

Abstract
Inflammation plays a crucial role in cardiac repair, but may also extend ischemic injury and contribute to post-infarction remodeling. This review manuscript discusses recent advances in our understanding of the cell biology of the post-infarction inflammatory response. Recently published studies demonstrated that the functional repertoire of inflammatory and reparative cells may extend beyond the roles suggested by traditional teachings. Neutrophils may play an important role in cardiac repair by driving macrophages toward a reparative phenotype. Subsets of activated fibroblasts have been implicated in protection of ischemic cardiomyocytes, in phagocytosis of apoptotic cells, and in regulation of inflammation. Dissection of the cellular effectors of cardiac repair is critical in order to develop new therapeutic strategies for patients with acute myocardial infarction.
Introduction
Implementation of early reperfusion strategies significantly reduced acute mortality in patients with myocardial infarction. However, this important therapeutic success resulted in expansion of the pool of patients who while surviving the acute event, exhibit adverse cardiac remodeling, and are susceptible to development of chronic heart failure. Following myocardial infarction, sudden death of up to a billion cardiomyocytes triggers an inflammatory response that plays a crucial role in repair of the infarcted heart, but is also implicated in the pathogenesis of adverse ventricular remodeling [1]. Excessive, prolonged, or dysregulated inflammatory responses following myocardial infarction are associated with accentuated dilation, infarct expansion and increased fibrosis and may worsen ventricular dysfunction [2]. Despite a growing understanding of the molecular signals regulating the post-infarction inflammatory response, therapeutic targeting of inflammatory mediators in patients with myocardial infarction has proved challenging [3]. This manuscript will discuss recent advances that contributed to our understanding of the role of inflammatory pathways in cardiac injury, repair and remodeling following myocardial infarction.
Activation of the post-infarction inflammatory response
Release of damage-associated molecular patterns (DAMPs) by dying cells represents the key molecular link between cardiomyocyte death and activation of the post-infarction inflammatory response. Experimental studies have suggested that a wide range of danger signals, such as high mobility group box-1 (HMGB1) [4,5], Interleukin (IL)-1α [6], and extracellular RNAs [7] are released by dying cells and activate innate immune pathways. Extracellular matrix protein fragments are rapidly generated in the infarcted heart and may also activate inflammatory cascades, linking injury of the interstitial matrix with the inflammatory response [8]. Recently published work has added several mediators to the list of alarmins, capable of activating inflammatory signaling following myocardial infarction. Mitochondrial DNA is released following cardiac injury and may activate innate immune pathways extending cardiomyocyte injury [9]. Fragments of sarcomeric proteins are generated following infarction and may also activate inflammatory cascades [10]. DAMPs have a wide range of likely cellular targets, including resident cardiac macrophages and mast cells, vascular endothelial cells, fibroblasts, and infiltrating leukocytes (Figure 1). The cellular specificity of the alarmins released in the infarcted myocardium has not been investigated.
Figure 1.
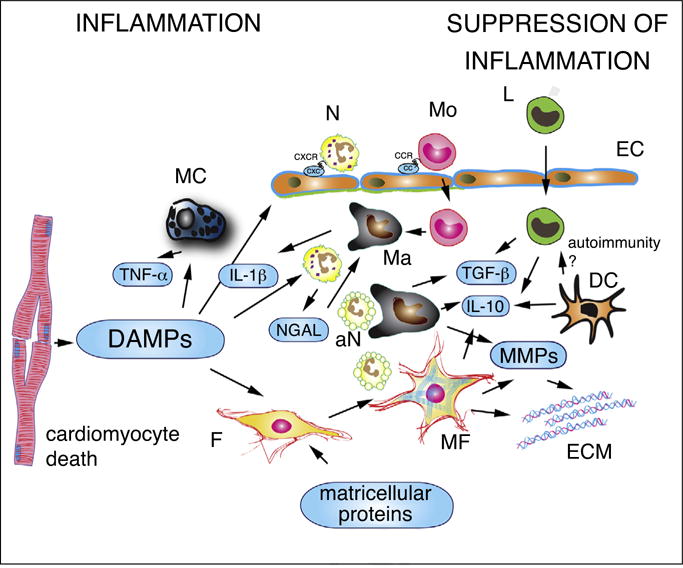
The cell biology of the inflammatory response following myocardial infarction. Dying cardiomyocytes release damage-associated molecular patterns (DAMPs), activating inflammatory signaling in resident macrophages (Ma), mast cells (MC) and fibroblasts (F) and in infiltrating leukocytes. Secretion of pro-inflammatory cytokines (such as IL-1β and TNF-α) stimulates expression of CXC and CC chemokines in endothelial cells (EC), promoting recruitment of neutrophils (N), monocytes (Mo) and lymphocytes (L). In addition to their pro-inflammatory actions, neutrophils may also trigger polarization of reparative macrophages. Phagocytotic macrophages clear the infarct from dead cells (including apoptotic neutrophils, aN) and acquire anti-inflammatory properties, releasing TGF-β and IL-10. Recently published experimental work suggests that myofibroblasts (MF) may also play a role in phagocytosis of dead cells and in negative regulation of the inflammatory reaction. Dendritic cells (DC) are also activated in the infarcted myocardium and have been suggested to inhibit pro-inflammatory signaling by secreting IL-10. Activation of autoimmune pathways by dendritic cells has been suggested, but remains poorly documented in myocardial infarction. Matricellular proteins play an important role in regulating inflammatory and reparative responses in the infarcted myocardium. Matrix metalloproteinases (MMPs) secreted by hematopoietic cells and fibroblasts are involved in extracellular matrix metabolism and regulate inflammation by modulating activity of inflammatory mediators.
Endothelial cells
Endothelial cells are the most abundant non-cardiomyocytes in the adult mouse myocardium [11] and represent a major source of pro-inflammatory chemokines [12,13] following myocardial infarction. Moreover, induction of adhesion molecules on the endothelial surface plays an important role in recruitment of inflammatory leukocytes in the healing infarct. The signals responsible for inflammatory activation of endothelial cells in the infarcted myocardium remain poorly understood. Recently published studies have suggested that in the infarcted myocardium, natriuretic peptides released by cardiomyocytes, may activate an inflammatory phenotype in endothelial cells, promoting leukocyte recruitment [14]. Moreover, activation of forkhead box protein O4 (FoxO4), a transcription factor involved in regulation ofmany cellular pathways has also been implicated in inflammatory activation of endothelial cells following myocardial infarction [15•]. The upstream signals responsible for FoxO4 activation in the endothelium have not been identified.
Neutrophils
Myeloid cells are rapidly recruited in the infarcted myocardium and have been implicated in clearance of dead cells, repair, fibrosis, and remodeling of the infarcted heart [16]. Abundant neutrophils infiltrate the infarcted myocardium in response to induction of chemotactic signals, including chemokines, complement and leukotrienes. Although early investigations demonstrated that neutrophils may exert cytotoxic effects on viable cardiomyocytes in the infarct border zone extending ischemic injury [17], current concepts suggest that neutrophils may also have reparative functions, by regulating recruitment and activation of mononuclear cells. Secretory products of neutrophils and platelets have been suggested to act synergistically in driving monocyte recruitment in the infarcted heart [18]. A recent study showed that neutrophil depletion using infusion of an Ly6G-specific monoclonal antibody did not affect infarct size but caused progressive deterioration in cardiac function following myocardial infarction. The beneficial effects of neutrophils were presumed due to macrophage polarization toward a reparative phenotype and were mediated, at least in part, through secretion of neutrophil gelatinase-associated lipocalin (NGAL), a key regulator of macrophage function [19•]. It should be emphasized that neutrophil depletion studies in experimental models of myocardial infarction have produced conflicting results [20,21,19•], likely reflecting differences in the effectiveness and specificity of various approaches and the wide range of functions of neutrophils in injured tissues. In addition to transducing reparative signals, neutrophils also secrete injurious mediators, such as myeloperoxidase, implicated in the pathogenesis of adverse remodeling and arrhythmogenesis following myocardial infarction [22]. Moreover, neutrophils infiltrating the infarcted heart may exhibit phenotypic heterogeneity. A recent study identified abundant early recruitment of pro-inflammatory N1 neutrophils in the infarcted heart, followed by late infiltration with N2 cells [23]. Activation of Toll-like receptor (TLR)4 signaling was implicated in N1 polarization, suggesting that DAMPs released in the infarct may stimulate a pro-inflammatory phenotype in neutrophils.
Macrophages in cardiac repair, remodeling and fibrosis
The adult mammalian heart contains a relatively small population of resident macrophages [24,25]; the role of these cells in cardiac homeostasis remains unclear. Recent studies in the mouse suggested that cardiac macrophages may play an important role in facilitating cardiac conduction [26•]; the significance of these observations in function of the human conduction system remains unknown. Following myocardial infarction, chemokine-driven recruitment of monocytes [27] results in marked expansion of the macrophage population in the infarcted region. Infarct macrophages exhibit phenotypic and functional heterogeneity and orchestrate the inflammatory and reparative response. Efferocytosis of apoptotic cardiomyocytes by macrophages suppresses expression of pro-inflammatory mediators and may drive resolution of inflammation following myocardial infarction [28]. Activation of negative regulators of the innate immune response, such as Interleukin receptor-associated kinase (IRAK)-M in macrophage subsets, suppresses inflammation and attenuates protease activation in the infarcted heart [29]. A subpopulation of alternatively activated M2-like macrophages activates a reparative program in fibroblasts, promoting repair of the infarcted heart [30]. Macrophages may also serve as an important source of Vascular endothelial growth factor (VEGF)-A in the infarcted heart, stimulating repair through activation of infarct angiogenesis [31•].
Which signals activate myeloid cells in the infarcted myocardium?
Myeloid cells respond to the dynamic changes in their microenvironment and acquire a wide range of functional phenotypes. Growth factors and cytokines, neurohumoral mediators, and components of the extracellular matrix network play an important role in modulating myeloid cell function. During the early pro-inflammatory phase of infarct healing, IL-1 promotes a pro-inflammatory macrophage phenotype inducing chemokine synthesis and protease activation [32]. Members of the bone morphogenetic protein family (BMP) may also stimulate pro-inflammatory signaling [33], whereas release of TGF-β by macrophages ingesting apoptotic cells may suppress inflammatory gene synthesis, marking the transition to the reparative phase. Maintenance and proliferation of macrophage populations in the infarcted myocardium and acquisition of an M2-like phenotype require activation of CSF-1 signaling cascades [34•,35]. Neurohumoral pathways and mechanosensitive signaling have been suggested as important modulators of macrophage activation [36,37]. Adrenergic signaling cascades may critically modulate leukocyte function following myocardial infarction. β2 adrenergic receptor signaling in leukocytes was found to be important for recruitment of leukocytes into the infarcted myocardium, and for repair following infarction [38,39•]. On the other hand, the β1 adrenergic receptor cascade has been suggested to exert detrimental effects, by promoting neutrophil-mediated injury following myocardial infarction [40]. Considering the wide range of cell types targeted by neurohumoral mediators, the relative significance of leukocyte-specific effects of catecholamines or angiotensin II in regulation of post-infarction injury and repair remains unknown.
Lymphocytes
Lymphocyte subpopulations are rapidly recruited in the infarcted myocardium and may downmodulate postinfarction inflammation by secreting inhibitory cytokines, such as IL-10 [41,42]. A growing body of evidence implicates regulatory T cells (Tregs) in suppression of postinfarction inflammation [43]. Although relatively small numbers of regulatory T cells (Tregs) infiltrate the infarcted myocardium, these cells seem to have important effects on macrophage and fibroblast phenotype [44,45]. A recent study suggested that epicardial activation of YAP and TAZ, two core Hippo pathway effectors, may suppress post-infarction inflammation and fibrosis by inducing Interferon (IFN)-γ and by promoting recruitment of Tregs [46]. The anti-fibrotic effects of IFN-γ may involve upregulation of the anti-fibrotic chemokine CXCL10/IFN-γ-inducible protein (IP)-10 [47].
Mast cells and dendritic cells
Early studies in a canine model of reperfused myocardial infarction demonstrated that in ischemic myocardial segments, cardiac mast cells degranulate, releasing histamine, mast cell-specific proteases and a wide range of cytokines and growth factors [48–50]. In comparison to large animals, mice have much lower numbers of cardiac mast cells and exhibit modest mast cell infiltration following myocardial infarction [51,52]. A recent study in a mouse model of myocardial infarction demonstrated that cardiac mast cells may be protective, enhancing cardiomyocyte contractility following myocardial infarction. Mast cell-mediated preservation of cardiomyocyte function was attributed to tryptase secretion and subsequent activation of protease-activated receptor (PAR)-2, leading to reduction in protein kinase A (PKA) activity and modulation of myofilament protein phosphorylation [53•]. Considering the wide range of mediators secreted by mast cells, and their diverse actions on many cell types involved in cardiac remodeling, the vivo role of this specific pathway remains unclear.
Dendritic cells are also activated following myocardial infarction; however, their role in regulation of post-infarction inflammation remains unclear. Depletion experiments in a mouse model of myocardial infarction suggested that dendritic cells may downmodulate post-infarction inflammation by secreting anti-inflammatory mediators, such as IL-10 [54]. In contrast, a recent study suggested that dendritic cells undergo activation following infarction and may prime cardiac-specific autoreactive CD4+ T cells [55]. However, the potential involvement of autoimmunity in extending or exacerbating injury following infarction has not been documented.
The cardiac fibroblasts as regulators of inflammation
The adult mammalian heart contains a significant population of resident fibroblast-like cells [11]. Following infarction, fibroblasts undergo myofibroblast conversion, express contractile proteins such as α-smooth muscle actin (α-SMA) and secrete large amounts of extracellular matrix proteins, serving as the main matrix-secreting cells in the healing scar [56–58]. Recent evidence suggests that infarct fibroblasts may have a diverse range of functions beyond matrix synthesis. During the early inflammatory phase of cardiac repair, fibroblasts may secrete inflammatory mediators and proteases, or modulate cardiomyocyte survival in the ischemic heart [59,60]. It has also been suggested that myofibroblasts may exhibit phagocytotic properties and, much like macrophages, may negatively regulate the inflammatory response as they engulf dead cells [61•]. Experiments in a mouse model of myocardial infarction suggested that expression of milk fat globule epidermal growth factor 8 (MFG-E8) may mediate engulfment of apoptotic cells, leading to acquisition of an anti-inflammatory myofibroblast phenotype. Quantitation of TUNEL+ signals in α-SMA+ myofibroblasts and in CD68+ macrophages suggested that the number of apoptotic cells engulfed by myofibroblasts in the infarcted heart was about 40% of that engulfed by macrophages. Fibroblast-specific loss-of-function approaches are needed to document the impact of the contribution of fibroblastmediated phagocytosis in healing infarcts.
Targeting inflammation in myocardial infarction
Despite extensive experimental evidence suggesting that targeting the inflammatory cascade may be effective in attenuating injury following infarction and in preventing adverse remodeling and heart failure, clinical translation has proved challenging [62–64]. Clearly, broad non-selective inhibition of post-infarction inflammation can be detrimental by perturbing the reparative response. The risks of broad immunomodulatory approaches are illustrated by recent observations showing that methotrexate administration in patients with ST elevation myocardial infarction (STEMI) worsened cardiac function [65]. However, several selective approaches targeting specific well-documented inflammatory signals appear to hold promise for clinical translation.
Considering the robust experimental evidence suggesting a crucial role for IL-1 signaling in dilative remodeling and dysfunction following myocardial infarction [66], targeting IL-1 represents a promising therapeutic approach for patients with myocardial infarction [67]. The promising effects of anakinra administration in early pilot studies in patients with myocardial infarction [68], and the effectiveness of IL-1β inhibition in attenuating inflammation and in reducing cardiovascular events in high-risk patients with atherosclerotic disease in the recently reported Canakinumab Antiinflammatory Thrombosis Outcome Study (CANTOS) [69,70] offer a rare air of optimism in a field marred by disappointments. Recent evidence suggesting that IL-1-driven inflammation may be implicated in arrhythmogenesis [71,72] may further strengthen the rationale for IL-1 inhibition in selected subpopulations of patients with myocardial infarction.
In addition to IL-1, several additional inflammatory targets have been suggested in patients with myocardial infarction. In human patients undergoing primary percutaneous coronary intervention (PCI) for acute myocardial infarction, intracoronary nitrite treatment decreased inflammatory activation of leukocytes; these effects were associated with attenuated microvascular obstruction and with a reduction in infarct size [73]. IL-6 inhibition through administration of the IL-6 receptor antagonist tocilizumab attenuated the systemic inflammatory response and reduced cardiomyocyte injury in patients with non-STEMI undergoing PCI [74]. In human patients with non-STEMI undergoing PCI, administration of the P-selectin antagonist inclacumab reduced myocardial injury [75]. Finally, administration of high dose ω-3 fatty acids in patients with acute myocardial infarction reduced adverse ventricular remodeling; the association of benefit with decreased levels of circulating inflammatory biomarkers may suggest that the protective effects may reflect attenuation of inflammation [76].
Conclusions
After decades of research in the field of myocardial inflammation, we may be closer than ever to therapeutic translation. Strategies targeting inflammatory cascades may exert beneficial actions in patients with myocardial infarction. Emerging evidence suggests that modulation of inflammation may mediate any protective effects of cell therapy with mesenchymal stem cells or with cardiosphere-derived cells [77,78]. Moreover, inflammatory cells may hold the key to the visionary goal of cardiac regeneration [79]. A lot remains to be done in order to advance the clinical implementation of strategies targeting inflammatory signals. First, understanding the cell biological mechanisms of myocardial inflammation, repair and fibrosis is crucial in order to design sound therapeutic strategies [3,80]. Second, identification of patient subpopulations with specific perturbations in inflammatory response is needed. Considering the pathophysiologic heterogeneity of myocardial infarction in the clinical context, identification of patients with excessive, prolonged or dysregulated inflammatory responses is critical in order to define patient subsets likely to benefit from targeted anti-inflammatory interventions.
Highlights.
In the infarcted myocardium, alarmins activate pro-inflammatory signals.
In addition to their injurious actions, neutrophils may orchestrate repair of the infarcted heart by modulating macrophage phenotype.
Infarct fibroblasts exhibit phenotypic and functional heterogeneity.
Selective therapeutic approaches targeting Interleukin-1-driven inflammation may hold promise for patients with myocardial infarction.
Acknowledgments
Dr Frangogiannis’ laboratory is supported by NIH Grants R01 HL76246 and R01 HL85440, and by Department of Defense Grants PR151134 and PR151029.
Footnotes
Conflicts of interest
None.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
- 1.Frangogiannis NG. Pathophysiology of myocardial infarction. Compr Physiol. 2015;5:1841–1875. doi: 10.1002/cphy.c150006. [DOI] [PubMed] [Google Scholar]
- 2.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11:255–265. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saxena A, Russo I, Frangogiannis NG. Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges. Transl Res. 2016;167:152–166. doi: 10.1016/j.trsl.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herzog C, Lorenz A, Gillmann HJ, et al. Thrombomodulin’s lectin-like domain reduces myocardial damage by interfering with HMGB1-mediated TLR2 signalling. Cardiovasc Res. 2014;101:400–410. doi: 10.1093/cvr/cvt275. [DOI] [PubMed] [Google Scholar]
- 5.Andrassy M, Volz HC, Igwe JC, et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. 2008;117:3216–3226. doi: 10.1161/CIRCULATIONAHA.108.769331. [DOI] [PubMed] [Google Scholar]
- 6.Lugrin J, Parapanov R, Rosenblatt-Velin N, et al. Cutting edge: IL-1alpha is a crucial danger signal triggering acute myocardial inflammation during myocardial infarction. J Immunol. 2015;194:499–503. doi: 10.4049/jimmunol.1401948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen C, Feng Y, Zou L, et al. Role of extracellular RNA and TLR3-Trif signaling in myocardial ischemia-reperfusion injury. J Am Heart Assoc. 2014;3:e000683. doi: 10.1161/JAHA.113.000683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frangogiannis NG. The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Invest. 2017;127:1600–1612. doi: 10.1172/JCI87491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bliksoen M, Mariero LH, Torp MK, et al. Extracellular mtDNA activates NF-kappaB via toll-like receptor 9 and induces cell death in cardiomyocytes. Basic Res Cardiol. 2016;111:42. doi: 10.1007/s00395-016-0553-6. [DOI] [PubMed] [Google Scholar]
- 10.Lipps C, Nguyen JH, Pyttel L, et al. N-terminal fragment of cardiac myosin binding protein-C triggers pro-inflammatory responses in vitro. J Mol Cell Cardiol. 2016;99:47–56. doi: 10.1016/j.yjmcc.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinto AR, Ilinykh A, Ivey MJ, et al. Revisiting cardiac cellular composition. Circ Res. 2016;118:400–409. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frangogiannis NG, Mendoza LH, Lewallen M, et al. Induction and suppression of interferon-inducible protein 10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB J. 2001;15:1428–1430. doi: 10.1096/fj.00-0745fje. [DOI] [PubMed] [Google Scholar]
- 13.Kumar AG, Ballantyne CM, Michael LH, et al. Induction of monocyte chemoattractant protein-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation. 1997;95:693–700. doi: 10.1161/01.cir.95.3.693. [DOI] [PubMed] [Google Scholar]
- 14.Chen W, Spitzl A, Mathes D, et al. Endothelial actions of ANP enhance myocardial inflammatory infiltration in the early phase after acute infarction. Circ Res. 2016;119:237–248. doi: 10.1161/CIRCRESAHA.115.307196. [DOI] [PubMed] [Google Scholar]
- 15•.Zhu M, Goetsch SC, Wang Z, et al. FoxO4 promotes early inflammatory response upon myocardial infarction via endothelial Arg1. Circ Res. 2015;117:967–977. doi: 10.1161/CIRCRESAHA.115.306919. The study provides evidence for an important role of endothelial-specific FoxO4 signaling in activation of the post-infarction inflammatory response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen B, Frangogiannis NG. Immune cells in repair of the infarcted myocardium. Microcirculation. 2017;24 doi: 10.1111/micc.12305. [DOI] [PubMed] [Google Scholar]
- 17.Entman ML, Youker K, Shoji T, et al. Neutrophil induced oxidative injury of cardiac myocytes. A compartmented system requiring CD11b/CD18-ICAM-1 adherence. J Clin Invest. 1992;90:1335–1345. doi: 10.1172/JCI115999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alard JE, Ortega-Gomez A, Wichapong K, et al. Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci Transl Med. 2015;7:317ra196. doi: 10.1126/scitranslmed.aad5330. [DOI] [PubMed] [Google Scholar]
- 19•.Horckmans M, Ring L, Duchene J, et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017;38:187–197. doi: 10.1093/eurheartj/ehw002. The study demonstrates a critical role for neutrophils in repair of the infarcted heart through modulation of macrophage phenotype. [DOI] [PubMed] [Google Scholar]
- 20.Romson JL, Hook BG, Kunkel SL, et al. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation. 1983;67:1016–1023. doi: 10.1161/01.cir.67.5.1016. [DOI] [PubMed] [Google Scholar]
- 21.Chatelain P, Latour JG, Tran D, et al. Neutrophil accumulation in experimental myocardial infarcts: relation with extent of injury and effect of reperfusion. Circulation. 1987;75:1083–1090. doi: 10.1161/01.cir.75.5.1083. [DOI] [PubMed] [Google Scholar]
- 22.Mollenhauer M, Friedrichs K, Lange M, et al. Myeloperoxidase mediates postischemic arrhythmogenic ventricular remodeling. Circ Res. 2017;121:56–70. doi: 10.1161/CIRCRESAHA.117.310870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Y, Yabluchanskiy A, Iyer RP, et al. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res. 2016;110:51–61. doi: 10.1093/cvr/cvw024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Epelman S, Lavine KJ, Beaudin AE, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mylonas KJ, Jenkins SJ, Castellan RF, et al. The adult murine heart has a sparse, phagocytically active macrophage population that expands through monocyte recruitment and adopts an ‘M2’ phenotype in response to Th2 immunologic challenge. Immunobiology. 2015 doi: 10.1016/j.imbio.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26•.Hulsmans M, Clauss S, Xiao L, et al. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169:510–522 e520. doi: 10.1016/j.cell.2017.03.050. A very interesting study suggesting that a cell population with macrophage characteristics may play a role in facilitating conduction of the electrical impulse in mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dewald O, Zymek P, Winkelmann K, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 28.Wan E, Yeap XY, Dehn S, et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res. 2013;113:1004–1012. doi: 10.1161/CIRCRESAHA.113.301198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen W, Saxena A, Li N, et al. Endogenous IRAK-M attenuates postinfarction remodeling through effects on macrophages and fibroblasts. Arterioscler Thromb Vasc Biol. 2012;32:2598–2608. doi: 10.1161/ATVBAHA.112.300310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shiraishi M, Shintani Y, Shintani Y, et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest. 2016;126:2151–2166. doi: 10.1172/JCI85782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31•.Howangyin KY, Zlatanova I, Pinto C, et al. Myeloid-epithelial-reproductive receptor tyrosine kinase and milk fat globule epidermal growth factor 8 coordinately improve remodeling after myocardial infarction via local delivery of vascular endothelial growth factor. Circulation. 2016;133:826–839. doi: 10.1161/CIRCULATIONAHA.115.020857. Demonstration of a critical role of infarct macrophages as a source of angiogenic mediators. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saxena A, Chen W, Su Y, et al. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol. 2013;191:4838–4848. doi: 10.4049/jimmunol.1300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanders LN, Schoenhard JA, Saleh MA, et al. BMP antagonist gremlin 2 limits inflammation after myocardial infarction. Circ Res. 2016;119:434–449. doi: 10.1161/CIRCRESAHA.116.308700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34•.Leblond AL, Klinkert K, Martin K, et al. Systemic and cardiac depletion of M2 macrophage through CSF-1R signaling inhibition alters cardiac function post myocardial infarction. PLOS ONE. 2015;10:e0137515. doi: 10.1371/journal.pone.0137515. The study shows the critical role of CSF-1 activated M2 macrophages in preservation of function following myocardial infarction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frangogiannis NG, Mendoza LH, Ren G, et al. MCSF expression is induced in healing myocardial infarcts and may regulate monocyte and endothelial cell phenotype. Am J Physiol Heart Circ Physiol. 2003;285:H483–H492. doi: 10.1152/ajpheart.01016.2002. [DOI] [PubMed] [Google Scholar]
- 36.Sager HB, Hulsmans M, Lavine KJ, et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ Res. 2016;119:853–864. doi: 10.1161/CIRCRESAHA.116.309001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hammer A, Stegbauer J, Linker RA. Macrophages in neuroinflammation: role of the renin-angiotensin-system. Pflugers Arch. 2017;469:431–444. doi: 10.1007/s00424-017-1942-x. [DOI] [PubMed] [Google Scholar]
- 38.Grisanti LA, Traynham CJ, Repas AA, et al. beta2-Adrenergic receptor-dependent chemokine receptor 2 expression regulates leukocyte recruitment to the heart following acute injury. Proc Natl Aoad Sci USA. 2016;113:15126–15131. doi: 10.1073/pnas.1611023114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39•.Grisanti LA, Gumpert AM, Traynham CJ, et al. Leukocyte-expressed beta2-adrenergic receptors are essential for survival after acute myocardial injury. Circulation. 2016;134:153–167. doi: 10.1161/CIRCULATIONAHA.116.022304. The study suggests a previously unappreciated role of β2 adrenergic receptor signaling in leukocyte-mediated repair of the infarcted heart. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia-Prieto J, Villena-Gutierrez R, Gomez M, et al. Neutrophil stunning by metoprolol reduces infarct size. Nat Commun. 2017;8:14780. doi: 10.1038/ncomms14780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frangogiannis NG, Mendoza LH, Lindsey ML, et al. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol. 2000;165:2798–2808. doi: 10.4049/jimmunol.165.5.2798. [DOI] [PubMed] [Google Scholar]
- 42.Hofmann U, Frantz S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ Res. 2015;116:354–367. doi: 10.1161/CIRCRESAHA.116.304072. [DOI] [PubMed] [Google Scholar]
- 43.Dobaczewski M, Xia Y, Bujak M, et al. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010;176:2177–2187. doi: 10.2353/ajpath.2010.090759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weirather J, Hofmann UD, Beyersdorf N, et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115:55–67. doi: 10.1161/CIRCRESAHA.115.303895. [DOI] [PubMed] [Google Scholar]
- 45.Saxena A, Dobaczewski M, Rai V, et al. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol Heart Circ Physiol. 2014;307:H1233–H1242. doi: 10.1152/ajpheart.00328.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramjee V, Li D, Manderfield LJ, et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J Clin Invest. 2017;127:899–911. doi: 10.1172/JCI88759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bujak M, Dobaczewski M, Gonzalez-Quesada C, et al. Induction of the CXC chemokine interferon-gamma-inducible protein 10 regulates the reparative response following myocardial infarction. Circ Res. 2009;105:973–983. doi: 10.1161/CIRCRESAHA.109.199471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frangogiannis NG, Perrard JL, Mendoza LH, et al. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Ciroulation. 1998;98:687–698. doi: 10.1161/01.cir.98.7.687. [DOI] [PubMed] [Google Scholar]
- 49.Frangogiannis NG, Lindsey ML, Michael LH, et al. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998;98:699–710. doi: 10.1161/01.cir.98.7.699. [DOI] [PubMed] [Google Scholar]
- 50.Somasundaram P, Ren G, Nagar H, et al. Mast cell tryptase may modulate endothelial cell phenotype in healing myocardial infarcts. J Pathol. 2005;205:102–111. doi: 10.1002/path.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gersch C, Dewald O, Zoerlein M, et al. Mast cells and macrophages in normal C57/BL/6 mice. Histochem Cell Biol. 2002;118:41–49. doi: 10.1007/s00418-002-0425-z. [DOI] [PubMed] [Google Scholar]
- 52.Dewald O, Ren G, Duerr GD, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–677. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53•.Ngkelo A, Richart A, Kirk JA, et al. Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J Exp Med. 2016;213:1353–1374. doi: 10.1084/jem.20160081. The study suggests that mast cell-derived proteases may regulate cardiomyocyte function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anzai A, Anzai T, Nagai S, et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation. 2012;125:1234–1245. doi: 10.1161/CIRCULATIONAHA.111.052126. [DOI] [PubMed] [Google Scholar]
- 55.Van der Borght K, Scott CL, Nindl V, et al. Myocardial infarction primes autoreactive T cells through activation of dendritic cells. Cell Rep. 2017;18:3005–3017. doi: 10.1016/j.celrep.2017.02.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shinde AV, Humeres C, Frangogiannis NG. The role of alpha-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim Biophys Acta. 2017;1863:298–309. doi: 10.1016/j.bbadis.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cleutjens JP, Verluyten MJ, Smiths JF, et al. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 58.Frangogiannis NG, Michael LH, Entman ML. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb) Cardiovasc Res. 2000;48:89–100. doi: 10.1016/s0008-6363(00)00158-9. [DOI] [PubMed] [Google Scholar]
- 59.Woodall MC, Woodall BP, Gao E, et al. Cardiac fibroblast GRK2 deletion enhances contractility and remodeling following ischemia/reperfusion injury. Circ Res. 2016;119:1116–1127. doi: 10.1161/CIRCRESAHA.116.309538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frangogiannis NG. The functional pluralism of fibroblasts in the infarcted myocardium. Circ Res. 2016;119:1049–1051. doi: 10.1161/CIRCRESAHA.116.309926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61•.Nakaya M, Watari K, Tajima M, et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J Clin Invest. 2017;127:383–401. doi: 10.1172/JCI83822. The first demonstration of a role for infarct myofibroblasts as phagocytotic cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sager HB, Dutta P, Dahlman JE, et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci Transl Med. 2016;8:342ra380. doi: 10.1126/scitranslmed.aaf1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Christia P, Frangogiannis NG. Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest. 2013;43:986–995. doi: 10.1111/eci.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seropian IM, Toldo S, Van Tassell BW, et al. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. J Am Coll Cardiol. 2014;63:1593–1603. doi: 10.1016/j.jacc.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 65.Moreira DM, Lueneberg ME, da Silva RL, et al. MethotrexaTE THerapy in ST-Segment Elevation MYocardial InfarctionS. J Cardiovasc Pharmacol Ther. 2017 doi: 10.1177/1074248417699884. http://dx.doi.org/10.1177/1074248417699884. [DOI] [PubMed]
- 66.Bujak M, Dobaczewski M, Chatila K, et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173:57–67. doi: 10.2353/ajpath.2008.070974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van Tassell BW, Toldo S, Mezzaroma E, et al. Targeting interleukin-1 in heart disease. Circulation. 2013;128:1910–1923. doi: 10.1161/CIRCULATIONAHA.113.003199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abbate A, Van Tassell BW, Biondi-Zoccai G, et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study] Am J Cardiol. 2013;111:1394–1400. doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ridker PM, Howard CP, Walter V, et al. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. 2012;126:2739–2748. doi: 10.1161/CIRCULATIONAHA.112.122556. [DOI] [PubMed] [Google Scholar]
- 70.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017 doi: 10.1056/NEJMoa1707914. http://dx.doi.org/10.1056/NEJMoa1707914. [DOI] [PubMed]
- 71.Monnerat G, Alarcon ML, Vasconcellos LR, et al. Macrophage-dependent IL-1beta production induces cardiac arrhythmias in diabetic mice. Nat Commun. 2016;7:13344. doi: 10.1038/ncomms13344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.De Jesus NM, Wang L, Lai J, et al. Antiarrhythmic effects of interleukin 1 inhibition after myocardial infarction. Heart Rhythm. 2017;14:727–736. doi: 10.1016/j.hrthm.2017.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jones DA, Khambata RS, Andiapen M, et al. Intracoronary nitrite suppresses the inflammatory response following primary percutaneous coronary intervention. Heart. 2017;103:508–516. doi: 10.1136/heartjnl-2016-309748. [DOI] [PubMed] [Google Scholar]
- 74.Kleveland O, Kunszt G, Bratlie M, et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: a double-blind, randomized, placebo-controlled phase 2 trial. Eur Heart J. 2016;37:2406–2413. doi: 10.1093/eurheartj/ehw171. [DOI] [PubMed] [Google Scholar]
- 75.Stahli BE, Gebhard C, Duchatelle V, et al. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention according to timing of infusion: insights from the SELECT-ACS trial. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.116.004255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heydari B, Abdullah S, Pottala JV, et al. Effect of omega-3 acid ethyl esters on left ventricular remodeling after acute myocardial infarction: the OMEGA-REMODEL randomized clinical trial. Circulation. 2016;134:378–391. doi: 10.1161/CIRCULATIONAHA.115.019949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luger D, Lipinski MJ, Westman PC, et al. Intravenously delivered mesenchymal stem cells: systemic anti-inflammatory effects improve left ventricular dysfunction in acute myocardial infarction and ischemic cardiomyopathy. Circ Res. 2017;120:1598–1613. doi: 10.1161/CIRCRESAHA.117.310599. [DOI] [PubMed] [Google Scholar]
- 78.de Couto G, Liu W, Tseliou E, et al. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Invest. 2015;125:3147–3162. doi: 10.1172/JCI81321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aurora AB, Porrello ER, Tan W, et al. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124:1382–1392. doi: 10.1172/JCI72181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spinale FG, Frangogiannis NG, Hinz B, et al. Crossing into the next frontier of cardiac extracellular matrix research. Circ Res. 2016;119:1040–1045. doi: 10.1161/CIRCRESAHA.116.309916. [DOI] [PMC free article] [PubMed] [Google Scholar]