichotomous classification of genes as either recombinant or nonrecombinant need not accurately depict the frequency or scale of recombination in a genome given that recombination can result in low, high or intermediate levels of polymorphism (Yahara etal. 2015). Multiple studies have shown that recombination events are smaller than the average size of genes (Dettman etal. 2014; Didelot and Maiden 2010; Joseph etal. 2012; Namouchi etal. 2012), averaging perhaps only 50 nucleotides in length (Touchon etal. 2009). Therefore, a genome in which every gene has been subject to recombination might actually have relatively few polymorphisms introduced by recombination.
To circumvent problems associated with the classification of recombinant regions, we employed a site-by-site approach that yielded the absolute numbers of polymorphisms attributable to mutation or recombination. We found that even in species where the vast majority of genes are impacted by recombination, most genes contain few polymorphic sites that can be ascribed to recombination. Although the evolution of such species has been influenced rather little by recombination, they would be viewed as highly recombining based on a scheme that categorizes genes as recombinant or non-recombinant. Additionally, such approaches tend to classify quickly evolving genes as recombinant (Yahara etal. 2015) and would likely introduce a bias, given that highly expressed genes evolve more slowly due to codon usage bias (Drummond and Wilke 2008; Gouy and Gautier 1982; Ikemura 1985; Sharp and Li 1987) and are typically of lower GC-content (Hildebrand etal. 2010).
Our site-by-site approach evaluated polymorphisms at 4-fold degenerate sites to impart neutral expectations; but since such sites can be under selective constraints due to adaptive codon usage, we also performed analyses restricted to sets of recent polymorphisms to search for effects on selection on such sites. When focusing only on recent events, our measures of mutational bias were both quantitatively and qualitatively similar to those reported in previous studies (Hershberg and Petrov 2010; Hildebrand etal. 2010). The differences in nucleotide changes attributed to recent vs. older alleles revealed that selection is acting both on mutations and on recombinant alleles (figs. 4B and 5), and confirmed that codon usage has contributed to the GC-content variation among bacteria (Hershberg and Petrov 2009, 2008). But because the GC-content of intergenic and noncoding DNA is positively associated with GC-content of the rest of the genome, codon usage cannot be the sole source of the compositional variation among bacterial genomes (Hershberg and Petrov 2010).
There is recent evidence showing that the environment and phylogeny (Foerstner etal. 2005; Reichenberger etal. 2015) shape the GC-contents of bacterial genomes, suggesting that base composition is a slow-evolving trait driven by environmental selection. In light of the many attempts to elucidate the basis of the variation in genomic base composition, it is likely that GC-content in bacteria is not attributable to a single force acting on all species but rather evolves through a complex combination of factors.
Materials and Methods
Species Sampling and Defining Core Genomes
We downloaded the complete set of genomes of each bacterial and archaeal species [according to species designations at the NCBI website (ftp.ncbi.nlm.nih.gov/genomes/) on June 2015] represented by at least 20 genomes, resulting in a total of 20,690 genomes for 105 named species. For each named species, the protein sequences of all pairs of strains were compared using Usearch Global (Edgar 2010), and orthologs were defined as reciprocal best hits having at least 70% sequence identity and 80% length conservation. Those gene families containing paralogs were excluded such that a gene family was only considered as part of the core genome if present exactly once in every strain within a species (i.e., the core genome represents the set of ubiquitous and unambiguous orthologs). Strains were removed from analyses when only few homologs could be identified, suggesting low sequence quality or annotation issues. In species represented by large numbers of sequenced strains (n ≥ 200), performing all pairwise comparisons becomes computationally intensive. In these cases, the core genomes were assembled in multiple steps such that core genomes were initially built for subgroups of up to 100 strains, then the composite core genome was based on one genome from each subgroup and those gene families present in every subgroup were assigned to the species’ core genome. The protein sequences of genes constituting the core genome of each species were aligned with MAFFT v7 (Katoh and Standley 2013) and reverse-translated into their corresponding nucleotide sequences. To avoid potential misalignments due to frameshifts that can occur at the extremities of the genes, we excluded the first ten and the last ten codons of each gene. We then merged these aligned core genes into a single concatenate from which we inferred pairwise distances D using RAxML v7 under a GTR + Γ model (Stamatakis 2006). In many species, sequenced strains were identical (or very nearly so), so we randomly excluded strains whose core genome sequences with highly similar (D < 0.00005) to remove redundancies from the data set.
Because several of the species included a very large number of strains (up to 4,221 genomes per species), our quality-filtering procedures sometimes resulted in a relatively small number of genes constituting the core genome of a species due to the cumulative effects of large numbers of sequences and uneven assembly and annotation qualities. Due to these factors, the core genomes of several species (e.g., Escherichia coli, Salmonella enterica, Vibrio parahaemolyticus) were rebuilt, and distance matrices recalculated, using only nonredundant strains, as determined by the initial core genomes. Information about the contents of the core genome of each species is provided in supplementary table S1, Supplementary Material online. After removal of redundant strains, the number of strains representing a given species could be greatly reduced from the original number of available genomes, and we confined our analyses to those species with at least 15 genomes after removing redundant strains. From the original set of 105 species, 93 passed this threshold and were retained for subsequent analyses. Analyzed genomes are listed in Data Set S1, Supplementary Material online.
Identification of Recombinant Alleles
Of the multiple methods that have been developed to identify recombination events, many are not applicable to the present study due to the size and scope of our data set (Martin etal. 2011). Similarly, methods that aim at delineating recombinant regions of genes or genomes are not suitable for determining the impact of recombination at individual sites since such an approach can result both in the inclusion of nonrecombinant alleles into recombinant tracts and in the omission of recombinant tracts that are supported by too few polymorphisms. Finally, some algorithms view highly polymorphic regions as imports from external sources, although such regions may result from diversifying selection, not recombination. To circumvent the problems associated with these methods, we employed a site-by-site approach that focuses only on those polymorphisms for which there is strong, direct evidence of recombination (i.e., recombinant alleles) and on those polymorphisms that show no evidence of recombination (i.e. mutations). Our study does not require the inclusion of all polymorphisms, and ambiguous alleles were systematically excluded from the analysis. We inferred recombinant alleles from the incidence of homoplasies in the concatenate of core genomes by expanding the approach developed in (Bobay etal. 2015). This approach has been found to be highly consistent with other recombination detection algorithms, such as recHMM and PHI (Bruen etal. 2006; Zhou etal. 2014), and we added additional criteria (described below) to detect homoplasies that can be confidently assigned as recombinant alleles. Because homoplasies can result either from recombination events internal to the data set or from mis-assignment and from convergent mutations occurring at the identical site in different strains, we required that polymorphic alleles meet four conditions to be considered as recombinant:
All homoplasic alleles were initially inferred from incongruencies in the genomic distances of the corresponding strains (“distance-based method” improved from (Bobay etal. 2015)) as follows: For each species, the matrix of maximum-likelihood distances D was used to infer the pairwise distances between the core genome of each pair of strains. Polymorphic sites can have up to four alleles, and in the simplest configuration, biallelic sites are composed of a major N0 (most frequent) allele and a minor N1 (least frequent) allele. (Major and minor alleles were assigned indiscriminately when at equal frequencies.) Each minor allele was considered homoplasic when max (DN1N1) > min (DN0N1), where max (DN1N1) represents the highest genome-wide distance between the strains containing the minor alleles, and min (DN0N1) represents the smallest genome-wide distance between the strains displaying the minor allele N1 and the strains possessing the major allele N0. For sites with three or four alleles, only one allele was defined as major and all others as minor. In such cases, each minor allele was considered homoplasic or nonhomoplasic by comparison of the genome-wide distances of the strains harboring the minor allele to the genome-wide distances of the strains displaying the major allele.
Recombinant alleles also needed to be inferred as homoplasic by a topology-based method. For each species, we built a maximum likelihood phylogenetic tree on the concatenate of the entire core genome using RAxML v7.2 with the GTR + Γ (Stamatakis 2006). The robustness of each tree was assessed with 100 bootstrap replicates performed under these same parameters, and the average bootstrap support across all nodes was calculated for each tree. Most trees displayed high bootstrap support, indicating that the topology was inferred robustly for most species (supplementary fig. S5, Supplementary Material online). Average bootstrap support for each species is listed in supplementary table S1, Supplementary Material online. Based on the topology of each tree, we defined groups of monophyletic strains. An allele was considered as homoplasic if strains containing this allele did not constitute a monophyletic group.
Because homoplasies result both from recombination events and from multiple convergent mutations, the latter must be excluded when analyzing recombinant alleles. To eliminate homoplasies caused by convergent mutations, we considered only recombination events that involved the transfer of two or more polymorphic sites (i.e., two or more homoplasic sites with the same distribution among strains) (fig. 1A). We stipulated that at least a second homoplasic site with the same allelic distribution (i.e., found in the same set of strains) must be present within the same gene. This condition makes it unlikely that we retained homoplasies that originated from multiple convergent mutations that independently gave rise to identical allelic distributions multiple times within the same gene.
Clusters of consecutive homoplasic sites with identical allelic distributions among strains likely correspond to alignment errors and were excluded if three or more homoplasies with the identical strain distributions were separated by a median distance of only 1 bp from one another.
In this context, the detection of recombinant alleles ultimately depends on the phylogenetic inference of each species. And in cases when species trees are not systematically well resolved (supplementary fig. S5, Supplementary Material online), it will potentially affect the quality of our data set. However, our stringent approach requires recombinant alleles to be inferred with both a distance-based and a topology-based method, and both methods are unlikely to infer identical recombinant alleles when species trees are poorly resolved. As a consequence, few recombinant alleles would be inferred for those species with poorly resolved topologies, and by limiting our analyses to species with a large number of inferred recombinant alleles (figs. 6 and supplementary fig. S6, Supplementary Material online), species with poorly resolved phylogenies were eliminated (supplementary table S1, Supplementary Material online).
After identifying homoplasies attributable to recombination, we determined the polarity of each recombination event by inferring which allele was exchanged and which allele was replaced at these recombinant sites. We reasoned that an allele must exhibit a sporadic distribution and be distributed among distantly related strains in the tree to be unambiguously considered the exchanged allele, and that a minor allele (one found in <50% of the strains and up to 20 strains) has been transferred if present in strains that are separated by at least four nodes in the unrooted tree of a species (fig. 1B). Such a scenario is more parsimonious than the transfer of a major allele, which would require at least three times more recombination events from strains with identical allelic patterns at these positions (based on our previous filter, in which only recombination events that exchanged two alleles or more are considered). This analysis identifies only those alleles that were transferred, not the donor strains. From the resulting data set, we defined those homoplasies present in only two terminal branches of the tree as “recent recombinants” in that they represent the subset of alleles that have been introduced most recently by mutation (as evident from their presence on one terminal branch) and that have been exchanged most recently relative to other recombinant alleles (as evident from their presence on the second branch).
Inference and Polarization of Mutations
All nonhomoplasic alleles, as inferred both by the distance- and topology-based methods, were considered as originating by mutations. As with homoplasies, we excluded those highly localized clusters of three or more polymorphic sites showing the identical strain distributions and separated by a median distance of only 1 bp from one another because they likely represent frameshifts and alignment errors. We determined the polarity of each mutation by a parsimony approach in which 1) the major allele—an allele present in ≥50% of the strains of a given species—was presumed to be that ancestral state of the site; 2) the phylogeny of each species was rooted at the midpoint and assumed to follow a molecular clock, and 3) only minor alleles whose last common ancestor is located at a minimal distance of four nodes from the root were considered. By excluding instances in which mutations arose in a branch near the root of the phylogeny, we eliminate those cases where there is uncertainty in localizing the true root of the trees, which could potentially lead to incorrect inferences about the ancestral state of the allele. From the resulting set of polymorphic sites, we defined as “recent” those mutations present only in a single strain.
Patterns of Mutation and Recombination
We determined the relative frequencies of nucleotides changed by recombination and by mutation after first correcting for the GC-content of each species, as in (Hershberg and Petrov 2010). We computed #GCsites and #ATsites, the number of G and C sites, and A and T sites, respectively, calculated over the entire core genome concatenate of a species, averaged among all strains, for each codon position independently. GC1, GC2, GC3, and GC4 (aka GC4fold) refer to the GC-content calculated on the first codon position, second codon position, the third codon position, and 4-fold-degenerate sites of codons, respectively. The six categories of nucleotide changes constitute two transitions (A/T to G/C and G/C to A/T) and four transversions (A/T to C/G, A/T to T/A, G/C to C/G, and G/C to T/A).
Counts of recombinant polymorphisms and mutations from A or T to any other nucleotide were normalized by #GCsites/#ATsites. The relative frequency of each of the six categories of nucleotide changes was calculated as the percent of all possible replacements (i.e., the sum of the six counts, with changes from A and T normalized by #GCsites/#ATsites) for each of the codon positions independently. This analysis was limited to species with ≥50 polymorphic sites. We then calculated the balance (B) between nucleotide changes that led to an enrichment in A or T relative to those that led to an enrichment in G or C, whereas correcting for the nucleotide composition of each core genome for each codon position independently:
We calculated GC-contents at equilibrium GCeq (Hershberg and Petrov 2010) based on the spectrum of polymorphisms attributable to mutations and to recombination, and for each codon position, separately:
Simulations
To assess the performance of our procedure for detecting recombinant alleles, we simulated the evolution of a 100-kb fragment composed of 100 genes, each 1,000 bp in length. The original test sequence corresponded to the first 100 kb of Escherichia coli K12 MG1655 genome, which was subsequently evolved in silico as in (Falush etal. 2006) under a constant population size of either N = 100 sequences or N = 500 sequences. Each simulated generation was formed by randomly selecting with replacement 100 or 500 sequences of the previous generation, and each sequence was subjected to random point mutations following a Poisson distribution of mean 1 or 10, corresponding to a mutation rate of 10−5 or 10−4 per generation per base pair, respectively. We set a mutation spectrum of kappa = 3 (i.e., transitions occurred three times more frequently than transversions) while maintaining a constant nucleotide composition. Simulations were conducted with different numbers of Poisson-distributed recombination events (10, 50, or 100 events per generation). Recombinant sequences, whose sizes were based on a normal distribution with a mean of 500 bp (±100 bp, standard deviation), were selected at random from the population and replaced at the corresponding positions of a randomly selected recipient. Simulations were run for 30,000 generations, and 50 sequences were randomly sampled at 10 time points (supplementary table S2, Supplementary Material online) for analysis.
Tests of Selection on Nucleotide Sites
Ratios of dN/dS were calculated separately for mutations and for recombinant alleles on the entire core genome of each species. Within each core genome, we considered only those codons with no allelic variants (i.e., codons identical among all strains) and those with a single allelic variant confirmed as a mutation or a recombinant allele as described above. For each species, numbers of changes at synonymous s and nonsynonymous n sites were calculated for recombinant and nonrecombinant alleles separately. The expected frequencies of changes at synonymous S and nonsynonymous N sites were calculated by simulating all possible changes at each codon position. The ratios of change are given as dS = s/S and dN = n/N. When estimating dN/dS for recombinant alleles, only the major codon was considered for the variable codons containing a mutation. Conversely, when estimating dN/dS of mutations, only the major codon was considered for the variable codons containing a recombinant allele.
Patterns of Codon Usage
For each species, we assembled a table of codon usage frequencies based on the genes included in the core genome. Codon usage frequencies were expressed as the fraction of the total times a codon was used to encode its corresponding amino acid. We then calculated δ, the shift in codon usage frequency introduced by synonymous changes, for the entire set of core genes of each species. For each synonymous allele, the shift in usage frequency from the codon inferred as ancestral was compared with the usage frequency of the derived codon using the usage tables to yield δ = fancestral− fderived, where fancestral represents the usage frequency of the ancestral codon and fderived represents the usage frequency of the derived codon. Thus, δ > 0 indicates the shift from a less frequently used to a more frequently used codon. Conversely, δ < 0 indicates that a more commonly used codon has been exchanged by a less frequent codon.
Related Data Sets
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health grants R01GM108657 and R35GM118038. We thank Kim Hammond for assistance in the preparation of figures.
References
- Bobay LM, Traverse CC, Ochman H.. 2015. Impermanence of bacterial clones. Proc Natl Acad Sci U S A. 112:8893–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruen TC, Philippe H, Bryant D.. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 172:2665–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettman JR, Rodrigue N, Kassen R.. 2014. Genome-wide patterns of recombination in the opportunistic human pathogen Pseudomonas aeruginosa. Genome Biol Evol. 7:18–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Maiden MC.. 2010. Impact of recombination on bacterial evolution. Trends Microbiol. 18:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO.. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N.. 2009. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 10:285–311. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. [DOI] [PubMed] [Google Scholar]
- Falush D, Torpdahl M, Didelot X, Conrad DF, Wilson DJ, Achtman M.. 2006. Mismatch induced speciation in Salmonella: model and data. Philos Trans R Soc Lond B Biol Sci. 361:2045–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foerstner KU, von Mering C, Hooper SD, Bork P.. 2005. Environments shape the nucleotide composition of genomes. EMBO Rep. 6:1208–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freese E, Strack HB.. 1962. Induction of mutations in transforming DNA by hydroxylamine. Proc Natl Acad Sci U S A. 48:1796–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Piganeau G, Mouchiroud D, Duret L.. 2001. GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics 159:907–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Gautier C.. 1982. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 10:7055–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA.. 2010. Evidence that mutation is universally biased towards AT in bacteria. PLoS Genet. 6:e1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA.. 2009. General rules for optimal codon choice. PLoS Genet. 5:e1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA.. 2008. Selection on codon bias. Annu Rev Genet. 42:287–299. [DOI] [PubMed] [Google Scholar]
- Hildebrand F, Meyer A, Eyre-Walker A.. 2010. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 6:e1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Robertson A.. 1966. The effect of linkage on limits to artificial selection. Genet Res. 8:269–294. [PubMed] [Google Scholar]
- Ikemura T. 1985. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 2:13–34. [DOI] [PubMed] [Google Scholar]
- Joseph SJ, Didelot X, Rothschild J, de Vries HJ, Morre SA, Read TD, Dean D.. 2012. Population genomics of Chlamydia trachomatis: insights on drift, selection, recombination, and population structure. Mol Biol Evol. 29:3933–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassalle F, Perian S, Bataillon T, Nesme X, Duret L, Daubin V.. 2015. GC-Content evolution in bacterial genomes: the biased gene conversion hypothesis expands. PLoS Genet. 11:e1004941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, Kucukyildirim S, Sung W, Williams E, Lee H, Ackerman M, Doak TG, Tang H, Lynch M.. 2015. Background mutational features of the radiation-resistant bacterium Deinococcus radiodurans. Mol Biol Evol. 32:2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Lemey P, Posada D.. 2011. Analysing recombination in nucleotide sequences. Mol Ecol Resour. 11:943–955. [DOI] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA.. 2010. Functional convergence in reduced genomes of bacterial symbionts spanning 200 My of evolution. Genome Biol Evol. 2:708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan CE, Gatherer D, McEwan NR.. 1998. Nitrogen-fixing aerobic bacteria have higher genomic GC content than non-fixing species within the same genus. Hereditas 128:173–178. [DOI] [PubMed] [Google Scholar]
- Musto H, Naya H, Zavala A, Romero H, Alvarez-Valin F, Bernardi G.. 2004. Correlations between genomic GC levels and optimal growth temperatures in prokaryotes. FEBS Lett. 573:73–77. [DOI] [PubMed] [Google Scholar]
- Muto A, Osawa S.. 1987. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc Natl Acad Sci U S A. 84:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namouchi A, Didelot X, Schock U, Gicquel B, Rocha EP.. 2012. After the bottleneck: Genome-wide diversification of the Mycobacterium tuberculosis complex by mutation, recombination, and natural selection. Genome Res. 22:721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya H, Romero H, Zavala A, Alvarez B, Musto H.. 2002. Aerobiosis increases the genomic guanine plus cytosine content (GC%) in prokaryotes. J Mol Evol. 55:260–264. [DOI] [PubMed] [Google Scholar]
- Raghavan R, Kelkar YD, Ochman H.. 2012. A selective force favoring increased G+C content in bacterial genes. Proc Natl Acad Sci U S A. 109:14504–14507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenberger ER, Rosen G, Hershberg U, Hershberg R.. 2015. Prokaryotic nucleotide composition is shaped by both phylogeny and the environment. Genome Biol Evol. 7:1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EP, Danchin A.. 2002. Base composition bias might result from competition for metabolic resources. Trends Genet. 18:291–294. [DOI] [PubMed] [Google Scholar]
- Rocha EP, Feil EJ.. 2010. Mutational patterns cannot explain genome composition: Are there any neutral sites in the genomes of bacteria? PLoS Genet. 6:e1001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH.. 1987. The rate of synonymous substitution in enterobacterial genes is inversely related to codon usage bias. Mol Biol Evol. 4:222–230. [DOI] [PubMed] [Google Scholar]
- Singer CE, Ames BN.. 1970. Sunlight ultraviolet and bacterial DNA base ratios. Science 170:822–825. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Sueoka N. 1962. On the genetic basis of variation and heterogeneity of DNA base composition. Proc Natl Acad Sci U S A. 48:582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SH, Wagner RD, Arakaki AK, Skolnick J, Kirby JR, Shimkets LJ, Sanford RA, Loffler FE.. 2008. The mosaic genome of Anaeromyxobacter dehalogenans strain 2CP-C suggests an aerobic common ancestor to the delta-proteobacteria. PLoS One 3:e2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, Bidet P, Bingen E, Bonacorsi S, Bouchier C, Bouvet O, et al. 2009. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 5:e1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M, Didelot X.. 2009. A comparison of homologous recombination rates in bacteria and archaea. ISME J. 3:199–208. [DOI] [PubMed] [Google Scholar]
- Yahara K, Didelot X, Jolley KA, Kobayashi I, Maiden MC, Sheppard SK, Falush D.. 2015. The landscape of realized homologous recombination in pathogenic bacteria. Mol Biol Evol. 33:456–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, McCann A, Weill FX, Blin C, Nair S, Wain J, Dougan G, Achtman M.. 2014. Transient Darwinian selection in Salmonella enterica serovar Paratyphi A during 450 years of global spread of enteric fever. Proc Natl Acad Sci U S A. 111:12199–12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.