

Abstract
The primary cilium is a ubiquitous organelle presented on most human cells. It is a crucial signaling hub for multiple pathways including growth factor and G-protein coupled receptors. Loss of primary cilia, observed in various cancers, has been shown to affect cell proliferation. Primary cilia formation is drastically decreased in glioblastoma (GBM), however, the role of cilia in normal astrocyte or glioblastoma proliferation has not been explored. Here we report that loss of primary cilia in human astrocytes stimulates growth rate in a lysophosphatidic acid (LPA)-dependent manner. We show that lysophosphatidic acid receptor 1 (LPAR1) is accumulated in primary cilia. LPAR1 signaling through Gα12/Gαq was previously reported to be responsible for cancer cell proliferation. We found that in ciliated cells, Gα12 and Gαq are excluded from the cilium, creating a barrier against unlimited proliferation, one of the hallmarks of cancer. Upon loss of primary cilia, LPAR1 redistributes to the plasma membrane with a concomitant increase in LPAR1 association with Gα12 and Gαq. Inhibition of LPA signaling with the small molecule compound Ki16425 in deciliated highly proliferative astrocytes or glioblastoma patient-derived cells/xenografts drastically suppresses their growth both in vitro and in vivo. Moreover, Ki16425 brain delivery via PEG-PLGA nanoparticles inhibited tumor progression in an intracranial glioblastoma PDX model. Overall, our findings establish a novel mechanism by which primary cilium restricts proliferation and indicate that loss of primary cilia is sufficient to increase mitogenic signaling, and is important for the maintenance of a highly proliferative phenotype. Clinical application of LPA inhibitors may prove beneficial to restrict glioblastoma growth and ensure local control of disease.
Keywords: cilia, proliferation, LPA, GBM, LPAR1
Introduction
The primary cilium is a ubiquitous, microtubule-based organelle which is built on top of a membrane-anchored basal body. Primary cilium is an important negative regulator of proliferation and a key sensory organelle. It serves as a hub for multiple signaling cascades including receptor tyrosine kinase (1), Sonic hedgehog (SHH) (2), and G-protein coupled receptors (GPCRs) (3). Disassembly of primary cilium leads to release of the basal body, also called the mother centriole, which is required for mitotic spindle formation and mitosis. Multiple mitotic kinases initiate cilium disassembly including AURKA, Plk1, and Nek2 (4-6). Centrosome sequestration is considered one of the primary mechanisms of negative regulation of proliferation by primary cilium. Nevertheless, cilium shortening and disassembly is often observed immediately after growth factor stimulation in interphase (4). The role of this short-term disassembly in cell proliferation is currently unknown. Recent publications suggest that in different types of cancer including breast, prostate, renal, and glioblastoma (GBM), cilia tend to be lost (7-10). However, the importance of this event on tumor maintenance and progression or treatment is not well understood.
Several studies report that loss of primary cilia in normal cells increases proliferation and supports attachment-independent growth (11, 12), which are common hallmarks of cancer. Attempts to restore primary cilia in cancer cells yield a significant inhibition on proliferation (12, 13). These observations suggest that cilia loss can promote/sustain a highly proliferative phenotype. However, in a subset of Sonic hedgehog (SHH)-dependent medulloblastomas, presence of cilium is mandatory (14) for cancer maintenance, therefore studies on specific cancer subtypes are warranted to establish cilia’s role in tumor biology and potential therapeutic applications. Astrocytoma is the most commonly diagnosed adult brain cancer (15), which often progresses to GBM. The majority of GBM patients succumb to the disease within 13-16 months (15). GBM is a highly proliferative disease with limited treatment options (16). Lysophosphatidic acid (LPA) is an abundant mitogen in brain tissue (17). LPA acts through binding of heterotrimeric G-protein coupled receptors (LPAR1-6). It was previously reported that LPAR1 can signal through Gαi, Gα12, and Gαq family members (18, 19). LPA stimulates cell proliferation in astrocytes (20) and cancer cells (21). Astrocytes are abundant glial cells and well known for their ability to proliferate, especially in the activated state (22), and were previously reported as the potential cells of origin for GBM (23).
In our current work, we establish that loss of primary cilia promotes proliferation of primary non-transformed human astrocytes, providing permissive conditions for transformation in an LPA-dependent manner. GBM primary cells, with a decreased occurrence of primary cilia, were also sensitive to LPA and LPAR1/3 inhibition. Mechanistically, we found that LPA-LPAR1-driven mitogenic signaling was restricted in cells with primary cilium due to compartmentalization of LPAR1 and its downstream effectors, Gα12 and Gαq, in cilia and in cytoplasm, respectively. LPAR1 was redistributed to the plasma membrane upon loss of primary cilium, thus enabling its binding to Gα12 and Gαq, and therefore suggesting that redistribution of LPAR1 is a key mechanism driving proliferation in a cilia-dependent manner.
Inhibition of LPAR1/3 with the small molecule inhibitor Ki16425 significantly reduces cell growth rate only in deciliated astrocytes. Likewise, patient-derived GBM proliferation was stimulated by LPA and abrogated by Ki16425 in a dose-dependent manner. Importantly, the growth of GBM patient-derived xenografts in vivo was drastically decreased upon Ki16425 administration as a monotherapy without significant side effects.
Taken together, our findings indicate that loss of primary cilia eliminates spatial barriers curbing proliferation, thus unlocking the potential for unlimited proliferation. LPA is one of the key mitogenic factors driving highly proliferative GBM with no or very low basal ciliation, therefore clinical interventions based on inhibiting LPA signaling may significantly improve GBM patient survival and local disease control.
Results
Loss of primary cilium promotes proliferation of astrocytes
Human astrocytes (HA) can form primary cilia. Incubation in serum-free media (SFM) promotes ciliation resulting in nearly 80% of cells having cilia (Fig.1A-B). This ciliation rate is similar to that observed in vivo (24). To allow for long term experiments, the primary human astrocytes (HA) were immortalized using SV40 large T antigen (HA-LTA) (25). Immortalization by LTA did not affect ciliation, which was similar to the parental astrocytes (Fig.S1A). To test how loss of primary cilia affects proliferation of primary (HA) or immortalized (HA-LTA) astrocytes, we utilized shRNA-driven knockdown of IFT88 or KIF3B, which are well characterized components of cilium assembly machinery (26, 27). Two shRNAs were used to target IFT88 or KIF3B resulting in up to a 90% knockdown (Fig.1C). Depletion of either IFT88 or KIF3B was sufficient to decrease ciliation to 5-15% (Fig.1D-E). These ciliation rates are similar to tissue biopsies from GBM patients (28).
Fig. 1. Loss of primary cilia promotes astrocyte proliferation in a growth factor-dependent manner.

(A) Representative image of the cilium formed by primary human astrocyte (HA), stained for acetylated α-tubulin (AcTub, cilium marker) and γ-tubulin (γTub, basal body marker); scale bar – 10μm. (B) Quantification of primary astrocytes forming cilium, as in (A), in regular serum-supplemented media (SSM) or upon 48h of serum starvation (SFM); 300 cells, 100 cells in each of 3 independent experiments; Student’s t-test, p<0.05. (C) Western blot of IFT88 and KIF3B in primary and immortalized astrocytes (HA-LTA) stably expressing non-targeting shRNA (Con) or shRNA against IFT88 or KIF3B. (D, E) Quantification of primary astrocytes (D) and immortalized astrocytes (E) forming primary cilium, as in (B) upon depletion of IFT88 or KIF3B, as in (C); 300 cells, 100 cells in each of 3 independent experiments; one-way ANOVA with Dunnett’s post hoc test, p<0.05. (F, G) Growth rates of primary astrocytes (F) and immortalized astrocytes (G) upon depletion of IFT88 or KIF3B in full media or in serum-free conditions (SFM); 3 independent experiments; one-way ANOVA with Dunnett’s post hoc test, p<0.05.
The loss of primary cilia resulted in a significant increase in growth rate of both immortalized and primary astrocytes (Fig1.F-G). Interestingly, this difference in growth rate was observed between ciliated (shCon) and deciliated (shKIF3B or shIFT88) cells only upon addition of serum-supplemented media (SSM), but not in SFM. Addition of SSM caused resorption of primary cilia in control cells in a biphasic wave pattern (Fig.S1B), which has been described previously for other cell types (4). These findings indicate that loss of primary cilia by itself does not promote proliferation, but rather increases the response to some mitogenic stimuli present in serum.
Loss of primary cilia changes spatio-temporal response to mitogen stimulation
To elucidate the potential mechanism underlying cilia-dependent changes in cell growth rate, a time course analysis of synchronized (SFM starved) astrocytes was performed. Phosphorylation of ERK1/2 (Thr202/Tyr204) and AKT (Ser473), common readouts for a variety of mitogen and pro-survival stimuli, was used to follow the serum-induced response. In agreement with previous reports, addition of 10% serum triggers a rapid (5-60min) increase in ERK1/2 and AKT phosphorylation followed by a gradual decrease (60-240min) in phosphorylation in all cell lines independent of ciliation status (Fig.2, Fig.3). Interestingly, the amount of phosphorylated ERK1/2 was twofold higher in deciliated cells (shKIF3B or shIFT88) than in control (shCon) based on immunofluorescent and western blot assays (Fig.2A-F). Contrary to ERK1/2, phosphorylation of AKT was twofold lower in deciliated cells than in control (Fig.3A-F), indicating that changes in the pattern and intensity of the signal initiation/propagation act in a cilia-dependent manner.
Fig. 2. Loss of cilium promotes increased ERK1/2 phosphorylation in response to serum stimulation.
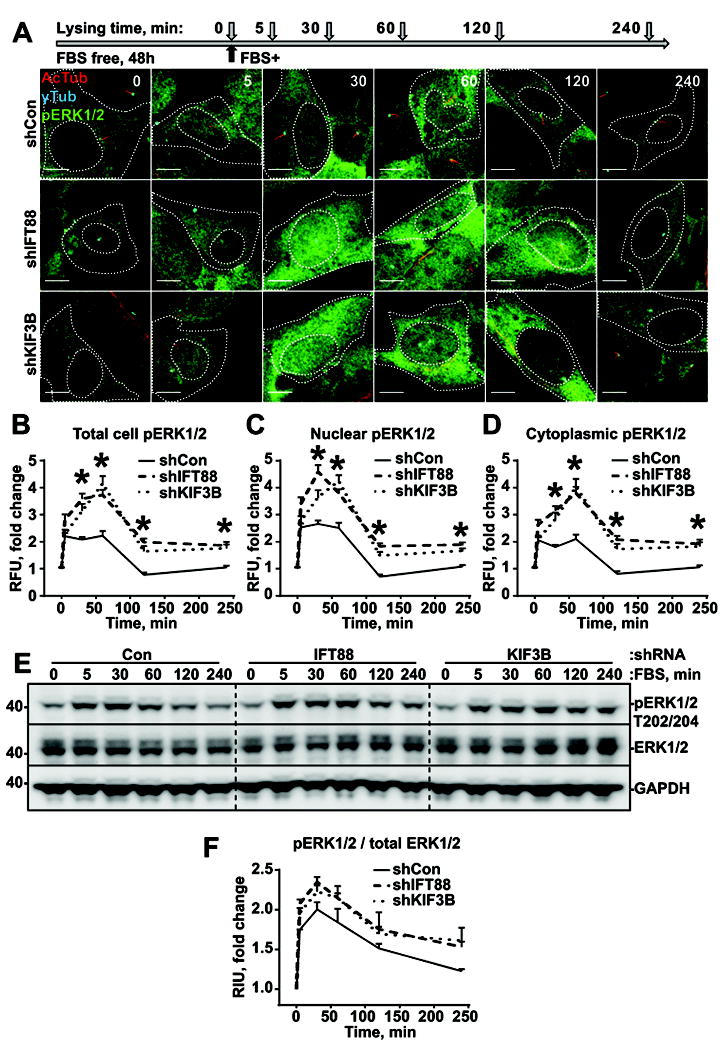
(A) Experiment schematics (top) and representative images of cells stably expressing non-targeting shRNA (Con) or shRNA against IFT88 or KIF3B, stained for acetylated α-tubulin (AcTub, cilium marker), γ-tubulin (γTub, basal body marker), and ERK1/2 phosphor-T202/Y204 (pERK1/2) ; scale bar – 10μm. (B, C, D) Quantifications of ERK1/2 phosphor-T202/Y204 intensities in whole cells (B), nuclei (C) and cytoplasm (D) as in (A); 100 cells in 3 independent experiments; two-way ANOVA with Dunnett’s post hoc test, p<0.05. (E) Representative western blot analysis of cells as in (A), stained for ERK1/2 phosphor-T202/Y204, total ERK1/2, and GAPDH. (F) Quantifications of ERK1/2 phosphor-T202/Y204 bands intensities, as in (E).
Fig. 3. Loss of cilium promotes decreased AKT phosphorylation in response to serum stimulation.
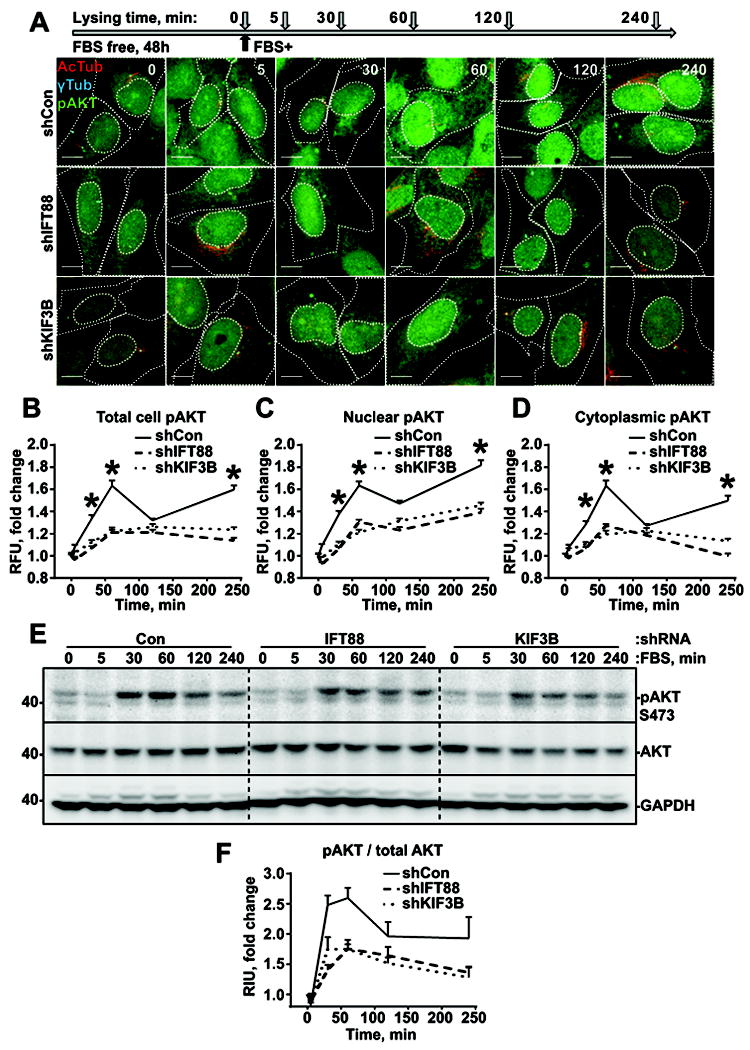
(A) Experiment schematics (top) and representative images of cells stably expressing non-targeting shRNA (Con) or shRNA against IFT88 or KIF3B, stained for acetylated α-tubulin (AcTub), γ-tubulin (γTub), and AKT phosphor-S473 (pAKT) ; scale bar – 10μm. (B, C, D) Quantifications of AKT phosphor-S473 intensities in whole cells (B), nuclei (C) and cytoplasm (D) as in (A); 100 cells in 3 independent experiments; two-way ANOVA with Dunnett’s post hoc test, p<0.05. (E) Representative western blot analysis of cells as in (A), stained for AKT phosphor-S473, total AKT, and GAPDH. (F) Quantifications of AKT phosphor-S473 bands intensities, as in (E).
Lysophosphatidic acid signaling is critical for increased proliferation in deciliated astrocytes
To define the mitogene/s potentially responsible for the hyperproliferative phenotype of deciliated astrocytes, charcoal-stripped serum was used. Charcoal stripping is an efficient way to deplete bioactive lipids, as well as hormones and some vitamins (29), but preserve protein-based growth factors. Surprisingly, charcoal-treatment of serum (cSSM) was sufficient to completely abrogate the increase in growth rate observed in deciliated astrocytes (Fig.4A). One of the most abundant lipid-based mitogens found in serum is lysophosphatidic acid (LPA), which binds to LPA receptors (LPARs) to elicit a response. To test if LPA is involved in deciliation-dependent stimulation of proliferation, a small molecule inhibitor of LPAR1-3, Ki16425 (30), was used. Similar to cSSM, addition of Ki16425 to SSM was sufficient to abrogate the deciliation-dependent increase in growth rates, without any effect on growth of ciliated astrocytes (SSM vs. SSM+Ki16425) (Fig.4B). Moreover, addition of LPA alone to the serum-free medium (SFM) was sufficient to stimulate proliferation of astrocytes, and partially recapitulate the difference observed between ciliated and deciliated cells in the presence of serum (Fig.4B). As expected, LPA addition resulted in a pattern of ERK1/2 and AKT phosphorylation similar to the one observed during serum stimulation (Fig.4C-E). Surprisingly, treatment with EGF, bFGF, PDGF-A/B, or HGF as single agents was neither sufficient to induce proliferation (Fig.S2A) nor able to recapitulate the difference in phosphorylation of both ERK1/2 and AKT (Fig.S2B-C, Fig.S3A-B) previously observed between ciliated and deciliated cells (Fig.2E, Fig.3E). Interestingly, addition of a cocktail of protein growth factors (GFs) including EGF, bFGF, and B27 supplement along with LPA was sufficient to fully recapitulate the phenotype observed with addition of serum (Fig.4F). These findings indicate that LPA signaling is responsible for the highly proliferative phenotype observed in astrocytes with disrupted ciliogenesis, but it requires additional growth factors to amplify its effect.
Fig. 4. Lysophosphatidic acid promotes proliferation in deciliated astrocytes.

(A) Growth rates of immortalized astrocytes stably expressing non-targeting shRNA (Con) or shRNA against IFT88 or KIF3B in serum-supplemented media (SSM), media supplemented with charcoal-stripped serum (cSSM), or in serum-free conditions (SFM); 3 independent experiments; one-way ANOVA with Dunnett’s post hoc test, p<0.05. (B) Growth rates of cells as in (A) in serum-supplemented media (SSM), serum-supplemented media supplemented with 10μmol/L Ki16425, serum-free media supplemented with 1μmol/L LPA (SFM LPA), or in serum-free conditions (SFM); 3 independent experiments; one-way ANOVA with Dunnett’s post hoc test, p<0.05. (C) Experiment schematics (top) and representative western blot analysis of cells as in (A) stained for ERK1/2 phosphor-T202/Y204, total ERK1/2, AKT phosphor-S473, total AKT, and α-tubulin. (D) Quantifications of ERK1/2 phosphor-T202/Y204 bands intensities, as in (C). (E) Quantifications of AKT phosphor-S473 bands intensities, as in (C). (F) Growth rates of cells as in (A) SSM, SFM supplemented with 20ng/ml EGF, 20ng/ml bFGF, and B27 supplement (SFM-GF), SFM supplemented with 20ng/ml EGF, 20ng/ml bFGF, B27 supplement and 1μmol/L LPA (SFM-GF LPA), or in serum-free conditions (SFM); 3 independent experiments; one-way ANOVA with Dunnett’s post hoc test, p<0.05.
LPAR1 localizes to primary cilia
To understand the mechanisms underlying cilia-dependent action of LPA, the subcellular localization of LPARs and Gα subunits participating in downstream signal transduction pathways was analyzed. A panel of LPARs fused with 3xFLAG-tag was exogenously expressed in astrocytes followed by immunofluorescent analysis using anti-FLAG antibodies. LPAR1 and LPAR3, but not LPAR6, were consistently localized in primary cilium (Fig.5A, S4A-B). Interestingly, in deciliated cells, both LPAR1 and LPAR3 were targeted to the plasma membrane (Fig.S5A-B). To exclude the possibility of overexpression-driven cilium targeting of LPAR1, we validated several anti-LPAR1 antibodies for immunofluorescent staining, using CRISPR-Cas9 driven LPAR1 depleted astrocytes as a control (Fig.S6). Utilizing validated antibodies we were able to confirm that endogenous LPAR1 localizes to primary cilium when it is present (Fig.S7).
Fig. 5. Intracellular localization of LPA signaling cascade components.

(A) Representative images of immortalized astrocytes expressing exogenous 3xFLAG-LPAR1, stained for acetylated α-tubulin (AcTub, cilium marker), γ-tubulin (γTub, basal body marker) and FLAG-tag, arrowheads indicate primary cilium; scale bar – 10μm (B) Representative images of immortalized astrocytes stained for acetylated α-tubulin (AcTub), γ-tubulin (γTub), Gαs, Gαq, Gα12 and Gαi1, arrowheads indicate primary cilium; scale bar – 10μm. (C) Immunoprecipitation of 3xFLAG tagged LPAR1 expressed in immortalized astrocytes stably expressing non-targeting shRNA (Con) or shRNA against IFT88 or KIF3B in serum-free conditions. (D) Schematic of potential mechanism of primary cilium restrictive action on the proliferative component of LPA signaling.
Analysis of the cellular localization of multiple Gα subunits shows that only Gαs consistently targeted to primary cilia, similar to previous reports (31). However, whether other Gα subunits enter primary cilia is currently unknown. We found that Gαi1, Gαq, and Gα12 did not display ciliary localization, but rather diffuse cytoplasmic and plasma membrane staining (Fig.5C). Such a pattern of compartmentalization suggests that LPARs may engage in interactions with different Gα subunits depending upon presence or absence of primary cilia.
LPAR1 directly binds to Gα12 and Gαq subunits in deciliated cells
To test this hypothesis, immunoprecipitation analysis was performed in shCon, shIFT88, and shKIF3B astrocytes transiently overexpressing 3xFLAG-LPAR1. Based on sequence similarity, LPAR1 is predicted to interact with Gαi, Gαq, and Gα12 (19). No interaction was detected between LPAR1 and Gαs. The robust co-immunoprecipitation of Gα12 and Gαq with LPAR1 was noted, being 2-3 times higher in deciliated astrocytes (Fig.5D). Previously, LPAR1 was reported to promote cancer cell proliferation specifically through Gα12 (18). Gαq was also reported to transmit pro-proliferative signals in cancer (32). We concluded that LPAR1 is sequestered in primary cilia, which prevents its interaction with cytoplasmic Gα12 and Gαq, restricting its proliferative signaling. Loss of cilia promotes LPAR1 interaction with Gα12 and Gαq, thus promoting the proliferative response to LPA (Fig.5D).
LPA signaling drives GBM proliferation both in vitro and in vivo
To evaluate our findings in disease-relevant settings, previously characterized GBM patient-derived cells and xenografts (33, 34) of two molecular subtypes (classical-GBM6 and mesenchymal-GBM12) were used. In agreement with previous reports (28), only 5-10% of GBM cells in vivo (GBM xenografts in mice, Fig.S8A-B) or in vitro (primary cells short term cultures, Fig.S8C-D) posess primary cilia. LPAR1 staining in GBM6 and 12 cells shows similar pattern to primary astrocytes (Fig.S8E). LPA was previously implicated in GBM progression and invasiveness (35), hence its effects on GBM cell proliferation was evaluated. Upon addition of LPA in serum-free GF-supplemented media, a significant increase of primary GBM cell growth rate in vitro was observed (Fig.S8F), while addition of the LPA inhibitor, Ki16425, to SSM decreases GBM cell growth rate in a dose-dependent manner (Fig.S8G).
Similar to in vitro studies, daily intraperitoneal administration of 30mg/kg Ki16425 as a monotherapy over 4-5 weeks significantly decreased the growth of subcutaneously transplanted GBM xenografts in immunodeficient mice (Fig 6A-E). In agreement with in vitro PDX cell line proliferation data (Fig.S8G), the number of mitotic figures in Ki16425-treated tumors (Fig 6D, E) was twofold lower. Overall, these findings indicate that LPA is an important mitogen in GBM and inhibition of LPA signaling is a viable option to improve anti-GBM therapy.
Fig. 6. Inhibition of LPA signaling suppresses proliferation of GBM PDXs in vivo.
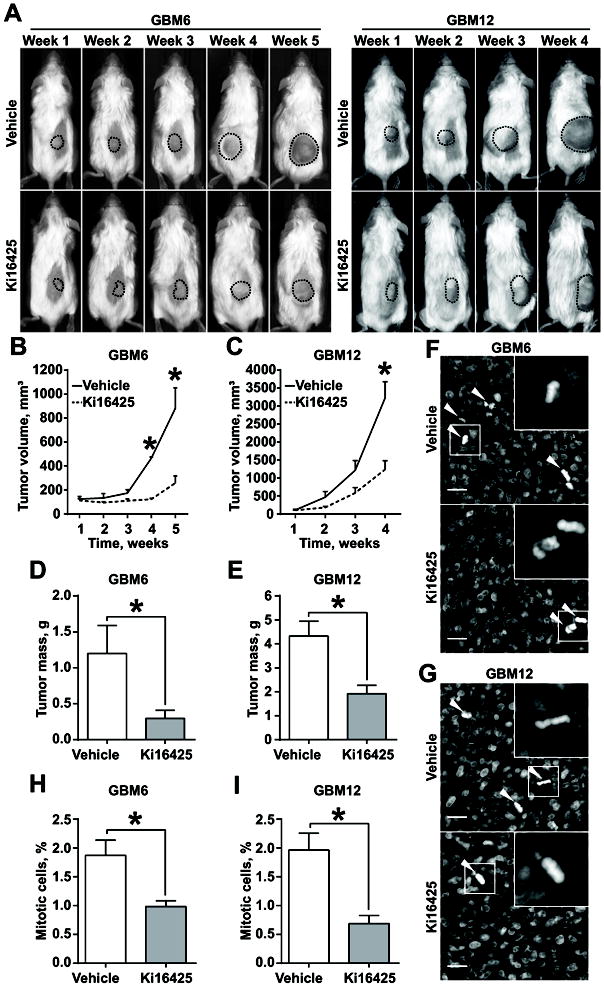
(A) Representative images of mice subcutaneously injected with GBM6 (left panel) and GBM12 (right panel) and administered with vehicle or 30mg/kg/day Ki16425. (B, C) Analysis of tumor growth as in (A) for GBM6 (B) and GBM12 (C); 5 mice per group; two-way ANOVA with Sidak’s post hoc test, p<0.05. (D, E) Analysis of GBM6 (D) and GBM12 (E) terminal tumor weight; 5 tumors per group; Student’s t-test, p<0.05. (F, G) Representative images of GBM6 (F) and GBM12 (G), stained with DAPI; arrowheads indicate mitotic cells; scale bar – 20μm. (H, I) Quantification of mitotic figures as in (F, G) for GBM6 (H) and GBM12 (I); at least 1000 cells within 10 random fields per group; Student’s t-test, p<0.05.
PEG-PLGA Ki16425 loaded nanoparticles slow down GBM growth in brain
To assess the feasibility of targeting GBM with Ki16425 in patients, its effect on GBM growth was evaluated in an intracranial mouse model. Preliminary studies indicated that the regimen used for the subcutaneous GBM PDX experiment was not efficient in the intracranial model (Fig.S9A-B), suggesting that Ki16425 is not capable of crossing the blood brain barrier. To overcome this issue, we used a PEG-PLGA nanoparticle-based delivery system (36) to allow for robust Ki16425 brain entry. Fluorescently labeled nanoparticles rapidly entered the brain upon IP injection and were gradually excreted/degraded over the next 12h period (Fig.S9C-D).To account for this decay, the regimen was modified to 30mg/kg Ki16425 loaded into nanoparticles and delivered every 12h. Mice were intracranially injected with 5×105 GBM12 cells and tumor growth was monitored weekly via MRI. Upon tumors reaching 5mm3, mice were randomly assigned to Ki16425 or a vehicle loaded nanoparticles control group. Over 2 weeks of treatment, the Ki16425 nanoparticle-treated group showed a twofold decrease in tumor progression, compared to control (Fig.7A-B). In agreement with our previous experiments, the number of mitotic cells was decreased twofold (Fig.7C-D). These findings show that Ki16425 with a proper delivery method can significantly suppress GBM progression, and potentially, in combination with the current standard of care, improve local disease control and GBM patient survival.
Fig. 7. Targeted brain delivery of Ki16425 suppresses GBM PDX growth in an intracranial model.
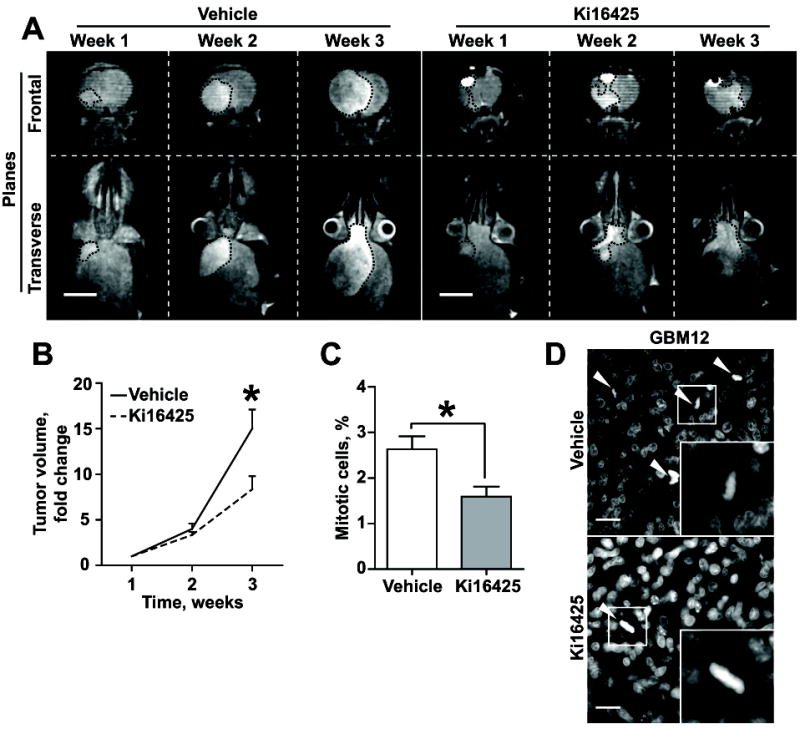
(A) Representative MRI images of mice bearing intracranially grafted GBM12 throughout the treatment with PEG-PLGA nanoparticles loaded with Ki16425 (30mg/kg twice a day); scale bar – 5mm. (B) Analysis of tumor growth as in (A); 5 mice per group; two-way ANOVA with Sidak’s post hoc test, p<0.05. (C) Quantification of mitotic figures as in (D); at least 1000 cells within 10 random fields per group; Student’s t-test, p<0.05. (D) Representative images of tumors from (A), stained with DAPI; arrowheads indicate mitotic cells; scale bar – 20μm.
Discussion
Primary cilium is well known for its role in multiple signaling cascades (1, 2, 11). Primary cilia length and the number of cells with cilium is significantly reduced or lost in multiple cancer types including GBM (7-10). Moreover, several studies have noted an increase in cell proliferation upon loss of primary cilia (11, 12). The proposed mechanism of cilia-driven proliferation control involves the sequestration of basal body (mother centriole) and the inability to form the mitotic spindle (37). Previously, it was shown that primary cilium disassembly is biphasic: a first wave of fast, transient disassembly within 1-2h after exposure to mitogenic stimuli (4), followed by a second wave of disassembly at 18-24h, which coincides with mitotic entry. The role of the first wave of cilium disassembly in mitogenic signaling is currently unknown, as is the mechanism/s underlying the increase in proliferation upon loss of primary cilia.
Several pro-proliferative signaling cascades are reported to require primary cilium for proper signal transduction. For instance, PDGF-AA signaling through PDGFRαα is lost in deciliated fibroblasts, therefore stimulation with PDGF-AA does not cause ERK1/2 or AKT activation (38); however, the overall proliferation effect of cilia loss was not evaluated in this work. SHH signaling is well-studied in conjunction with primary cilium. Loss of cilia ablates SHH signaling and SHH-driven proliferation (39), and correspondingly, SHH-driven cancers have a tendency to maintain high ciliation rates (14, 40). On the other hand, ciliary localization of IGF1R (41) and Notch (11) signaling is needed for differentiation and restriction of cell proliferation. Importantly, cilium can selectively sequester signaling components like GPR88 (42) and LPAR1/3 (Fig.5, S4) from their interaction with cilia-excluded co-factors, thus conferring selectivity on signal propagation/amplification through the lateral segregation of receptors. This strongly argues that the effect of primary cilia on proliferation is context-dependent, with pro-proliferative effects during development through SHH signaling and anti-proliferative effects in more differentiated cells.
In GBM, the complete inhibition of ciliogenesis seems to have minimal and non-coherent effects on overall proliferation and tumor progression (43). Nevertheless, ciliation compared to normal brain cells is drastically decreased (9), supporting an anti-mitogenic function of primary cilium in GBM. Our current findings suggest that the engineered loss of primary cilia in normal human astrocytes phenotypically closely mimics transient cilium disassembly (Fig.S1B), and also resembles cilia loss in GBM cells (Fig.S8B, D). The increase in growth rate observed in deciliated cells (Fig.1F, G) suggests that transient cilia disassembly may be a key event augmenting mitogenic signaling. Mechanistically, we found that loss of primary cilia in human non-transformed astrocytes results in the amplification of ERK1/2 phosphorylation and promotes proliferation in an LPA-dependent manner (Fig.2, Fig.4). Interestingly, phosphorylation of AKT in response to LPA or serum stimulation was decreased in deciliated astrocytes (Fig.3). This can be attributed to inactivation of PDGFRαα (38) and IGF1R (41) driven signaling, and adds to the understanding of increased stress sensitivity in deciliated astrocytes (44). These findings support the idea that loss of primary cilia changes the pattern of the cellular response to mitogen stimulation, resulting in higher, more sustainable ERK1/2 activation thus explaining the increase in proliferation, but revealing the potential vulnerability of deciliated cells to stress via a decrease in pro-survival signaling (pAKT). Since GBMs usually have high pAKT levels (45) and we did not observe the transformation of deciliated astrocytes, we conclude that additional hit/s such as those well-known for GBM including RTK amplification/PTEN inactivation/PI3K activation, are required for overcoming the decrease in AKT activation.
Previously, LPA was implicated in the regulation of cell migration and proliferation (21, 35), however, the role of primary cilia in these signaling pathways was not explored. The LPA receptors (LPAR1/3) are specifically localized to primary cilium, but the LPAR downstream effectors, Gα12 and Gαq, are excluded from it (Fig.5, S4). Both Gα12 and Gαq activation is known to be implicated in cancer cell proliferation and cancer progression (46, 47). For Gα12, activation through LPAR1 specifically was reported in ovarian cancer cells (18). Our findings support the notion that in the absence of primary cilia, LPAR1 localizes to the plasma membrane and interacts with Gα12 and Gαq (Fig.5D, S5, S7). However, further research is required to pinpoint the intracellular compartment for Gα12 and Gαq and endogenous LPAR1 interaction. Hence, loss of primary cilia could be responsible for increased proliferation in a subset of cancers, and thus cilia restoration or the manipulation of cilia-dependent signaling such as LPA could be used to develop new therapeutic approaches to fight cancer.
GBM cells seem to be highly dependent on LPA as a mitogen, since inhibition of LPA signaling with Ki16425 abrogates their growth in vitro, and in both subcutaneous and intracranial GBM models (Fig.S8, Fig.6, Fig.7). Interestingly, the magnitude of the Ki16425 effect was the same between two in vivo models (Fig.6, Fig.7), suggesting that LPA signaling is equally engaged in both of them. LPA is known to be highly abundant in brain (3.7-35 pmol/mg) and serum concentration of LPA was reported to be close to this range (4-15.5 μM) (17). In addition, GBMs were reported to increase LPA production by secreting autotaxin (48). However, further studies are required for comprising the LPA levels in GBM PDXs grown in subcutaneous versus brain settings.
GBM6 and GBM12 used in the current study are of classical and mesenchymal molecular subtypes respectively, and are highly aggressive in mouse models (33, 34). Both of these subtypes are associated with rapid disease progression and poor prognosis. Our in vivo experiments clearly show that targeting LPA signaling yields a twofold reduction of tumor growth, specifically through a decrease in the proliferation rate of tumor cells (Fig.6, Fig.7). Moreover, utilizing a PEG-PLGA nanoparticles delivery system confirmed the effectiveness of targeting LPA signaling in intracranial settings. The median survival of GBM patients with the current standard of care including aggressive surgery, radiation, and chemotherapy (49) is about 12 months (15), with the majority of patients experiencing recurrence within 32-36 weeks (16). Targeting LPA signaling can prove to be highly beneficial in addition to standard care, since LPA is implicated in proliferation and migration/invasion (21, 35). Further studies are required, but based on our findings, we expect a substantial increase in recurrence-free survival upon inhibition of LPA signaling.
Overall, our data supports the role of LPA signaling in cancer cell proliferation, and for the first time, highlights primary cilia as a switch for the interpretation of LPA as a mitogen.
Materials and Methods
Cell lines and reagents
Human astrocytes isolated from human cortex (1800) were obtained from ScienCell Research Labs and maintained in DMEM/F12, supplemented with 10% heat-inactivated FBS, Antibiotic-Antimycotic (ThermoFisher) and 10 μg/ml of gentamycin (MP Biomedicals). Cells were propagated and cryopreserved at passage 2, for all studies cells were not passaged more than 10 times or 8 weeks, no authentication or mycoplasma testing were performed. For immortalization, primary astrocytes at passage 5 or 6 were cultured until 50–75% confluency and transfected with SV40 large T-antigen construct (25), followed by selection with 500μg/ml G418 (Sigma) until resistant colonies were formed. GBM6 and 12 PDXs (33) were kindly provided by Dr. Jann Sarkaria (Mayo Clinic, Rochester, MN) through a shared MTA agreement, no authentication or mycoplasma testing were performed. PDXs were maintained and used for intracranial injections or primary cell culture isolation as previously described (34). A list of antibodies and their used applications is outlined in Table S1. shRNA constructs were obtained from Dharmacon and are outlined in Table S2. All primers were purchased from IDT Technologies or Invitrogen, the sequences of which are reported in Table S3. The following reagents were used: Lysophosphatidic acid (LPA, Cayman Chemical), EGF and PDGF-AB (Sigma), HGF (R&D Technologies), bFGF (Peprotech), B27 supplement (ThermoFisher), Ki16425 (ApexBio). For lysophosphatidic acid stimulation experiments, bovine serum albumin (BSA; BP1600, Fisher Scientific) was used as a carrier and was added to all cells to a final concentration of 0.1%.
Western blotting
Western blotting was performed using standard procedures (50). Primary antibodies used are outlined in Table S1. Secondary anti-mouse and anti-rabbit HRP-conjugated antibodies (Jackson ImmunoResearch Labs) were diluted 1:10,000 followed by chemiluminescence-based detection with HyGLO™ (Denville Scientific). Bands were quantified using the digital electrophoresis documentation and image analysis software GeneTools (Syngene Corp.), with signal intensity normalized to either α-tubulin or GAPDH.
Immunofluorescent cell analysis
Cells were processed as previously described (4). Primary antibodies used are outlined in Table S1. Secondary antibodies included AlexaFluor 488, 555, and 647 (ThermoFisher). Images were captured using a standard setting by an LSM510 confocal microscope (Zeiss) (50). All images represent whole-cell 3D reconstructed projections with 0.4μm steps. All quantifications were done using ImageJ (NIH).
Cell growth/proliferation analysis
Cell growth rate was determined using semi-automated cell counting in ImageJ (NIH). Cells were plated at 1×104 cells per well and grown in the specified conditions for five days, fixed with methanol, and stained with Hoechst33342. Four random 10x fields per well were analyzed with at least three replicates per independent experiment; each graph represents at least three independent experiments.
Fluorescent immunohistochemistry (F-IHC)
Deparaffinization and rehydration of 4-5μm thin sections was performed as following: 1) three times for 3 min in xylene, 2) three times for 2 min in 100% ethanol, 3) 2 min in 95% ethanol, 4) 2 min in 80% ethanol, 5) 2 min in 70% ethanol, and 6) 5 min in 1XTBS. Antigen retrieval was done using 98°C citrate buffer, pH 6.0 for 20 minutes. Sections were blocked for 60 min with 5% BSA, 1XTBS solution and stained with the indicated primary antibodies. Secondary antibodies included AlexaFluor-488, 555, and 647 (ThermoFisher) and sections were mounted with ProLong Gold DAPI-containing media (ThermoFisher). Images were captured using LSM510 confocal microscope as previously described (50) (Zeiss). All images represent whole-cell 3D reconstructed projections with 0.4μm steps. All quantifications were done using ImageJ (NIH).
LPAR cloning
For robust expression and detection of LPARs, pcDNA3.1 vector was modified as previously described (51). Briefly, the insert containing the cleavable ER-targeting sequence followed by a 3xFLAG-tag and multiple cloning site (Table S3) was synthesized through String™ service (ThermoFisher) and cloned into pcDNA3.1 between HindIII and SalI restriction sites. LPAR1, 3, 6 cDNA was amplified from a Human Mammary Epithelial Cells (HMECs) cDNA library kindly provided by Dr. Alexey Ivanov (West Virginia University, Morgantown, WV); primers are shown in Table S3. The generated inserts were cloned into pcDNA3.1 ER-3xFLAG between BamHI and XhoI restriction sites. All constructs were validated by sequencing.
Immunoprecipitation
For LPAR1 immunoprecipitation, 3xFLAG-LPAR1 was transiently overexpressed in cells pre-incubated for 24h in serum-free media. Cells were lysed as previously described (52). 3xFLAG-LPAR1 was precipitated with anti-FLAG M2 affinity gel (Sigma) and used for subsequent Western blot analysis.
Animal experiments
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) immunodeficient male mice (The Jackson Laboratory) were housed in the West Virginia University Animal Facility under pathogen-free conditions with an approved Institutional Animal Care and Use Committee protocol. For subcutaneous injections, 100μl of GBM6 or GBM12 tumor mash mixed 1:1 with Matrigel (Corning) was injected into the mice flanks. For intracranial injections, transient primary cell cultures were established as previously described (34), and 5×105 cells in 5μl of Ca/Mg-free Dulbecco modified Phosphate Buffer Saline (DPBS) were administrated into the mouse cortex via stereotactic device-guided injection (34). Subcutaneous tumor growth was assessed weekly via caliper measurements; treatment was initiated upon tumor volume reaching 100mm3. Intracranial tumor volume was assessed weekly via contrast enhanced MRI (53) or twice a week via bioluminescence imaging (54); treatment was initiated upon tumor volume exceeding 5mm3. Animals were randomly assigned to the treatment group by simple randomization, inversigator was single blinded during group allocation. Mice bearing subcutaneous tumors were intraperitoneally injected with 30mg/kg Ki16425 in 95% corn oil/5% DMSO or vehicle alone daily. For the intracranial model, mice were administered 30mg/kg Ki16425 loaded into PEG-PLGA nano-particles in PBS or given nano-particles alone twice daily. All animals were euthanized upon reaching a moribund condition, according to the WVU IACUC Tumor Development and Tumor Scoring in Rodents policy.
Contrast enhanced Magnetic Resonance Imaging (MRI)
To visualize the intracranial tumors, mice were intraperitoneally injected with gadolinium-DTPA (contrast reagent, BioPAL) to a final concentration of 2mmol/kg (55), and imaged with a compact MRI system (ICON, Bruker). Images were taken with T1 weighted RARE sequence (echo time: 26.23ms; repetition time 1984.158ms; averages: 8; rare factor: 6). Overall time of scan was 25min and final resolution was 0.125mm for all axes. Stack images were reconstructed and analyzed using ImageJ (NIH).
Animal bioluminescence imaging
Mice were imaged twice a week for quantitative evaluation of tumor growth as previously described (56). Images were obtained using the IVIS Lumina-II Imaging System and Living Image-4.0 software (PerkinElmer).
PEG-PLGA nano-particles
Nano-particles were prepared as previously described (36). Briefly, PEG-PLGA and Ki16425 were dissolved in acetone and added dropwise into water. The nano-particles were stirred for 2 h at 40°C before being collected by centrifugation (4000g for 90 min). After discarding the supernatant, the nanoparticles were resuspended in Phosphate-Buffered Saline (PBS).
Statistical analysis
Statistical comparisons were made using two-tailed Student’s t-test. When more than two groups were analyzed, one-way or two-way analysis of variance (ANOVA) was used. P<0.05 was considered to be significant (*) as indicated in figure legends. All treatment groups were compared to vehicle/control unless mentioned otherwise. Experimental values were reported as the means with +/-S.E.M (standard error of mean). All calculations of statistical significance were made using GraphPad software. Sampe sizes and statistical analysis chosen were based on our previous experience and recommendations of biostatistician (4, 50, 56).
Supplementary Material
Acknowledgments
The authors thank Dr. Alexey Ivanov for sharing of the HMLE cDNA library and Brandon Jones for invaluable help with manuscript preparation.
Financial support: This research was supported by grants R21CA208875 (E.N.P), R01CA148671 (E.N.P) from the NIH/NCI, U54GM104942 (W.J.G) from the NIGMS/NIH, WVU Cancer Institute Undergraduate Research Fellowship (C.L.G), and “Let the Journey Begin” fund established by Erin Dunmire, WVU Cancer Institute. Andrey Bobko is supported by startup funding from WVCTSI. Naira V. Margaryan is supported by U54GM104942 from WVCTSI.
West Virginia University Microscope Imaging Facility is supported by the WVU Cancer Institute and NIH grants P20RR016440, P20 RR016477and P30RR032138/P30GM103488.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
Disclaimers: This manuscript contains original work only and has not been published or submitted elsewhere. All of the authors have directly participated in the planning, execution and analysis of this study and have approved the submitted version of this manuscript.
Bibliography
- 1.Christensen ST, Clement CA, Satir P, Pedersen LB. Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. The Journal of pathology. 2012;226(2):172–84. doi: 10.1002/path.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Science signaling. 2012;5(246):re6. doi: 10.1126/scisignal.2002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schou KB, Pedersen LB, Christensen ST. Ins and outs of GPCR signaling in primary cilia. EMBO reports. 2015;16(9):1099–113. doi: 10.15252/embr.201540530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129(7):1351–63. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim S, Lee K, Choi JH, Ringstad N, Dynlacht BD. Nek2 activation of Kif24 ensures cilium disassembly during the cell cycle. Nature communications. 2015;6:8087. doi: 10.1038/ncomms9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang G, Chen Q, Zhang X, Zhang B, Zhuo X, Liu J, et al. PCM1 recruits Plk1 to the pericentriolar matrix to promote primary cilia disassembly before mitotic entry. Journal of cell science. 2013;126(Pt 6):1355–65. doi: 10.1242/jcs.114918. [DOI] [PubMed] [Google Scholar]
- 7.Basten SG, Willekers S, Vermaat JS, Slaats GG, Voest EE, van Diest PJ, et al. Reduced cilia frequencies in human renal cell carcinomas versus neighboring parenchymal tissue. Cilia. 2013;2(1):2. doi: 10.1186/2046-2530-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hassounah NB, Nagle R, Saboda K, Roe DJ, Dalkin BL, McDermott KM. Primary cilia are lost in preinvasive and invasive prostate cancer. PloS one. 2013;8(7):e68521. doi: 10.1371/journal.pone.0068521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moser JJ, Fritzler MJ, Rattner JB. Primary ciliogenesis defects are associated with human astrocytoma/glioblastoma cells. BMC cancer. 2009;9:448. doi: 10.1186/1471-2407-9-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan K, Frolova N, Xie Y, Wang D, Cook L, Kwon YJ, et al. Primary cilia are decreased in breast cancer: analysis of a collection of human breast cancer cell lines and tissues. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2010;58(10):857–70. doi: 10.1369/jhc.2010.955856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ezratty EJ, Stokes N, Chai S, Shah AS, Williams SE, Fuchs E. A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell. 2011;145(7):1129–41. doi: 10.1016/j.cell.2011.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gradilone SA, Radtke BN, Bogert PS, Huang BQ, Gajdos GB, LaRusso NF. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer research. 2013;73(7):2259–70. doi: 10.1158/0008-5472.CAN-12-2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y, Roine N, Makela TP. CCRK depletion inhibits glioblastoma cell proliferation in a cilium-dependent manner. EMBO reports. 2013;14(8):741–7. doi: 10.1038/embor.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A. Dual and opposing roles of primary cilia in medulloblastoma development. Nature medicine. 2009;15(9):1062–5. doi: 10.1038/nm.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ries LAG SEER Program (National Cancer Institute (U.S.)) Cancer survival among adults US SEER program, 1988-2001 : patient and tumor characteristics. Bethesda, MD: U.S. Dept. of Health and Human Services, National Institutes of Health, National Cancer Institute; 2007. Available from: http://seer.cancer.gov/publications/survival/seer_survival_mono_highres.pdf http://purl.fdlp.gov/GPO/gpo808 http://seer.cancer.gov/publications/survival/ Freely available online. [Google Scholar]
- 16.Birk HS, Han SJ, Butowski NA. Treatment options for recurrent high-grade gliomas. CNS oncology. 2016 doi: 10.2217/cns-2016-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yung YC, Stoddard NC, Chun J. LPA receptor signaling: pharmacology, physiology, and pathophysiology. Journal of lipid research. 2014;55(7):1192–214. doi: 10.1194/jlr.R046458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldsmith ZG, Ha JH, Jayaraman M, Dhanasekaran DN. Lysophosphatidic Acid Stimulates the Proliferation of Ovarian Cancer Cells via the gep Proto-Oncogene Galpha(12) Genes & cancer. 2011;2(5):563–75. doi: 10.1177/1947601911419362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez-Gil I, Zian D, Vazquez-Villa H, Ortega-Gutierrez S, Lopez-Rodriguez ML. The status of the lysophosphatidic acid receptor type 1 (LPA(1)R) Medchemcomm. 2015;6(1):13–23. [Google Scholar]
- 20.Shano S, Moriyama R, Chun J, Fukushima N. Lysophosphatidic acid stimulates astrocyte proliferation through LPA1. Neurochemistry international. 2008;52(1-2):216–20. doi: 10.1016/j.neuint.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gschwind A, Prenzel N, Ullrich A. Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer research. 2002;62(21):6329–36. [PubMed] [Google Scholar]
- 22.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta neuropathologica. 2010;119(1):7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338(6110):1080–4. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kasahara K, Miyoshi K, Murakami S, Miyazaki I, Asanuma M. Visualization of astrocytic primary cilia in the mouse brain by immunofluorescent analysis using the cilia marker Arl13b. Acta medica Okayama. 2014;68(6):317–22. doi: 10.18926/AMO/53020. [DOI] [PubMed] [Google Scholar]
- 25.Schuermann M. An expression vector system for stable expression of oncogenes. Nucleic acids research. 1990;18(16):4945–6. doi: 10.1093/nar/18.16.4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, et al. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95(6):829–37. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- 27.Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. The Journal of cell biology. 2000;151(3):709–18. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarkisian MR, Siebzehnrubl D, Hoang-Minh L, Deleyrolle L, Silver DJ, Siebzehnrubl FA, et al. Detection of primary cilia in human glioblastoma. Journal of neuro-oncology. 2014;117(1):15–24. doi: 10.1007/s11060-013-1340-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao Z, West C, Norton-Wenzel CS, Rej R, Davis FB, Davis PJ, et al. Effects of resin or charcoal treatment on fetal bovine serum and bovine calf serum. Endocrine research. 2009;34(4):101–8. doi: 10.3109/07435800903204082. [DOI] [PubMed] [Google Scholar]
- 30.Ohta H, Sato K, Murata N, Damirin A, Malchinkhuu E, Kon J, et al. Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Molecular pharmacology. 2003;64(4):994–1005. doi: 10.1124/mol.64.4.994. [DOI] [PubMed] [Google Scholar]
- 31.Barzi M, Kostrz D, Menendez A, Pons S. Sonic Hedgehog-induced proliferation requires specific Galpha inhibitory proteins. The Journal of biological chemistry. 2011;286(10):8067–74. doi: 10.1074/jbc.M110.178772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel BR, Tall GG. Ric-8A gene deletion or phorbol ester suppresses tumorigenesis in a mouse model of GNAQ(Q209L)-driven melanoma. Oncogenesis. 2016;5(6):e236. doi: 10.1038/oncsis.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlson BL, Grogan PT, Mladek AC, Schroeder MA, Kitange GJ, Decker PA, et al. Radiosensitizing effects of temozolomide observed in vivo only in a subset of O6-methylguanine-DNA methyltransferase methylated glioblastoma multiforme xenografts. International journal of radiation oncology, biology, physics. 2009;75(1):212–9. doi: 10.1016/j.ijrobp.2009.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carlson BL, Pokorny JL, Schroeder MA, Sarkaria JN. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Current protocols in pharmacology / editorial board, SJ Enna. 2011;Chapter 14(Unit 14.16) doi: 10.1002/0471141755.ph1416s52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manning TJ, Jr, Parker JC, Sontheimer H. Role of lysophosphatidic acid and rho in glioma cell motility. Cell motility and the cytoskeleton. 2000;45(3):185–99. doi: 10.1002/(SICI)1097-0169(200003)45:3<185::AID-CM2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 36.Geldenhuys W, Wehrung D, Groshev A, Hirani A, Sutariya V. Brain-targeted delivery of doxorubicin using glutathione-coated nanoparticles for brain cancers. Pharmaceutical development and technology. 2015;20(4):497–506. doi: 10.3109/10837450.2014.892130. [DOI] [PubMed] [Google Scholar]
- 37.Basten SG, Giles RH. Functional aspects of primary cilia in signaling, cell cycle and tumorigenesis. Cilia. 2013;2(1):6. doi: 10.1186/2046-2530-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, et al. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Current biology : CB. 2005;15(20):1861–6. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Barzi M, Berenguer J, Menendez A, Alvarez-Rodriguez R, Pons S. Sonic-hedgehog-mediated proliferation requires the localization of PKA to the cilium base. Journal of cell science. 2010;123(Pt 1):62–9. doi: 10.1242/jcs.060020. [DOI] [PubMed] [Google Scholar]
- 40.Li L, Grausam KB, Wang J, Lun MP, Ohli J, Lidov HG, et al. Sonic Hedgehog promotes proliferation of Notch-dependent monociliated choroid plexus tumour cells. Nature cell biology. 2016;18(4):418–30. doi: 10.1038/ncb3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu D, Shi S, Wang H, Liao K. Growth arrest induces primary-cilium formation and sensitizes IGF-1-receptor signaling during differentiation induction of 3T3-L1 preadipocytes. Journal of cell science. 2009;122(Pt 15):2760–8. doi: 10.1242/jcs.046276. [DOI] [PubMed] [Google Scholar]
- 42.Marley A, Choy RW, von Zastrow M. GPR88 reveals a discrete function of primary cilia as selective insulators of GPCR cross-talk. PloS one. 2013;8(8):e70857. doi: 10.1371/journal.pone.0070857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoang-Minh LB, Deleyrolle LP, Siebzehnrubl D, Ugartemendia G, Futch H, Griffith B, et al. Disruption of KIF3A in patient-derived glioblastoma cells: effects on ciliogenesis, hedgehog sensitivity, and tumorigenesis. Oncotarget. 2016;7(6):7029–43. doi: 10.18632/oncotarget.6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshimura K, Kawate T, Takeda S. Signaling through the primary cilium affects glial cell survival under a stressed environment. Glia. 2011;59(2):333–44. doi: 10.1002/glia.21105. [DOI] [PubMed] [Google Scholar]
- 45.McDowell KA, Riggins GJ, Gallia GL. Targeting the AKT pathway in glioblastoma. Current pharmaceutical design. 2011;17(23):2411–20. doi: 10.2174/138161211797249224. [DOI] [PubMed] [Google Scholar]
- 46.Kalinec G, Nazarali AJ, Hermouet S, Xu N, Gutkind JS. Mutated alpha subunit of the Gq protein induces malignant transformation in NIH 3T3 cells. Molecular and cellular biology. 1992;12(10):4687–93. doi: 10.1128/mcb.12.10.4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gan CP, Patel V, Mikelis CM, Zain RB, Molinolo AA, Abraham MT, et al. Heterotrimeric G-protein alpha-12 (Galpha12) subunit promotes oral cancer metastasis. Oncotarget. 2014;5(20):9626–40. doi: 10.18632/oncotarget.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoelzinger DB, Nakada M, Demuth T, Rosensteel T, Reavie LB, Berens ME. Autotaxin: a secreted autocrine/paracrine factor that promotes glioma invasion. Journal of neuro-oncology. 2008;86(3):297–309. doi: 10.1007/s11060-007-9480-6. [DOI] [PubMed] [Google Scholar]
- 49.PDQ Cancer Information Summaries. Bethesda (MD): 2002. Adult Central Nervous System Tumors Treatment (PDQ(R)): Health Professional Version. [Google Scholar]
- 50.Kozyreva VK, Kiseleva AA, Ice RJ, Jones BC, Loskutov YV, Matalkah F, et al. Combination of Eribulin and Aurora A Inhibitor MLN8237 Prevents Metastatic Colonization and Induces Cytotoxic Autophagy in Breast Cancer. Molecular cancer therapeutics. 2016;15(8):1809–22. doi: 10.1158/1535-7163.MCT-15-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guan XM, Kobilka TS, Kobilka BK. Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide. The Journal of biological chemistry. 1992;267(31):21995–8. [PubMed] [Google Scholar]
- 52.Pal K, Badgandi H, Mukhopadhyay S. Studying G protein-coupled receptors: immunoblotting, immunoprecipitation, phosphorylation, surface labeling, and cross-linking protocols. Methods in cell biology. 2015;127:303–22. doi: 10.1016/bs.mcb.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 53.Koutcher JA, Hu X, Xu S, Gade TP, Leeds N, Zhou XJ, et al. MRI of mouse models for gliomas shows similarities to humans and can be used to identify mice for preclinical trials. Neoplasia. 2002;4(6):480–5. doi: 10.1038/sj.neo.7900269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA, et al. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Molecular cancer therapeutics. 2007;6(3):1167–74. doi: 10.1158/1535-7163.MCT-06-0691. [DOI] [PubMed] [Google Scholar]
- 55.Portnoy S, Bishop J, Dazai J, Spring S, Henkelman R. Characterization of signal enhancement following the intraperitoneal injection of Gadolinium based contrast agents [Google Scholar]
- 56.Loskutov YV, Kozyulina PY, Kozyreva VK, Ice RJ, Jones BC, Roston TJ, et al. NEDD9/Arf6-dependent endocytic trafficking of matrix metalloproteinase 14: a novel mechanism for blocking mesenchymal cell invasion and metastasis of breast cancer. Oncogene. 2015;34(28):3662–75. doi: 10.1038/onc.2014.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.