

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
Genetic barcoding of HSPCs in aged macaques reveals impaired long-term clonal output from multipotent HSPCs.
Aged macaques showed prolonged contributions from lineage-biased HSPCs and late clonal expansions.
Abstract
Age-associated changes in hematopoietic stem and progenitor cells (HSPCs) have been carefully documented in mouse models but poorly characterized in primates and humans. To investigate clinically relevant aspects of hematopoietic aging, we compared the clonal output of thousands of genetically barcoded HSPCs in aged vs young macaques after autologous transplantation. Aged macaques showed delayed emergence of output from multipotent (MP) clones, with persistence of lineage-biased clones for many months after engraftment. In contrast to murine aging models reporting persistence of myeloid-biased HSPCs, aged macaques demonstrated persistent output from both B-cell and myeloid-biased clones. Clonal expansions of MP, myeloid-biased, and B-biased clones occurred in aged macaques, providing a potential model for human clonal hematopoiesis of indeterminate prognosis. These results suggest that long-term MP HSPC output is impaired in aged macaques, resulting in differences in the kinetics and lineage reconstitution patterns between young and aged primates in an autologous transplantation setting.
Visual Abstract
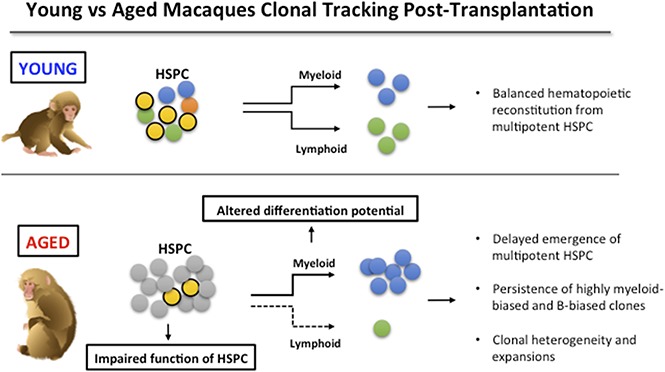
Introduction
Aging has been associated with a number of changes in the hematopoietic system, including diminished regenerative potential, skewed lineage differentiation, increased incidence of anemia, decline in adaptive immunity, and higher rates of neoplastic transformation.1-3 These changes have important clinical consequences, for instance, in the ability of an aging individual to tolerate myelosuppressive therapies or recover blood counts and immunity after stem cell transplantation. A retrospective analysis of donor characteristics after allogeneic hematopoietic stem cell transplantation from unrelated donors suggested that donor age was the only characteristic that significantly affected overall and disease-free survival.4 In human autologous hematopoietic stem and progenitor cell (HSPC) transplantation, one study focusing on the impact of age demonstrated a significantly higher percentage of aged compared with young patients with abnormal recovery of blood count, which suggests that clinically relevant aspects of long-term HSPC regeneration and function are affected by age, particularly under the chemotoxic and replicative stress associated with autologous transplantation for malignancies.5 Recently, the revolution in high-throughput sequencing has uncovered a striking increase in clonal hematopoiesis as humans age, even in the absence of cytopenias or other signs of dysregulated hematopoiesis.6
HSPCs represent a functionally heterogeneous population that differs in self-renewal potential, differentiation pathways, cycling kinetics, and clonal lifespan.7-9 Multiparameter antibody staining of cell surface proteins, followed by flow cytometric sorting and in vitro culture or in vivo engraftment assays, have been used to delineate functional subsets of murine HSPCs and human HSPCs transplanted in immunodeficient mice.10,11 To track the clonal output of individual HSPCs, limiting-dilution murine transplantation assays have been considered the gold standard assay to define behavior at a single cell or well-defined population level. However, limiting-dilution transplantation analyses likely do not reflect physiologic polyclonal hematopoiesis, given the extraordinary replicative stress applied to individual HSPCs, and this strategy depends on a priori knowledge of HSPC phenotypes.12,13
Semi-random replication-incompetent retroviral integration sites can be used as clonal tags via restriction fragment length polymorphisms or vector integration site retrieval and is a powerful approach to tracking clonal output of individual HSPCs in murine, nonhuman primates and, more recently, studies on human gene therapy.14-16 However, vector integration site retrieval methodologies are inefficient and semi-quantitative at best,17,18 and they are difficult to use for quantitative analysis of clonal HSPC output. Most human gene therapy trials to date have enrolled primarily children, and obtaining sufficiently large volumes of blood and bone marrow at frequent sequential time points as optimal for robust clonal tracking is clinically unacceptable. In addition, many of the patients enrolled into human HSPC gene therapy trials to date have had underlying abnormal hematopoiesis or immune function, potentially adversely affecting HSPC behavior. Furthermore, to our knowledge no elderly patients have been enrolled in gene therapy trials to date, so there is no information available on the clonal output of aged human HSPCs.
Most investigations of the impact of aging on hematopoiesis and the immune system have used murine models. However, murine HSPCs have evolved very different properties from human HSPCs in characteristics relevant to aging, for instance, organismal lifespan, HSPC cycling and frequency, telomere length, and transformation susceptibility.19 It is unclear whether some of the properties linked to aged murine HSPCs are shared by aged human HSPCs. Thus, although murine models may provide mechanistic insights into some pathways affecting hematopoietic aging, many human findings, such as naturally occurring clonal hematopoiesis of elderly people, are difficult to investigate experimentally in mice. Previous studies identifying age-related changes in human HSPCs have almost exclusively relied on indirect evaluations such as in vitro phenotypic characterization or functional assays.20,21 The rhesus macaque is a potentially more instructive species in which to model human hematopoiesis and aging, based on the close phylogenetic relationship between macaques and humans, and similar telomere lengths, lifespans, and aging phenotypes.22,23
Our group developed genetic barcoding technology combining lentiviral delivery of high-diversity short genetic barcodes with high-throughput sequencing in order to quantitatively retrieve and track the clonal output from thousands of individual HSPCs in vivo after autologous transplantation of rhesus macaque HSPCs.11,24 We now apply this approach to investigate the clonal histories, quantitative output, and lineage characteristics of young vs aged rhesus macaque HSPCs. We observed a number of distinct differences in clonal HSPC output in aged rhesus macaques, providing further insights into primate HSPC aging.
Materials and methods
Information regarding production of lentiviral barcoded libraries, HSPC transduction, autologous transplantation, and hematopoietic cell processing can be found in supplemental Methods (available on the Blood Web site).
DNA barcode retrieval and analysis
We performed low-cycle polymerase chain reaction to amplify the DNA barcode region followed by multiplexed sequencing (HiSeq 2500/3000; illumina, San Diego, CA), as previously described, on 200- to 500-ng DNA samples from hematopoietic cells of various lineages sorted by flow cytometry at least 95% purity.24,25 Approximately 4 million sequencing reads were obtained per sample. Using custom Python processing, we filtered reads for the presence of a 6-bp library ID at the beginning of the read, followed by the putative barcode. Putative barcodes with read counts of 100 or less per fastq file were discarded. We then combined barcodes within 2-bp indels/mismatches to produce a final master barcode list; only barcodes representing a fractional contribution of at least 0.05% in at least 1 sample were considered valid, a conservative threshold taking into account both sequencing and sampling constraints as described in detail.24,25 Barcode retrieval in replicate samples confirmed reproducible barcode retrieval using these parameters24 (supplemental Figure 1). Data analysis, Pearson correlations, P values, and plots were generated using custom R code (Foundation for Statistical Computing, Vienna, Austria) and Prism (GraphPad Software, La Jolla, CA). Python code and associated R functions used for all analyses are freely available at www.github.com/dunbarlabNIH/.
Results
Autologous transplantation of HSPCs in aged rhesus macaques
To track in vivo hematopoiesis at a clonal level in aged vs young rhesus macaques, we used genetic barcoding to clonally tag CD34+ HSPCs prior to myeloablative autologous transplantation.24,25 By targeting the heterogeneous CD34+ population, we could monitor hematopoietic output of individual short- and long-term engrafting cells, without a priori assumptions regarding their phenotype. We transplanted 2 otherwise-healthy macaques 18 years of age (RQ3600) and 25 years of age (RQ859), considered “aged” based on lifespans in captivity of 20 to 30 years. Clonal hematopoietic patterns in these aged macaques were compared with young adult macaques, aged 3 to 5 years at the time of transplantation, that were transplanted with similar cell doses of autologous HSPCs transduced with the same barcoded lentiviral vector libraries (results in young macaques have, in part, been previously reported).24,25 We used libraries of sufficient diversity and controlled transduction efficiency to ensure that each barcode would tag only a single engrafting HSPC and its progeny.24-26 We performed quantitative tracking of the output of individual HSPCs across time and in various lineages after transplantation via high-throughput sequencing, followed by data processing to identify valid barcodes and accurately quantify the fractional contribution of each barcode to a sample (Figure 1A).
Figure 1.

Experimental design and clonal characteristics of hematopoiesis. (A) Schematic diagram of the rhesus macaque autologous transplantation model. CD34+ cells were collected following mobilization and transduced with a lentiviral vector containing a 6 base-pair library ID followed by a highly diverse 27 or 35 base-pair randomly generated barcode library. After total body irradiation, the transduced CD34+ cells were reinfused back into the autologous macaque, and T cells (T), B cells (B), monocytes (Mo), and granulocytes (Gr) were purified after transplantation. Barcode retrieval was performed by polymerase chain reaction of genomic DNA and illumina sequencing. (B) Transplantation characteristics of 2 aged (RQ3600, RQ859) and 3 young (ZH33, ZG66, ZK22) macaques. (C) Percentage of GFP+ cells in hematopoietic lineages across time after transplantation. (D) Cumulative number of independent barcoded clones detected above the threshold at a minimum of 1 time point is shown for each hematopoietic lineage and the total for all lineages across time. Clone numbers were calculated after applying a threshold of a clone achieving a fractional read abundance of at least 0.05% in at least 1 cell type at a minimum of at least 1 time point. The plateaus indicate highly sensitive capture of contributing clones. (E) Number of unique barcoded clones detected at each time point in each hematopoietic lineage and in all lineages. (F) Shannon diversity of each lineage and for all lineages. The Shannon diversity index encompasses both the number of clones and the evenness of their distribution.
Figure 1B summarizes transplantation parameters for all animals. Before transplantation, both aged macaques had normal blood counts, and we collected similar doses of CD34+ cells in aged and young macaques, mobilized with either granulocyte colony-stimulating factor/stem cell factor (G-CSF)/SCF or G-CSF/AMD3100, equivalent mobilizing regimens in our model. Both aged macaques engrafted promptly after autologous transplantation, without significant transplantation-related complications. Granulocytes (Gr) and monocytes (Mo) recovered at similar rates in aged vs young animals, but recovery to normal numbers of circulating T and B cells were delayed in the oldest macaque, RQ859 (supplemental Figure 2).
In vivo marking levels and clonal diversity
Although marking levels in transduced CD34+ cells prior to infusion were successfully targeted at 30% to 35%, in vivo green fluorescent protein (GFP) marking levels in circulating cells varied between macaques (5%-50%), likely because of variation in the degree of endogenous hematopoietic recovery from HSPCs surviving irradiation, and/or the engrafting fitness of the transplanted cells (Figure 1C). There were no clear relationships among animal age, transduction efficiency, or marking levels in vivo. By 2 to 4 months after transplantation, the contribution of transduced cells, measured by GFP expression, stabilized in B-cell and myeloid lineages in both young and aged animals. As previously noted in this ablative rhesus macaque autologous transplantation model, the fraction of T cells originating from transduced HSPCs lagged behind other lineages, likely because of thymic radiation damage and requirement of 5 to 9 months to approach the levels achieved in other lineages, even in young animals.24,25 However, aged macaques had even slower recovery of T cells derived from transduced HSPCs, with the fraction of GFP+ T cells still below other lineages, even at 28 to 32 months, likely reflecting more severe impairment of thymic function in the aged animals.
Thousands of barcodes were retrieved from blood lineages, each representing an individual HSPC clone, with the cumulative number of clones detected as contributing to hematopoiesis generally plateauing by 3 to 6 months in both young and aged macaques (Figure 1D). Aged macaques, particularly RQ859, had lower retrieved clone numbers in T cells compared with other lineages, in contrast to no differences in clone numbers between T cells and other lineages in young macaques. Figure 1E depicts the number of clones contributing at each individual time point. Both aged animals had large drop-offs in the number of clones contributing at the earliest time points through 6 months. We have previously shown that more abundant short-term repopulating clones are replaced by long-term multipotent (MP) clones by 3 to 6 months in young animals.27 These short-term clones appear to be more abundant in the aged animals. Both aged animals continued to show gradual decline in the number of clones detected as contributing at later time points, particularly the oldest animal RQ859. The Shannon index, a measure of clonal diversity that accounts for both overall number and evenness of distribution, was similar in young and aged Gr, Mo, and B cells in the first 6 months after engraftment, but lagged in T cells of both aged macaques, reflecting the slower recovery of T cells originating from the autologous graft (Figure 1F). In contrast to stable clonal diversity in all blood lineages long-term in the young macaques, both aged macaques had a gradual decrease in Shannon diversity in all lineages across time, most marked in the 25-year-old animal RQ859. This loss of diversity may reflect clonal exhaustion with loss of HSPC clones in the aged animals, as documented in Figure 1E, and/or individual clonal expansions, as investigated below.
Delayed emergence of contributions from MP HSPCs in aged macaques
We assessed the quantitative patterns of clonal contributions to the various hematopoietic lineages after engraftment in the young vs aged macaques. As we previously reported in young macaques,24,27 initial engraftment was supported by output primarily from lineage-restricted HPSCs, reflected by low pairwise Pearson correlations between lineages at 1 to 2 months after transplantation in both young and aged animals, as measured at a population level taking into account all clonal contributions (Figure 2A-B). In both young and aged animals, Gr/Mo became closely correlated by 2 months and remained stable across time as we have previously reported for young animals,24,25 other than an unexplained temporary drop in RQ859 at 12 to 14 months. However, correlations lagged between Gr, B-cell, and T-cell lineages in the aged macaques compared with young macaques. These patterns also can be visualized at an individual clonal level using heat maps of top contributing clones, clustering clones with similar characteristics, showing a lag in emergence of clones contributing to more than 1 lineage in the aged animals (Figure 2C). Triangle plots visualize the contribution of each clone to Gr, B cells, and T cells, with the location of the dot corresponding to relative clonal abundance in each of the 3 cell lineages. Figure 2D demonstrates large numbers of highly lineage-skewed clones in aged macaques vs many multilineage-contributing clones in young macaques contributing by 4 to 5 months.
Figure 2.

Short-term clonal contributions and emergence of MP clones. (A) Summary of clonal relationships at a population level as depicted by Pearson correlation coefficients of fractional contributions from all valid barcodes for all samples through 6.5 m. P values were all highly significant based on thousands of clones analyzed. (B) Pearson correlations between lineages plotted across time. Dashed lines represent the aged animals and solid lines, the young animals. (C) Heat maps of contributions from top contributing clones across time and across lineages during initial hematopoietic reconstitution through 6.5 m, ordered by unsupervised hierarchical clustering of the Euclidean distance between barcodes’ log fractional abundances in the samples. Each row in the heat map corresponds to a barcode and each column to a sample. The top 10 most abundant barcodes in at least one of the depicted samples are plotted across time in all samples. Color gradient depicts the log fractional contribution of individual barcodes to each sample. (D) Ternary (triangle) plots of the clonal contributions to T cells, B cells, and Gr in young and aged macaques at 4- to 5-m time points. Each dot within the triangle represents an individual barcode (clone), and the location of the dot within the triangle corresponds to the ratio of barcode abundance in each of the 3 cell lineages. Kernel density of dots within triangle regions is represented by colors and represents the density of dots (barcodes); red depicts high density and green depicts lower density.
Most previous studies have used arbitrary definitions of lineage bias. We plotted the distribution of each clone’s relative contributions to the various hematopoietic lineages in order to better define multilineage vs “biased” HSPC clones and allow quantitative assessment of their relative contributions across time. Given the close correlation between Gr and Mo, we used Gr to represent myeloid output. We developed bias distribution histograms (supplemental Figure 3) by calculating the ratio of each clone’s contribution to 1 lineage vs its contribution to all other lineages at a time point, and assigning the clone to a bias “bin” for that lineage based on this ratio. We identified a ratio of 10-fold contribution to a single lineage vs other lineages as the most distinct cutoff point between clones with distinctly different contributions patterns. Ratios of less than 10-fold fell generally within normal distributions of lineage contributions of the population of HPSC clones. Clones of all sizes fell into each bias bin; thus, lineage bias was a property independent of overall clone size (supplemental Figure 3).
We defined multilineage (MP) clones as those contributing to all 3 lineages at a time point, with less than 10-fold bias toward any single lineage compared with the other 2 lineages. Unilineage-biased clones contributed with 10-fold bias toward 1 lineage compared with both other lineages. Bilineage-biased clones contributed to 2 lineages with less than 10-fold bias toward each other, but more than 10-fold bias toward the third lineage. Indeterminate clones falling outside of these categories made up a relatively small percentage of hematopoiesis in all samples; they were clones falling in between definitions of unilineage-biased and bilineage-biased clones.
We compared the contributions of each clone “type” with total hematopoietic output across time in young vs aged animals. MP clonal contributions to circulating Gr, B, and T cells emerged more slowly in both aged animals (Figure 3A-C). Besides the level of contribution, the number of MP clones was profoundly reduced in the oldest animal, RQ859, with MP contributions derived from a small number of clones (Figure 3D). Contributions from myeloid-biased clones persisted at levels of 12% to 15% in the aged macaques at more than 2 years after transplantation, in comparison with less than 5% in all 3 young macaques at longest follow-up (Figure 3A). B-biased clones continued to contribute substantially to B-cell production in aged compared with young macaques, comprising more than 25% of B-cell output in RQ859, with corresponding lower contributions from MP clones to B cells in the aged animals (Figure 3B). Both young and aged animals retained output from bilineage B/myeloid-biased clones and, to a lesser extent, B/T-biased clones long-term, with no clear relationship to age in terms of the level of contribution (Figure 3A-C). A relationship between the relative contributions of clone types to T cells and age was less clear. Homotypic peripheral expansion of long-lived T cells complicates interpretation of clonal composition of the circulating T-cell lineage, compared with much shorter-lived blood myeloid and B cells. The aged animal RQ859 had a very large persistent contribution from T-biased clones through 9 months, but at later time points, there was no clear segregation of patterns between the young and aged animals.
Figure 3.

Contributions of MP vs biased clones to hematopoiesis. The relative contributions of all MP, myeloid-biased (Gr), B-biased, T-biased, Gr/B-biased, Gr/T-biased, B/T-biased, and indeterminate clones (falling between unilineage and bilineage biased) across time after transplantation to Gr (panel A), B cells (panel B), and T cells (panel C). Each barcoded clone was categorized, and its fractional contribution to the lineage was summed to create the stacked bars. MP: ≤10-fold ratio of contributions between Gr, T, and B lineages (black); myeloid (Gr)-biased: >10-fold ratio of Gr vs T and B contributions (yellow); B-biased: >10-fold ratio of B vs Gr and T contributions (blue); T-biased: >10-fold ratio of B vs Gr and T contributions (red); T/myeloid-biased: T and Gr both >10-fold biased vs B and ≤10-fold biased toward each other (orange); B/myeloid-biased: B and Gr both >10-fold biased vs T and ≤10-fold biased toward each other (green); T/B-biased: T and B both >10-fold biased vs Gr and ≤ 10-fold biased toward each other (purple); indeterminate: falling in between unilineage and bilineage definitions (gray). (D) Number of MP clones detected as contributing at each time point for young (solid lines) and aged (dotted lines) animals.
In summary, aged macaques showed a distinct pattern of clonal HSPC output compared with young macaques, with persistence of major contributions from both myeloid and B-lineage–biased clones even years after transplantation, and slower recovery of contributions from MP clones.
Long-term tracking of individual clonal stability
To investigate the behavior of individual clones in aged vs young macaques, we plotted heat maps of contributions from the largest contributing clones in each lineage and at each time point from 6 months through 28 to 32 months for both aged macaques and the 2 young macaques with equivalent length of follow-up (Figure 4). The persistence of myeloid-biased and B-biased clones in aged vs young animals can be appreciated, with a range of clone sizes. These longitudinal heat maps also demonstrated very stable output from individual MP clones across time in the young animals, with relatively homogeneous clone sizes, in contrast to more heterogeneous clone sizes and much less stable contribution patterns in the aged animals from both MP and biased HSPC types, with evidence for clonal expansions occurring in all lineages. Correlation plots quantitating the degree of overall clonal stability across time are provided in supplemental Figure 4.
Figure 4.

Long-term tracking of clones in aged vs young animal populations. Heat maps showing the log fractional contributions of individual top clones to each lineage for time points between 6 months and longest follow-up for the 2 aged macaques and for the young animals with follow-up >2 years. Barcodes that are the top 10 most abundant barcodes in at least one of the depicted samples are plotted over all samples. Heat maps are ordered by unsupervised hierarchical clustering of the Euclidean distance between barcodes’ log fractional abundances in the samples. Each row in the heat map corresponds to a barcode and each column to a sample. Color gradient depicts the log fractional contribution of individual barcodes to each sample. Colored bars on the left of each heat map reflect the clone bias “type” of each clustered group of clones as defined in the text and in Figure 3. MP: black; myeloid (Gr)-biased: yellow; B-biased: blue; T-biased: red; T/B-biased: purple; T/myeloid-biased: orange; B/myeloid-biased: green; indeterminate: gray; short-term: white. TP: transplantation.
To elucidate clonal expansions, we focused on the 10 highest contributing clones in each lineage at the latest time point for each animal and mapped their contributions across time to that lineage (Figure 5). By 6 to 12 months in both young animals, the largest clones contributing to Gr, B, or T cells were all MP type and remained stable in contribution levels across time through the latest time points, without evidence of expansion after clonal stability was reached by 6 months. Even these “large” individual clones contributed at levels of no more than 5% and, generally, only 1% to 2%. In contrast, a subset of much larger and continuously expanding clones existed in the aged animals. Both myeloid-biased and MP-expanding clones were contributed to Gr, and both MP and B-biased–expanding clones contributed to B cells in the aged animals, with some individual clones reaching contribution levels of greater than 10% to 15%. No clones of equivalent sizes arose or expanded in the young animals. Similar clonal expansions originating from MP clones were also observed in T cells from the aged animals. The combination of these pronounced clonal expansions and loss of total number of contributing clones across time (Figure 1E) explain the decline in Shannon diversity observed in the aged macaques (Figure 1F).
Figure 5.

Clonal expansion across time in aged vs young macaques. (A-C) Clonal contributions from individual clones were plotted for Gr (A), B cells (B), and T cells (C) across time. Each line represents a clone, and the y-axis indicates the percent clonal contribution. The 10 most abundant clones in each lineage at the latest time point were tracked across time. Line colors reflect clone type definitions and match those in Figure 3. MP: black lines; myeloid (Gr)-biased: yellow lines; B-biased: blue lines; T-biased: red lines; T/B-biased: purple lines; T/myeloid-biased: orange lines; B/myeloid-biased: green lines; indeterminate: gray lines.
Discussion
HSPCs are rare cells residing in the marrow niche and are responsible for maintenance of hematopoietic output throughout life. The defining self-renewal abilities of at least a subset of HSPCs were initially thought exempt from aging. However, numerous lines of evidence have suggested that age-dependent perturbations result in the decline of regenerative capacity, an increased risk of clonal transformation, and deficits in responding to infectious challenges and recovery after chemotherapy or transplantation.4,28 Despite an increase in the frequency of phenotypically defined HSCs in aged mice, homing and the functional frequency of engrafting HSCs are significantly reduced.29
One of the most prominent changes in hematopoietic aging as studied in mice is preferential production of myeloid vs lymphoid cells from aged HSPCs.30-33 A clonal composition shift model has been proposed, based on mouse models suggesting that intrinsically myeloid-biased HSPCs accumulate with age at the expense of lymphoid-biased and balanced HSPCs, and that this program is intrinsically maintained by individual HSPCs across time or after serial transplantation, likely through epigenetic reprogramming associated with aging.32,34-37
The frequency of phenotypically defined human HSCs also increases with aging; however, calculation of the absolute number of HSCs is difficult given the decreased and heterogeneous marrow cellularity of aged humans and lack of quantitative, physiologic in vivo assays.21 Transplantation of aged HSPCs into immunodeficient mice demonstrated loss of B-cell lineage compared with myeloid engraftment, but no clonal tracking has been performed on aged human HSPCs; thus, lack of information exists on whether the intrinsic myeloid bias of murine aged HSPCs is also present in individual human aged HSPCs.21,38
The rhesus macaque is a robust model to study clinically relevant aspects of human hematopoietic aging due to its similar physiology, lifespan, inferred HSPC dynamics, and aging phenotype.39,40 Given the average lifespan of ∼27 years in captivity, the aged rhesus macaques used in our study corresponded to humans of 60 to 80 years of age.23,41 With similar doses of transplanted barcoded CD34+ HSPCs per kilogram given to the young and aged animals, there was no difference in time to recovery of blood counts or other complications of transplantation, but there were important differences in lineage reconstitution patterns and clonality. We observed delayed emergence of contributions from MP clones and more prolonged and higher-level persistence of clones highly biased toward either B-cell or myeloid lineages in the aged animals. Both aged animals showed a gradual decrease in numbers of contributing clones across time, suggesting a degree of clonal exhaustion, in contrast to stable clone numbers contributing long-term in the young animals. In the oldest animal, there was a marked decrease in the number of MP clones at all time points. Our data are most consistent with a model of decreased numbers and/or slower cycling of long-term MP repopulating HSPCs, resulting in a significant delay in emergence of hematopoiesis from these clones in aged macaques. Our recent study used barcoding of murine HSCs to study the impact of HSC dose on the recovery of multilineage vs lineage-biased hematopoiesis. Low HSC doses generated engraftment with more biased clones, whereas high HSC doses resulted in a greater proportion of balanced clones, suggesting that transplantation dose determines HSC differentiation dynamics, not solely intrinsic HSPC features.42
A recent study suggested that murine HSCs have the ability to count the number of symmetric divisions and are limited to a maximum of 4 such divisions.43 Once HSCs from aged mice passed this limit, they lost the capacity for self-renewal and robust hematopoietic contributions. The loss of clonal diversity and MP clone numbers and function across time perhaps reflects a running out of the “HSC clock” and/or impaired function of remaining aged HSPCs not yet at their cycling limit. These observations in diverse murine experimental systems all lend support to our model attributing persistent lineage bias to loss or slowing of contributions from MP HSPCs with aging.
Recent population-based genetic analyses of blood cells from large cohorts of aged humans have revealed a surprising incidence of oligoclonal or even clonal hematopoiesis, including evidence for specific clonal expansion of cells derived from progenitors with mutations in genes known to be associated with myelodysplastic syndromes or acute myeloid leukemia, such as DNMT3A and TET2.6 Studies to date have sequenced whole peripheral blood; thus, it is unknown whether the clonal expansions are MP or lineage restricted, but it is interesting to note that later malignancies were both lymphoid and myeloid in nature.44 In our macaques, the clones that began to expand late after engraftment included MP, B-biased, and myeloid-biased clones. In the case of MP-expanding clones, perhaps these clones were the few that retained some proliferative potential, and they began to Alternatively, they may have acquired a mutation conferring fitness. The appearance of expanding clones and loss of clonal diversity in the aged animals are then not surprising. Our recapitulation of loss of clonal diversity across time in aged macaques should provide a relevant model for studying the characteristics of clonal expansions related to human HSPC aging.
Although our data successfully demonstrated the impact of aging on the dynamics of HSPC behavior at a clonal level, a number of caveats and additional questions are raised. The constraints of primate studies, including cost, complexity, and lack of availability of aged animals, limit experiments to small numbers of animals; therefore, it is possible that some findings reflect individual differences not related to age. However, the tracking methodology used allowed analysis of many thousands of individual HSPC clones, even in this small number of animals, increasing the likelihood that the findings reflect general properties of aged vs young HSPC. Because of the lack of availability of pharmaceutical grade SCF, one of the young animals and both aged animals were mobilized with G-CSF/AMD3100, in contrast to a G-CSF/SCF mobilization regimen used in the 2 young animals transplanted previously. In our experience to date, cells collected with these 2 regimens behave identically in vivo, but this change in mobilization regimen could have had some impact on our findings. The autologous transplantation design precludes distinguishing between intrinsic vs microenvironmental extrinsic factors associated with hematopoietic aging, although murine studies have generally found cell-intrinsic factors to be dominant.32,34-37 Allogeneic HSPC transplantation in rhesus macaques is feasible, although identification of aged vs young matched or haploidentical donors and recipients would be very challenging, and very few aged, specific pathogen-free aged macaques exist that are suitable for transplantation. Our finding may be in part specific to the transplantation setting, where changes in homing and response to marked replicative stress by aged HSPCs may not be relevant to steady-state hematopoiesis. Given the appearance of clonal expansions in the aged macaques, whole-exome sequencing to analyze the acquisition of clonally restricted somatic mutations such as DNMT3A would be of interest. Our current data already provide insight into the impact of aging on hematopoiesis, which is relevant for designing transplantation and other therapies in elderly patients.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Janis L. Abkowitz for constructive discussion; the DNA Sequencing and Genomics Core, Flow Cytometry Core, and Primate Facility of the National Heart, Lung, and Blood Institute; and Keyvan Keyvanfar, Stephanie Sellers, and Lemlem Alemu for technical assistance.
This work was supported by the Korean Visiting Scientist Training Award (KVSTA) (K.-R.Y.) and the Intramural Research Program of the National Institutes of Health, National Heart, Lung, and Blood Institute.
Footnotes
Presented in abstract form at the 57th annual meeting of the American Society of Hematology, Orlando, FL, 5-8 December 2015.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: K.-R.Y., C.W., and C.E.D. performed conceptualization of this work; R.L. provided resources; D.A.E., S.K., L.T., and R.L. provided software; D.A.E., S.K., and L.T. conducted formal analysis; D.A.E. and L.T. performed visualization; K.-R.Y., C.W., T.-H.S., X.F., I.M.Y., S.P., S.C., S.G.H., A.B., A.K., and M.M. conducted the investigations; C.E.D. and R.E.D. provided supervision; and K.-R.Y. and C.E.D. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Cynthia E. Dunbar, Molecular Hematopoiesis Section, Hematology Branch, Building 10 CRC Room 4E-5132, National Heart, Lung and Blood Institute, National Institutes of Health, 9000 Rockville Pike, Bethesda, MD 20892; e-mail: dunbarc@nhlbi.nih.gov.
REFERENCES
- 1.Gazit R, Weissman IL, Rossi DJ. Hematopoietic stem cells and the aging hematopoietic system. Semin Hematol. 2008;45(4):218-224. [DOI] [PubMed] [Google Scholar]
- 2.Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol. 2013;13(5):376-389. [DOI] [PubMed] [Google Scholar]
- 3.Geiger H, Denkinger M, Schirmbeck R. Hematopoietic stem cell aging. Curr Opin Immunol. 2014;29:86-92. [DOI] [PubMed] [Google Scholar]
- 4.Kollman C, Howe CW, Anasetti C, et al. Donor characteristics as risk factors in recipients after transplantation of bone marrow from unrelated donors: the effect of donor age. Blood. 2001;98(7):2043-2051. [DOI] [PubMed] [Google Scholar]
- 5.Woolthuis CM, Mariani N, Verkaik-Schakel RN, et al. Aging impairs long-term hematopoietic regeneration after autologous stem cell transplantation. Biol Blood Marrow Transplant. 2014;20(6):865-871. [DOI] [PubMed] [Google Scholar]
- 6.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao JL, Ma C, O’Connell RM, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell. 2014;14(4):445-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrison SJ, Wandycz AM, Hemmati HD, Wright DE, Weissman IL. Identification of a lineage of multipotent hematopoietic progenitors. Development. 1997;124(10):1929-1939. [DOI] [PubMed] [Google Scholar]
- 9.Guenechea G, Gan OI, Dorrell C, Dick JE. Distinct classes of human stem cells that differ in proliferative and self-renewal potential. Nat Immunol. 2001;2(1):75-82. [DOI] [PubMed] [Google Scholar]
- 10.Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med. 2010;2(6):640-653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu R, Neff NF, Quake SR, Weissman IL. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat Biotechnol. 2011;29(10):928-933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szilvassy SJ, Humphries RK, Lansdorp PM, Eaves AC, Eaves CJ. Quantitative assay for totipotent reconstituting hematopoietic stem cells by a competitive repopulation strategy. Proc Natl Acad Sci USA. 1990;87(22):8736-8740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Purton LE, Scadden DT. Limiting factors in murine hematopoietic stem cell assays. Cell Stem Cell. 2007;1(3):263-270. [DOI] [PubMed] [Google Scholar]
- 14.Dick JE, Magli MC, Huszar D, Phillips RA, Bernstein A. Introduction of a selectable gene into primitive stem cells capable of long-term reconstitution of the hemopoietic system of W/Wv mice. Cell. 1985;42(1):71-79. [DOI] [PubMed] [Google Scholar]
- 15.Jordan CT, Lemischka IR. Clonal and systemic analysis of long-term hematopoiesis in the mouse. Genes Dev. 1990;4(2):220-232. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt M, Zickler P, Hoffmann G, et al. Polyclonal long-term repopulating stem cell clones in a primate model. Blood. 2002;100(8):2737-2743. [DOI] [PubMed] [Google Scholar]
- 17.Berry CC, Ocwieja KE, Malani N, Bushman FD. Comparing DNA integration site clusters with scan statistics. Bioinformatics. 2014;30(11):1493-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bushman F, Lewinski M, Ciuffi A, et al. Genome-wide analysis of retroviral DNA integration. Nat Rev Microbiol. 2005;3(11):848-858. [DOI] [PubMed] [Google Scholar]
- 19.Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10(2):120-136. [DOI] [PubMed] [Google Scholar]
- 20.Kuranda K, Vargaftig J, de la Rochere P, et al. Age-related changes in human hematopoietic stem/progenitor cells. Aging Cell. 2011;10(3):542-546. [DOI] [PubMed] [Google Scholar]
- 21.Pang WW, Price EA, Sahoo D, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci USA. 2011;108(50):20012-20017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larochelle A, Dunbar CE. Hematopoietic stem cell gene therapy: assessing the relevance of preclinical models. Semin Hematol. 2013;50(2):101-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roth GS, Mattison JA, Ottinger MA, Chachich ME, Lane MA, Ingram DK. Aging in rhesus monkeys: relevance to human health interventions. Science. 2004;305(5689):1423-1426. [DOI] [PubMed] [Google Scholar]
- 24.Wu C, Li B, Lu R, et al. Clonal tracking of rhesus macaque hematopoiesis highlights a distinct lineage origin for natural killer cells. Cell Stem Cell. 2014;14(4):486-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koelle SJ, Espinoza DA, Wu C, et al. Quantitative stability of hematopoietic stem and progenitor cell clonal output in rhesus macaques receiving transplants. Blood. 2017;129(11):1448-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verovskaya E, Broekhuis MJ, Zwart E, et al. Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood. 2013;122(4):523-532. [DOI] [PubMed] [Google Scholar]
- 27.Koelle SJ, Espinoza DA, Wu C, et al. Quantitative stability of hematopoietic stem and progenitor cell clonal output in rhesus macaques receiving transplants. Blood. 2017;129(11):1448-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geiger H, Van Zant G. The aging of lympho-hematopoietic stem cells. Nat Immunol. 2002;3(4):329-333. [DOI] [PubMed] [Google Scholar]
- 29.Florian MC, Nattamai KJ, Dörr K, et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature. 2013;503(7476):392-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med. 2000;192(9):1273-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rossi DJ, Bryder D, Zahn JM, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci USA. 2005;102(26):9194-9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho RH, Sieburg HB, Muller-Sieburg CE. A new mechanism for the aging of hematopoietic stem cells: aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood. 2008;111(12):5553-5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beerman I, Bhattacharya D, Zandi S, et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci USA. 2010;107(12):5465-5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dykstra B, Kent D, Bowie M, et al. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell. 2007;1(2):218-229. [DOI] [PubMed] [Google Scholar]
- 35.Benz C, Copley MR, Kent DG, et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell. 2012;10(3):273-283. [DOI] [PubMed] [Google Scholar]
- 36.Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med. 2011;208(13):2691-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wahlestedt M, Erlandsson E, Kristiansen T, et al. Clonal reversal of ageing-associated stem cell lineage bias via a pluripotent intermediate. Nat Commun. 2017;8:14533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pang WW, Schrier SL, Weissman IL. Age-associated changes in human hematopoietic stem cells. Semin Hematol. 2017;54(1):39-42. [DOI] [PubMed] [Google Scholar]
- 39.Shepherd BE, Kiem HP, Lansdorp PM, et al. Hematopoietic stem-cell behavior in nonhuman primates. Blood. 2007;110(6):1806-1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Catlin SN, Busque L, Gale RE, Guttorp P, Abkowitz JL. The replication rate of human hematopoietic stem cells in vivo. Blood. 2011;117(17):4460-4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Didier ES, Sugimoto C, Bowers LC, Khan IA, Kuroda MJ. Immune correlates of aging in outdoor-housed captive rhesus macaques (Macaca mulatta). Immun Ageing. 2012;9(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brewer C, Chu E, Chin M, Lu R. Transplantation dose alters the differentiation program of hematopoietic stem cells. Cell Reports. 2016;15(8):1848-1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bernitz JM, Kim HS, MacArthur B, Sieburg H, Moore K. Hematopoietic stem cells count and remember self-renewal divisions. Cell. 2016;167(5):1296.e10-1309.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.