

Abstract
Iron(II)- and 2-(oxo)-glutarate-dependent oxygenases catalyze diverse oxidative transformations that are often initiated by abstraction of hydrogen from carbon by iron(IV)-oxo (ferryl) complexes. Control of the relative orientation of the substrate C–H and ferryl Fe–O bonds, primarily by direction of the oxo group into one of two cis-related coordination sites (termed inline and offline), may be generally important for control of the reaction outcome. Neither the ferryl complexes nor their fleeting precursors have been crystallographically characterized, hindering direct experimental validation of the offline hypothesis and elucidation of the means by which the protein might dictate an alternative oxo position. Comparison of high-resolution x-ray crystal structures of the substrate complex, an Fe(II)-peroxysuccinate ferryl precursor, and a vanadium(IV)-oxo mimic of the ferryl intermediate in the L-arginine 3-hydroxylase, VioC, reveals coordinated motions of active site residues that appear to control the intermediate geometries to determine reaction outcome.
Graphical Abstract
INTRODUCTION
Oxygen-mediated modification of biomolecules enables a significant proportion of the structural diversity present in natural products, molecules of unquestioned medicinal and industrial importance.1–2 All biological catalysts that rely on dioxygen as a source of oxidative power must exert tight spatial and temporal control over reactive O2-derived adducts.3 X-ray crystallographic characterization of such intermediates via reaction initiation in single crystals has been important in understanding these mechanisms of control in many classes of O2-reactive enzymes, including the extradiol ring-cleaving dioxygenases,4 copper amine oxidases,5 cytochrome P450s,6 and diiron hydroxylases.7 Crystallographic analysis of these reaction pathways has also unmasked new intermediates and provided important structural context for states characterized by other methods.
Fe(II)- and 2OG oxygenases constitute one of the primary enzymatic routes to O2-mediated functionalization of natural products and are overrepresented in biosynthetic pathways owing to their unique ability to selectively functionalize otherwise chemically inert C-H bonds.8 More than 100,000 sequences in prokaryotes and eukaryotes have been annotated as probable Fe/2OG oxygenases on the basis of their conservation of an iron-binding motif, H-X-(D/E)-Xn-H (Fig. 1).9–10 Fe/2OG enzymes are functionally diverse, catalyzing both simple –O atom insertions and more complex transformations that include industrially-relevant halogenation and cyclization reactions.8 All members of the superfamily use their FeII cofactor and a common metabolite, 2OG, to form an FeIV-oxo (ferryl) intermediate;11 addition of O2 to the complete preformed enzyme•substrate complex results in oxidation of the iron and decarboxylation of 2OG to succinate. X-ray crystal structures of the O2-reactive enzyme•FeII•2OG•substrate complexes, as well as inhibitor and product-bound states, have been reported for many Fe/2OG enzymes,12 and the ferryl intermediates have been characterized by spectroscopic methods11, 13–18 in a smaller number of cases. Importantly, neither the key FeIV=O state nor any of its fleeting precursors have been structurally characterized in any member of this enzyme superfamily.
Figure 1.

Proposed mechanism of Fe/2OG hydroxylases.
In the majority of cases, the ferryl oxidant initiates transformation of the primary substrate by abstracting a hydrogen atom (H•),13 yielding an intermediate with a carbon-centered radical and FeIII-OH cofactor form. Different pathways emerging from this key branch-point intermediate lead to the known divergent reaction outcomes in the superfamily.8 The most common transformation is hydroxylation, accomplished by attack of the substrate radical upon the coordinated oxygen ligand, a step thought to have a low activation barrier. The product is a new R-OH group and an FeII-succinate complex. Although the precise fates of the initial substrate radical in the alternative transformations are less well understood, it is crucial to avoid facile coupling to the oxygen ligand. For some systems, distinct ligand geometries of the ferryl state have been proposed18–19 as a mechanism to suppress this step, but direct evidence for this notion is lacking.
RESULTS AND DISCUSSION
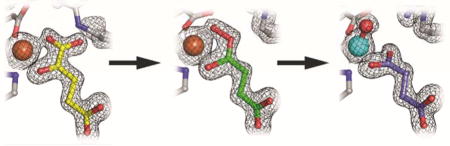
VioC from Streptomyces vinaceus 1326 is an Fe/2OG oxygenase that hydroxylates C3 of L-arginine on the biosynthetic pathway to the tuberactin antibiotic viomycin,20 which was originally included in multi-drug cocktails targeting Mycobacterium tuberculosis infections.21 In the study elucidating its role in the viomycin pathway, multiple structures of VioC complexes were solved to near-atomic resolution by x-ray crystallography.22 However, the x-ray datasets provide only limited insight into the reaction mechanism because the complexes characterized are not on the catalytic pathway. In one, 2OG was replaced by tartrate; in a second, the 3-hydroxy L-Arg product was bound along with succinate; and in a third, the iron cofactor was absent (fig. S1). However, this initial study indicated that VioC crystals routinely diffract to high resolution (< 1.8 Å) and might therefore be amenable to crystallographic characterization of intermediates in the reaction cycle. Reproduction of the previously reported crystallization conditions but with O2 excluded and 2OG replacing tartrate or succinate as the precipitant yielded a 1.6 Å resolution structure of the complete O2-reactive reactant complex with highly reproducible crystallization and x-ray diffraction (Fig. 2A) (Supporting Information).
Figure 2.

Crystal structures of VioC with L-Arg and 2OG (A), 3-OH-L-Arg and 2OG (B), L-Arg and peroxysuccinate (C), 3-OH-L-Arg and succinate (D), L-Arg and succinate (E), and the photoreduced vanadyl complex (F). A 2Fo-Fc composite electron density map is shown in black mesh and contoured to 1.5 σ. Selected amino acids and active site components are shown in stick format. Iron (orange) and vanadium (teal) ions are shown as space-filling models.
With the goal of accumulating the H• abstracting ferryl intermediate, crystals of the reactant complex prepared anoxically with d7-L-Arg were briefly exposed to O2 (for less than 1 min) and flash frozen. Deuterium substitution slows the H-atom transfer (HAT) step, which is rate-limiting in solution.23–24 Initial efforts yielded many data-sets representative of the 3-S-OH-L-Arg/2OG complex (Fig. 2B), suggesting that the reaction proceeds in crystallo on this timescale, even with the heavy-isotope challenge. Despite evidence of L-Arg turnover, in no case was the succinate coproduct also present; the electron density was instead consistent with the larger cosubstrate 2OG (present as a precipitant at ~1.0 M concentration), suggestive of rapid diacid ligand exchange without dissociation of the primary 3-OH-L-Arg product.
A few datasets, however, yielded a different and surprising result. In these structures, the electron density map is consistent with the presence of the L-Arg substrate; no electron density for the 3-OH group of the product is apparent. New electron density bridging the iron cofactor and the organic cosubstrate is inconsistent with the targeted ferryl-succinate complex that accumulates in solution (Fig. 2C), nor could this density be modeled as an FeII-succinate adduct or the photoreduced and protonated [FeII-O(H)x] form of the ferryl complex (fig. S2). Attempts to do so consistently yielded a large and elongated peak of positive Fo-Fc difference electron density between the metal and cosubstrate that is not fully accounted for by these models (fig. S2). By contrast, a model of a computationally-predicted precursor,25–26 in which the C1 carboxylate of 2OG has departed and an O2-derived peroxide adduct bridges the metal and the succinate coproduct with the O-O bond still intact (Fig. 2C), fits remarkably well into the observed electron density. This complex is one of two postulated ferryl precursors that have yet to be detected (Fig. 1) by any experimental method. The other option is a bicyclic FeIV-peroxyhemiketal complex27 with the C1 carboxylate intact (Fig. 1) but this structure is clearly too large to fit into the observed density. Refinement of a model of the FeII-peroxysuccinate intermediate against the electron density from these datasets gave Fe-O-O bond distances and angles that agree with computational predictions (fig. S3).
The observation that multiple sidechain motions associated with conversion of the FeII•2OG•L-Arg reactant complex to the FeII•succinate•3-S-OH-L-Arg product complex (vide infra) have already occurred in the putative intermediate state lends further credence to its assignment as an on-pathway FeII-peroxysuccinate complex. Prior to O2 exposure, the FeII center in VioC exhibits a distorted 5-coordinate geometry in which the C1 carboxylate of 2OG is shifted ~35° out of the equatorial plane defined by His168, Glu170, and the 2OG ligand (figs. S4–S6). C3 of L-Arg is positioned 4.8 Å from the cofactor, a distance suitable for HAT, and clear electron density is visible for all protein side chains in the vicinity of the active site with the notable exceptions of Arg334 and Gln137 (fig. S4). These side chains are best modeled as rotated away from the active site to avoid clash with C1 of 2OG, but occupancy refinement (~33% for the terminal heteroatoms) suggests that the amino acids are partially disordered in the reactant complex.
To obtain the product complex via reaction in single crystals, we exposed VioC co-crystallized with 2OG to O2 for a longer period of time (~4 hours) in crystallization solution containing > 1.0 M succinate as the precipitant. Electron density maps obtained from these crystals reveal a new oxygen substituent at C3 of L-Arg and succinate bound in the co-substrate site (Fig. 2D). The coordination of the new oxygen of the 3-S-OH-L-Arg product to the FeII cofactor trans to His316 suggests that it is not protonated. The presence of the alkoxide is somewhat surprising, given the high pKa of the hydroxyl group (~ 17). The observation may, however, be consistent with proposals of initial rebound of the deprotonated oxo unit28 and could have important implications for bifunctional Fe/2OG enzymes that first hydroxylate their substrates and then effect distinct transformations of the alcohol (or alkoxide) intermediates.8
The product complex shows that, upon cleavage of the C1–C2 bond of 2OG, the resulting succinate ligand relaxes into the equatorial plane perpendicular to the Fe-His168 axis, yielding a more idealized octahedral coordination geometry. The side chain of the 3-OH-L-Arg product contorts significantly to enable coordination of the hydroxyl group, and the guanidinium moiety rotates, accommodated by accompanying protein side chain shifts within the L-Arg binding pocket. The α-NH3+ group of L-Arg moves away from the Glu170 ligand to interact with Gln137, which can now be modeled unequivocally because it becomes well-ordered upon conversion of substrate to product. Furthermore, the Glu170 carboxylate rotates toward the sidechain of Arg334, which also becomes ordered as it moves into the space vacated by C1 of 2OG to anchor an H-bonding network involving the L-Arg carboxylate and succinate O1 (Fig. 3). The motions of these residues are enabled by the additional space in the active site made available by departure of CO2. The observations indicate that the coordination mode of 2OG and the positions of Arg334 and Gln137 are markedly different in the reactant and product states.
Figure 3.

An H-bonding network involving Arg334 orients the oxygen-derived ligand in the FeII-peroxysuccinate complex (A) and vanadyl-bound state of VioC (C). An octahedral base geometry in each state (B, D) leaves a second site open trans to His168. Selected bonding interactions are indicated with distances listed in angstroms (Å) and amino acid side chains and substrate molecules are shown in stick format. Hydrogen bonding interactions and weak coordination bonds are illustrated as black dashed lines. Metal ions and oxygen adducts are shown as space-filling models.
X-ray structures of the 3-OH-L-Arg/2OG and L-Arg/succinate VioC complexes also corroborate this model for second sphere side chain motions associated with 2OG decarboxylation in progression to intermediate states (Figs. 2B and 2E). In these structures, Arg334 remains ordered and in the inward-facing position, anchored by interaction with the product 3-OH group and/or O2 of 2OG/succinate. The 2OG coordination mode in the 3-OH-L-Arg/2OG complex is distinct from that of the reactant complex: the C1 carboxylate is significantly displaced to accommodate Arg334, a binding mode that is more reminiscent of the structures with succinate bound and is probably driven by coordination of the product C3 oxygen (fig. S5A).
In the structure of the posited L-Arg/peroxysuccinate complex, Arg334 and Gln137 are both well-ordered and found in their product-state conformations (fig. S4). This observation provides support for the assignment of the structure as a new intermediate and not simply a mixture of reactant and product states. In the latter scenario, increased disorder of the Arg334 and Gln137 side chains, manifested as weaker electron density, would be anticipated. We additionally ruled out the possibility of a mixture by modeling and refinement of 2OG and the 3-OH-L-Arg product into the observed electron density in the intermediate structure (figs. S2, S6), yielding negative difference density for the 3-OH group and weak density for the C1 2OG carboxylate. These observations are consistent with a state that accumulates after loss of C1 as CO2 but prior to substrate hydroxylation. Early movement of Arg334 and Gln137 may also explain the surprising accumulation of the peroxysuccinate complex in VioC crystals. Full ordering of the side chains and/or formation of H-bonds and other n0n-covalent interactions with oxygen-derived ligand may be important for progression to the ferryl-succinate complex and these steps could be disproportionately impeded in the crystal lattice. Our crystallographic observation of the peroxysuccinate intermediate implies mechanistic relevance of this state,29–30 here as a transient on-pathway intermediate to the substrate-targeting ferryl. The structure bears resemblance to a metastable acylperoxo-FeIII intermediate that forms upon incubation of biomimetic mononuclear FeII complexes with peracetic acid.31–33 In those systems, the acylperoxide ligand undergoes reversible O-O bond cleavage to yield a C-H oxidizing FeV(O) species. It is also interesting to consider the possibility that the FeII-peroxysuccinate complex could undergo alternative (non-ferryl-generating) transformations in other Fe/2OG enzymes, which would serve to significantly expand the catalytic diversity in the superfamily.
To understand the macromolecular context of the key H• abstracting intermediate, we implemented a structural mimic of the ferryl state, in which the transition metal component was exchanged for vanadium.34 High-valent Fe centers in enzymes are notoriously fleeting and highly reactive,24 but early transition metal oxycations are known to be significantly more stable.34–35 The vanadyl ion (VIV-oxo) has been shown to occupy divalent cation binding sites in proteins, and, in a recent study, to mimic the H•-abstracting ferryl intermediate (FeIV=O) in Fe/2OG enzymes.36 Vanadium(IV) is a d1 ion with a ground-state total electron spin quantum number (S) of 1/2, and the VioC•vanadyl•succinate•L-Arg complex therefore exhibits an electron paramagnetic resonance (EPR) spectrum with octet splitting due to hyperfine coupling with the 51V nucleus (100% natural abundance, I = 7/2; fig. S7). Moreover, structural information can be extracted by examining hyperfine couplings to nearby nuclei using hyperfine sublevel correlation (HYSCORE) spectroscopy. Field-dependent 2H-HYSCORE spectra collected in the presence of 3,3-d2-arginine can be simulated (Fig. 4A) to furnish the disposition of the target hydrogen atom relative to the vanadyl (fig. S8). Features of the spectra arising from deuterium (fig. S9) can be simulated by a single axial hyperfine coupling tensor with magnitude A = 0.33 ± 0.04 MHz (Euler angles [0, 37, 217] ± 10°) and quadrupole coupling [−0.06, −0.06, 0.12] ± 0.01 MHz (Euler angles [0, 70, 30] ± 10°). The hyperfine coupling can be interpreted according to a point-dipole model, indicating the presence of a deuterium nucleus 3.3 ± 0.2 Å from the vanadium nucleus, at an angle of 37 ± 10° relative to the V-oxo bond (Fig. 4B).
Figure 4.

(A) Field-dependent HYSCORE spectra (middle) of vanadyl-VioC collected in the presence of 3,3-d2-L-Arg, collected at 282.0 (i), 297.5 (ii), and 342.5 (iii) mT and corresponding simulations (bottom). Ori-entation selectivities that were probed by each magnetic field position (top) are color-coded using an RGB color scheme (red - fully excited; blue, not excited). Spectra were collected with microwave frequency 9.46 GHz and temperature 35 K. (B) Comparison of crystallographic (black) and HYSCORE (red) models of the C-H → V-oxo angle and distance.
To obtain global structural information for this ferryl-mimicking complex, a 1.55-Å resolution structure was solved from crystals obtained after incubation of VioC with vanadyl ion, L-Arg, and succinate. Clear electron density is observed for all components of the active site (Fig. 2F), and the high degree of order is both notable and consistent with significant stabilization of the ferryl intermediate by VioC. The V-oxo bond distance refines to 1.87 Å, noticeably longer than would be expected from the values of 1.58–1.63 Å reported for inorganic models.37 This deviation indicates that, although the crystals formed and were frozen with VIV-oxo at the cofactor site, exposure to x-rays during data collection resulted in photoreduction of the vanadium to an oxidation state ≤ +III. Overlay of the first coordination sphere with geometry-optimized models of the VioC and TauD ferryl complexes,38 obtained by density functional theory calculations, shows that the remaining coordinating atoms can be superimposed almost exactly (fig. S10). The VioC DFT model was validated by calculation of the ferryl Mössbauer parameters, which resulted in excellent agreement with those obtained experimentally on a sample enriched in the intermediate complex by rapid-freeze quench methods (fig. S11) [0.29 (exp) and 0.24 (calc) mm/s for the isomer shift and ±0.96 (exp) and −0.95 (calc) mm/s for ΔEQ]. In sum, the analysis indicates that elongation of the metal-oxo bond, which is unrestrained by covalent bonds to the protein or substrate/product, is likely to be the only major structural perturbation resulting from x-ray irradiation at 100 K. Therefore, we conclude that the photoreduced vanadyl-VioC structure is a valid and stable structural mimic of the ferryl state, as previously proposed.34
In the structure of the vanadium VioC complex with substrate, the V-O vector undergoes a minor shift of ~25° from its location in the peroxysuccinate complex such that it is now ~180° away from His316 (Fig 3B, 3D). Of the two atoms ultimately derived from dioxygen, the O2 atom of succinate undergoes a much more significant conformational change, rotating 33° and moving ~2 Å after O-O bond cleavage. These observations suggest that such motions may represent the largest activation barrier in the crystal lattice. The vanadium oxo ligand is positioned appropriately to accept the C3 pro-S hydrogen of L-Arg in the HAT step (if it were the actual ferryl complex), as anticipated from the stereochemistry of the observed product and consistent with the C3-H…VIV-oxo distance and angle measurements obtained in the HYSCORE analysis (Fig. 4B). The metal ion can be viewed as six-coordinate (Fig. 3D) but is severely distorted from idealized octahedral symmetry toward a more nearly square-pyramidal geometry. Five of the metal-ligand distances are normal for such complexes (1.9–2.3 Å), and there is clear continuous electron density along the internuclear axes of these bonds (Fig. 2F). By contrast, the sixth site appears open, because O2 of succinate is positioned > 2.3 Å from the vanadium, and there is a distinct break in the electron density along the bond axis. As in the peroxysuccinate structure, the succinate coproduct and Arg334/Gln137 side chains in the vanadium complex are in their product conformations. This arrangement locates the terminal nitrogen atoms of Arg334 within 3.1 Å of both the O2 atom of succinate and the vanadyl (derived) oxygen. The proximity of a competing H-bond donor to the coproduct opposite the vanadium ion could be responsible for the weaker coordination by succinate O2. Although the marked distortion from idealized octahedral symmetry may result, at least in part, from photoreduction of the vanadyl complex, it nevertheless highlights the potential vacancy of a symmetry-equivalent coordination site, cis to the observed oxo position and trans to the other His ligand (His168), that H-atom abstracting ligand could potentially occupy. The structures of the vanadyl and peroxysuccinate intermediates suggest a key role for outer sphere residue Arg334 in stabilization of Fe-O adducts in certain coordination positions. Direct interaction between the O-O unit of the FeII-peroxysuccinate complex and the side chain of Arg334 in its newly ordered position suggests a mechanistic role for the second-sphere residue, perhaps in favoring the different charge distribution of the nascent peroxide adduct in order to promote decarboxylation and C2–O-bond formation. In Fe/2OG halogenases, second sphere H-bonding to the oxo ligand has been implicated in stabilization of alternative ferryl geometries, inferred from computational modeling of the active site.18–19 The VioC structure with the vanadium mimic offers the opportunity to fully map all of the H-bonding interactions in the active site immediately prior to the HAT step (figs. S12–S14), including the bonds between active site side chains and the oxo ligand. Surprisingly, this analysis reveals that Arg334 does not H-bond directly to the vanadium oxygen ligand. It is not clear if H-bond stabilization of the oxo is necessary in hydroxylation reactions but, in VioC, the α-ammonium group of the L-Arg substrate is instead optimally oriented for this role. The likely H-bonding partners for the Arg334 side chain are the succinate O2 atom and the L-Arg carboxylate, although the latter interaction indirectly stabilize the oxo via H-bonding by control of substrate position. In VioC, Arg334 therefore promotes an inline oxo configuration via a combination of electrostatic interactions, steric block of ligand dynamics, and maintenance of an extended active site H-bonding network.
Analysis of the available Fe/2OG oxygenase structures (Table S1) reveals that an Arg is often conserved near the active site, consistent with a general role in directing the location of the oxygen ligand. The residue can be mapped to the last beta strand in the core fold and it is located 4–6 residues C-terminal to another strictly conserved Arg implicated in ionic bonding with the C5 carboxylate of 2OG/succinate (fig. S15A). In most structures, the side chain resides in the second sphere and is positioned similarly to its counterpart in VioC. In a minority of cases, the site is occupied instead by a hydrophobic amino acid (fig. S15B). Strikingly, these enzymes often catalyze transformations other than hydroxylation. A recent structure of an Fe/2OG chlorinase, WelO5, which harbors a Phe at the site occupied by Arg334 in VioC, suggests use of a geometrically distinct ferryl located in the equatorial plane to achieve selective halogenation in this system.19 For this reason, the coordination geometry observed in the VioC vanadium complex, in which a site cis to the present location of the oxo is accessible for its possible migration during or after ferryl formation and controlled by interaction of the ligand (or lack thereof) with outer-sphere residues, is intriguing. Notably, the amino acid hydroxylase, SadA, which has Phe at the position equivalent to Arg334 in VioC, can be modified to halogenate its substrate, 39 demonstrating successful reprogramming of function via site-directed mutagenesis in an Fe/2OG enzyme. Family-wide bioinformatic analysis for Arg334 substitutions could be a viable strategy to identification of enzyme platforms that enable different outcomes, natively or following modification.
CONCLUSIONS
The structures reported here provide a complete and fully contextualized view of the reaction cycle in Fe/2OG-dependent oxygenase VioC. The study reveals a previously unobserved FeII-peroxysuccinate intermediate and an x-ray structure of a stable vanadyl-substituted enzyme-substrate complex, representative of the substrate-targeting ferryl intermediate that accumulates in solution. The vanadyl probe is a viable crystallographic tool that may be applied more generally to other enzymes that deploy high-valent iron species in order to map the structural changes that accompany intermediate formation and control its reactivity. The structures of VioC at intermediate states in the reaction illustrate how second-sphere residues in the active site might control geometric configuration of ligands at key points, providing new routes to enzyme engineering and functional prediction in the larger Fe/2OG enzyme superfamily.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM119707 to A.K.B. and GM113106 to J.M.B. and C.K.), a National Science Foundation Graduate Research Fellowship Program (Grant No. DGE1255832) to R.J.M, and an NIH National Research Service Award to C.J.P. (GM113389). We gratefully acknowledge the resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). The Eiger 16M detector was funded by an NIH–Office of Research Infrastructure Programs, High-End Instrumentation Grant (1S10OD012289-01A1).
Footnotes
Experimental section, SI figures S1–S17, and tables S1–S4.
The Supporting Information is available free of charge on the ACS Publications website.
Supporting information (PDF)
References
- 1.Tang MC, Zou Y, Watanabe K, Walsh CT, Tang Y. Chem Rev. 2016;117:5226–5333. doi: 10.1021/acs.chemrev.6b00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh CT. Nat Chem Biol. 2015;11:620–4. doi: 10.1038/nchembio.1894. [DOI] [PubMed] [Google Scholar]
- 3.Kovaleva EG, Lipscomb JD. Nat Chem Biol. 2008;4:186–93. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kovaleva EG, Lipscomb JD. Science. 2007;316:453–7. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilmot CM, Hajdu J, McPherson MJ, Knowles PF, Phillips SE. Science. 1999;286:1724–8. doi: 10.1126/science.286.5445.1724. [DOI] [PubMed] [Google Scholar]
- 6.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–22. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 7.Acheson JF, Bailey LJ, Brunold TC, Fox BG. Nature. 2017;544:191–195. doi: 10.1038/nature21681. [DOI] [PubMed] [Google Scholar]
- 8.Bollinger JM, Jr, Chang W-c, Matthews ML, Martinie RJ, Boal AK, Krebs C. Mechanisms of 2-oxoglutarate-dependent oxygenases: the hydroxylation paradigm and beyond. In: Hausinger RP, Schofield CJ, editors. 2-oxoglutarate-dependent oxygenases. The Royal Society of Chemistry; London: 2015. pp. 95–122. [Google Scholar]
- 9.Que L., Jr Nat Struct Biol. 2000;7:182–4. doi: 10.1038/73270. [DOI] [PubMed] [Google Scholar]
- 10.Koehntop KD, Emerson JP, Que L., Jr J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 11.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497–508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 12.Aik W, McDonough MA, Thalhammer A, Chowdhury R, Schofield CJ. Curr Opin Struct Biol. 2012;22:691–700. doi: 10.1016/j.sbi.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM., Jr J Am Chem Soc. 2003;125:13008–9. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 14.Grzyska PK, Appelman EH, Hausinger RP, Proshlyakov DA. Proc Natl Acad Sci U S A. 2010;107:3982–7. doi: 10.1073/pnas.0911565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2004;126:8108–9. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 16.Neidig ML, Brown CD, Light KM, Fujimori DG, Nolan EM, Price JC, Barr EW, Bollinger JM, Jr, Krebs C, Walsh CT, Solomon EI. J Am Chem Soc. 2007;129:14224–31. doi: 10.1021/ja074557r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM., Jr Biochemistry. 2009;48:4331–43. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong SD, Srnec M, Matthews ML, Liu LV, Kwak Y, Park K, Bell CB, 3rd, Alp EE, Zhao J, Yoda Y, Kitao S, Seto M, Krebs C, Bollinger JM, Jr, Solomon EI. Nature. 2013;499:320–3. doi: 10.1038/nature12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchell AJ, Zhu Q, Maggiolo AO, Ananth NR, Hillwig ML, Liu X, Boal AK. Nat Chem Biol. 2016;12:636–640. doi: 10.1038/nchembio.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ju J, Ozanick SG, Shen B, Thomas MG. Chembiochem. 2004;5:1281–5. doi: 10.1002/cbic.200400136. [DOI] [PubMed] [Google Scholar]
- 21.Schroeder R, Waldsich C, Wank H. EMBO J. 2000;19:1–9. doi: 10.1093/emboj/19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helmetag V, Samel SA, Thomas MG, Marahiel MA, Essen LO. FEBS J. 2009;276:3669–82. doi: 10.1111/j.1742-4658.2009.07085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujimori DG, Krebs C, Walsh CT, Bollinger JM., Jr Acc Chem Res. 2007;40:484–493. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bollinger JM, Jr, Krebs C. J Inorg Biochem. 2006;100:586–605. doi: 10.1016/j.jinorgbio.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 25.Ye S, Riplinger C, Hansen A, Krebs C, Bollinger JM, Jr, Neese F. Chemistry. 2012;18:6555–67. doi: 10.1002/chem.201102829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wójcik A, Radon M, Borowski T. J Phys Chem A. 2016;120:1261–74. doi: 10.1021/acs.jpca.5b12311. [DOI] [PubMed] [Google Scholar]
- 27.Borowski T, Bassan A, Siegbahn PE. Chemistry. 2004;10:1031–41. doi: 10.1002/chem.200305306. [DOI] [PubMed] [Google Scholar]
- 28.Proshlyakov DA, McCracken J, Hausinger RP. J Biol Inorg Chem. 2017;22:367–379. doi: 10.1007/s00775-016-1406-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cicero G, Carbonera C, Valegård K, Hajdu J, Andersson I, Ranghino G. Int J Quantum Chem. 2007;107:1514–1522. [Google Scholar]
- 30.Valegard K, Terwisscha van Scheltinga AC, Dubus A, Ranghino G, Oster LM, Hajdu J, Andersson I. Nat Struct Mol Biol. 2004;11:95–101. doi: 10.1038/nsmb712. [DOI] [PubMed] [Google Scholar]
- 31.Serrano-Plana J, Oloo WN, Acosta-Rueda L, Meier KK, Verdejo B, Garcia-Espana E, Basallote MG, Munck E, Que L, Jr, Company A, Costas M. J Am Chem Soc. 2015;137:15833–42. doi: 10.1021/jacs.5b09904. [DOI] [PubMed] [Google Scholar]
- 32.Oloo WN, Meier KK, Wang Y, Shaik S, Munck E, Que L. Nat Commun. 2014;5:3046. doi: 10.1038/ncomms4046. [DOI] [PubMed] [Google Scholar]
- 33.Furutachi H, Hashimoto K, Nagatomo S, Endo T, Fujinami S, Watanabe Y, Kitagawa T, Suzuki M. J Am Chem Soc. 2005;127:4550–1. doi: 10.1021/ja0427202. [DOI] [PubMed] [Google Scholar]
- 34.Ballhausen CJ, Gray HB. Inorganic Chemistry. 1962;1:111–122. [Google Scholar]
- 35.Winkler JR, Gray HB. Struct Bond. 2012;142:17–28. [Google Scholar]
- 36.Martinie, et al. [Google Scholar]
- 37.Krakowiak J, Lundberg D, Persson I. Inorg Chem. 2012;51:9598–609. doi: 10.1021/ic300202f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinnecker S, Svensen N, Barr EW, Ye S, Bollinger JM, Jr, Neese F, Krebs C. J Am Chem Soc. 2007;129:6168–79. doi: 10.1021/ja067899q. [DOI] [PubMed] [Google Scholar]
- 39.Mitchell AJ, Dunham NP, Bergman JA, Wang B, Zhu Q, Chang W-c, Liu X, Boal AK. Biochemistry. 2017;56:441–444. doi: 10.1021/acs.biochem.6b01173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.