

Abstract
Many genetic variants affect complex traits through gene expression, which can be exploited to boost statistical power and enhance interpretation in genome-wide association studies (GWASs) as demonstrated by the transcriptome-wide association study (TWAS) approach. Furthermore, due to polygenic inheritance, a complex trait is often affected by multiple genes with similar functions as annotated in gene path-ways. Here we extend TWAS from gene-based analysis to pathway-based analysis: we integrate public pathway collections, expression quantitative trait locus (eQTL) data and GWAS summary association statistics (or GWAS individual-level data) to identify gene pathways associated with complex traits. The basic idea is to weight the SNPs of the genes in a pathway based on their estimated cis-effects on gene expression, then adaptively test for association of the pathway with a GWAS trait by effectively aggregating possibly weak association signals across the genes in the pathway. The p-values can be calculated analytically and thus fast. We applied our proposed test with the KEGG and GO pathways to two schizophrenia (SCZ) GWAS summary association data sets, denoted SCZ1 and SCZ2 with about 20,000 and 150,000 subjects respectively. Most of the significant pathways identified by analyzing the SCZ1 data were reproduced by the SCZ2 data. Importantly, we identified 15 novel pathways associated with SCZ, such as GABA receptor complex (GO:1902710), which could not be uncovered by the standard single SNP-based analysis or gene-based TWAS. The newly identified pathways may help us gain insights into the biological mechanism underlying SCZ. Our results showcase the power of incorporating gene expression information and gene functional annotations into pathway-based association testing for GWAS.
Keywords: aSPU, aSPUpath, aSPUpath2, gene expression, TWAS
Introduction
Although genome-wide association studies (GWASs) have been remarkably successful in identifying genetic variants associated with complex traits and diseases, only a small to modest proportion of heritability for most complex traits and diseases can be explained by the identified genetic variants (Manolio et al., 2009). Furthermore, since the majority of identified variants are found in non-coding regions that are not in linkage disequilibrium (LD) with coding exons, a mechanistic understanding of how these variants influence traits is generally lacking (Locke et al., 2015; Albert and Kruglyak, 2015). However, it is now known that an important class of variants, termed expression quantitative trait loci (eQTLs), affect complex traits by regulating gene expression levels; there is an enrichment of eQTLs among the GWAS trait-associated variants (Lappalainen et al., 2013; Albert and Kruglyak, 2015). Accordingly, transcriptome-wide association study (TWAS) and related methods (Gusev et al., 2016a; Gamazon et al., 2015; Xu et al., 2017b) were proposed to integrate eQTL data with GWAS data to identify the genes associated with a complex trait. These methods may iimprove statistical power to detect associations relative to traditional SNP-based GWAS and gene-based tests that ignore information on gene expression regulation. Nevertheless, due to the limited sample sizes of eQTL data and GWAS data, they may fail to identify some more weakly associated genes with smaller effect sizes. On the other hand, genes do not work in isolation; instead, a group of functionally related genes as annotated in a biological pathway are often involved in the same disease susceptibility and progression (Heinig et al., 2010). Gene-based analysis testing each gene one-by-one may miss an important pathway if each gene in the pathway has only a small effect size, but in aggregation they contribute substantially. Hence, association analysis of a group of functionally related genes, called pathway-based analysis, has been proposed and applied in practice to boost statistical power and improve interpretability over gene-based analysis for GWAS (Wang et al., 2007; Chen et al., 2010; Peng et al., 2010; Wei et al., 2012; Schaid et al., 2012; Pan et al., 2015; Bakshi et al., 2016; Li et al., 2016, 2017).
Here, we extend integrative gene-based testing like TWAS to integrative pathway-based association analysis to identify pathways associated with complex traits and diseases. Specifically, we propose a new self-contained test that integrates eQTL-derived weights, GWAS individual-level or summary data, SNP LD information, and gene functional annotations as public pathway collections to identify pathways associated with a complex trait (Figure 1). As in TWAS, we first estimate the cis-effects of the SNPs in each gene on its expression level, then adaptively test for association between a pathway and a trait by effectively aggregating possibly weak association signals across the genes in the pathway.
Figure 1.

Workflow of pathway-based analysis.
We note that our methodology differs from existing approaches. In principle, existing pathway-based analysis methods can be applied in a two-step approach. After obtaining the p-value for each gene by applying TWAS or a related method, an existing pathway analysis method, such as gene set enrichment analysis (GSEA; Subramanian et al. (2005)) or DAVID (Huang et al., 2009), can be applied to identify significant pathways. As to be shown later, a two-step approach, critically depending on the output of a gene-based test, may lose power as compared to our integrated single-step method. Furthermore, many existing pathway methods, including GSEA and DAVID, belong to the category of competitive tests, which compare the p-values of the genes in a given pathway with the p-values of other background genes to determine the significance level, while our method is a self-contained test with a null hypothesis that none of any genes in the pathway is associated with the disease; it is known that a self-contained test is often more powerful (Goeman and Bühlmann, 2007). In addition, all the existing pathway analysis methods are only for GWAS data alone while failing to take advantage of eQTL information, leading to power loss and difficulties in interpreting the findings.
Our study was motivated by analyses of schizophrenia (SCZ) GWAS summary data. SCZ is a major chronic and severe mental disorder that is associated with considerable morbidity and mortality (Tiihonen et al., 2009) and affects about 1% of the population. Although the high heritability of SCZ has been demonstrated by previous studies (Sullivan et al., 2012), to date, one of the largest GWAS meta-analyses, conducted by the Schizophrenia Working Group of the Psychiatric Genomics Consortium (PGC), has only identified 128 independent associations spanning 108 conservatively defined loci (Schizophrenia Working Group, 2014). To improve the statistical power and interpretability of the results, Gusev et al. (2016a) applied TWAS to the PGC GWAS summary data and identified 157 significant genes, of which 35 did not overlap with a genome-wide significant locus within 500 kb. However, the pathophysiology of SCZ remains largely unknown and thus it is hard to develop new drugs with high efficacy and low side effects. Identifying SCZ-associated pathways is a crucial step for mechanistic understanding of SCZ and thus developing new drugs. Here, we performed gene- and pathway-based analyses to identify SCZ-associated genes and pathways, providing insights into the underlying mechanism of SCZ.
We reanalyzed two SCZ GWAS summary data sets, which were downloaded from the PGC website (see URLs): a meta-analyzed SCZ GWAS data set with 8,832 cases and 12,067 controls, denoted as SCZ1 (Ripke et al., 2013), and a more recent and larger one with 36,989 cases and 113,075 controls, denoted as SCZ2 (Schizophrenia Working Group, 2014). First, we focused on gene-based analysis. By noting that TWAS is the same as the weighted Sum test with gene expression derived weights (Xu et al., 2017b), we applied some more powerful tests, such as the weighted sum of squared score (SSU) test and the weighted adaptive sum of powered score (aSPU) test (Pan et al., 2014). We analyzed the SCZ1 data and identified 51, 108, and 87 significant genes by applying TWAS, (weighted) SSU, and (weighted) aSPU, respectively. Among these identified genes, about 90% genes contained genome-wide significant SNPs within 500 kb in the SCZ2 data, constituting a highly significant and intuitive support for the identified loci. We then applied these tests to the SCZ2 data and identified 75 novel SCZ genes, of which 50 have not been reported in the literature yet. These results further confirm that both weighted SSU and weighted aSPU can improve statistical power to identify more associated genes over that of TWAS. Second, we conducted pathway-based analysis by applying our proposed approach with the Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa and Goto (2000)) and Gene Ontology (GO; Consortium et al. (2004)) candidate pathways to the SCZ1 and SCZ2 data. Most of the significant pathways identified by analyzing the SCZ1 data were confirmed by the SCZ2 data. When analyzing the SCZ2 data, a two-step approach combining TWAS and an existing pathway method, DAVID, identified only one significant pathway, sequence-specific DNA binding (GO:0003700), which was also identified by our proposed method. Importantly, by analyzing the SCZ2 data we identified 15 novel significant SCZ-associated pathways, such as pathway GABA receptor complex (GO:1902710), which were missed by the gene-based TWAS or aSPU analysis. Hence, pathway-based analysis, as a complementary tool to gene-based analysis, may identify some pathways in which individual genes may have only too weak effects to be detected but their aggregated effects are strong. Overall, our results showcase the increased power of integrating GWAS summary data, eQTL data, reference LD information, and gene functional annotations to gain insights into the genetic basis of complex traits.
Material and Methods
Data Sets
We downloaded two publicly available SCZ GWAS summary data sets from the PGC website (see URLs): the SCZ1 data, which contains the meta-analyzed summary statistics based on 20,899 individuals (Ripke et al., 2013), and the SCZ2 data based on 150,064 individuals (Schizophrenia Working Group, 2014). The sets of gene expression-derived weights and the 1000 Genomes Project reference panel were downloaded from the TWAS website (see URLs). Following the TWAS set-up, we removed the SNPs with the strand-ambiguous alleles (A/T, G/C) from the GWAS summary data. Two pathway collections, GO and KEGG, were downloaded from the Molecular Signatures Database (see URLs).
Review of TWAS and Related Methods
We review TWAS and its related methods, which take GWAS summary statistics, a set of gene expression-derived weights, and SNP LD information as input. Since all the methods are gene-based by testing the genes one by one, for the purpose of presentation we only need to consider a single gene.
For a given gene, we only consider a region around it (i.e. its coding region extended by a certain distance, say ±500 kb, upstream and downstream from its TSS and TES respectively) for its cis-effects. Let Z = (Z1, …,Zp)′ be a vector of z-scores of the SNPs for the gene based on the GWAS summary data, or constructed from the GWAS individual-level data. The null hypothesis H0 to be tested is that the SNPs in a given SNP set (of a gene or a pathway) are not associated with a GWAS trait. With W = (ŵ1, …,ŵp)′, a vector of the estimated cis-effects of the SNPs on gene expression based on a reference eQTL data set, TWAS tests on H0 using the weighted z-scores. Note that, with GWAS individual-level data, TWAS can be interpreted as testing for association between imputed gene expression and the GWAS trait; however, with GWAS summary data, W′Z may be regarded as an imputed z–score for the gene, but not imputed expression level. It turns out that TWAS is equivalent to the weighted Sum test (Pan, 2009; Xu et al., 2017b). Because the Sum test implicitly assumes that all variants have an equal effect size and the same effect direction, the Sum test and thus TWAS, as discussed in the previous studies (Pan, 2009; Wu et al., 2011; Pan et al., 2014), may lose statistical power if the true association effects are sparse (i.e. with many 0s) or the effect directions are different. Note that, due to the usually small sample size of the eQTL dataset, there are always estimation errors with the estimated cis-effects W. More generally, any more powerful tests, such as the weighted SSU test or the weighted aSPU test, can be applied (Xu et al., 2017b). In particular, the SPU(γ) tests are possible candidates to use, covering some existing ones as special cases (Pan et al., 2014). For example, SPU(1) equals to the Sum test, while SPU(2) equals to SSU and a kernel machine regression-based test (also known as SKAT (Wu et al., 2011) in rare variant analysis) with a linear kernel. As to be confirmed later, the SPU(2) test may yield higher statistical power than TWAS (or SPU(1)). Generally, the SPU(γ) tests with γ ∈ Γ = {1, 2, …, 6,∞} can be applied, and their results can be combined by the adaptive aSPU test (Pan et al., 2014).
Since not all SNPs with non-zero weights (derived from the reference eQTL data set) were presented in the GWAS summary data, we used the ImpG-Summary software (Pasaniuc et al., 2014) to impute missing z-scores to the 1000 Genomes Project reference panel accordingly. Because the correlations among Z can be approximated by LD among the SNPs (Kwak and Pan, 2016; Gusev et al., 2016b), we used the 1000 Genomes Project reference panel (European ancestry) (or other panels for other ethnic/racial groups) to estimate the LD and thus the correlation matrix for Z. In this study, we used five sets of gene expression reference weights that were based on the following four eQTL data sets: microarray gene expression data measured in peripheral blood from 1,245 unrelated subjects from the Netherlands Twin Registry (NTR), microarray expression array data measured in blood from 1,264 individuals from the Young Finns Study (YFS), RNA-seq measured in adipose tissue from 563 individuals from the Metabolic Syndrome in Men study (METSIM), and RNA-seq measured in the dorsolateral prefrontal cortex from 621 individuals from CommonMind Consortium (CMC) (Gusev et al., 2016b). The weights for differentially spliced introns were further constructed by analyzing CMC data (CMC-introns) (Gusev et al., 2016b). All these weights were downloaded from the TWAS website (see URLs). To account for multiple testing, we applied the Bonferroni correction for each set of weights to maximize the consistency with the previously published results (Gusev et al., 2016b) and not to over-penalize the use of additional (and often highly correlated) gene expression-derived weights. Specifically, we reported the number of significant genes after correcting for the number of genes tested within the use of each of the five gene expression sets (YFS, NTR, METSIM, CMC, and CMC-introns; 5004 genes on average with none-zero weights and being tested).
A New Pathway-based Test
Given a pathway, we would like to test the null hypothesis H0 that none of the genetic variants in the pathway is associated with a trait. We introduce a new pathway-based test to integrate gene functional annotations and a reference eQTL dataset with GWAS data. Figure 1 illustrates the workflow of our new pathway-based analysis. As a comparison, we also describe a two-step approach combining an existing integrative gene-based test (like TWAS) and an existing pathway analysis method (like DAVID), in which a gene-based p-value is calculated for each gene before they are combined in pathway analysis.
Given a pathway S*, we first remove the genes whose gene expression-derived SNP weights are all 0, resulting in a subset S containing n genes. We partition its z-score vector into the z-score sub-vectors for the genes, say for gene g (with kg SNPs) as Zg· = (Zg1, …,Zgkg)′. For each gene g, we standardize the gene expression derived weights Wg· by such that the weights of the genes are in a similar scale to avoid one or few genes (e.g. with large expression levels) dominate. The standardized weights for the gene set S are with . We propose the following test statistics:
where PPathSPU(γ)is the p–value of the PathSPU(γ) test. Because PathSPU(1) and Path-SPU(2) are independent (Derkach et al., 2014), we can obtain the p-value of aSPUpath2 via the following steps:
Calculate the p-values, p1 = PPathSPU(1) and p2 = PPathSPU(2), based on the theory that PathSPU(1) and PathSPU(2) asymptotically follow a normal distribution and a mixture of χ2 distribution under H0, respectively (Pan, 2009).
Take the minimum p-value of PathSPU(1) and PathSPU(2), that is pmin = min(p1, p2).
By the asymptotic independence of PathSPU(1) and PathSPU(2), the p-value for the aSPUpath2 is paSPUpath2 = 1 − (1 − pmin)2.
The aSPUpath2 test is new in two aspects: first, unlike many other pathway-based methods aggregating information from only SNP data (Kwak and Pan, 2015; Bakshi et al., 2016), aSPUpath2 incorporates information in a reference eQTL data set, thus increasing the power and providing mechanistic insights; second, unlike many other methods, for example fastBAT (Bakshi et al., 2016), which are non-adaptive and thus only powerful under some specific alternatives, aSPUpath2 adaptively combines information and thus can maintain relatively high power across a wider range of situations. Finally, we note that aSPUpath2 is a special case of a more general and adaptive pathway-based test called aSPUpath (Pan et al., 2015; Kwak and Pan, 2015), motivated by the following two considerations. First, unlike aSPUpath, the p-value of aSPUpath2 can be calculated analytically and thus fast, though a simulation-based method can be equally applied; as to be demonstrated in the results section, the analogical method provides a good approximation to the simulation-based method. Second, aSPUpath2 is tailored to identifying pathways containing many associated genes or SNPs with only weak effects that cannot be detected by single SNP- or single gene-based analysis, for which it is more powerful. Hence, aSPUpath2 can be used either alone or as a fast screening procedure for the more time-consuming and more general aSPUpath test.
We extracted candidate pathways from two gene functional annotation sources, KEGG and GO, which were downloaded from the MSigDB database (Subramanian et al. (2005); see URLs). Because a small pathway gives results not much different from a gene-based analysis, whereas the biological function of a large pathway is not specific, we restricted our analyses to the pathways containing between 10 and 200 genes, which is widely adopted in pathway-based analysis (Network and of the Psychiatric Genomics Consortium, 2015; Pan et al., 2015). Supplementary Table 1 shows the summary statistics for the candidate pathways. On average, we analyzed 4,220 gene sets for each set of weights. To account for multiple testing, we applied the Bonferroni correction within each set of weights and used a slightly conservative cutoff 0.05/5000 = 1×10−5. Owing to the non-independence nature of many pathways, the Bonferroni correction might be over-conservative here.
Other Existing Pathway-based Tests
In principle, an existing pathway analysis method, in couple with a gene-based test, can be applied in a two-step approach. We compared our new method with this two-step approach using two popular pathway analysis methods, i-GSEA4GWAS (Zhang et al., 2010) and DAVID (Huang et al., 2009), to further illustrate the power of our proposed test. Specifically, for i-GSEA4GWAS, we uploading the p-values for the genes (calculated by TWAS or SSU or aSPU) for a given pathway to the i-GSEA4GWAS server (see URLs). For DAVID, we uploaded to the DAVID server (see URLs) the significant genes identified by TWAS or SSU or aSPU as the gene list and used the genes we analyzed as the background.
Results
TWAS and Related Methods Identify Known and Novel SCZ-associated Genes
First we applied TWAS (i.e. the weighted Sum test), the (weighted) SSU and (weighted) aSPU tests (that integrate gene expression-derived weights) to the SCZ1 data (Ripke et al., 2013) of 20,899 individuals to identify SCZ-associated genes. Then we looked for genome-wide significant SNPs around these genes in the larger SCZ2 data (Schizophrenia Working Group, 2014) of 150,064 individuals for partial validation. Table 1 summarizes the numbers of the significant genes identified by the methods with the SCZ1 data. TWAS, SSU, and aSPU identified 51, 108, 87 significant genes (after taking the union of the results using the five sets of weights), respectively. Among these 87 significant genes identified by aSPU, 64 (around 70%) and 79 (around 90%) contained the genome-wide significant SNPs (p-value < 5×10−8) within 500 kb in the SCZ1 data and the SCZ2 data respectively, offering a highly significant validation of the identified loci. For TWAS and SSU, we have the similar proportions of the genes containing the genome-wide significant SNPs in both the SCZ1 and SCZ2 data. Clearly, SSU and aSPU identified more associated genes than TWAS. Compared to TWAS, SSU and aSPU can still maintain high power if many of the weighted SNPs in a gene are not associated with a trait or their associations are in different directions. Since we do not know the sparsity level and association directions of the underlying association patterns, we used the adaptive aSPU test. Here, perhaps due to the denser association patterns (i.e. with many associated SNPs), SSU identified a larger number of SCZ-associated genes than aSPU.
Table 1.
The numbers of the significant genes identified by analyzing the SCZ1 data for each single set of the weights and their union across these weights. The numbers a/b/c in each cell indicate the numbers of (a) the significant genes; (b) the significant genes covering at least one genome-wide significant SNP within ±500 kb in the SCZ1 data; (c) the significant genes covering at least one genome-wide significant SNP within ±500 kb in the SCZ2 data.
| YFS | NTR | METSIM | CMC-introns | CMC | Combined | |
|---|---|---|---|---|---|---|
| TWAS | 14/11/14 | 13/8/13 | 8/5/7 | 18/10/13 | 16/10/13 | 51/31/43 |
| SSU | 31/25/26 | 27/19/26 | 24/14/23 | 27/17/23 | 39/25/34 | 108/67/95 |
| aSPU | 29/26/26 | 23/16/22 | 21/16/21 | 26/18/21 | 28/22/25 | 87/64/79 |
Supplementary Table 2 shows the significant gene sets identified by TWAS or SSU or aSPU based on the SCZ1 data, and Supplementary Figures 1–5 present the Manhattan plots for the methods with the different sets of weights. The strongest gene association identified by TWAS and SSU was NT5C2 (MIM: 600417), which was also reported by other studies (Guan et al., 2016). This analysis also provides additional in silico support for some reported SCZ-associated genes, including SDCCAG8 (MIM: 613524), ITIH4 (MIM: 600564), and NISCH (MIM: 615507), and many other genes.
Then, we applied TWAS, SSU, and aSPU to the SCZ2 data, listing the number of significant genes identified by each method in Table 2. The quantile-quantile (Q-Q) and Manhattan plots for different sets of weights are shown in Supplementary Figures 6–11, respectively. Here, we analyzed the whole SCZ2 data, which were based on 36,989 cases and 113,075 controls, while Gusev et al. (2016b) analyzed the non-overlapping case-control samples with 34,241 cases and 45,604 controls. This data difference led to our findings slightly different from their published ones (Gusev et al., 2016b): applying TWAS to the SCZ2 data, we identified 202 significant genes, while Gusev et al. (2016b) identified 157 significant genes. Because the sample size of the SCZ2 is much larger than that of the SCZ1, applying to the SCZ2 data identified a much larger number of significant genes by each method. Again, SSU and aSPU appeared to be more powerful than TWAS in terms of the number of the identified significant associations. However, because under different scenarios different tests may be more powerful, each test identified some unique genes missed by the other tests.
Table 2.
The numbers of the significant genes identified by analyzing the SCZ2 data for each single set of the weights and their union across these weights. The numbers a/b/c in each cell indicate the numbers of (a) the significant genes; (b) the significant genes covering at least one genome-wide significant SNP within ±500 kb in the SCZ1 data; (c) the significant genes covering at least one genome-wide significant SNP within ±500 kb in the SCZ2 data.
| YFS | NTR | METSIM | CMC-introns | CMC | Combined | |
|---|---|---|---|---|---|---|
| TWAS | 63/19/46 | 49/22/39 | 43/11/32 | 56/17/37 | 69/21/50 | 202/63/142 |
| SSU | 127/40/94 | 78/32/59 | 108/32/76 | 100/22/61 | 124/32/85 | 381/108/255 |
| aSPU | 105/40/83 | 69/34/60 | 87/33/72 | 85/24/55 | 110/34/82 | 314/110/234 |
Overall, we identified 410 significant (and unique) genes by the three methods based on analyzing the SCZ2 data (Supplementary Table 3), of which 142 did not overlap with any genome-wide significant SNPs within ±500 kb in the SCZ2 data. Next, to consider the effects of different sets of weights (25,018 tests in total), we used a more stringent cutoff (0.05=25, 018 = 2 × 10−6) to report the highly significant genes. We report the new associations that are more than 500 kb away from any genome-wide significant SNPs in the SCZ2 data. Supplementary Table 4 lists 75 highly significant genes identified by the three methods; TWAS, SSU, and aSPU identified 23, 68, and 32 highly significant genes, respectively, showcasing the increased discovery power of applying other tests over TWAS. Table 3 reports 32 highly significant genes identified by aSPU. We searched the NHGRI-EBI GWAS Catalog (MacArthur et al. (2017); see URLs) to determine if these significant genes have been reported by other studies. Among these 32 genes, 10 have been reported by other studies. On the other hand, among the 75 significant genes identified by any method, 20 genes, such as FOXN2 (MIM: 143089; Cross-Disorder Group (2013)), MSRA (MIM: 601250; Ma et al. (2011), and PAX5 (MIM: 167414; Loo et al. (2012)), have been reported by other studies. Overall, these 75 newly identified genes represent a class of discoveries that would have been missed by the standard single SNP-based test, due to not only their power differences, but also the distal locations of the genome-wide significant SNPs.
Table 3.
The significant and novel genes overlapping with no known GWAS risk variants within ±500 kb as identified by aSPU applied to the SCZ2 data. The validated gene-trait associations appeared in the following references: [1] Goes et al. (2015); [2] Schizophrenia Working Group (2014).
| Weight | Gene | CHR | P0 | P1 | aSPU | TWAS | SSU | Most sig. SNP | Validation |
|---|---|---|---|---|---|---|---|---|---|
| YFS/NTR | MAP7D1 | 1 | 36621801 | 36646448 | 5.0E-07 | 5.2E-07 | 6.7E-07 | 3.3E-07 | |
| YFS | CNN3 | 1 | 95362507 | 95392834 | 9.0E-07 | 7.1E-02 | 7.4E-08 | 9.4E-07 | |
| CMC-introns | GABPB2 | 1 | 151043079 | 151091007 | 7.0E-07 | 4.5E-07 | 4.5E-07 | 5.6E-08 | |
| CMC-introns | TBC1D5 | 3 | 17198653 | 17784240 | 1.7E-06 | 3.1E-06 | 1.7E-06 | 5.5E-08 | [1] |
| YFS | IK | 5 | 140026643 | 140042064 | 1.4E-06 | 6.4E-07 | 4.3E-07 | 3.6E-07 | |
| CMC-introns | CXXC5 | 5 | 139028300 | 139062680 | 1.0E-07 | 2.1E-09 | 1.9E-08 | 1.5E-06 | |
| YFS | TMCO6 | 5 | 140019012 | 140024993 | 1.8E-06 | 1.1E-04 | 4.2E-07 | 3.6E-07 | |
| METSIM | DND1 | 5 | 140050379 | 140053171 | 8.0E-07 | 3.6E-06 | 1.1E-06 | 3.6E-07 | |
| CMC | ZMAT2 | 5 | 140080031 | 140086239 | 1.5E-06 | 1.6E-05 | 1.9E-07 | 3.6E-07 | |
| YFS | ABCB1 | 7 | 87133175 | 87342611 | 1.8E-06 | 5.5E-04 | 2.5E-07 | 1.4E-07 | [1] |
| METSIM/CMC-introns | ZDHHC2 | 8 | 17013538 | 17082308 | 2.0E-07 | 5.0E-06 | 7.3E-08 | 1.1E-07 | [1] |
| CMC-introns | FGFR1 | 8 | 38268655 | 38326352 | 3.0E-07 | 8.5E-07 | 7.6E-07 | 2.3E-07 | |
| YFS | ENDOG | 9 | 131580753 | 131584956 | 1.2E-06 | 6.4E-07 | 9.2E-07 | 1.9E-06 | |
| METSIM | PKN3 | 9 | 131464802 | 131483197 | 6.0E-07 | 8.2E-01 | 2.2E-08 | 1.9E-06 | |
| CMC | TEK | 9 | 27109146 | 27230172 | 4.0E-07 | 1.1E-07 | 6.4E-08 | 4.7E-07 | [1] |
| YFS | ZDHHC5 | 11 | 57435219 | 57468659 | 2.0E-07 | 2.2E-07 | 1.2E-07 | 6.7E-08 | [2] |
| NTR/METSIM | CLIP1 | 12 | 122755979 | 122907179 | 1.7E-06 | 1.9E-04 | 9.2E-08 | 3.8E-06 | [1] |
| CMC | CCDC92 | 12 | 124420954 | 124457163 | 1.0E-06 | 6.8E-03 | 6.5E-06 | 4.1E-07 | |
| YFS/NTR | PPP2R3C | 14 | 35554678 | 35591519 | 6.0E-07 | 2.7E-07 | 3.2E-07 | 1.5E-07 | |
| METSIM | KIAA0391 | 14 | 35591052 | 35743271 | 5.0E-07 | 6.6E-07 | 3.0E-07 | 1.5E-07 | [1] |
| METSIM | PCNX | 14 | 71374122 | 71582099 | 1.0E-06 | 3.8E-06 | 1.8E-07 | 1.6E-07 | [1] |
| CMC-introns | AP3B2 | 15 | 83328032 | 83378635 | 1.0E-07 | 1.7E-06 | 8.5E-08 | 5.5E-08 | |
| METSIM/CMC-introns | NMRAL1 | 16 | 4511694 | 4524896 | 5.0E-07 | 1.6E-07 | 9.3E-06 | 2.8E-07 | |
| CMC | CORO7 | 16 | 4404542 | 4466962 | 6.0E-07 | 5.2E-07 | 4.8E-07 | 2.8E-07 | |
| CMC | CPNE7 | 16 | 89642175 | 89663654 | 1.0E-07 | 5.0E-08 | 4.4E-07 | 1.1E-07 | |
| YFS/CMC | CHMP1A | 16 | 89710838 | 89724193 | 1.4E-06 | 1.5E-02 | 9.1E-08 | 1.1E-07 | |
| CMC-introns | TCF25 | 16 | 89939993 | 89977792 | 5.0E-07 | 1.4E-02 | 6.8E-08 | 1.1E-07 | |
| CMC-introns | CDK10 | 16 | 89753075 | 89762772 | 1.7E-06 | 2.1E-01 | 9.4E-08 | 1.1E-07 | |
| CMC | RPL13 | 16 | 89627064 | 89633237 | 1.5E-06 | 7.8E-04 | 1.6E-07 | 1.1E-07 | |
| YFS | PRPSAP2 | 17 | 18743398 | 18834581 | 1.0E-07 | 5.6E-08 | 5.3E-07 | 7.8E-07 | |
| METSIM | KCNG2 | 18 | 77623668 | 77660184 | 1.9E-06 | 4.5E-03 | 5.1E-08 | 2.2E-07 | [1] |
| CMC | SNRNP70 | 19 | 49588464 | 49611870 | 1.0E-07 | 2.1E-03 | 1.7E-08 | 2.2E-07 |
New Pathway Method Identifies Known and Novel SCZ-associated Pathways
We applied the new pathway test aSPUpath2 to both the SCZ1 and SCZ2 data. Figure 2 compares its p-values from the asymptotics- and Monte Carlo simulation-based methods, showing that the asymptotics gave a good approximation to the gold standard but time-consuming simulation-based method. The correlation of −log10 p-values between these two methods for PathSPU(1), PathSPU(2), and aSPUpath were 0.9989, 0.9981, and 0.9972, respectively. Because the simulation-based method is computationally demanding while the asymptotics-based method is accurate and much faster, we used the asymptotics-based method to calculate the p-values of aSPUpath2 for the subsequent analysis.
Figure 2.

Comparison between the asymptotics- and simulation-based p-values of Path-SPU(1) (left), PathSPU(2) (middle), and aSPUpath (right) based on the SCZ2 data with the GO Biological Process pathways.
Supplementary Tables 5 and 6 show the significant pathways identified by aSPUpath2 with the CMC- and YFS-based weights when applied to the SCZ1 data, respectively. We gave the gene sets in the Supplementary Tables 2 and 3 as the SCZ1- and SCZ2-based significant gene sets. For simplicity, we denote them as the SCZ1 and SCZ2 gene sets, respectively. Our new method aSPUpath2 with the CMC-based weights identified 33 significant pathways, of which 24 (around 80%) contained the significant genes in the SCZ1 gene set while 31 (around 94%) contained the significant ones in the SCZ2 gene set. In particular, aSPUpath2 with the CMC-based weights identified six significant pathways that contained at least one significant gene in the SCZ2 gene set but no significant genes in the SCZ1 gene set, such as pathways synapse organization (GO:0050808, p-value = 1.14×10−6), response to transforming growth factor beta (GO:0071559, p-value = 1.83 × 10−6), transforming growth factor beta receptor signaling pathway (GO:0007179, p-value = 4.28×10−6), and positive regulation of transforming growth factor beta production (GO:0071636, p-value = 5.65 × 10−6). There exist some biological findings partially supporting these identified pathways that would be otherwise missed by gene-based analysis. Multiple members of transforming growth factor (TGF) beta superfamily play some roles in the developing nervous system (Kapelski et al., 2016). Alteration in TGF-β1 expression has been observed in SCZ patients (Kim et al., 2004). Synapse is an important component in the nervous system and SCZ patients were found to have enriched mutations in the genes belonging to the postsynaptic density at glutamatergic synapses (Hall et al., 2015). In contrast, aSPUpath2 with the YFS-based weights identified 19 significant pathways, all of which contained at least one significant gene in both the SCZ1 and SCZ2 gene sets. Perhaps due to that the CMC-based gene expression was measured from the brain tissue and were more closely related to SCZ, while the YFS-based ones from the blood, the CMC-based weights were more informative. Overall, it was confirmed that pathway-based analysis is useful as a complementary tool to gene-based analysis, offering insights into the genetic basis of complex traits.
As an adaptive test, aSPUpath2 can maintain high power under various scenarios. For example, based on the SCZ1 data, for pathway nuclear speck (GO:0016607) with the CMC-based weights, there were 300 marginally significant and negatively associated SNPs (z-score < −1.96) and 309 marginally and positively associated SNPs (z-score > 1.96) among 5741 SNPs with non-zero weights. The varying association directions among marginally significant SNPs led to a non-significant p-value= 3.0 × 10−3 of PathSPU(1). In contrast, because PathSPU(2) was robust to varying association directions, it yielded a significant p-value= 2.1×10−8. By combining the results of PathSPU(1) and PathSPU(2), aSPUpath2 yielded a significant p-value= 4.1 × 10−8. Furthermore, this pathway contained at least two significant genes in both the SCZ1 and SCZ2 gene sets, supporting the significance of the pathway. For pathway regulation of cellular senescence (GO:2000772) with the CMC-based weights, there were 86 marginally and negatively associated SNPs (z-score < −1.96) and 45 marginally but positively associated SNPs (z-score > 1.96) among 1516 SNPs with non-zero weights. The associations in different directions were not completely canceled out since the number of the negatively associated SNPs was almost twice as that of the positively associated SNPs. PathSPU(1) yielded a significant p-value (= 1.9×10−7), while PathSPU(2) yielded a non-significant p-value (= 2.4 × 10−3). Again by combining information from the two tests, aSPUpath2 yielded a significant p-value (= 3.8 × 10−7). This pathway also contained at least one significant gene in both the SCZ1 and SCZ2 gene sets. Generally, as any non-adaptive test, PathSPU(1) or PathSPU(2) may lose statistical power under different situations; however, by contrast, aSPUpath2 that data-adaptively aggregates information can maintain relatively high power across a wide range of situations.
Then we analyzed the SCZ2 data. The new test aSPUpath2 with the CMC- and YFS-based weights identified 235 and 242 significant pathways, respectively (see Supplementary Table 6 and 7 for details). Table 4 shows the 6 significant KEGG pathways identified by aSPUpath2 with the CMC-based weights. All of these significant pathways covered at least one significant gene in the SCZ2 gene set while three pathways, Alzheimer’s disease (hsa05010, p-value = 2.4 × 10−8), systemic lupus erythematosus (hsa05322, p-value = 0.0), and hypertrophic cardiomyopathy (hsa05410, p-value = 2.3 × 10−9), have been reported by other studies to be associated with SCZ (Wu et al., 2016; Santarelli et al., 2011).
Table 4.
The significant KEGG pathways identified by aSPUpath2 with the CMC-based weights for the SCZ2 data.
| ID | Pathway name | PathSPU(1) | PathSPU(1) | aSPUpath2 | # sig. genes |
|---|---|---|---|---|---|
| hsa05322 | Systemic lupus erythematosus | 2.6E-04 | 5.5E-10 | 1.1E-09 | 16 |
| hsa05410 | Hypertrophic cardiomyopathy | 8.3E-02 | 1.5E-09 | 2.9E-09 | 2 |
| hsa05414 | Dilated cardiomyopathy | 4.0E-01 | 3.1E-08 | 6.3E-08 | 2 |
| hsa04120 | Ubiquitin mediated proteolysis | 6.1E-02 | 2.9E-07 | 5.8E-07 | 5 |
| hsa05010 | Alzheimer’s disease | 6.7E-01 | 9.1E-07 | 1.8E-06 | 5 |
| hsa05016 | Huntington’s disease | 4.7E-01 | 2.3E-06 | 4.5E-06 | 5 |
Table 5 shows the significant and novel pathways containing no significant genes in the SCZ2 gene set but detected by aSPUpath2 with either the CMC- or the YFS-based weights. Perhaps due to that the CMC-based weights were derived from the brain tissue and thus more relevant to SCZ than the YFS-based weights, using the CMC-based weights identified 12 significant and novel pathways, while using the YFS-based identified only three. Some existing studies partially supported the newly identified pathways. For example, GABA system plays an important role in orchestrating the synchronicity of local networks and affects cognitive and emotional behavior (Rudolph and Möhler, 2014). Further, cognitive symptoms in SCZ are attributed to a cortical GABAergic deficit (Rudolph and Möhler, 2014), partially supporting that pathway GABA receptor complex (GO:1902710) is possibly related to SCZ. Overall, these 15 newly identified pathways represent a class of discoveries that would have been missed by gene-based analysis.
Table 5.
The significant and novel gene sets containing no significant genes as identified by aSPUpath2 with the CMC- or YFS-based weights.
| ID | Description | # genes | PathSPU(1) | PathSPU(2) | aSPUpath2 | Weights |
|---|---|---|---|---|---|---|
| GO:1902710 | GABA receptor complex | 18 | 9.6E-03 | 0.0E+00 | 0.0E+00 | CMC |
| GO:1901661 | quinone metabolic process | 29 | 7.4E-01 | 1.0E-08 | 2.0E-08 | YFS |
| GO:0043162 | ubiquitin-dependent protein catabolic process | 18 | 5.8E-01 | 4.4E-08 | 8.8E-08 | CMC |
| GO:0016339 | calcium-dependent cell-cell adhesion | 27 | 5.7E-01 | 1.1E-07 | 2.2E-07 | CMC |
| GO:0030315 | T-tubule | 45 | 7.3E-02 | 1.1E-07 | 2.3E-07 | CMC |
| GO:0007528 | neuromuscular junction development | 36 | 4.7E-01 | 2.9E-07 | 5.7E-07 | CMC |
| GO:0003143 | embryonic heart tube morphogenesis | 62 | 5.5E-03 | 4.7E-07 | 9.5E-07 | CMC |
| GO:0007569 | cell aging | 67 | 2.0E-04 | 8.1E-07 | 1.6E-06 | CMC |
| GO:0035050 | embryonic heart tube development | 73 | 2.3E-02 | 8.8E-07 | 1.8E-06 | CMC |
| GO:0004181 | metallo-carboxypeptidase activity | 27 | 7.3E-01 | 9.7E-07 | 1.9E-06 | CMC |
| hsa00590 | Arachidonic acid metabolism | 56 | 3.0E-01 | 1.2E-06 | 2.5E-06 | YFS |
| GO:0051279 | regulation of release of sequestered calcium ion into cytosol | 75 | 2.7E-06 | 2.2E-05 | 5.3E-06 | CMC |
| GO:0072665 | protein localisation to vacuole | 46 | 3.5E-01 | 3.0E-06 | 6.1E-06 | CMC |
| GO:0010880 | regulation of release of sequestered calcium ion into cytosol by sarcoplasmic reticulum | 25 | 5.6E-06 | 3.0E-06 | 6.1E-06 | CMC |
| GO:1901800 | positive regulation of proteasomal protein catabolic process | 98 | 1.5E-03 | 4.4E-06 | 8.7E-06 | YFS |
Comparisons Between aSPUpath2 and Other Methods
With the application to the SCZ2 data with the CMC-based weights, we compared our proposed method with the two-step approach combining a gene-based test and an existing pathway analysis method, including the popular DAVID (Huang et al., 2009) or i-GSEA4GWAS (Zhang et al., 2010). We also compared it with the more general and standard aSPUpath (Pan et al., 2015).
We applied DAVID (Huang et al., 2009) with the CMC-based weights and identified one significant pathway: transcription factor activity, sequence-specific DNA binding (GO:0003700, Benjamini-corrected p-value = 4.2×10−3). This pathway was excluded in our earlier analysis because it contained more than 200 genes; when applied, aSPUpath2 could identify this pathway as well (p-value = 4.5 × 10−7). We also applied i-GSEA4GWAS (Zhang et al., 2010) but failed to identify any significant pathways. In addition to the two-step nature of the above two pathway methods (thus depending on the output or performance of the gene-based testing in the first step), in contrast to the one-step approach of aSPUpath2, they also differ with respect to their null hypotheses being tested: both DAVID and i-GSEA4GWAS belong to the category of “competitive tests” testing for the enrichment of the associated genes in the pathway being tested as compared to other pathways, while our aSPUpath2 method is a “self-contained test” as a global test for identifying whether there is (are) any significant gene(s) in the pathway; due to the difference between the null hypotheses being tested, a self-contained test is in general more powerful than a corresponding competitive test.
Figure 3 shows the running times for aSPUpath2 and aSPUpath. Due to the computational constraint, we ran at most B = 106 simulations to calculate the p-values for aSPUpath. For the simulation-based method, the running time increased rapidly with the number of simulations, for which a larger value is required for a more significant p-value. In contrast, since the p-values of aSPUpath2 was calculated by the asymptotics-based method, the running time was invariant to the p-values. Supplementary Table 9 shows the 179 significant pathways identified by aSPUpath with the CMC-based weights, of which 139 (around 80%) were also identified by applying aSPUpath2 with the CMC-based weights, constituting a highly significant overlap between their results. Furthermore, aSPUpath2 identified a total of 235 significant pathways, showcasing possibly higher statistical power over aSPUpath for the SCZ2 data. In summary, aSPUpath2 is several orders faster than aSPUpath, more so for large and highly significant pathways, and can be more powerful for densely associated pathways (i.e. those containing many associated SNPs/genes), thus we recommend using aSPUpath2 either alone or as a fast screening procedure for the more time-consuming and more general aSPUpath test.
Figure 3.
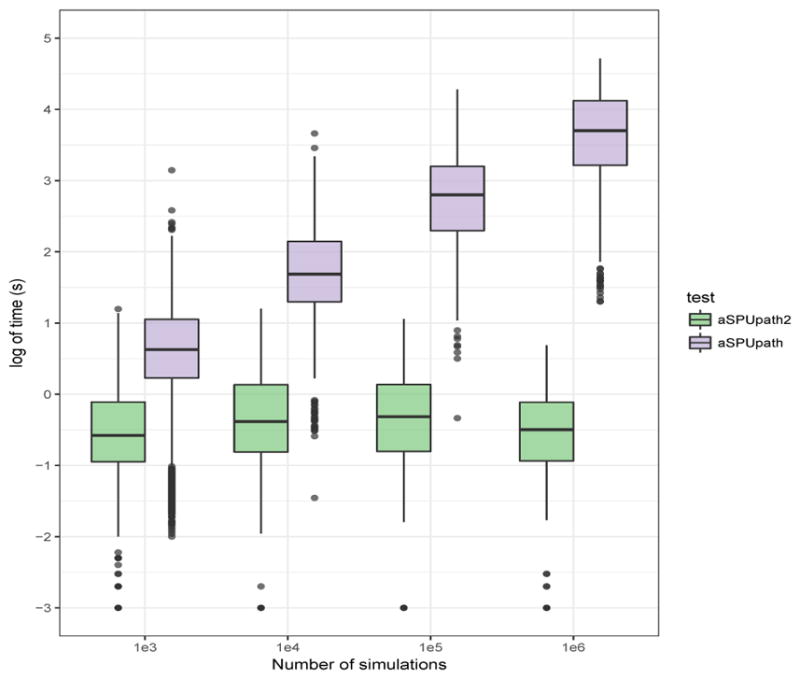
Comparison between running times of aSPUpath2 and aSPUpath for the SCZ2 data with the pathways in the GO Biological Process.
Simulations
We conducted simulation studies to evaluate and compare the performance of our proposed new aSPUpath2 test with the aSPUpath test. We generated simulated data to mimic real data: we used the GO Biological Process pathways and CMC-derived SNP weights, and simulated z-scores as GWAS summary statistics for SNPs. Specifically, for a given pathway S* in the GO Biological Process pathway database, we first removed the genes whose CMC-derived SNP weights were all 0, resulting in a subset S containing n genes and p SNPs with none-zero weights. We generated a z-score vector from a multivariate normal distribution, Z ~ N(μ, Σ), where μ = (μ1, …, μp)′ was the mean and Σ was the LD matrix based on the 1000 Genomes Project reference panel (European ancestry), respectively. Note that z-scores are expected to have a multivariate normal distribution asymptotically. To save computing time, we assumed that the SNPs from different chromosomes were independent and only considered the pathways with less than 2000 SNPs. In total, we considered 1905 pathways. Further, we defined SNP j was associated or informative with the corresponding μj = sign(Wj)c, where Wj was the CMC-derived weight for SNP j, c ≠ 0 was some positive constant, and sign(a) gave the sign of a; in contrast, SNP j was non-informative with μj = 0. Note that we also considered non-constant |μj| for associated SNPs. To evaluate type I error rates, we considered the null case (set-up A) with no informative SNP (μ = 0). To evaluate power, we further considered the following four set-ups under different situations: set-up B, 50% SNPs in each gene were informative; set-up C, 10% SNPs in each gene were informative; set-up D, only one SNP in each gene was informative; and set-up E, only one SNP in 20% of the genes in the pathway was informative. Other SNPs were set to non-informative and we varied the true association strength c to generate power curves for set-up B to E. After generating a z-score vector for each pathway, we applied both the aSPUpath2 and aSPUpath tests. The entire procedure was repeated about 38,000 times (i.e. 20 per pathway) for set-up A. For other set-ups, with different c, we repeated the entire procedure about 1,900 times (1 per pathway) and fixed the nominal significance level at α = 0.05.
Table 6 shows the empirical type I error rates, indicating that the PathSPU(1), Path-SPU(2), and aSPUpath2 could control their type I rates satisfactorily under various nominal significance levels.
Table 6.
Empirical type I error rates of our proposed pathway-based tests with some varying nominal significance levels α under simulation set-up A.
| α | 0.05 | 0.01 | 0.001 |
|---|---|---|---|
| PathSPU(1) | 4.9 × 10−2 | 9.8 × 10−3 | 1.2 × 10−3 |
| PathSPU(2) | 5.3 × 10−2 | 1.1 × 10−2 | 1.3 × 10−3 |
| aSPUpath2 | 4.4 × 10−2 | 1.0 × 10−2 | 1.2 × 10−3 |
Figure 4 shows statistical power under set-ups B to E. In set-up B, because 50% of the SNPs in the pathway were informative with dense association signals, PathSPU(1) was expected to be most powerful as confirmed in Figure 4; since aSPUpath2 combined the information from both the PathSPU(1) and PathSPU(2), aSPUpath2 also achieved high power close to PathSPU(1). When the association signals were less dense with only 10% of the SNPs as informative (set-up C), all the tests performed similarly, though aSPUpath2 and PathSPU(1) had a slight edge over aSPUpath and PathSPU(2) respectively. When most SNPs (set-up D) or most genes were not associated with the trait (set-up E), aSPU-path was expected to be more powerful than aSPUpath2 because aSPUpath2 is tailored to identifying dense associations of pathways containing many associated SNPs/genes with only weak effects. In other simulation set-ups with varying |μj| for associated SNPs and/or different proportions of associated SNPs/genes, we obtained similar results as shown in Supplementary Figure 12. Note that, by theory, there is no uniformly most powerful test for pathway analysis; aSPUpath is more general and thus expected to be high powered across a wider range of scenarios than aSPUpath2, which is tailored for and more powerful for detecting dense association signals like in set-up A. However, aSPUpath2 is much faster than aSPUpath. Hence, as mentioned earlier, we recommend using aSPUpath2 either alone to detect densely associated pathways, or as a fast screening procedure for aSPUpath if one is interested in both densely and sparsely associated pathways.
Figure 4.

Empirical power at α = 0.05 under different simulation set-ups (B–E).
Discussion
In this work, we have presented a powerful and adaptive method that integrates genetic and transcriptional variations to identify pathways associated with a complex trait. Using gene expression to construct weights and then adaptive weighting to identify significant pathways has some potential advantages. First, a pathway may be a more interpretable biological unit than a single SNP or gene, and may shed light into biological mechanisms underlying a trait or disease. Second, pathway-based analysis, complementary to gene-based analysis, and as demonstrated here, can identify important pathways that may be missed by gene-based analysis. Since different tests will be powerful under different underlying true association patterns, in particular, our proposed test may maintain relatively high statistical power across a wider range of situations due to its adaptive nature of aggregating association information across the genes in a pathway. Third, our proposed method is similar to other integrative gene-based methods, such as TWAS (Gusev et al., 2016a), PrediXcan (Gamazon et al., 2015) and aSPU (Xu et al., 2017b), that incorporate eQTL information into GWAS analysis. However, differing from that the above integrative methods are gene-based, our method aggregates information across the genes to identify significant pathways. Importantly, unlike TWAS and PrediXcan, which use a simple weighted linear combination of genetic variants (or their z-scores) to construct test statistics, our approach adaptively (and non-linearly) weights the genetic variants and thus aggregates information based on the underlying association patterns to increase discovery power. As shown in our applications, our method could identify some important pathways that were missed by the above integrative gene-based tests, even followed with a standard pathway analysis. Finally, we note that our proposed approach is in the category of “self-contained tests”, in which we are interested in identifying any pathway containing one or more genes or SNPs associated with a trait. This is different from the “competitive tests”, such as DAVID and GSEA, that would detect pathways enriched with associated genes or SNPs as compared to background pathways.
Application of our proposed and other integrative gene-based methods to two SCZ summary data not only recapitulated many known genes or pathways but also identified many new ones. Specifically, we identified 75 significant genes without any known associated SNPs within 500 kb, of which 50 have not been reported in any studies yet. It is possible that some of these significant genes represent new findings that have been missed due to the lower statistical power in other standard single SNP- or gene-based test without incorporating gene expression data. Furthermore, some pathways may contain only genes with small effect sizes, which may not be detected even by integrative gene-based tests like TWAS, but may be by our proposed pathway test. Here, we identified 15 novel significant pathways associated with SCZ, such as pathway GABA receptor complex (GO:1902710), which could be missed by gene-based TWAS or aSPU. Taken together, our results showcase the power of incorporating reference gene expression data into gene-based or pathway-based association testing for GWAS. The newly identified genes and pathways may help us gain insights into the biological mechanism underlying SCZ.
Although in this study we have mainly focused on SCZ and applied the various methods to two GWAS summary data sets, it is natural to apply our method to other complex traits with either individual-level or summary data. We expect that applying our proposed and other integrative methods like TWAS to other existing GWAS data may identify more novel associations and shed more light on the underlying biological mechanisms. We note that our proposed methodology can be applied with other endophenotype-derived weights (Xu et al., 2017a) or even without weights (i.e. all SNPs with an equal weight).
Finally we comment on our view that TWAS is a weighted Sum test and its related issues, which are also discussed by Wainberg et al. (2017) and in http://hakyimlab.org/post/vulnerabilities/. Although TWAS was originally proposed to identify GWAS associations through gene expression, any such discovery based on a single eQTL/GWAS dataset is at most only suggestive to mediating effects of gene expression. As discussed in Xu et al. (2017b), in spite of the connections of TWAS with two-stage least squares and Mendelian randomization (MR), due to the adopted strong assumptions that are likely to be violated in practice, cautions should be taken to avoid extrapolating any discovered GWAS associations to causal effects mediated through gene expression. Hence, we simply regard TWAS as a special case of weighted association testing. In this view, we yield a few benefits while avoiding possible over-interpretation of an association as a causal effect. First, due to some well-known limitations of the Sum test and inherent errors in estimating the cis-effects (i.e. weights) of genetic variants with usually small eQTL datasets, modifications to TWAS may lead to more powerful analysis methods, such as based on the SSU/SPU(2) and aSPU tests (Xu et al 2017a). Other tests, like aSPU, with a more flexible weighting scheme, may also identify associations through other non-gene expression-mediated mechanisms. Second, in addition to gene expression, other molecular or clinical intermediate phenotypes can be used to construct weights for weighted GWAS association analysis (Xu et al., 2017a).
The proposed statistical tests are implemented in R package aSPU2 that is currently publicly available on GitHub (and will be put on CRAN); the online manual and example computer code are publicly available at wuchong.org/aspupath2.html.
Supplementary Material
Acknowledgments
The authors thank the reviewers for many helpful comments. This research was supported by NIH grants R21AG057038, R01HL116720, R01GM113250 and R01HL105397, and by the Minnesota Supercomputing Institute.
Footnotes
Web Resources
- DAVID server: https://david.ncifcrf.gov;
- iGSEA4GWAS server: http://gsea4gwas.psych.ac.cn;
- NHGRI-EBI GWAS Catalog: http://www.ebi.ac.uk/gwas/home;
- PGC summary data: https://www.med.unc.edu/pgc/downloads;
- TWAS website: http://gusevlab.org/projects/fusion.
References
- Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nature Reviews. Genetics. 2015;16(4):197–212. doi: 10.1038/nrg3891. [DOI] [PubMed] [Google Scholar]
- Bakshi A, Zhu Z, Vinkhuyzen AA, Hill WD, McRae AF, Visscher PM, Yang J. Fast set-based association analysis using summary data from GWAS identifies novel gene loci for human complex traits. Scientific Reports. 2016;6:32894. doi: 10.1038/srep32894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LS, Hutter CM, Potter JD, Liu Y, Prentice RL, Peters U, Hsu L. Insights into colon cancer etiology via a regularized approach to gene set analysis of GWAS data. The American Journal of Human Genetics. 2010;86(6):860–871. doi: 10.1016/j.ajhg.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, G. O. et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Research. 2004;32(Database issue):D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Disorder Group PGC. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. The Lancet. 2013;381(9875):1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach A, Lawless JF, Sun L, et al. Pooled association tests for rare genetic variants: a review and some new results. Statistical Science. 2014;29(2):302–321. [Google Scholar]
- Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, Eyler AE, Denny JC, Nicolae DL, Cox NJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nature Genetics. 2015;47(9):1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeman JJ, Bühlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics. 2007;23(8):980–987. doi: 10.1093/bioinformatics/btm051. [DOI] [PubMed] [Google Scholar]
- Goes FS, McGrath J, Avramopoulos D, Wolyniec P, Pirooznia M, Ruczinski I, Nestadt G, Kenny EE, Vacic V, Peters I, et al. Genome-wide association study of schizophrenia in ashkenazi jews. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2015;168(8):649–659. doi: 10.1002/ajmg.b.32349. [DOI] [PubMed] [Google Scholar]
- Guan F, Zhang T, Li L, Fu D, Lin H, Chen G, Chen T. Two-stage replication of previous genome-wide association studies of as 3mt-cnnm2-nt5c2 gene cluster region in a large schizophrenia case–control sample from han chinese population. Schizophrenia Research. 2016;176(2):125–130. doi: 10.1016/j.schres.2016.07.004. [DOI] [PubMed] [Google Scholar]
- Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, Jansen R, De Geus EJ, Boomsma DI, Wright FA, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nature Genetics. 2016a;48(3):245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Mancuso N, Finucane HK, Reshef Y, Song L, Safi A, Oh E, Mc-Caroll S, Neale B, Ophoff R, et al. Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. bioRxiv. 2016b:067355. doi: 10.1038/s41588-018-0092-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Trent S, Thomas KL, O’Donovan MC, Owen MJ. Genetic risk for schizophrenia: convergence on synaptic pathways involved in plasticity. Biological Psychiatry. 2015;77(1):52–58. doi: 10.1016/j.biopsych.2014.07.011. [DOI] [PubMed] [Google Scholar]
- Heinig M, Petretto E, Wallace C, Bottolo L, Rotival M, Lu H, Li Y, Sarwar R, Langley SR, Bauerfeind A, et al. A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature. 2010;467(7314):460–464. doi: 10.1038/nature09386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nature Protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapelski P, Skibińska M, Maciukiewicz M, Zaremba D, Jasiak M, Hauser J. Family association study of transforming growth factor beta1 gene polymorphisms in schizophrenia. Psychiatr Pol. 2016;50(4):761–770. doi: 10.12740/PP/61273. [DOI] [PubMed] [Google Scholar]
- Kim YK, Myint AM, Lee BH, Han CS, Lee HJ, Kim DJ, Leonard BE. Th1, th2 and th3 cytokine alteration in schizophrenia. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2004;28(7):1129–1134. doi: 10.1016/j.pnpbp.2004.05.047. [DOI] [PubMed] [Google Scholar]
- Kwak IY, Pan W. Adaptive gene-and pathway-trait association testing with GWAS summary statistics. Bioinformatics. 2015;32(8):1178–1184. doi: 10.1093/bioinformatics/btv719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak IY, Pan W. Adaptive gene-and pathway-trait association testing with GWAS summary statistics. Bioinformatics. 2016;32(8):1178–1184. doi: 10.1093/bioinformatics/btv719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, AC’t Hoen P, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501(7468):506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wei Z, Chang X, Cardinale CJ, Kim CE, Baldassano RN, Hakonarson H, et al. Consortium IIG. Pathway-based genome-wide association studies reveal the association between growth factor activity and inflammatory bowel disease. Inflammatory Bowel Diseases. 2016;22(7):1540–1551. doi: 10.1097/MIB.0000000000000785. [DOI] [PubMed] [Google Scholar]
- Li L, Wang X, Xiao G, Gazdar A. Integrative gene set enrichment analysis utilizing isoform-specific expression. Genetic Epidemiology. 2017;41:498–510. doi: 10.1002/gepi.22052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo SK, Shtir C, Doyle AE, Mick E, McGough JJ, McCracken J, Biederman J, Smalley SL, Cantor RM, Faraone SV, et al. Genome-wide association study of intelligence: additive effects of novel brain expressed genes. Journal of the American Academy of Child & Adolescent Psychiatry. 2012;51(4):432–440. doi: 10.1016/j.jaac.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Ma X, Deng W, Liu X, Li M, Chen Z, He Z, Wang Y, Wang Q, Hu X, Collier D, et al. A genome-wide association study for quantitative traits in schizophrenia in china. Genes, Brain and Behavior. 2011;10(7):734–739. doi: 10.1111/j.1601-183X.2011.00712.x. [DOI] [PubMed] [Google Scholar]
- MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, Junkins H, McMahon A, Milano A, Morales J, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog) Nucleic Acids Research. 2017;45(Database issue):D896–D901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network T of the Psychiatric Genomics Consortium PAS. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nature Neuroscience. 2015;18(2):199–209. doi: 10.1038/nn.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W. Asymptotic tests of association with multiple SNPs in linkage disequilibrium. Genetic Epidemiology. 2009;33(6):497–507. doi: 10.1002/gepi.20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W, Kim J, Zhang Y, Shen X, Wei P. A powerful and adaptive association test for rare variants. Genetics. 2014;197(4):1081–1095. doi: 10.1534/genetics.114.165035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W, Kwak IY, Wei P. A powerful pathway-based adaptive test for genetic association with common or rare variants. The American Journal of Human Genetics. 2015;97(1):86–98. doi: 10.1016/j.ajhg.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasaniuc B, Zaitlen N, Shi H, Bhatia G, Gusev A, Pickrell J, Hirschhorn J, Strachan DP, Patterson N, Price AL. Fast and accurate imputation of summary statistics enhances evidence of functional enrichment. Bioinformatics. 2014;30(20):2906–2914. doi: 10.1093/bioinformatics/btu416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G, Luo L, Siu H, Zhu Y, Hu P, Hong S, Zhao J, Zhou X, Reveille JD, Jin L, et al. Gene and pathway-based second-wave analysis of genome-wide association studies. European Journal of Human Genetics. 2010;18(1):111–117. doi: 10.1038/ejhg.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kähler AK, Akterin S, Bergen SE, Collins AL, Crowley JJ, Fromer M, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nature Genetics. 2013;45(10):1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Möhler H. Gabaa receptor subtypes: Therapeutic potential in down syndrome, affective disorders, schizophrenia, and autism. Annual Review of Pharmacology and Toxicology. 2014;54:483–507. doi: 10.1146/annurev-pharmtox-011613-135947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli DM, Beveridge NJ, Tooney PA, Cairns MJ. Upregulation of dicer and microrna expression in the dorsolateral prefrontal cortex brodmann area 46 in schizophrenia. Biological psychiatry. 2011;69(2):180–187. doi: 10.1016/j.biopsych.2010.09.030. [DOI] [PubMed] [Google Scholar]
- Schaid DJ, Sinnwell JP, Jenkins GD, McDonnell SK, Ingle JN, Kubo M, Goss PE, Costantino JP, Wickerham DL, Weinshilboum RM. Using the gene ontology to scan multilevel gene sets for associations in genome wide association studies. Genetic Epidemiology. 2012;36(1):3–16. doi: 10.1002/gepi.20632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group t. P. G. C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Daly MJ, O’donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nature Reviews Genetics. 2012;13(8):537–551. doi: 10.1038/nrg3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiihonen J, Lönnqvist J, Wahlbeck K, Klaukka T, Niskanen L, Tanskanen A, Haukka J. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study) The Lancet. 2009;374(9690):620–627. doi: 10.1016/S0140-6736(09)60742-X. [DOI] [PubMed] [Google Scholar]
- Wainberg M, Sinnott-Armstrong N, Knowles D, Golan D, Ermel R, Ruusalepp A, Quertermous T, Hao K, Bjorkegren JL, Rivas MA, et al. Vulnerabilities of transcriptome-wide association studies. bioRxiv. 2017:206961. [Google Scholar]
- Wang K, Li M, Bucan M. Pathway-based approaches for analysis of genomewide association studies. The American Journal of Human Genetics. 2007;81(6):1278–1283. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P, Tang H, Li D. Insights into pancreatic cancer etiology from pathway analysis of genome-wide association study data. PloS One. 2012;7(10):e46887. doi: 10.1371/journal.pone.0046887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Green MJ, Gardiner EJ, Tooney PA, Scott RJ, Carr VJ, Cairns MJ. Altered neural signaling and immune pathways in peripheral blood mononuclear cells of schizophrenia patients with cognitive impairment: A transcriptome analysis. Brain, Behavior, and Immunity. 2016;53:194–206. doi: 10.1016/j.bbi.2015.12.010. [DOI] [PubMed] [Google Scholar]
- Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. The American Journal of Human Genetics. 2011;89(1):82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wu C, Pan W, Initiative ADN, et al. Imaging-wide association study: Integrating imaging endophenotypes in GWAS. NeuroImage. 2017a;159:159–169. doi: 10.1016/j.neuroimage.2017.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wu C, Wei P, Pan W. A powerful framework for integrating eqtl and GWAS summary data. Genetics. 2017b;207(3):893–902. doi: 10.1534/genetics.117.300270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Cui S, Chang S, Zhang L, Wang J. i-GSEA4GWAS: a web server for identification of pathways/gene sets associated with traits by applying an improved gene set enrichment analysis to genome-wide association study. Nucleic Acids Research. 2010;38(Web Server issue):W90–W95. doi: 10.1093/nar/gkq324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.