

Abstract
Background
Imaging features derived from MRI scans can be used for not only breast cancer detection and measuring disease extent, but can also determine gene expression and patient outcomes. The relationships between imaging features, gene/protein expression, and response to therapy hold potential to guide personalized medicine. We aim to characterize the relationship between radiologist-annotated tumor phenotypic features (based on MRI) and the underlying biological processes (based on proteomic profiling) in the tumor.
Methods
Multiple-response regression of the image-derived, radiologist-scored features with reverse-phase protein array expression levels generated association coefficients for each combination of image-feature and protein in the RPPA dataset. Significantly-associated proteins for features were analyzed with Ingenuity Pathway Analysis software. Hierarchical clustering of the results of the pathway analysis determined which features were most strongly correlated with pathway activity and cellular functions.
Results
Each of the twenty-nine imaging features was found to have a set of significantly correlated molecules, associated biological functions, and pathways.
Conclusions
We interrogated the pathway alterations represented by the protein expression associated with each imaging feature. Our study demonstrates the relationships between biological processes (via proteomic measurements) and MRI features within breast tumors.
Keywords: breast invasive carcinoma, MRI, protein expression, signaling pathway analysis, TCGA
INTRODUCTION
Breast cancer is the most common cancer in women [1], with incidence rates rising since the 1990s [2]. Molecular expression profiling of tumors has been effective in allowing for individualized therapy plans in certain types of breast cancer [3]. Expression of three receptors—estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor (HER2)—are routinely used to determine optimal treatment plans for breast cancer patients [4]. PR and ER expression are associated with luminal A and B subtypes of breast cancer, with a lower proliferation index and pathological grade [5]. Disease-free and overall survival is lower in HER2 over-expression and triple negative breast cancers when compared to luminal A and B subtypes [5].
Despite obtaining multiple specimens from percutaneous biopsies as well as analysis of surgical specimens, the temporal and spatial heterogeneity of tumor gene and protein expression cannot be adequately determined [6, 7, 8]. Readily available imaging databases such as The Cancer Imaging Archive (TCIA) are leveraged in order to address the problem of tumor heterogeneity and to predict gene expression and patient responses to therapy based on imaging data [9]. MRI, as well as other modalities, is now used by researchers for extraction of features which correlate with patient responses and gene expression [10-12]. Breast cancer radiomic signatures can potentially predict recurrence when compared with multi-gene assays [13]. Deciphering the associations between imaging features, breast tumor gene/protein expression levels, and patient outcomes holds the potential to guide personalized medicine [12, 14].
High-dimensional variable selection (Supplementary Table S1) is commonly used to analyze relationships between multiple modalities (copy number, expression, etc.) in genomic data. To avoid generating spurious correlations, a number of Bayesian and frequentist approaches have been devised. Bayesian approaches use a sparsity-inducing prior, such as spike-and-slab [15, 16], double-exponential [17], horseshoe [18], horseshoe+[19], or generalized double Pareto prior [20]. Frequentist approaches use penalized regression models: l1 [18], horseshoe+ [19], generalized double Pareto prior [20], L1-norm penalty of the LASSO [21], combined l1/l2 penalty of elastic net [21], or combined L1 and L2-norm penalty of elastic net [22]. These regression models allow us to ignore the loss of statistical efficiency that occurs through correlation structures because they treat all variables as independent [23]. Several approaches to high-dimensional variable selection in highly-correlated datasets have been taken [24-26]. In this study, we used a Bayesian approach to model the correlation structure as previously described [27].
Analyzing a cohort of 82 breast cancer patients included in the TCGA database, we built a model correlating MRI-derived imaging features with proteomics data using a high-dimensional regression approach. Though a previous study of 353 breast cancer patients assessed correlations between 21 imaging traits and mRNA transcript levels [28], to our knowledge our approach has not yet been applied to proteomics data for breast cancer.
RESULTS
Molecules were found to be significantly correlated to each imaging feature (Supplementary Table S2) with the exception of clumped non-mass internal enhancement. These molecules were obtained through high dimensional regression of the RPPA protein expression data on the imaging features set. For example, the axillary lymphadenopathy feature was found to be directly correlated with expression of EIF4EBP1 and PRDX1, and inversely correlated with RAB25, SHC1, XRCC1, and PARK7. Cell surface receptors associated with imaging features are EGFR, KDR, and PDK1.
IPA analysis was implemented to determine the functional implications of the molecules. The IPA software generated p-values and Z-scores for the IPA Canonical Pathways of each feature (Figure 2), as well as scores for the IPA Diseases and Biological Functions of each feature (Figure 3). The Canonical Signaling Pathways most strongly associated with each imaging feature are summarized in Table 2. The results show that the same proteins are found to be correlated with a specific feature, irrespective of whether the data sets were separated into global or primary features.
Figure 2. Representative pattern of associations between BRCA imaging features and IPA Canonical Pathways based on (A) p-values and (B) activation Z-scores.

A subset of the p-values and Z-scores are shown. Values shown are -log(p-value).
Figure 3. Representative pattern of associations between BRCA imaging features and IPA Diseases and Bio-Functions based on (A) p-values and (B) activation Z-scores.

A subset of the p-values and Z-scores are shown. Values shown are -log(p-value).
Table 2. Radiological features are associated with unique pathway alterations in breast invasive carcinoma.
Lists of molecules (proteins and post-translational modifications) were analyzed in IPA. Top pathways for each feature are shown with the associated –log (p-value) computed by IPA demonstrating the strength of the association of each imaging feature to each pathway.
| 3 greatest P-values per imaging feature | 1 | 2 | 3 |
|---|---|---|---|
| T2 Signal Intensity | Pancreatic Adenocarcinoma Signaling 6.177 | Melanoma Signaling 4.405 | Non-Small Cell Lung Cancer Signaling 4.111 |
| T2 Heterogeneity | UVB-Induced MAPK Signaling 6.245 | EGF Signaling 6.206 | ErbB Signaling 5.725 |
| Skin Thickening | Epithelial Adherens Junction Signaling 8.957 | Regulation of the Epithelial-Mesenchymal Transition Pathway 8.282 | Pancreatic Adenocarcinoma Signaling 5.692 |
| Skin Invasion | 14-3-3-mediated Signaling 10.378 | Cell Cycle: G2/M DNA Damage Checkpoint Regulation 7.914 | UVB-Induced MAPK Signaling 7.385 |
| Irregular Shape | UVC-Induced MAPK Signaling 6.845 | EGF Signaling 6.206 | STAT3 Pathway 6.112 |
| Rim Enhancement | ATM Signaling 8.26 | AMPK Signaling 6.385 | Cell Cycle: G2/M DNA Damage Checkpoint Regulation 5.037 |
| Pectoral Invasion | PI3K/AKT Signaling 12.834 | Neuregulin Signaling 10.295 | p70S6K Signaling 9.069 |
| Non-Mass Heterogeneous Internal Enhancement | ILK Signaling 7.812 | PI3K/AKT Signaling 6.646 | Endometrial Cancer Signaling 5.511 |
| Non-Mass Clustered Ring Internal Enhancement | ATM Signaling 4.078 | CDK5 Signaling 3.892 | B Cell Receptor Signaling 3.349 |
| Non-Mass Clumped Internal Enhancement | DNA Double-Strand Break Repair by Homologous Recombination 3.181 | DNA Double-Strand Break Repair by Non-Homologous End Joining 3.181 | DNA damage-induced 14-3-3σ Signaling 3.049 |
| Regional Non-Mass Distribution | Acute Myeloid Leukemia Signaling 3.745 | Cancer Drug Resistance By Drug Efflux 1.94 | autophagy 1.853 |
| Multiple Regions Non-Mass Distribution | PI3K/AKT Signaling 4.471 | IL-8 Signaling 4.068 | CD27 Signaling in Lymphocytes 2.311 |
| Linear Non-Mass Distribution | ErbB2-ErbB3 Signaling 6.883 | ErbB Signaling 6.421 | Relaxin Signaling 5.845 |
| Focal Non-Mass Distribution | UVC-Induced MAPK Signaling 11.037 | Cancer Drug Resistance By Drug Efflux 10.613 | AMPK Signaling 10.337 |
| Diffuse Non-Mass Distribution | DNA Double-Strand Break Repair by Homologous Recombination 5.395 | Role of BRCA1 in DNA Damage Response 3.879 | ATM Signaling 3.857 |
| Nipple Retraction | PI3K/AKT Signaling 6.112 | UVB-Induced MAPK Signaling 4.246 | FLT3 Signaling in Hematopoietic Progenitor Cells 4.025 |
| Nipple Invasion | Estrogen-mediated S-phase Entry 2.346 | Induction of Apoptosis by HIV1 1.949 | Lymphotoxin β Receptor Signaling 1.901 |
| Margin | Molecular Mechanisms of Cancer 3.746 | DNA damage-induced 14-3-3σ Signaling 2.205 | GADD45 Signaling 2.205 |
| Lesion Size | Hereditary Breast Cancer Signaling 7.625 | PI3K/AKT Signaling 5.976 | Insulin Receptor Signaling 5.753 |
| Heterogeneous Enhancement Intensity | Prolactin Signaling 6.64 | Th1 Pathway 6.001 | Th1 and Th2 Activation Pathway 5.588 |
| Fibroglandular | UVC-Induced MAPK Signaling 13.629 | UVB-Induced MAPK Signaling 12.176 | Neuregulin Signaling 11.271 |
| Extent Heterogeneity | Prostate Cancer Signaling 7.076 | UVB-Induced MAPK Signaling 4.546 | FLT3 Signaling in Hematopoietic Progenitor Cells 4.325 |
| Extent - Multi-focal | CNTF Signaling 4.587 | UVB-Induced MAPK Signaling 4.546 | EGF Signaling 4.52 |
| Extent - Multi-centric | Hepatic Fibrosis / Hepatic Stellate Cell Activation 3.359 | Tumoricidal Function of Hepatic Natural Killer Cells 2.346 | Coagulation System 2.182 |
| Edema | Hereditary Breast Cancer Signaling 4.797 | AMPK Signaling 4.425 | Endometrial Cancer Signaling 3.607 |
| Dark Internal Septum | Huntington's Disease Signaling 3.418 | Glucocorticoid Receptor Signaling 3.267 | Parkinson's Signaling 2.646 |
| Background | Insulin Receptor Signaling 5.753 | Molecular Mechanisms of Cancer 5.539 | NF-κB Signaling 5.331 |
| Axillary Lymphadenopathy | ERK/MAPK Signaling 4.8 | EGF Signaling 3.824 | Erythropoietin Signaling 3.693 |
| Associated Non-Mass Enhancement | Pancreatic Adenocarcinoma Signaling 7.206 | UVC-Induced MAPK Signaling 6.399 | Cancer Drug Resistance By Drug Efflux 6.194 |
Figure 1. Sagittal T1 post-contrast MRI of a 48-year-old female patient diagnosed with infiltrating ductal carcinoma (ER-, PR-, HER2-) shows an oval rim enhancing mass.
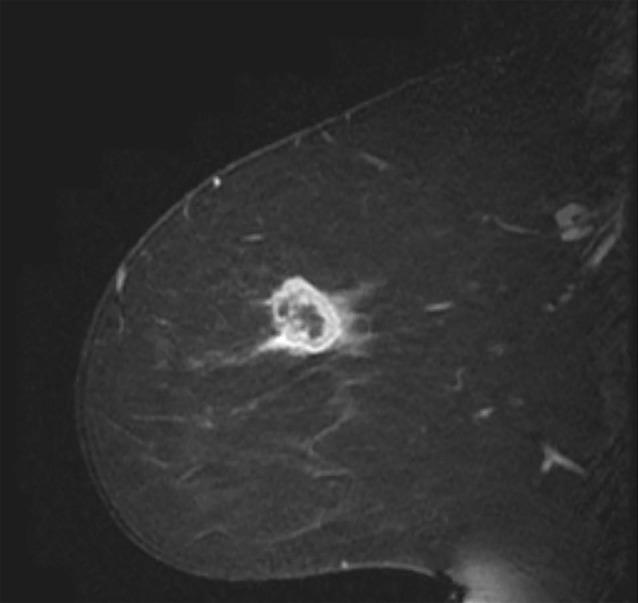
MRI sequences were obtained from The Cancer Imaging Archive [37].
Table 1. Patient demographic information.
Demographics are given for the 82 patients included in this study.
| Statistic | |
|---|---|
| Mean Age at Diagnosis (Range) | 53.2(29 - 82) |
| Median Overall Survival (Months) | 41.72 |
| Median Disease-Free Survival (Months) | 42.015 |
| Estrogen Receptor (ER) Status (Positive / Negative) | 67/ 15 |
| Progesterone Receptor (PR) Status (Positive / Negative) | 59/ 23 |
| Infiltrating Lobular Carcinoma | 9 |
| Infiltrating Ductal Carcinoma | 69 |
| Medullary Carcinoma | 1 |
| Other | 3 |
In order to determine which features were most strongly associated with functional alterations to signaling pathways, agglomerative unsupervised hierarchical clustering was performed on the p-values and Z-scores (Figures 2 and 3). This analysis separated the features into groups based on the strength of their correlations with altered pathway activity and disease functions. The most strongly deregulated IPA Diseases and Biological functions featured activation Z-scores between -3.5 and +3.5 (Supplementary Table S3).
DISCUSSION
The strength of the associations between imaging features and protein expression, signaling pathways, and biological functions was computed using a sequential analysis of the protein expression data found through RPPA analysis of MRI scans of the TCGA patients. Correlation coefficients for each possible combination of imaging feature and protein were computed using a high-dimensional regression with a Bayesian selection of covariates. Corrected p-values were computed for each correlation coefficient in order to minimize the false discovery rate (FDR). Only the strongest ten percent of significantly-correlated molecules were analyzed using the standardized Core Analysis workflow in IPA, using correlation coefficients in lieu of gene expression values. The IPA analysis provides associations with pathway activity and pathobiology, allowing for hypotheses regarding the relationship between pathway activity at the cellular level and the manifestations of the alterations at the macroscopic, imaging levels. The activation Z-scores computed from the correlation coefficients indicate whether each pathway (or function) is up- or down-regulated by upstream transcription factor activity. A similar approach integrated breast cancer transcriptomics data with imaging features and extended the interpretation with gene set enrichment analysis to identify metagene signatures such as wound response and hypoxia [28]. Our study extends this approach by leveraging the IPA Knowledge Base to interpret the patterns of protein expression associated with each imaging feature.
In our study, we used a stringent two-step method to select the correlations least likely to result from chance association, overcoming a common issue with high dimensional regression analysis. Despite this, the approach we have described is essentially a hypothesis-generation pipeline, and should be interpreted carefully, following in-vivo perturbation experiments in appropriate model systems.
We found that enhancing rim fraction score, a quantitative MRI feature, was shown to be significantly associated with the expression of the long, non-coding RNA HOTAIR [29]. This expression is known to be associated with breast cancer progression and metastasis [30]. The results of the high dimensional regression method used hints at the molecular underpinnings of macroscopic imaging phenotypes. It is known that MRI features correlate with pathologic stage and lymph node involvement [31]. The results found in this study point to multiple significant associations between molecular expression patterns in the tumor cells and how these manifest as MRI phenotypes [32].
METHODS
TCGA patient datasets
Eighty-two patients from multiple institutions with de-identified MRIs and reverse-phase protein array (RPPA) expression data were included in this study. All subject data was de-identified prior to the study through inclusion in The Cancer Genome Atlas (TCGA), and was thus exempt from requiring institutional review board approval, following the terms of the TCGA data use agreement. Imaging data was obtained through The Cancer Imaging Archive (TCIA) database. RPPA protein expression data was obtained from the TCGA through Firehose (https://gdac.broadinstitute.org/).
Scores of twenty-nine MRI semantic features were defined by the TCGA Breast Phenotype Research Group [33]. We used the imaging features as defined by the TCGA group to include mass- and non-mass associated features as shown in Table 3. These feature groups include background features, tumor related features, tumor dimensional features, features associated with the morphology of the non-mass enhancing lesion, and T2-weighted MR acquisition features.
Table 3. List of imaging features.
| Feature Group | Features |
|---|---|
| Background | Background Enhancement Fibroglandular |
| Tumor Features | Irregular Shape Heterogeneous Enhancement Intensity Dark Internal Septum Rim Enhancement Margin |
| Tumor Dimensions | Lesion Size Multicentric Extent Multifocal Extent Heterogeneity Extent |
| Associated Features | Pectoral Invasion Nipple Invasion Skin Invasion Axillary Lymphadenopathy Edema Skin Thickening Nipple Retraction |
| Morphology of Non-Mass Enhancing Lesions | Associated Non-Mass Enhancement Non-mass Clumped Internal Enhancement Non-mass Clustered Ring Internal Enhancement Non-mass Heterogeneous Internal Enhancement Diffuse Non-Mass Distribution Focal Non-Mass Distribution Linear Non-Mass Distribution Multiple Regions Non-Mass Distribution Regional Non-Mass Distribution |
| Associated with T2 Weighted MR Acquisition | Heterogeneity Signal Intensity |
In order to ensure that the effects of each individual feature were appropriately described, the feature set was split into three subsets: one set with only the 8 mass-associated features, one with only the 21 global features, and an aggregate set with all 29 features. The features were isolated in order to determine if there were any significant proteins, associated pathways, or biological functions that appeared in purely global or mass-associated-only feature sets.
Statistical analysis
High dimensional regression
High dimensional regression was done in Matlab using the joint Bayesian selection of covariates developed by Bhadra and Mallick [27]. In this analysis, the independent variables were the imaging features, and the molecules (proteins and phospho-proteins) were the response variables. This arrangement allowed the expression of each protein to be correlated with the expression of many other proteins.
Multiple-testing correction
Multiple testing correction was employed to control the false-discovery rate (FDR) by sequentially designating p-value thresholds [34]. First, the posterior probabilities of the covariates were thresholded at an FDR of 0.25, giving a sparse set of predictors (imaging variables). Second, t-tests were performed using “no-association” as the null hypothesis and “non-zero association” as the alternative hypothesis. The t-tests were computed between each combination of imaging features and molecules in the RPPA dataset. Correlation coefficients with p-values in the 10th percentile and that were less than 0.05 after adjustment for multiple comparisons were considered statistically significant. This approach is similar to that used to discern the relative impact of copy number alterations on messenger RNAs and microRNAs in glioblastoma [30].
Pathway analysis
Pathway analysis was performed on each of the three data sets (based on the image feature subsets) using the “Core Analysis” feature in the IPA software [35]. For the purposes of this analysis, regression correlation coefficients served as expression values. P-values and activation Z-scores were computed internally in IPA as previously described.
Hierarchical clustering
Agglomerative unsupervised hierarchical clustering of p-values and activation Z-scores was carried out the using the “Stats” package in R. Euclidean distance matrices were computed and Ward's method was minimized within-cluster variance [36].
SUPPLEMENTARY MATERIALS TABLES
ACKNOWLEDGMENTS AND FUNDING
A.R. was supported by CCSG Bioinformatics Shared Resource P30 CA016672, an Institutional Research Grant from The University of Texas MD Anderson Cancer Center (MD Anderson), CPRIT RP170719, CPRIT RP150578, NCI R01CA214955-01A1, a Career Development Award from the MD Anderson Brain Tumor SPORE, and a Research Scholar Grant from the American Cancer Society (RSG-16-005-01).
A.B. was supported by grant no. DMS-1613063 from the National Science Foundation. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
E.S and E.M were funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748. The sponsor had no involvement in the study design; the collection, analysis and interpretation of data; the writing of the report; and the decision to submit the article for publication.
Footnotes
Author contributions
Conception and design: A.R, A.B Development of methodology: A.R, A.B, M.L
Acquisition of data: M.L, A.B, S.A, V.R, Y.Z, B.D, E.B, E.S.B, E.M, E.S, G.J.W, J.N, K.B, M.G, M.Z, A.R
Analysis and interpretation of data: M.L, S.A, A.B, A.R,
Writing, review, and/or revision of manuscript: M.L, A.B, S.A, A.R
Administrative, technical, or material support (i.e., reporting or organizing data, constructing database): A.R, E.S, E.M, E.S.B
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
REFERENCES
- 1.Howlader N, Noone A, Krapcho M, Garshell J, Miller D, Altekruse S, Kosary C, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis L, Chen H, et al. Based on November 2013 SEER data submission, posted to the SEER web site, April 2014. National Cancer Institute; Bethesda, MD: 2014. SEER Cancer Statistics Review, 1975-2011. [Google Scholar]
- 2.Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends—an update. Cancer Epidemiol Biomarkers Prev. 2016;25:16–27. doi: 10.1158/1055-9965.EPI-15-0578. [DOI] [PubMed] [Google Scholar]
- 3.Van ‘t Veer LJ, Dai H, Van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, Van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 4.Nattinger AB, Mitchell JL. Breast cancer screening and prevention. Ann Intern Med. 2016;164:ITC81–96. doi: 10.7326/AITC201606070. [DOI] [PubMed] [Google Scholar]
- 5.Zardavas D, Irrthum A, Swanton C, Piccart M. Clinical management of breast cancer heterogeneity. Nat Rev Clin Oncol. 2015;12:381–94. doi: 10.1038/nrclinonc.2015.73. [DOI] [PubMed] [Google Scholar]
- 6.Voduc KD, Cheang MC, Tyldesley S, Gelmon K, Nielsen TO, Kennecke H. Breast cancer subtypes and the risk of local and regional relapse. J Clin Oncol. 2010;28:1684–91. doi: 10.1200/JCO.2009.24.9284. [DOI] [PubMed] [Google Scholar]
- 7.Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature. 2013;501:355–64. doi: 10.1038/nature12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davnall F, Yip CS, Ljungqvis G, Selmi M, Ng F, Sanghera B, Ganeshan B, Miles KA, Cook GJ, Goh V. Assessment of tumor heterogeneity: an emerging imaging tool for clinical practice? Insights Imaging. 2012;3:573–89. doi: 10.1007/s13244-012-0196-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orel SG, Schnall MD. MR imaging of the breast for the detection, diagnosis, and staging of breast cancer. Radiology. 2001;220:13–30. doi: 10.1148/radiology.220.1.r01jl3113. [DOI] [PubMed] [Google Scholar]
- 11.Houssami N, Ciatto S, Macaskill P, Lord SJ, Warren RM, Dixon JM, Irwig L. Accuracy and surgical impact of magnetic resonance imaging in breast cancer staging: systematic review and meta-analysis in detection of multifocal and multicentric cancer. J Clin Oncol. 2008;26:3248–58. doi: 10.1200/JCO.2007.15.2108. [DOI] [PubMed] [Google Scholar]
- 12.Yip SS, Aerts HJ. Applications and limitations of radiomics. Phys Med Biol. 2016;61:R150–66. doi: 10.1088/0031-9155/61/13/R150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Zhu Y, Burnside ES, Drukker K, Hoadley KA, Fan C, Conzen SD, Whitman GJ, Sutton EJ, Net JM, Ganott M, Huang E, Morris EA, et al. MR imaging radiomics signatures for predicting the risk of breast cancer recurrence as given by research versions of MammaPrint, Oncotype DX, and PAM50 gene assays. Radiology. 2016:152110. doi: 10.1148/radiol.2016152110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gillies RJ, Kinahan PE, Hricak H. Radiomics: Images are more than pictures, they are data. Radiology. 2016;278:563–77. doi: 10.1148/radiol.2015151169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell TJ, Beauchamp JJ. Bayesian variable selection in linear-regresion. Journal of the American Statistical Association. 1988;83:1023–32. [Google Scholar]
- 16.George EI, McCulloch RE. Variable selection via gibbs sampling. Journal of the American Statistical Association. 1993;88:881–9. [Google Scholar]
- 17.Park T, Casella G. The Bayesian lasso. Journal of the American Statistical Association. 2008;103:681–6. [Google Scholar]
- 18.Carvalho CM, Polson NG, Scott JG. The horseshoe estimator for sparse signals. Biometrika. 2010;97:465–80. [Google Scholar]
- 19.Bhadra A, Datta J, Polson NG, Willard B. The horseshoe+ estimator of ultra-sparse signals. arXiv. 2016:1502. 00560. [Google Scholar]
- 20.Armagan A, Dunson DB, Lee J. Generalized double pareto shrinkage. Statistica Sinica. 2013;23:119–43. [PMC free article] [PubMed] [Google Scholar]
- 21.Tibshirani R. Regression shrinkage and selection via the lasso. Journal of the Royal Statistical Society Series B-Methodological. 1996;58:267–88. [Google Scholar]
- 22.Zou H, Hastie T. Regularization and variable selection via the elastic net. Journal of the Royal Statistical Society Series B-Statistical Methodology. 2005;67:301–20. [Google Scholar]
- 23.Zellner A. An efficient method of estimating seemingly unrelated regressions and tests for aggregation bias. Journal of the American Statistical Association. 1962;57:348. [Google Scholar]
- 24.Rothman AJ, Levina E, Zhu J. Sparse multivariate regression with covariance estimation. Journal of Computational and Graphical Statistics. 2010;19:947–62. doi: 10.1198/jcgs.2010.09188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin J, Li H. A sparse conditional gaussian graphical model for analysis of genetical genomics data. Annals of Applied Statistics. 2011;5:2630–50. doi: 10.1214/11-AOAS494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai TT, Li H, Liu W, Xie J. Covariate-adjusted precision matrix estimation with an application in genetical genomics. Biometrika. 2013;100:139–56. doi: 10.1093/biomet/ass058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhadra A, Mallick BK. Joint high-dimensional Bayesian variable and covariance selection with an application to eQTL analysis. Biometrics. 2013;69:447–57. doi: 10.1111/biom.12021. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto S, Maki DD, Korn RL, Kuo MD. Radiogenomic analysis of breast cancer using MRI: a preliminary study to define the landscape. American Journal of Roentgenology. 2012;199:654–63. doi: 10.2214/AJR.11.7824. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto S, Han W, Kim Y, Du L, Jamshidi N, Huang D, Kim JH, Kuo MD. Breast cancer: radiogenomic biomarker reveals associations among dynamic contrast-enhanced MR imaging, long noncoding RNA, and metastasis. Radiology. 2015;275:384–92. doi: 10.1148/radiol.15142698. [DOI] [PubMed] [Google Scholar]
- 30.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–6. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burnside ES, Drukker K, Li H, Bonaccio E, Zuley M, Ganott M, Net JM, Sutton EJ, Brandt KR, Whitman GJ, Conzen SS, Lan L, Ji Y, et al. Using computer-extracted image phenotypes from tumors on breast magnetic resonance imaging to predict breast cancer pathologic stage. Cancer. 2016;122:748–57. doi: 10.1002/cncr.29791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu Y, Li H, Guo W, Drukker K, Lan L, Giger M.L, Ji Y. Deciphering genomic underpinnings of quantitative MRI-based radiomic phenotypes of invasive breast carcinoma. Sci Rep. 2015;5:17787. doi: 10.1038/srep17787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo W, Li H, Zhu Y, Lan L, Yang S, Drukker K, Morris E, Burnside E, Whitman E, Giger ML, Ji Y, TCGA Breast Phenotype Research Group Prediction of clinical phenotypes in invasive breast carcinomas from the integration of radiomics and genomics data. J Med Imaging (Bellingham) 2015;2:041007. doi: 10.1117/1.JMI.2.4.041007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhadra A, Baladandayuthapani V, IEEE Integrative sparse Bayesian analysis of high-dimensional multi-platform genomic data in glioblastoma. 2013 IEEE International Workshop on Genomic Signal Processing and Statistics (Gensips 2013); 2013; pp. 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krämer A, Green J, Pollard J, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–30. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ward J. Hierarchical grouping to optimize an objective function. Journal of the American Statistical Association. 1963;58:236. [Google Scholar]
- 37.The Cancer Imaging Archive. 2016 https://public.cancerimagingarchive.net/ncia/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.