

Abstract
Previous findings on the association of genetic factors and pancreatic cancer survival are limited and inconsistent. In a two-stage study, we analyzed the existing genome-wide association study dataset of 868 pancreatic cancer patients from MD Anderson Cancer Center in relation to overall survival using Cox regression. Top hits were selected for replication in another 820 patients from the same institution using the Taqman genotyping method. Functional annotation, pathway analysis, and gene expression analysis were conducted using existing software and databases. We discovered genome-wide significant associations of patient survival with three imputed SNPs which, in complete LD (r2=1), were intronic SNPs of the PAIP2B (rs113988120) and DYSF genes (rs112493246 and rs138529893) located on chromosome 2. The variant alleles were associated with a 3.06-fold higher risk of death (95% confidence interval [CI]=2.10-4.47, P = 6.4 × 10−9) after adjusting for clinical factors. Eleven SNPs were tested in the replication study and the association of rs113988120 with survival was confirmed (HR: 1.57, 95%CI: 1.13-2.20, P = 0.008). In silico analysis found rs1139988120 might lead to altered motif. This locus is in LD (D′=0.77) with 3 eQTL SNPs near or belong to the NAGK and MCEE genes. According to The Cancer Genome Atlas data and our previous RNA-sequencing data, the mRNA expression level of PAIP2B but not NAGK, MCEE or DYSF was significantly lower in pancreatic tumors than in normal adjacent tissues. Additional validation efforts and functional studies are warranted to demonstrate whether PAIP2B is a novel tumor suppressor gene and a potential therapeutic target for pancreatic cancer.
Keywords: pancreatic cancer, survival, GWAS, prognosis, SNP
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer which is the fourth leading cause of cancer death in the United States. PDAC has a dismal prognosis; the average 5-year survival rate was 6.7% according to cumulative statistics from 2004 to 2010 1. The poor outcome of this disease is due mainly to late diagnosis and the high invasiveness and profoundly drug-resistant nature of the tumor. The recent advances in the understanding of the molecular and genetic alterations in pancreatic tumor have not yet been translated into significant improvement in patient survival or reduced mortality. There is a critical need for novel therapeutic targets and molecular markers in personalized PDAC management to achieve better treatment efficacy.
Emerging evidence suggests that germline genetic variations influence survival in PDAC. Studies using the candidate-gene approach focusing on genes involved in DNA repair 2, cell cycle regulation 3, drug metabolism 4, and signaling pathways 5, 6 have observed many nominally significant associations. Genome-wide association studies (GWAS) on overall survival (OS) in PDAC have also identified a number of interesting loci, but the findings are inconsistent and none of these loci have reached genome-wide significance7-10. Identifying genetic variants associated with PDAC survival may lead to the discovery of novel therapeutic targets for the development of new strategies in patient treatment.
Many of the previous studies on genetic factors in PDAC survival suffered the limitations of insufficient study power or heterogeneous study population. Taking advantage of the existing GWAS data and the large patient population recruited in a case-control study on PDAC conducted at The University of Texas MD Anderson Cancer Center, we performed a two-stage study to identify genetic variants that are associated with survival in 1,688 patients with pathologically diagnosed PDAC.
Materials and Methods
Study population, GWAS dataset, and discovery study
The study population was drawn from a hospital-based case-control study of patients with pathologically confirmed PDAC conducted at The University of Texas MD Anderson Cancer Center from June 2002 through May 200911. All patients were recruited consecutively with the exclusion criteria of non-US resident or having a prior history of cancer. All patients signed an informed consent for interview and a blood sample. DNA was extracted from peripheral lymphocytes and GWAS was conducted as previously described by the Pancreatic Cancer Cohort Consortium (PanScan) and the Pancreatic Cancer Case-Control Consortium (PanC4)12, 13. A total of 903 MD Anderson patients with sufficient amount of DNA samples were genotyped in the PanScan II and PamC4 GWASs.
Prior to analysis, we carried out further quality control of the GWAS data. We extracted 11 duplicate samples that were intentionally preset to check the genotyping concordance between PanScan II and PanC4. Comparison of the duplicate samples showed 100% concordance. Then, we excluded 15 patients who had later been diagnosed with diseases other than PC or who did not have complete clinical data. Identity-by-descent analyses did not show evident familial relationships between any of the patients. A total of 877 samples remained for population structure analysis. Using the International HapMap Project genotype data (phase 3 release #3, National Center for Biotechnology Information [NCBI] build 36, SNP Database (dbSNP) b126, 2010-05-28, minor allele frequency [MAF]>5%) for CEU, JPT/CHB, and YRI 14, we seeded 10,195 high-quality markers from our dataset (r2 < 0.004) in STRUCTURE analysis 15. A total of 868 individuals of European descent (0.82-1.00 similarity to CEU) were identified for the survival analysis in the discovery study. We also derived five principal components for population substructure based on the 868 patients using Genome-wide Complex Trait Analysis software16.
Replication study
The replication study was conducted in 820 MD Anderson patients who were mostly enrolled in the case-control study after the conduction of the GWAS in 2009. The only selection criteria we applied are: 1) have not been involved in GWAS and 2) being self-reported non-Hispanic whites. We selected the highest-ranked single-nucleotide polymorphisms (SNPs), which had the smallest P values in Cox regression analysis, high imputation quality scores (≥0.7), and MAFs of >0.15. We also preferentially considered the nonsynonymous SNPs or SNPs of known PDAC-related genes. Genotyping was performed using the TaqMan method in an Applied Biosystems 7900HT Fast Real-time PCR System (Foster City, CA). Five percent of the samples were analyzed in duplicates, and a 99% agreement rate was achieved. The inconsistency was resolved by further genotyping.
Statistical methods
Imputation
Because PanScan II and PanC4 used different genotyping platforms, imputation was conducted to generate a common dataset for both studies. The GWAS data were first pre-phased in SHAPEIT2 17, then imputed in IMPUTE218, 19 with the 1000 Genomes Project (phase1_release_v3.20101123) as the reference 20. Because SNPs in PanScan II were originally mapped to an older genome assembly (NCBI build 36 [hg18]), we systematically converted their genome positions to genome assembly NCBI build 37 (hg19) using the liftOver tool by the University of California, Santa Cruz, (http://genome.ucsc.edu). SNPs not listed in NCBI build 37 were removed, with the RsMergeArch.bcp.gz database as the reference (ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606/database/organism_data/). Moreover, we removed SNPs that had a MAF of less than 1%, deviated from Hardy–Weinberg equilibrium (P < 0.0004), or had a high number of missing genotypes (>2%). Because the PanScan II and PanC4 datasets had only ∼320,000 SNPs in common, imputation was performed in separate dataset. After imputation, we pruned out SNPs with quality scores of <0.3, MAFs of <0.01, missing rates of >0.02, or deviating from Hardy–Weinberg equilibrium (P < 0.0001), resulting in 7,738,399 bi-allelic SNPs for further analysis.
Survival analysis
OS was calculated from the date of cancer diagnosis to the date of death or to the last follow-up date. The maximum follow-up time was 5 years. Patients who were alive at the last follow-up date were censored. Median survival times were estimated and compared using the Kaplan–Meier plot and log-rank test. A Cox proportional hazards model was fitted to assess risk of death using hazard ratios (HRs) and 95% confidence intervals (95% CIs). Genotype data were analyzed using an additive inheritance model with adjustment for demographics such as age (continuous) and sex and for clinical predictors such as tumor stage (localized, locally advanced, or metastatic), tumor resection (performed or not performed) and chemotherapy (given or not given). In the discovery study, five principal components accounting for population substructure were also adjusted.
Statistical analysis used the R package version 3.1.0. We took P < 5.0 × 10−8 as genome-wide significant in the GWAS analysis and took P < 5.0 × 10−2 as nominally significant and P < 4.5 × 10−3 (P/the number of SNP tested) as Bonferroni-corrected significant in analyses of the validation and the combined GWAS and validation dataset.
Functional annotation and network analysis
We conducted functional annotation and linkage disequilibrium (LD) analysis of the top hit SNP using HaploReg version 4 21, RegulomeDB22 and PolyPhen-223. The association of selected SNPs and gene expression levels was evaluated using Genotype-Tissue Expression (GTEx) and NCBI eQTL database and the RNASeq level 3 data from TCGA. A previously conducted RNA-Seq study in paired normal and tumor tissues from 10 patients with resected PDAC tumors24 was also considered.
To explore functionally enriched biological pathways, we conducted network analysis on SNPs with a P value of <0.0005 using Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA [www.ingenuity.com]). SNPs were assigned to relevant genes using the University of California Santa Cruz (UCSC) Table Browser data retrieval tool25. For each gene region, SNPs within 20 kb upstream or downstream of the gene were included.
Replication analysis of SNPs reported in association with OS of PDAC
As a validation effort, we examined SNPs that were associated with PDAC survival in previous GWASs (P<10−5)7-10 in association with survival in the current MD Anderson GWAS dataset.
Results
Patient characteristics and OS
The distributions of sex, age, and tumor stage were similar between patients in the discovery and validation sets (P > 0.15 for each) (Table 1). However, more patients in the discovery set received chemotherapy than did those in the validation set (73.73% vs. 66.34%, P = 0.002), and more patients underwent tumor resection in the validation set than did those in the discovery set (34.15% vs. 29.26%, P = 0.031). As expected, disease stage and tumor resection status were strong predictors for OS (Table 1).
Table 1. Patient characteristics and overall survival.
| Variable | Discovery set | Validation set | Combined set | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||
| No. (%) | MSTa | P valueb | No. (%) | MST | P value | No. (%) | MST | P value | |
| Vital Status | |||||||||
| Dead | 800 (92.2) | 630 (76.8) | 1430 (84.7) | ||||||
| Alive | 68 (7.8) | 190 (23.27) | 258 (15.3) | ||||||
| Sex | |||||||||
| Male | 517 (59.6) | 14.7 | 492 (60.0) | 17.6 | 1009 (59.8) | 15.4 | |||
| Female | 351 (40.4) | 14.5 | 0.84 | 328 (40.0) | 17.9 | 0.70 | 679 (40.2) | 16.2 | 0.72 |
| Age (years)c | |||||||||
| ≤63 | 467 (53.8) | 15.2 | 407 (49.6) | 18.9 | 874 (51.8) | 15.9 | |||
| >63 | 401 (46.2) | 13.8 | 0.55 | 413 (50.4) | 16.6 | 0.18 | 814 (48.2) | 15.7 | 0.69 |
| Stage | |||||||||
| Local | 266 (30.7) | 32.1 | 277 (33.8) | 32.9 | 543 (32.2) | 32.1 | |||
| Locally advanced | 215 (24.8) | 13.9 | 214 (26.1) | 14.8 | 429 (25.4) | 14.7 | |||
| Metastatic | 387 (44.6) | 9.5 | <0.0001 | 329 (40.1) | 11.1 | <0.0001 | 716 (42.4) | 10.0 | <0.0001 |
| Chemotherapy | |||||||||
| Yes | 640 (73.73) | 14.9 | 545 (66.95) | 18 | 1185 (70.45) | 16.2 | |||
| No | 228 (26.27) | 13.9 | 0.74 | 269 (33.05) | 17.2 | 0.74 | 497 (29.55) | 14.8 | 0.28 |
| Resection | |||||||||
| Yes | 254 (29.26) | 33.7 | 280 (34.15) | 33.7 | 534 (31.64) | 33.7 | |||
| No | 614 (70.74) | 11.2 | <0.0001 | 540 (65.85) | 12.8 | <0.0001 | 1154 (68.36) | 12.0 | <0.0001 |
MST: median survival time in months.
P Values were derived from log-rank test.
Median age for all patients.
Discovery study
In the discovery study, with the use of the GWAS data, 528,088 SNPs were nominally associated with OS, and three SNPs in complete linkage disequilibrium reached genome-wide significance (Figure 1). Detailed information on the top 214 SNPs with a P value of less than or equal to 1 × 10−5 is given in STable 1. The top ten SNPs with the smallest P values in this analysis were all imputed, and their quality scores ranged from 0.556 to 0.995 (STable 1). To check the quality of imputation, we re-genotyped the top SNP, rs113988120, in the 868 GWAS samples using the Taqman method, and a concordance rate of 99.7% was observed. We estimated the genomic control inflation factor λ using the imputed genotype dosage after adjusting for the leading five principal components in the survival analysis. The quantile-quantile plot showed a slightly inflated λ value of 1.063 (SFigure 2). It is unlikely the inflation is due to population stratification because we have adjusted the five principal components accounting for population substructure in the analysis. It's more likely due to the survival analysis itself.
Fig. 1.

Manhattan plot for P values in survival analysis of the GWAS data.
The top three SNPs that showed a genome-wide significant association with survival are intronic SNPs (rs113988120) of the PAIP2B (poly(A) binding protein interacting protein 2B) and rs112493246 and rs138529893 of the DYSF (dysferlin) gene located on chromosome 2. The minor alleles of these three SNPs were significantly associated with shorter survival by a margin of 5.2 months (log-rank P = 1.6 × 10−5) (Figure 2A). Cox regression analysis with adjustment for demographic and clinical factors demonstrated a genome-wide significant (P ≤ 6.4 × 10−9) association of the three SNPs with increased risk of death (HR: 3.06, 95% CI: 2.10-4.47).
Fig. 2.
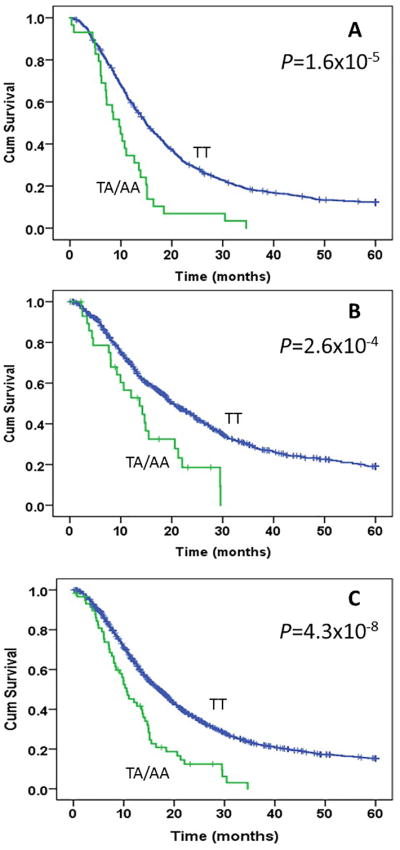
Kaplan-Meier plot for overall survival of patients with pancreatic cancer by rs113988120 genotype. Panels A, B, and C present survival curve in the discovery, validation, and combined datasets, respectively. The median survival time was 14.87 vs 9.67 months, 19.7 vs 13.3 months, and 16.9 vs 10.6 months for TT vs TA/AA genotype carriers and the number of patients with the at-risk (TA/AA) genotypes are 29, 31 and 60 in the discovery, replication and combined datasets, respectively. P values are from log-rank test.
Replication study and combined dataset
For the replication study, we selected 11 SNPs located in eight genes coding for PAIP2B, RAB6B, ZBTB2O, ROBO1, B4GALT4, UPK1B, LRRC15, and SLIT3; in a non-coding RNA (LOC101927026); and in an intergenic region (Table 2). The top hit rs113988120 was included in the replication study even though the MAF (0.015) was quite low (0.015). The allele frequencies of the 11 SNPs tested in the replication study were similar to those in the discovery dataset (Table 2). The variant allele of rs113988120 was associated with a 6.4 months and 6.3 months shorter survival in the replication and combined discovery and replication datasets, respectively (Figure 2B and 2C). A HR (95% CI) of 1.57 (1.13-2.20, P = 0.08) in the replication dataset and 1.86 (1.46-2.37, P = 4.6 × 10−7) in the combined dataset was observed for this variant allele in Cox proportion hazards regression analysis with adjustment for demographics, tumor stage, and resection status, and chemotherapy (Table 3). Other SNPs in the replication study were not significantly associated with OS (Table 3).
Table 2. Eleven SNPs tested in the validation and combined datasets.
| SNP | Gene | Quality | RegulomeDB | MAF | Discovery | Validation | Combined | |||
|---|---|---|---|---|---|---|---|---|---|---|
| (chromosome) | scorea | scoreb | N (Dead|Alive) | MSTd | N (Dead|Alive) | MST | N (Dead|Alive) | MST | ||
| rs113988120 (2) | PAIP2B | 0.9 | 6 | 0.015 | ||||||
| TT | 721|118 | 14.9 | 505|248 | 19.7 | 1226|366 | 16.3 | ||||
| TA | 29|0 | 9.7 | 20|5 | 14.7 | 49|5 | 10.3 | ||||
| AA | 0|0 | - | 4|1 | 13.3 | 4|1 | 13.7 | ||||
| rs4780973 (16) | LOC101927026 | 0.99 | 5 | 0.31 | ||||||
| CC | 377|53 | 12.9 | 267|109 | 17.6 | 644|162 | 14.7 | ||||
| CT | 315|50 | 15.07 | 228|119 | 17.6 | 543|169 | 16.2 | ||||
| TT | 58|15 | 21.7 | 44|27 | 22.4 | 102|42 | 21.93 | ||||
| rs9815265 (2) | RAB6B | 0.9 | 3 | 0.403 | ||||||
| TT | 279|36 | 13.4 | 165|86 | 19.5 | 444|122 | 14.8 | ||||
| TC | 362|55 | 14.6 | 264|108 | 19.1 | 626|163 | 15.6 | ||||
| CC | 109|27 | 22 | 126|65 | 18.7 | 235|92 | 18.8 | ||||
| rs6438217 (3) | ZBTB20 | 1 | 6 | 0.164 | ||||||
| TT | 492|93 | 15.7 | 367|173 | 19.5 | 859|266 | 16.6 | ||||
| TC | 234|22 | 13.9 | 166|80 | 19.5 | 400|102 | 14.8 | ||||
| CC | 24|3 | 10.5 | 19|5 | 13.3 | 43|8 | 12.4 | ||||
| rs12638565 (3) | ROBO1 | 0.93 | No data | 0.233 | ||||||
| CC | 448|72 | 16 | 310|143 | 20.7 | 758|215 | 17.3 | ||||
| CT | 264|43 | 13.1 | 180|87 | 17.6 | 444|130 | 14.4 | ||||
| TT | 38|3 | 9.5 | 60|27 | 19.7 | 98|30 | 13.7 | ||||
| rs4568126 (3) | B4GALT4 | 0.99 | 6 | 0.421 | ||||||
| CC | 266|35 | 13.4 | 120|59 | 18.4 | 386|94 | 14.5 | ||||
| CA | 361|53 | 14.3 | 249|123 | 18.8 | 610|176 | 15.4 | ||||
| AA | 123|30 | 18.6 | 187|77 | 20 | 310|107 | 18.9 | ||||
| rs12186112 (3) | UPK1B | 0.99 | 6 | 0.405 | ||||||
| AA | 286|40 | 12.9 | 199|82 | 19.5 | 485|122 | 14.7 | ||||
| AC | 342|52 | 14.8 | 250|122 | 19.5 | 592|174 | 16.3 | ||||
| CC | 122|26 | 18.3 | 93|49 | 18.1 | 215|75 | 17.4 | ||||
| rs13060627 (3) | LRRC15 | 1 | V264I | 0.264 | ||||||
| CC | 423|58 | 13.1 | 304|144 | 19 | 727|202 | 14.7 | ||||
| CT | 281|46 | 15.7 | 207|95 | 20.3 | 488|141 | 17.2 | ||||
| TT | 46|14 | 20 | 37|18 | 15 | 83|32 | 18.4 | ||||
| rs923936 (3) | LRRC15 | 1 | L133L | 0.234 | ||||||
| GG | 458|62 | 13.5 | 29|13 | 18.5 | 487|75 | 13.8 | ||||
| GA | 254|43 | 15.2 | 203|92 | 19.5 | 457|135 | 16.9 | ||||
| AA | 38|13 | 20.7 | 313|150 | 19 | 351|163 | 17.6 | ||||
| rs13173842 (5) | SLIT3 | 0.79 | 4 | 0.321 | ||||||
| CC | 363|59 | 15.1 | 246|116 | 18.9 | 609|175 | 16 | ||||
| CT | 305|52 | 14.8 | 237|112 | 20.1 | 542|164 | 16.3 | ||||
| TT | 82|7 | 12.4 | 60|27 | 17.3 | 142|34 | 13.7 | ||||
| rs10926274 (1) | NA | 0.72 | 4 | 0.483 | ||||||
| AA | 189|22 | 12.4 | 127|50 | 19.1 | 316|72 | 14.2 | ||||
| AG | 393|57 | 14.8 | 262|131 | 19.9 | 655|188 | 15.8 | ||||
| GG | 168|39 | 17 | 162|72 | 18.4 | 330|111 | 17 |
Imputation quality score.
Minor allele frequency based on imputation of the GWAS data.
Score 3a, 4,5, and 6 indicate TF binding + any motif + DNase peak, TF binding + DNase peak, TF binding or DNase peak, or other, respectively.
Median survival time in months.
Table 3. Cox regression on 11 SNPs in the discovery, validation and combined dataset.
| SNP | Discovery | Validation | Combined | |||
|---|---|---|---|---|---|---|
| HR (95%CI) | P value | HR (95%CI) | P value | HR (95%CI) | P value | |
| rs113988120 | 3.06 (2.10-4.47) | 6.40E-09 | 1.44 (1.02-2.02) | 0.037 | 1.79 (1.40-2.29) | 2.70E-06 |
| rs4780973 | 0.77 (0.68-0.86) | 2.60E-06 | 0.90 (0.79-1.02) | 0.11 | 0.82 (0.76-0.89) | 4.40E-06 |
| rs9815265 | 0.75 (0.68-0.83) | 6.70E-08 | 1.02 (0.92-1.14) | 0.70 | 0.87 (0.81-0.94) | 2.70E-04 |
| rs6438217 | 1.40 (1.23-1.59) | 4.40E-07 | 1.01 (0.86-1.17) | 0.94 | 1.19 (1.08-1.31) | 5.80E-04 |
| rs12638565 | 1.35 (1.19-1.52) | 1.80E-06 | 1.04 (0.92-1.17) | 0.57 | 1.14 (1.05-1.24) | 2.10E-03 |
| rs4568126 | 0.77 (0.70-0.85) | 3.10E-07 | 0.97 (0.87-1.09) | 0.64 | 0.85 (0.79-0.91) | 6.10E-06 |
| rs12186112 | 0.78 (0.70-0.86) | 6.00E-07 | 0.99 (0.88-1.12) | 0.92 | 0.87 (0.81-0.94) | 3.60E-04 |
| rs13060627 | 0.79 (0.70-0.88) | 4.50E-05 | 0.94 (0.82-1.07) | 0.33 | 0.85 (0.78-0.93) | 2.40E-04 |
| rs923936 | 0.79 (0.70-0.88) | 5.90E-05 | 1.08 (0.94-1.24) | 0.27 | 0.88 (0.82-0.94) | 1.30E-04 |
| rs13173842 | 1.27 (1.14-1.42) | 1.30E-05 | 0.97 (0.86-1.10) | 0.61 | 1.11 (1.02-1.20) | 1.40E-02 |
| rs10926274 | 0.83 (0.75-0.91) | 2.20E-04 | 1.02 (0.91-1.14) | 0.72 | 0.91 (0.84-0.98) | 1.50E-02 |
Cox regression analysis was under additive genetic model with adjustment for age, sex, tumor stage, resection, and chemotherapy in validation and combined datasets and with additional adjustment for five principal components accounting for population structure in the discovery set.
Next we assessed the impact of rs113988120 on survival by disease stage. The median survival time was 34.3 vs 14.9 months (Plog–rank <0.001), 15.7 vs 10.3 months (Plog–rank <0.001), and 10.6 vs 8.0 months (Plog–rank =0.318) for TT vs TA/AA genotype carriers for patients with localized, locally advanced and metastatic disease, respectively (SFigure 2).
Functional annotation and gene expression analysis
RegulomeDB analysis suggest that it may have an undefined altered mortif with a score of 6, and it is located at a DNAseI hypersensitive site and an enhancer region of the chromatin, which make it more accessible for the binding of proteins such as transcription factors. HaploReg analysis demonstrated that rs113988120 was in LD (r2=0.49, D′=0.77) with three eQTL SNPs (rs34634781, rs35098046 and rs34022557) near or belong to the NAGK (N-acetylglucosamine kinase) and MCEE (methylmalonyl-CoA epimerase) gene on chromosome 2, in addition to the two SNPs of the DYSF gene (STable 2). Cox regression analysis of the GWAS data showed that rs34634781 had an HR of 2.44 (95% CI: 1.70 -3.49, P = 1.10 × 10-6) and s35098046/rs34022557 had an HR of 2.43 (95% CI: 1.71 -3.46 P = 8.50 × 10-7) (STable 1).
Using TCGA RNA-seq level 3 data from four PDAC patients, we found that PAIP2B expression was downregulated in tumors (mean=90.58) compared with paired normal tissues from the same patient (mean = 150.22) (P = 0.23, paired t-test). This observation agreed with data from our previous RNA-seq study on paired tumor-normal tissues of 10 PDAC patients, which showed a 7.8-fold lower expression of PAIP2B in tumors than in normal adjacent tissues (P = 0.00032)24. Notably, the expression of PAIP2B was significantly reduced in tumors compared with that in normal adjacent tissues of other gastrointestinal cancers according to TCGA RNA-seq data (Ps ≤ 8 × 10−5) (Table 4). We did not detect a significant association of PAIP2B expression with patient survival in the entire TCGA dataset of 178 tumor samples (P = 0.30). Among patients with stage II or earlier disease (n=48), those with lower (below median) PAIP2B expression exhibited a non-significant shorter survival (by 39 months, Plog–rank = 0.10) and increased risk of death than those with higher (at or above median) PAIP2B expression (HR=2.45, 95%CI: 0.90-6.80, P = 0.081). The mRNA expression levels of NAGK, MCEE and DYSF genes were not significantly different between tumor and normal adjacent tissues and were not related to survival (data not shown).
Table 4. Expression of PAIP2B in gastrointestinal cancers in TCGA databases.
| Cancer type | No. of patients | Mean expression (tumor|normal) | P valuea |
|---|---|---|---|
| Pancreatic cancer | 4 | 90.58|150.22 | 2.30E-01 |
| Cholangiocarcinoma | 9 | 107.93|950.49 | 1.40E-07 |
| Colon adenocarcinoma | 26 | 166.67|253.97 | 5.30E-05 |
| Colorectal adenocarcinoma | 32 | 171.43|264.07 | 2.40E-06 |
| Hepatocellular carcinoma | 50 | 569.4|1009.75 | 8.00E-05 |
| Stomach adenocarcinoma | 32 | 116.79|414.84 | 4.90E-08 |
Derived from paired t-test
Ingenuity Pathway Analysis (IPA)
Next, we conducted IPA on 162 genes whose tagging SNPs had P < 0.0005 in Cox regression analysis. The top four most significant pathways identified were G beta-gamma signaling (P = 1.77 × 10−5, q value = 0.012), CXCR4 signaling (P = 8.82 × 10−5, q value = 0.03), CREB signaling in neurons (P = 1.99 × 10−4, q value = 0.07), and CCR5 signaling in macrophages (P = 4.51 × 10−4, q value = 0.08) and Agrin interactions at neuromuscular junction (P = 4.47 × 10−4, q value = 0.08) (STable 3). When we reran IPA on genes selected at various P value thresholds ranging from 0.008 to 0.00001, the top four pathways remained almost unchanged (data not shown).
Replication analysis of SNPs previously associated with OS or risk of PC
We analyzed the top SNPs associated with PC survival in previous GWAS studies 7-10 and found that 11 of 131 such SNPs (P<10−5) in the study by Wu et al. 7 were nominally significant in our study (P < 0.05) (STable 4).
Discussion
In this GWAS study, we discovered a significant association of a polymorphic variant (rs113988120) of the PAIP2B gene with shorter survival in patients with PADC and confirmed this association in a replication study. This is, to our knowledge, the first report of a genome-wide association of germline genetic variation and PDAC survival in a GWAS dataset.
The rs113988120 SNP is located in the intron region of the PAIP2B gene. In silico analysis suggested weak evidence for possible adverse functional consequences of this SNP but it is not related to gene expression. Furthermore, this SNP is located in a DNAseI hypersensitive and chromatin enhancer region, which suggests more accessibility for transcription factor binding. Furthermore, rs113988120 is in LD with five SNPs, including three eQTLs, near or located at the NAGK, MCEE, and DYSF genes and all five SNPs were among the top hits identified from the GWAS dataset with P values less than or equal to 1.1 × 10-6. Thus, which gene or SNP is truly responsible for the observed association with PDAC survival is unclear at present. Further fine mapping and functional studies are required to identify the causal allele and to understand the mechanisms involved.
At the gene level, the gene expression data support a tumor suppressor role of PAIP2B and possible link with patient survival. While a significantly reduced expression of PAIP2B mRNA was observed in pancreatic cancer24 and several other gastrointestinal cancers (TCGA data), the mRNA expression level of NAGK, MCEE, and DYSF genes were not significantly different between tumor and normal tissues. In addition, the reduced expression of PAIP2B was related to shorter survival in PADC patients with early-stage disease (N=48) even though the difference was not statistically significant. Because of the sample size is small, this association needs further investigation.
PAIP2B is a translational inhibitor26 that regulates poly(A) binding protein activity. Poly(A) binding protein enhances translation by circularizing mRNA through its interaction with the translation initiation factor EIF4G1 and the poly(A) tail27. PAIP2B regulates the translation of many genes that have important biological significance in cancer28, 29. For example, it is a strong regulator of vascular endothelial growth factor30 and is an anti-proliferative factor31. Although we have no evidence to link SNP rs113988120 with the expression of the PAIP2B gene, it is conceivable that the polymorphic variants may result in altered poly(A) binding protein binding activity, which in turn leads to upregulated expression of many pro-proliferative or metastatic genes that contribute to reduced patient survival. Further replication studies in other GWAS datasets and functional studies on PAIP2B in pancreatic carcinogenesis are warranted to confirm the observed association and to understand the mechanisms underlying the association.
DYSF is a skeletal muscle protein, which plays a role in calcium-mediated membrane fusion events, suggesting that it may be involved in membrane regeneration and repair32. Mutation of these genes have been associated with sarcolemma in animal model33, 34. The NAGK gene encodes N-acetylhexosamine kinase that catalyzes the conversion of N-acetyl-D-glucosamine to N-acetyl-D-glucosamine 6-phosphate, and is the major mammalian enzyme which recovers amino sugars. NAGK has been associated with speckle, paraspeckle and general transcription factor suggesting its regulatory roles in gene expression 35. NAGK gene mutation has been related to inclusion body myopathy 36. MCEE catalyzes the interconversion of D- and L-methylmalonyl-CoA during the degradation of branched chain amino acids, odd chain-length fatty acids, and other metabolites. Mutations in this gene result in methylmalonyl-CoA epimerase deficiency, which is presented as mild to moderate methylmalonic aciduria37. Whether these two genes play any role in cancer is currently unknown.
Network analysis identified several signaling pathways that are significantly associated with survival, such as the G beta-gamma and CXCR4 signaling pathways. The CXCL12/CXCR4 axis may promote dissociation of the G beta-gamma complex (Gβγ), then activate PI3K-AKT and Rho-ROCK-MLC pathways to promote cancer cell survival and migration38. A high level of CXCL12/CXCR4 expression was reported to be significantly associated with metastasis and low OS in many types of cancer, including PDAC39, 40. Suppression of the CXCL12/CXCR4 axis contributed to immune control of pancreatic ductal cancer growth41. Our findings on the CXCL12/CXCR4 pathway genes may have potential value in future PDAC therapy targeting this particular pathway.
The strengths of the current study include a large sample size and a relatively homogeneous study population from the same institution. As part of a hospital-based study, the clinical data are relatively accurate and complete. The existing large databases such as TCGA and GTEx have facilitated the functional annotation of the GWAS top hits. However, the number of SNPs selected for validation was limited because of cost constraints. We may have missed some important ones. The low MAF of the identified SNP and the small number of samples available for PC gene expression analysis limited our ability to fully characterize this gene variant.
Overall, our study has reported a low-frequency SNP of the translation inhibitor gene PAIP2B with a significant association with PC survival. These data need further validation in other datasets. If confirmed, they may open a new research avenue in illustrating the molecular mechanisms underlying the clinical phenotypes and offer a potential tool in identifying therapeutic targets for future individualized cancer treatment.
Supplementary Material
SFigure 1. Quantile-Quantile (QQ) plot of observed vs. expected P values for the discovery set. The genomic control inflation factor λ was calculated using the imputed genotype dosage after adjusting for the leading five principal components account for population structure in the survival analysis
SFigure 2. Kaplan-Meier plot for overall survival of patients with pancreatic cancer by rs113988120 genotype. Panels A, B, and C present survival curve for patients with localized, locally advanced and metastatic disease, respectively. The median survival time was 34.3 vs 14.9 months, 15.7 vs 10.3 months, and 10.6 vs 8.0 months for TT vs TA/AA genotype carriers and the number of patients with the at-risk (TA/AA) genotypes are 15, 16 and 29 for patients with localized, locally advanced and metastatic disease, respectively. P values are from log-rank test.
STable 1. Top SNPs (P < 0.00001) associated with overall survival of pancreatic cancer patients in PanScanII + PanC4
STable 2. SNP: rs113988120 and variants with r2 ≥ 0.4
STable 3. IPA analysis of significant canonical pathways in the GWAS dataset
STable 4. Top SNPs associated with survival of pancreatic cancer by previous GWAS (P<1.0 × 10-5).
Novelty and Significance.
We found a significant association of a single nucleotide polymorphic variant and pancreatic cancer survival in a two stage study. This finding may help to identify novel tumor suppressor gene and therapeutic target for pancreatic cancer.
Acknowledgments
We thank all our collaborators in the PanScan II and PanC4 GWASs. We appreciate all participants who took part in this research and the funders and technical staff who made this study possible. We thank Ms. Sarah Bronson for editing this manuscript.
Grant sponsor: Supported by National Institutes of Health grant R01CA169122 (to P.W.) and the Sheikh Ahmed Bin Zayed Al Nahyan Center for Pancreatic Cancer Research Funds (to D.L.). The University of Texas MD Anderson Cancer Center is supported in part by the National Institutes of Health through Cancer Center Support Grant P30CA016672.
Abbreviations
- CI
confidence interval
- eQTL
expression quantitative trait locus
- GWAS
genome wide association study
- GTEx
Genotype-Tissue Expression
- HR
hazard ratio
- LD
linkage disequilibrium
- MAF
minor allele frequency
- OS
overall survival
- PDAC
pancreatic ductal adenocarcinoma
- SNP
single nucleotide polymorphism, TCGA, The Cancer Genome Atlas
References
- 1.SEER Stat Fact Sheets: Pancreas Cancer. http://seer.cancer.gov/statfacts/html/pancreas.html.
- 2.Li D, Liu H, Jiao L, Chang DZ, Beinart G, Wolff RA, Evans DB, Hassan MM, Abbruzzese JL. Significant effect of homologous recombination DNA repair gene polymorphisms on pancreatic cancer survival. Cancer Res. 2006;66:3323–30. doi: 10.1158/0008-5472.CAN-05-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Couch FJ, Wang X, Bamlet WR, de Andrade M, Petersen GM, McWilliams RR. Association of mitotic regulation pathway polymorphisms with pancreatic cancer risk and outcome. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2010;19:251–7. doi: 10.1158/1055-9965.EPI-09-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okazaki T, Javle M, Tanaka M, Abbruzzese JL, Li D. Single nucleotide polymorphisms of gemcitabine metabolic genes and pancreatic cancer survival and drug toxicity. Clin Cancer Res. 2010;16:320–9. doi: 10.1158/1078-0432.CCR-09-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong X, Javle M, Hess KR, Shroff R, Abbruzzese JL, Li D. Insulin-like growth factor axis gene polymorphisms and clinical outcomes in pancreatic cancer. Gastroenterology. 2010;139:464–73. 73 e1–3. doi: 10.1053/j.gastro.2010.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong X, Tang H, Hess KR, Abbruzzese JL, Li D. Glucose metabolism gene polymorphisms and clinical outcome in pancreatic cancer. Cancer. 2011;117:480–91. doi: 10.1002/cncr.25612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu C, Kraft P, Stolzenberg-Solomon R, Steplowski E, Brotzman M, Xu M, Mudgal P, Amundadottir L, Arslan AA, Bueno-de-Mesquita HB, Gross M, Helzlsouer K, et al. Genome-wide association study of survival in patients with pancreatic adenocarcinoma. Gut. 2014;63:152–60. doi: 10.1136/gutjnl-2012-303477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willis JA, Olson SH, Orlow I, Mukherjee S, McWilliams RR, Kurtz RC, Klein RJ. A replication study and genome-wide scan of single-nucleotide polymorphisms associated with pancreatic cancer risk and overall survival. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:3942–51. doi: 10.1158/1078-0432.CCR-11-2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Innocenti F, Owzar K, Cox NL, Evans P, Kubo M, Zembutsu H, Jiang C, Hollis D, Mushiroda T, Li L, Friedman P, Wang L, et al. A genome-wide association study of overall survival in pancreatic cancer patients treated with gemcitabine in CALGB 80303. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:577–84. doi: 10.1158/1078-0432.CCR-11-1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizzato C, Campa D, Talar-Wojnarowska R, Halloran C, Kupcinskas J, Butturini G, Mohelnikova-Duchonova B, Sperti C, Tjaden C, Ghaneh P, Hackert T, Funel N, et al. Association of genetic polymorphisms with survival of pancreatic ductal adenocarcinoma patients. Carcinogenesis. 2016 doi: 10.1093/carcin/bgw080. [DOI] [PubMed] [Google Scholar]
- 11.Li D, Morris JS, Liu J, Hassan MM, Day RS, Bondy ML, Abbruzzese JL. Body mass index and risk, age of onset, and survival in patients with pancreatic cancer. JAMA. 2009;301:2553–62. doi: 10.1001/jama.2009.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Childs EJ, Mocci E, Campa D, Bracci PM, Gallinger S, Goggins M, Li D, Neale RE, Olson SH, Scelo G, Amundadottir LT, Bamlet WR, et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat Genet. 2015;47:911–6. doi: 10.1038/ng.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, Arslan AA, Bueno-de-Mesquita HB, Gallinger S, Gross M, Helzlsouer K, Holly EA, et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010;42:224–8. doi: 10.1038/ng.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, et al. A second generation human haplotype map of over 3.1 million. SNPs Nature. 2007;449:851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for Genome-wide Complex Trait Analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nature methods. 2012;9:179–81. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 18.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–13. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 19.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nature genetics. 2012;44:955–9. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic acids research. 2012;40:D930–4. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–7. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mao Y, Shen J, Lu Y, Lin K, Wang H, Li Y, Chang P, Walker MG, Li D. RNA sequencing analyses reveal novel differentially expressed genes and pathways in pancreatic cancer. Oncotarget. 2017 doi: 10.18632/oncotarget.16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karolchik DHA, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32(Database issue):D493–6. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berlanga JJ, Baass A, Sonenberg N. Regulation of poly(A) binding protein function in translation: Characterization of the Paip2 homolog, Paip2B. Rna. 2006;12:1556–68. doi: 10.1261/rna.106506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Derry MC, Yanagiya A, Martineau Y, Sonenberg N. Regulation of poly(A)-binding protein through PABP-interacting proteins. Cold Spring Harbor symposia on quantitative biology. 2006;71:537–43. doi: 10.1101/sqb.2006.71.061. [DOI] [PubMed] [Google Scholar]
- 28.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 2015;162:425–40. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimura K, Wakamatsu A, Suzuki Y, Ota T, Nishikawa T, Yamashita R, Yamamoto J, Sekine M, Tsuritani K, Wakaguri H, Ishii S, Sugiyama T, et al. Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome research. 2006;16:55–65. doi: 10.1101/gr.4039406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Onesto C, Berra E, Grepin R, Pages G. Poly(A)-binding protein-interacting protein 2, a strong regulator of vascular endothelial growth factor mRNA. J Biol Chem. 2004;279:34217–26. doi: 10.1074/jbc.M400219200. [DOI] [PubMed] [Google Scholar]
- 31.Ezzeddine N, Chang TC, Zhu W, Yamashita A, Chen CY, Zhong Z, Yamashita Y, Zheng D, Shyu AB. Human TOB, an antiproliferative transcription factor, is a poly(A)-binding protein-dependent positive regulator of cytoplasmic mRNA deadenylation. Mol Cell Biol. 2007;27:7791–801. doi: 10.1128/MCB.01254-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Redpath GM, Woolger N, Piper AK, Lemckert FA, Lek A, Greer PA, North KN, Cooper ST. Calpain cleavage within dysferlin exon 40a releases a synaptotagmin-like module for membrane repair. Molecular biology of the cell. 2014;25:3037–48. doi: 10.1091/mbc.E14-04-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmidt WM, Uddin MH, Dysek S, Moser-Thier K, Pirker C, Hoger H, Ambros IM, Ambros PF, Berger W, Bittner RE. DNA damage, somatic aneuploidy, and malignant sarcoma susceptibility in muscular dystrophies. PLoS Genet. 2011;7:e1002042. doi: 10.1371/journal.pgen.1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hosur V, Kavirayani A, Riefler J, Carney LM, Lyons B, Gott B, Cox GA, Shultz LD. Dystrophin and dysferlin double mutant mice: a novel model for rhabdomyosarcoma. Cancer genetics. 2012;205:232–41. doi: 10.1016/j.cancergen.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharif SR, Lee H, Islam MA, Seog DH, Moon IS. N-acetyl-D-glucosamine kinase is a component of nuclear speckles and paraspeckles. Molecules and cells. 2015;38:402–8. doi: 10.14348/molcells.2015.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mori-Yoshimura M, Monma K, Suzuki N, Aoki M, Kumamoto T, Tanaka K, Tomimitsu H, Nakano S, Sonoo M, Shimizu J, Sugie K, Nakamura H, et al. Heterozygous UDP-GlcNAc 2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE gene result in a less severe GNE myopathy phenotype compared to homozygous N-acetylmannosamine kinase domain mutations. Journal of the neurological sciences. 2012;318:100–5. doi: 10.1016/j.jns.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 37.Gradinger AB, Belair C, Worgan LC, Li CD, Lavallee J, Roquis D, Watkins D, Rosenblatt DS. Atypical methylmalonic aciduria: frequency of mutations in the methylmalonyl CoA epimerase gene (MCEE) Human mutation. 2007;28:1045. doi: 10.1002/humu.9507. [DOI] [PubMed] [Google Scholar]
- 38.Debnath B, Xu S, Grande F, Garofalo A, Neamati N. Small molecule inhibitors of CXCR4. Theranostics. 2013;3:47–75. doi: 10.7150/thno.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang JJ, Zhu S, Bruggeman R, Zaino RJ, Evans DB, Fleming JB, Gomez HF, Zander DS, Wang H. High levels of expression of human stromal cell-derived factor-1 are associated with worse prognosis in patients with stage II pancreatic ductal adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2010;19:2598–604. doi: 10.1158/1055-9965.EPI-10-0405. [DOI] [PubMed] [Google Scholar]
- 40.Krieg A, Riemer JC, Telan LA, Gabbert HE, Knoefel WT. CXCR4--A Prognostic and Clinicopathological Biomarker for Pancreatic Ductal Adenocarcinoma: A Meta-Analysis. PloS one. 2015;10:e0130192. doi: 10.1371/journal.pone.0130192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, Teichmann SA, Janowitz T, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:20212–7. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SFigure 1. Quantile-Quantile (QQ) plot of observed vs. expected P values for the discovery set. The genomic control inflation factor λ was calculated using the imputed genotype dosage after adjusting for the leading five principal components account for population structure in the survival analysis
SFigure 2. Kaplan-Meier plot for overall survival of patients with pancreatic cancer by rs113988120 genotype. Panels A, B, and C present survival curve for patients with localized, locally advanced and metastatic disease, respectively. The median survival time was 34.3 vs 14.9 months, 15.7 vs 10.3 months, and 10.6 vs 8.0 months for TT vs TA/AA genotype carriers and the number of patients with the at-risk (TA/AA) genotypes are 15, 16 and 29 for patients with localized, locally advanced and metastatic disease, respectively. P values are from log-rank test.
STable 1. Top SNPs (P < 0.00001) associated with overall survival of pancreatic cancer patients in PanScanII + PanC4
STable 2. SNP: rs113988120 and variants with r2 ≥ 0.4
STable 3. IPA analysis of significant canonical pathways in the GWAS dataset
STable 4. Top SNPs associated with survival of pancreatic cancer by previous GWAS (P<1.0 × 10-5).