

Abstract
Expression of transposable elements in the germline is controlled by Piwi-interacting (pi) RNAs produced by genomic loci termed piRNA clusters and associated with Rhino, a heterochromatin protein 1 (HP1) homolog. Previously, we have shown that transgenes containing a fragment of the I retrotransposon form de novo piRNA clusters in the Drosophila germline providing suppression of I-element activity. We noted that identical transgenes located in different genomic sites vary considerably in piRNA production and classified them as “strong” and “weak” piRNA clusters. Here, we investigated what chromatin and transcriptional changes occur at the transgene insertion sites after their conversion into piRNA clusters. We found that the formation of a transgenic piRNA cluster is accompanied by activation of transcription from both genomic strands that likely initiates at multiple random sites. The chromatin of all transgene-associated piRNA clusters contain high levels of trimethylated lysine 9 of histone H3 (H3K9me3) and HP1a, whereas Rhino binding is considerably higher at the strong clusters. None of these chromatin marks was revealed at the “empty” sites before transgene insertion. Finally, we have shown that in the nucleus of polyploid nurse cells, the formation of a piRNA cluster at a given transgenic genomic copy works according to an “all-or-nothing” model: either there is high Rhino enrichment or there is no association with Rhino at all. As a result, genomic copies of a weak piRNA transgenic cluster show a mosaic association with Rhino foci, while the majority of strong transgene copies associate with Rhino and are hence involved in piRNA production.
Keywords: Drosophila, retrotransposon, transgene, germline, piRNA, HP1, Rhino, nascent RNA
INTRODUCTION
Small RNAs are central elements in a nucleic acid-based immune system that targets transposable elements (TEs). In the animal germline, the major pool of TE-specific piRNAs (Piwi-interacting RNA) is generated by distinct genomic regions termed piRNA clusters (Aravin et al. 2006; Brennecke et al. 2007). Both genomic strands of endogenous dual-strand piRNA clusters in Drosophila are transcribed to produce piRNA precursors. The assembly of a specialized protein complex is essential for transcription of piRNA clusters as well as for the channeled transport of its transcripts to the particular cytoplasmic compartment, called nuage, where they are processed into small RNAs (Mohn et al. 2014; Zhang et al. 2014). Rhino (Rhi) protein, which belongs to heterochromatin protein 1 (HP1) family and is expressed in the germline (Volpe et al. 2001), is considered as a key factor of piRNA cluster definition. In the nuclei of nurse cells, piRNA-producing loci are colocalized with Rhi foci (Mohn et al. 2014), which, in turn, interact with the TREX complex containing UAP56 RNA helicase involved in transcription of piRNA precursors and their nuclear export (Zhang et al. 2012; Hur et al. 2016). As a result, piRNA precursors are cotranscriptionally recognized and directed to the perinuclear piRNA processing machinery. One of the essential questions in the field is what conditions are necessary to form de novo piRNA clusters?
In Drosophila ovarian somatic cells (OSC), reporter constructs bearing piRNA targets serve as a powerful tool for understanding requirements for efficient recognition of piRNA targets (Post et al. 2014). Determinants of piRNA production have thus been identified for uni-strand piRNA clusters specific to OSCs (Homolka et al. 2015; Ishizu et al. 2015). In both fly and mouse germline, transgenes inserted into dual-strand piRNA clusters have shown that the prerequisites for piRNA production lay within piRNA clusters themselves (Josse et al. 2007; Muerdter et al. 2012; Le Thomas et al. 2013). However, in the germline, such studies have been rendered difficult because endogenous piRNA clusters are typically very large and enriched for repeats, furthermore, no Drosophila germline culture cell line has been established. Therefore, the discovery of transgene-associated dual-strand piRNA clusters significantly simplified piRNA cluster research in the Drosophila germline. The phenomenon of paramutation, consisting of a stable production of small RNAs from a transgenic locus targeted by homologous small RNAs and accompanied by chromatin reorganization, provides one of the transgenic models of de novo piRNA cluster establishment in the germline (de Vanssay et al. 2012). In this transgenic system, maternally inherited piRNAs induce piRNA production and installment of the trimethylated lysine 9 of histone H3 (H3K9me3) mark on the homologous locus, and H3K9me3 is considered as a determinant of the piRNA production (Le Thomas et al. 2014).
By studying transgene-mediated cosuppression of I-element activity (Jensen et al. 1999), we have shown that transgenes, containing a 2.3 kb fragment of the LINE-like retrotransposon I-element (designated as I-TG) and inserted in different euchromatic positions, form piRNA clusters in the Drosophila germline (Olovnikov et al. 2013). piRNAs originating from the endogenous piRNA clusters and complementary to the I-element fragment likely drive this transformation leading to the formation of secondary transgene-associated piRNA clusters. We have shown that these transgenic piRNA clusters generate small RNAs from all transgenic fragments as well as from adjacent genomic regions. In addition, we found that the same I-TG transgenes, located at different genomic loci, are not equivalent in their ability to produce piRNAs; they can be classified as “strong” and “weak” transgenic piRNA clusters according to the amount of transgene-mapped piRNAs.
This transgenic model provides a unique opportunity for investigating the role of the genomic context, allowing conversion of euchromatic loci complementary to endogenous piRNAs into de novo piRNA clusters. In this study, we address a broad range of questions surrounding the molecular mechanisms of Drosophila piRNA cluster establishment. The transcriptional status and chromatin structure of the transgene appear to be key factors in this process. At the same time, we show here that piRNA-mediated transcriptional silencing accompanied by HP1 and H3K9me3 deposition at the target transgenes is not sensitive to the genomic position. Using DNA fluorescence in situ hybridization (FISH) combined with Rhi immunostaining, we show that the transformation of a transgene that is a piRNA target into a piRNA cluster in polyploid nurse cell nuclei is a stochastic process determined by an “all-or-nothing” model. Thus, the majority of “strong” transgene genomic copies are involved in piRNA production, while for weak transgenes, the portion of transgenic copies transformed into piRNA clusters is much lower. These observations reveal a bimodal fashion of de novo piRNA cluster formation and an essential role of genomic context in successful realization of this process.
RESULTS
Transgenic piRNA clusters identical in structure but located within different genomic sites can be classified as strong or weak
According to our previous results, only 20% of natural TE insertions in euchromatin lead to the formation of secondary piRNA clusters (Shpiz et al. 2014b). Similarly, transgenes containing a transcribed I-element fragment (I-TG constructs) inserted into different genomic loci vary greatly in their ability to produce small RNAs (Olovnikov et al. 2013). In order to address the nature of such differences, we have chosen some of the previously characterized I-TG transgenes that differ in their ability to produce piRNAs (Olovnikov et al. 2013). The level of transgenic piRNA production was independent of the sense or antisense orientation of the I-element fragment in the transgene (Olovnikov et al. 2013).
According to the level and profile of piRNA production along the transgenic constructs, transgene-associated piRNA clusters may be defined as weak and strong (Table 1). In addition, we sequenced small RNAs from the ovaries of a transgenic strain 2.4, which is capable of strong I-element suppression (Jensen et al. 1999). In this strain, the I-TG transgene, being inserted in an intergenic euchromatic region, which normally does not produce small RNAs, becomes a strong dual-strand piRNA cluster (Table 1; Supplemental Fig. S1), which explains I-element suppression activity of this transgenic strain. Euchromatic transgenes 2.1 and 2.4 produce abundant piRNAs similar to 3.1, which is located within an endogenous piRNA cluster at telomere associated sequences (TAS), and these three transgenes can be defined as strong piRNA clusters. In contrast, the 1.9 and 3.6 transgenes form weak piRNA clusters. The most prominent difference between strong and weak transgenic clusters is the number of transgenic piRNAs homologous to the mini-white gene. Indeed, production of piRNAs spreads beyond the I-element fragment, which is the direct target of endogenous piRNAs within the transgene. Thus, mini-white-specific piRNAs serve as an indicator of de novo production of transgenic piRNAs. Furthermore, accumulation of I-specific piRNAs appears to be mainly driven by the so-called ping-pong piRNA amplification mechanism (Brennecke et al. 2007; Gunawardane et al. 2007) between endogenous I-specific piRNAs and transgenic I-element transcripts. Previously, we have shown that the characteristic nucleotide bias of secondary piRNAs (10A-bias) is observed only for small RNAs specific to the I-element fragment but not for other transgenic piRNAs, suggesting that transgenic mini-white primary piRNAs are not involved in the ping-pong amplification loop (Olovnikov et al. 2013). Indeed, we do not observe considerable variation in the transgenic I-element fragment piRNA content between weak and strong transgenes. At the same time, the amount of piRNAs mapped to mini-white is 10- to 30-fold lower in weak strains and almost absent at antisense strand (Table 1). Thus, despite the identical amount of endogenous I-element piRNAs in all transgenic strains, not all transgenes are able to produce the same levels of novel piRNAs.
TABLE 1.
Characteristics of weak and strong transgenic piRNA clusters: small RNA production and chromatin state

Our study addresses the question of why these transgenes, which are identical in their structure, are so different in their ability to form de novo piRNA clusters.
I-element containing transgenes differ in their chromatin structure
In order to address the role of chromatin in the formation of strong and weak piRNA clusters, we analyzed the association of transgene insertion sites with HP1a, H3K9me3, and Rhi, before and after the integration of the I-TG transgene. It is firmly established that any sequence inserted into endogenous piRNA clusters starts to produce piRNAs and acquires piRNA-cluster-specific chromatin marks (Muerdter et al. 2012; Le Thomas et al. 2014). Transgene 3.1, which is integrated in 3R TAS, produces abundant piRNAs from both genomic strands and, as expected, is enriched for the chromatin modification H3K9me3, HP1a, and Rhi proteins (Fig. 1A).
FIGURE 1.

Chromatin status of the transgene-associated piRNA clusters. (A) H3K9me3, HP1a, and Rhi ChIP-qPCR on ovaries of transgenic strains. The normalized amounts of precipitated 5′P-element transgenic regions are shown for transgenic strains. “Empty” sites are analyzed by primers corresponding to insertion sites (see Supplemental Table 1). (B) Distribution of chromatin marks and piRNA profile along 2.4 transgene and its flanking regions. The structure of the genomic region of transgene insertion in strain 2.4 is diagrammed. Position and orientation of transgene is shown. Plots of unique small RNAs density, in a 30 bp window, for genomic plus (blue) and minus (brown) strand, in transgenic strains 2.4 and R-strain wK (genomic positions according to dm3 assembly are indicated). Read numbers were normalized to sequencing depths of libraries (RPM, reads per million). Length distribution of small RNAs mapping to corresponding flanking genomic regions is shown to the right. Genomic regions analyzed by ChIP-qPCR are indicated from a to e. H3K9me3, HP1a, and Rhi ChIP-qPCR were done on ovaries of 2.4 strain using primers corresponding to the indicated regions. Error bars represent SEM of at least three biological replicates.
We therefore asked whether the euchromatic genomic regions before the insertion of I-TG transgenes were associated with some of these chromatin components. ChIP assays were performed on chromatin of lines carrying or not carrying the transgene. 5′P primers corresponding to the P-element sequence were used to test chromatin protein binding to the corresponding transgene. No H3K9me3, HP1a, or Rhi enrichment was observed at the “empty” sites for weak and strong transgenic strains (Fig. 1A). These heterochromatic hallmarks are revealed only after transgene insertion. The 67.2.1 promoter-less I-TG transgene that does not produce small RNAs (Olovnikov et al. 2013) was used as a control. It was devoid of these chromatin components. Of note, HP1a and H3K9me3 enrichment is similar at strong and weak transgenes, indicating that all these transgenes acquire a repressive chromatin state guided by piRNAs (Fig. 1A). In contrast, Rhi enrichment varies depending on the line; where it is higher at 2.1 and 2.4 strong transgenic piRNA clusters than at 1.9 and 3.6 weak clusters. These data show that the efficiency of piRNA production by transgene-associated clusters is in relationship with the level of Rhi enrichment but is not correlated with the presence of HP1a and H3K9me3.
Thus, H3K9me3, HP1a, and Rhi are not observed at the “empty” sites of euchromatic transgene insertions but accompany the formation of transgenic double-stranded piRNA clusters when the transgenes are inserted.
Next, we asked how H3K9me3, HP1a, and Rhi are distributed along the transgenes and whether these chromatin components spread into the surrounding genomic areas. We chose to analyze the 2.4 transgene, which is a strong dual-strand cluster (Table 1). Gradually decreasing peaks of piRNAs mapped to one genomic strand are revealed in transgene flanking regions (Fig. 1B). High levels of Rhi, HP1, and H3K9me3 are observed at different regions of the transgene itself (Fig. 1B), which is in accordance with the high level of transgenic piRNAs. The transgene flanking region adjacent to the 3′P-element arm is also enriched by Rhi, HP1, and H3K9me3, indicating the spreading of silencing chromatin from the transgene (Fig. 1B). Similar nonsymmetric distribution of piRNAs was observed in the TE flanking regions (Shpiz et al. 2014b). In the genomic flank adjacent to the 3′ end of 2.4, Rhi binds the genomic region, in which piRNAs are mapped to only one genomic strand, but this Rhi binding is not sufficient to induce formation of double-strand piRNA cluster.
Activation of convergent transcription accompanies transgenic piRNA cluster formation
We have previously shown that I-transgene-associated small RNA production was extended into the flanking regions that did not produce small RNAs before the insertion (Olovnikov et al. 2013). Comparing the transcriptional status of the locus in the presence or absence of a transgene provides a unique opportunity to understand which transcriptional changes have occurred during de novo piRNA cluster formation. For such comparison, global run-on sequencing (GRO-seq) allowing measurement of nascent RNA synthesis was performed on ovaries from both weak and strong transgenic strains. Two series of independent GRO-seq experiments were performed, comparing 1.9 and 3.1 strains and 1.9 and 2.4 strains, respectively. 2.4 and 3.1 transgenes are related to strong piRNA clusters. In strain 1.9, the I-TG transgene is inserted in the region immediately upstream of the cwo gene on chromosome 3R. Despite the fact that 1.9 produces a low level of piRNAs, its insertion causes transformation of the flanking region into a dual-strand mixed si/piRNA cluster, which provides an opportunity to compare the transcription state of a unique genomic site before and after cluster formation.
First, we compared the distribution of GRO-seq reads at the genomic regions flanking transgene 1.9 with the same genomic regions in the 3.1 and 2.4 strains, which do not contain a transgene in this site (“empty” site) (Fig. 2A,B). We observed no significant changes in the number of nascent transcripts of the cwo gene, which is located downstream from the 1.9 transgene (Fig. 2A; Supplemental Fig. S2A). In the upstream flanking region, which contains two 3.5 kb tandem repeats, transcription of both genomic strands significantly increased after the 1.9 transgene insertion in both GRO-seq experiments (Fig. 2A,B). Transcriptional up-regulation is accompanied by the production of pi- and siRNAs, indicating that transcripts generated from this region serve as small RNA precursors (Fig. 2C). Well-defined GRO-seq peaks, generally observed at the transcription start sites (TSS) of coding genes including cwo, are not revealed in the 1.9 upstream small RNA-producing region (Fig. 2A). This observation is most likely due to the initiation of transcription at multiple random sites.
FIGURE 2.
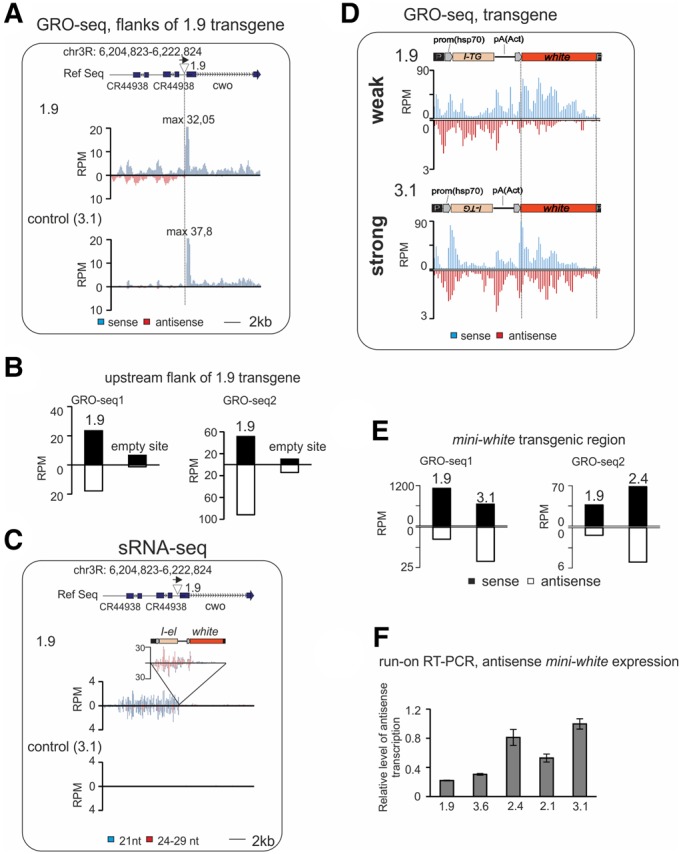
Transcriptional changes accompany piRNA cluster formation. Analysis of GRO-seq made on ovaries of transgenic strains. (A) Normalized GRO-seq densities mapping to the 1.9 transgene insertion site (indicated by triangle above the plots) in control (3.1) and in 1.9 strains (no mismatches allowed). Schema of genomic region is shown above; genome coordinates are given according to Drosophila R5 release. (B) GRO-seq read counts at the 9 kb region upstream of the 1.9 insertion site in 1.9 and 3.1 (GRO-seq1) and in 1.9 and 2.4 (GRO-seq2) strains. (C) Mapping of normalized small RNA reads (no mismatches allowed) to the 1.9 transgene insertion region in control (3.1) and in 1.9 strains. Sense and antisense reads are shown above or under the x-axis, respectively. Bars corresponding to endo-si RNA (21 nt) and piRNA (24–29 nt) fractions are colored in blue and red, respectively. (D) Mapping of normalized (RPM) GRO-seq reads to the transgene calculated for 100 bp window size in strong (3.1) and in weak 1.9 strains (no mismatches allowed). Schemes of transgenes are shown above. (E) GRO-seq read counts at mini-white transgene region calculated for 1.9 and 3.1 (GRO-seq1) and 1.9 and 2.4 (GRO-seq2) strains. (F) The level of nascent mini-white antisense RNA is higher in ovaries of strong transgenic strains. Strand-specific RT-qPCR was done on ovarian run-on RNAs from indicated strains. Relative levels of normalized values are shown. Error bars represent SEM of three technical replicates.
We then compared the number of GRO-seq reads mapping to the transgenic constructs in strains 1.9, 2.4, and 3.1. In all strains, the amount of transgenic sense GRO-seq reads was much higher than antisense ones (Fig. 2D; Supplemental Fig. S2B). This difference is observed for all transgene-mapped reads and also for single-mapped transgenic reads (Supplemental Fig. S2C). The most noticeable difference between 1.9 and 3.1 or between 1.9 and 2.4 GRO-seq profiles concerns the amounts of antisense mini-white reads. In contrast to the strong 3.1 and 2.4 transgenes, 1.9 produces a negligible amount of antisense mini-white nascent RNAs (Fig. 2D,E; Supplemental Fig. S2B). This is in agreement with the lack of mini-white piRNAs in the 1.9 strain (Table 1). To better support these observations, we have done strand-specific RT-qPCR using mini-white primers on ovarian run-on RNAs from 1.9, 3.6, 2.1, 2.4, and 3.1 strains (Fig. 2F) and observed activation of mini-white antisense transcription in strong transgenes. This result is in accordance to the GRO-seq data. Thus, launching of even a low level of antisense transcription, as detected in 3.1 and 2.4 strains, provides generation of piRNA precursors and formation of strong dual-strand piRNA clusters. In contrast to the comparable amounts of sense and antisense mini-white piRNAs in strong strains, the amount of antisense GRO-seq reads is much reduced compared to sense (Fig. 2E) suggesting that the level of antisense precursor transcripts limits the generation of piRNAs in dual-strand piRNA clusters. As in the case of the 1.9 upstream genomic region (Fig. 2A), we did not observe distinct GRO-seq peaks for the transgenic antisense strand suggesting that transcription initiated at multiple random sites of the transgene.
To confirm that white antisense RNAs are transcribed by transgenes, but not by the endogenous white gene, we compared the steady-state level of this RNA by strand-specific RT-qPCR in the ovaries of wK and transgenic 3.1 and 2.4 strains. A negligible level of antisense white RNA was revealed in wK, indicating that these transcripts are indeed of transgenic origin (Supplemental Fig. S3A).
The steady-state level of transgenic mini-white transcripts, detected by RT-qPCR, was greater in strain 1.9 than in 3.1 or 2.4 (Supplemental Fig. S3B). Evidently, nascent transcripts synthesized at the TAS region, where the 3.1 transgene is localized, and in the euchromatic site of the 2.4 insertion are subject to more stringent post-transcriptional degradation by the piRNA biogenesis machinery.
Comparison of the nascent transcription at the 2.4 insertion site with the empty site using GRO-seq libraries or RT-qPCR on run-on RNAs reveals an increased transcription at the 2.4 3′P-flanking region (Supplemental Fig. S2D–F), where small RNAs are mapped only to the negative genomic strand. Notably, Rhi, HP1, and H3K9me3 are also associated with this region (Fig. 2B). Most likely, transcripts initiated apart from the transgene avoid being recognized by Rhi as piRNA precursors thus restricting propagation of the dual-strand piRNA cluster beyond the transgene.
To summarize, de novo formation of the transgene-associated piRNA cluster is accompanied by activation of bidirectional transcription in the transgenic region and in flanking regions that provides precursors for si- and piRNA production. However, we did not detect canonical TSSs suggesting that transcription in piRNA clusters instead initiates at multiple random sites.
Weak transgenic piRNA clusters show discrete association with Rhino foci according to bimodal Rhino deposition
Expression of the transgenes studied here occurs in highly polyploid nurse cells of the ovaries. Thus, two models can explain why weak transgenes produce low level of piRNAs (Fig. 3A). According to a gradual model, each transgenic copy of a weak strain produces a low amount of piRNAs and binds a low level of Rhi in the nurse cell nucleus. According to an all-or-nothing model, most weak transgenic copies are not associated with Rhi at all and therefore, are not involved in piRNA production, with only a few transgenic copies producing abundant piRNAs. According to the latter model, in the polyploid nucleus, weak transgenes represent a mix of strong transgenic piRNA copies and transgenic copies not involved in piRNA production at all (Fig. 3A). In strong clusters, where each transgenic copy is highly productive, either model can fit. We have shown that the level of Rhi enrichment at the transgenes is in accordance with the small RNA production (Fig. 1A). Thus, to distinguish between these two scenarios, we studied the colocalization of weak and strong I-TG transgenes with Rhi using DNA FISH combined with Rhi immunostaining.
FIGURE 3.

Formation of piRNA cluster at single transgenic site occurs according to an “all-or-nothing” model. (A) Two models explaining why weak transgenic piRNA clusters produce a low level of piRNAs. According to the gradual model, every genomic copy of a weak transgenic cluster produces a lower amount of piRNA (arrowheads) compared with strong transgenes, each copy of which produces abundant piRNAs. According to the bimodal model, most weak transgenic copies are not involved in piRNA production at all, and only a few of them produce abundant piRNAs comparable to strong transgenes. Polyploid (here, tetraploid) nuclei of nurse cells are outlined by rectangles. (B,C) Nuclei of germ cells from stages X of oogenesis are stained for Rhi (red) in combination with DNA FISH with BAC probes corresponding to 2.1 (B) and 1.9 (C) insertion regions in wK strain (control) and in corresponding transgenic strain. DNA is stained by DAPI (blue). The “all-or-nothing” model (Fig. 3A) is supported by the immunofluorescence data, shown in B and C (arrow).
Genomic fragments corresponding to euchromatic regions of the 1.9, 2.1, 2.4, and 3.6 insertions were used as DNA probes. We ascertained that these probes were never colocalized with Rhi foci in the nurse cell nuclei of the nontransgenic wK strain (Fig. 3B; Supplemental Fig. S4). In the 2.1 and 2.4 strong transgenic strains, the majority of BAC signals totally or partially overlap with Rhi (Table 2).
TABLE 2.
Colocalization of transgenes with Rhino in polyploid nuclei of nurse cells
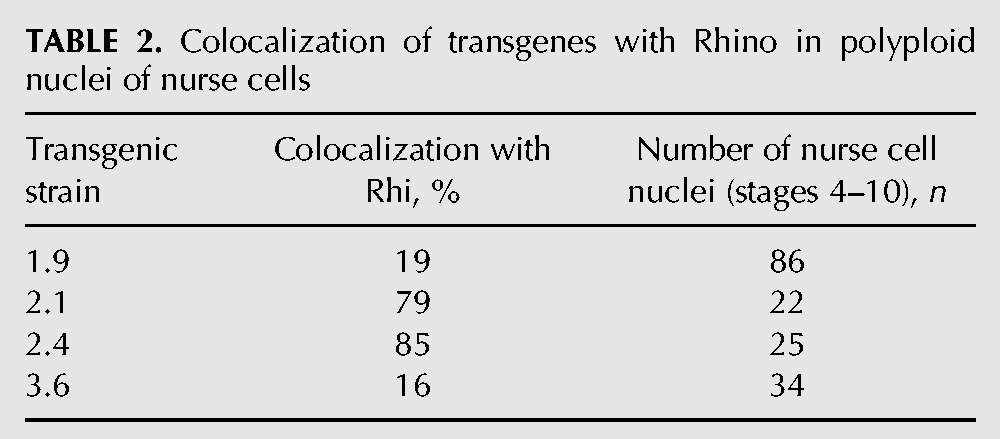
For the weak 1.9 and 3.6 transgenes, DNA FISH signal colocalization with Rhi was significantly reduced (Table 2); with most DNA FISH signals evidently distant from Rhi foci (Fig. 3C; Supplemental Fig. S4). This observation is in accordance with the poor enrichment of the 1.9 and 3.6 by Rhi that we observed in ChIP experiments. Thus, the formation of a piRNA cluster at a single transgenic site in the polyploid nucleus seems to work according to an “all-or-nothing” model (Fig. 3A). This bimodal fashion of piRNA cluster formation means that the potency of the piRNA cluster is a matter of statistics: It is defined by the number of transgene copies involved in piRNA production in the nuclei of polyploid nurse cells.
It was recently observed that Rhi and HP1a are largely colocalized in primordial germ cell nuclei of Drosophila third instar larval gonads (Marie et al. 2017). At the same time, we observed that all I-TG transgenes associate with HP1a but only some of them bind Rhi. HP1a immunostaining was more sensitive to the high temperature treatment during FISH than Rhi; therefore, we were unable to perform HP1a immunostaining combined with DNA FISH. To address the question of mutual localization of these proteins in nurse cell nuclei, we performed HP1a and Rhi immunostaining on adult ovaries. Rhi signals tend to cluster into sharp foci, while HP1a staining forms areas resembling clouds (Fig. 4) that likely reflects the recently discovered ability of HP1a to form phase-separated droplets (Larson et al. 2017; Strom et al. 2017). We found that Rhi foci begin to colocalize with HP1a staining in the germ cells of germarium region 2b; at later stages of oogenesis, Rhi foci are always positioned within more diffuse HP1a-positive nuclear domains.
FIGURE 4.

HP1a and Rhi have different but partially overlapped localization patterns in the nurse cell nuclei. Ovaries of wK strain are immunostained for Rhi (red) and HP1a (green). Nuclei of germ cells at indicated stages of oogenesis are shown. DNA is stained by DAPI (blue). Schema of ovariole is shown above; germarium is enlarged. Bars, 5 μm.
piRNA-mediated chromatin silencing requires H3K9 methyltransferase activity. In Drosophila ovarian somatic cells, methyltransferase dSETDB1 is required for initiation of piRNA-mediated silencing of TE insertions, while another histonemethyltransferase, Su(var)3-9, provides local spreading of the H3K9me3 marks (Sienski et al. 2015). In the germline, dSETDB1 is required for piRNA cluster transcription (Rangan et al. 2011). dSETDB1 acts at earlier stages of oogenesis; its function is taken over by Su(var)3-9 from stage III of oogenesis (Yoon et al. 2008). Therefore, we were interested to find out whether Rhi associates with Su(var)3-9 methyltransferase during oogenesis. Immunostaining of both Rhi and Su(var)3-9 did not reveal colocalization of these proteins in the nuclei of germ cells at any stage of oogenesis (Supplemental Fig. S5). This observation is in accordance with the ovarian Su(var)3-9 DamID-seq data showing no overlapping between Su(var)3-9 enriched regions and piRNA-producing loci (Maksimov et al. 2017).
Thus, from immunostaining combined with DNA FISH and ChIP data one may deduce that strong transgenes associated with Rhi foci appear to be positioned within HP1a domains, while weak transgenes also localized within HP1a zones are more resistant to the Rhi deposition.
DISCUSSION
Using a transgenic model, we studied the molecular mechanisms of de novo dual-strand piRNA cluster establishment in the germline. An obvious advantage provided by these transgenes is the ability to compare the status of different genomic loci before and after transgene insertion. This transgenic model shows that requirements for de novo piRNA cluster formation are more complex than piRNA-mediated chromatin assembly and include changes of the transcriptional status and chromatin dynamics in a given genomic context.
We have shown that the transformation of genomic sites into dual-strand piRNA clusters is accompanied by the activation of transcription in both genomic strands without any appearance of well-defined promoters suggesting that transcription in dual-strand piRNA clusters initiates at multiple random sites. In parallel to this observation, widespread promoter-independent initiation of Pol II transcription within heterochromatic dual-strand piRNA clusters was shown (Andersen et al. 2017). We also argued that even subtle activation of read-through antisense transcription of the transgene, as observed in the 2.1, 2.4, and 3.1 strains, is sufficient to transform a transgene into a potent dual-strand piRNA cluster. GRO-seq analysis of 1.9 and 2.4 flanking regions as well as of transgenic mini-white clearly shows that the number of nascent transcripts is not dramatically changed upon piRNA cluster formation. What we detected in all cases is the appearance of traces of transcription from the opposite genomic strand. Our data suggest that the setting of convergent transcription is the main factor promoting formation of a dual-strand piRNA cluster.
We found that the same constructs, located at different genomic loci, are not equivalent in their ability to produce piRNAs and defined them as weak and strong piRNA clusters. All transgenes are targeted by similar amounts of endogenous I-element-specific piRNAs and form repressed chromatin enriched by HP1a and H3K9me3. Nevertheless, transgenes differ in their ability to attract Rhi to insertion sites indicating that the chromatin of genomic loci may be refractory to piRNA cluster formation. Thus, all piRNA targets are silenced at transcription level but not all of them become germline piRNA clusters. Our transgenic model allows to differentiate between these scenarios and conclude that HP1 and H3K9me3 binding is not enough for the locus to gain piRNA-producing capacity. Le Thomas et al. (2014) also addressed the question regarding the prerequisites of the piRNA production using two transgenic piRNA targets, both capable of stable piRNA production and enriched by H3K9me3 chromatin mark. This fact led to a suggestion that H3K9me3 could be a factor sufficient for launching piRNA biogenesis, which is debatable, as we show here.
H3K9me3 is a substrate for the binding of both HP1-like proteins, HP1a and Rhi (Schotta et al. 2002; Le Thomas et al. 2014). Our data indicate that Rhi and HP1a cooperate rather than compete in the chromatin complex of piRNA clusters because we do not observe a significant decrease in HP1a binding for strong transgenes that are much more enriched in Rhi as compared with weak transgenes. One could therefore assume that a competitive relationship for H3K9me3 binding between HP1a and Rhi is not the main rule that determines the potency of piRNA production. Indeed, HP1a and Rhi immunostaining show that, in nurse cell nuclei sharp Rhi foci are embedded into the HP1a diffuse domains. However, some of the piRNA target loci are resistant to the Rhi binding. As well as HP1a and H3K9me3 enrichment, 21-nt RNA content is not considerably different between weak and strong transgenic piRNA clusters indicating that siRNA production and HP1a binding are stable features of the I-TG transgenes that do not depend on their involvement in the piRNA production (Table 1).
A further important question is how propagation of dual-strand piRNA clusters is regulated. Gradually decreasing peaks of piRNAs mapping to one genomic strand are observed at regions flanking dual-strand piRNA clusters formed by individual TEs (Shpiz et al. 2014b), by some I-TG transgenes (Olovnikov et al. 2013) and by endogenous piRNA clusters (Mohn et al. 2014). Remarkably, even in the case of the strong 2.4 transgene that produces abundant piRNAs from both genomic strands, the double-strand piRNA cluster is not extended beyond the transgene suggesting that the spreading of piRNA clusters is limited. It is believed that Rhi serves as a marker of dual-strand clusters and defines cluster transcripts as the piRNA precursors (Mohn et al. 2014). Surprisingly, Rhi associates with uni-strand piRNA tracks flanking both TEs (Mohn et al. 2014) and the transgene 2.4 insertion (Fig. 1B). At the same time, transcripts from both genomic strands are detected at the 2.4 flanking region (Supplemental Fig. S2D) suggesting that Rhi binding at a particular locus is not sufficient to define all transcripts mapping to this locus as piRNA precursors. Most likely, such recognition is crucial at the initial step of transcription. Thus, RNAs initiated within a transgene, appear to be identified as piRNA complementary targets and directed to piRNA processing. In the absence of complementary piRNAs, transcripts initiated at a distant TSS apart from the transgene are not recognized by the piRNA system. Either mechanism appears to be necessary to restrict uncontrolled propagation of dual-strand piRNA clusters beyond the direct piRNA targets.
Combined DNA FISH/immunostaining has shown that the efficiency of piRNA production by a particular transgene is determined by the number of Rhi positive transgenic genomic copies in the polyploid nucleus of the nurse cell. In contrast to the polytene chromosomes of salivary glands, chromatids of highly polyploid (up to 1024 haploid DNA content) nurse cells are only partially conjugated which leads to the detection of multiple DNA FISH signals. That means that each genomic copy of strong transgenes binds Rhi and produces abundant piRNAs within a single nucleus. Indeed, the major endogenous dual-strand piRNA cluster at the 42AB locus strongly colocalizes with Rhi foci in the nuclei of nurse cells (Mohn et al. 2014).
Our data show that weak transgenic piRNA clusters are not weak due to the low efficiency of piRNA production by each transgenic copy, but rather due to the low frequency of formation of Rhi-enriched chromatin at transgenic copies. As a result, only a few of the transgenic DNA signals coincide with Rhi (and hence form piRNA clusters) but the majority of transgenic copies in the same nucleus do not colocalize with Rhi. This results in mosaic transgenic DNA FISH—Rhi staining in the polyploid nuclei of nurse cells. Notably, in fission yeast, siRNAs initiate in trans gene silencing in an “all-or-nothing” fashion in Paf1C (RNA polymerase-associated factor 1 complex) mutant cells (Kowalik et al. 2015). In yeast, RNAi-directed heterochromatin formation is also accompanied by the production of novel siRNAs in target loci (Simmer et al. 2010; Kowalik et al. 2015) demonstrating a striking similarity to Drosophila transgenic piRNA cluster formation. Remarkably, in a wild-type fission yeast, the ability of siRNAs to direct heterochromatin formation in trans is limited and strongly depends on the genomic location of the target genes (Simmer et al. 2010) suggesting that general mechanisms control small RNA-mediated heterochromatin propagation in eukaryotic genomes.
MATERIALS AND METHODS
Drosophila transgenic strains
Transgenic strains 1.9, 2.1, 3.1, 3.6 bearing the I-retrotransposon fragment cloned in pW8-hsp-pA vector have previously been described (Jensen et al. 1999; Olovnikov et al. 2013). Additionally, strain 2.4 (Jensen et al. 1999) was included in this study. Transgene 2.4 is located at chr2L:12010762-63 (BDGP assembly R5/dm3) in minus orientation relative to genomic strands as determined by inverse PCR on genomic DNA.
RT-PCR analysis
RNA was isolated from ovaries of 3-d-old females. cDNA was synthesized using random or strand-specific primers and SuperScriptII reverse transcriptase (Invitrogen) according to the manufacturer's protocol. cDNA samples were analyzed using quantitative PCR (qPCR). Primers used for RT-qPCR are listed in Supplemental Material.
Small RNA library preparation and analysis
Small RNAs, 19–29 nt in size, from total ovarian RNA extract of transgenic strain 2.4 were cloned as previously described in Muerdter et al. (2012) and sequenced using the Illumina HiSeq-2000 system. After clipping the Illumina 3′-adapter sequence, small RNA reads that passed quality control and minimal length filter (>18 nt) were mapped (allowing zero mismatches) to the Drosophila melanogaster genome (April 2006, BDGP assembly R5/dm3) or transgenes by Bowtie (Langmead et al. 2009). Small RNA libraries were normalized to the library depth. The plotting of size distributions and read coverage, and measuring nucleotide biases were performed by custom scripts. Ovarian small RNA-seq data for 2.4 strain are available at the Gene Expression Omnibus (GEO), accession number GSE88774.
Small RNAs from the ovaries of 1.9, 2.1, 3.1, 3.6, and 67.2.1 strains were described previously (Olovnikov et al. 2013). rRNA reads were excluded from the small RNA-seq data before analysis.
GRO-seq library preparation and analysis
Run-on transcription and preparation of ovarian GRO-seq libraries was performed as previously described (Shpiz et al. 2011; Rozhkov et al. 2013). Reads were mapped to the R5/dm3 genomic assembly or transgene sequence by bowtie2 and de-duplexed. GRO-seq data for 1.9, 2.4, and 3.1 strains are deposited at GEO, accession number GSE88774. rRNA reads were excluded from the GRO-seq data before analysis.
Chromatin immunoprecipitation
One hundred and twenty pairs of ovaries were dissected and homogenized in buffer A (60 mM KCl, 15 mM NaCl, 15 mM HEPES [pH 7.6], 4 mM MgCl2, 0.5% Triton-X-100, 0.5 mM DTT, complete EDTA-free protease inhibitor cocktail, Roche) using Dounce homogenizer with type B pestle (14 strokes) in the presence of 1.8% formaldehyde on ice. Crosslinking was performed at room temperature for 15 min on a rotating wheel and was stopped by addition of glycine (final concentration 225 mM) with subsequent incubation on ice for 5 min. All further procedures were done at 4°C unless otherwise specified. Nuclei were pelleted by centrifugation for 5 min, at 4000g and washed with 1 mL buffer A three times, then incubated for 10 min in 1 mL lysis buffer (140 mM NaCl, 15 mM HEPES [pH 7.6], 1 mM EDTA, 0.5 mM EGTA, 1% Triton-X-100, 0.5 mM DTT, 0.1% sodium deoxycholate, complete EDTA-free protease inhibitor cocktail [Roche]) with rotation at 4°C. After centrifugation at 4000g at 4°C, supernatant was discarded and the pellet was incubated for 15 min in 500 µL of lysis buffer in the presence of 0.1% SDS and 0.5% N-lauroylsarcosine (lysis-SDS buffer) on a rotation wheel at 4°C. Chromatin was fragmented by sonication on Bioruptor Next Gen (Diagenode SA) during eight cycles (15″ON, 40″ OFF), cooled on ice for 15 min, then sonication was resumed for additional six cycles (15″ON, 40″ OFF). The size of DNA fragments obtained was about 100–500 bp. Sonication was followed by centrifugation at 16,000g for 15 min and supernatant was aliquoted and stored at −70°C.
For immunoprecipitation, 2.5 µg of chromatin was diluted up to 1 mL with dilution buffer (0.1% SDS, 1.1% Triton-X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl [pH 8.1], 167 mM NaCl, and complete EDTA-free protease inhibitor cocktail, Roche), 20 µL were taken for the input probe, and the remaining material was incubated with antibodies for 5 h at 4°C with rotation. Then samples were incubated overnight at 4°C with 50 µL Protein G Agarose/Salmon sperm DNA (Millipore). Agarose beads were subsequently washed with low salt buffer (0.1% SDS, 1% Triton-X-100, 2 mM EDTA, 20 mM Tris-HCl [pH 8.1], 150 mM NaCl), high salt buffer (0.1% SDS, 1% Triton-X-100, 2 mM EDTA, 20 mM Tris-HCl [pH 8.1], 500 mM NaCl), LiCl wash buffer (0.25 M LiCl, 1% NP40, 1% deoxycholic acid, 1 mM EDTA, 10 mM Tris-HCl [pH 8.1]), and twice with TE buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA). Elution was done twice with 250 µL elution buffer (1% SDS, 0.1 M NaHCO3) for 10 min at 65°C in a thermomixer; eluates were combined. To revert cross-linking, 20 μL of 5 M NaCl was added and the probe was incubated overnight at 65°C with shaking. Then, 10 μL of 0.5 M EDTA and 20 μL of 1 M Tris-HCl (pH 7.5) were added and chromatin was digested with Proteinase K (final concentration 3.7 μg/mL) for 1 h at 50°C, phenol–chloroform was extracted and ethanol precipitated in the presence of glycogen. The resulting pellet was dissolved in 120 µL of 10 mM Tris-HCl (pH 8). Chromatin was immunoprecipitated with the following antibodies: anti-HP1a (Covance or Developmental Studies Hybridoma Bank, C1A9), anti-trimethyl-histone H3 Lys9 (Millipore), and Rhi antiserum (Radion et al. 2017). qPCR was performed with primers corresponding to control and specific loci that are listed in the Supplemental Material. Percent input was calculated using the formula: % input = (2(Ct input–Ct IP)) × Fd × 100, where % input is the ChIP efficiency expressed in percent when compared with total DNA; Ct IP and Ct input are threshold cycles for ChIP and input samples, respectively; Fd is the dilution factor. Occupancy was calculated according to the formula: %input (specific loci)/%input (background loci). Standard error of the mean (SEM) of triplicate PCR measurements for three–five biological replicates was calculated in each experiment.
DNA FISH combined with immunostaining
DNA FISH combined with immunostaining on whole mount ovaries was performed according to a described protocol (Shpiz et al. 2014a). Treatment of tissues with RNase A, DNase and protease-free (ThermoFisher) in a final concentration of 100 µg/mL was used. Bacterial artificial chromosome (BAC) clones BACR03L12, BACR21A02, and CH321-84A9 corresponding to “empty” sites of 1.9, 2.1, and 2.4 insertions, respectively, were used for the DNA probe preparation. Probes were generated by direct labeling of BAC DNA, digested with Sau3AI, by Spectrum Green dUTP or Cy3 using the Nick Translation Kit (Roche). PCR fragments amplified with primers listed in Supplemental Table S1 (3.6 flanks) and labeled with a DIG DNA Labeling Kit (Roche) were used to detect the transgene 3.6 insertion region. The primary antibodies used were Rhi antiserum (1:200) (Radion et al. 2017), anti-HP1a (1:500, Covance), and anti-Su(var)3-9 (1:100, Abcam). Alexa 546 anti-rat IgG and Alexa 488 anti-rabbit IgG secondary antibodies (Invitrogen) were used at dilution 1:500. After washing, samples were mounted in Vectashield Antifade Mounting Medium with DAPI (Vector Laboratories). Images were captured using a Zeiss LSM 510 Meta confocal microscope.
DATA DEPOSITION
The data have been deposited in the NCBI GEO under accession number GSE88774.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Emilie Brasset for methodological advice on ChIP and DNA FISH and Nikolay Rozhkov for the helpful recommendations on GRO-seq. This work was supported by the grant from the Russian Foundation for Basic Research 16-54-150003 to A.K., by a Skoltech Systems Biology Fellowship to S.R., by the Centre National de la Recherche Scientifique (CNRS) grant “PRC Russie” no. 1051 to S.J., by the grant “plasTiSiPi” from the Agence Nationale de la Recherche to C.V. (ANR-13-BSV2-0007), and by the Russian Academy of Sciences program for Molecular and Cell Biology to A.K.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.062851.117.
REFERENCES
- Andersen PR, Tirian L, Vunjak M, Brennecke J. 2017. A heterochromatin-dependent transcription machinery drives piRNA expression. Nature 549: 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, et al. 2006. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442: 203–207. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128: 1089–1103. [DOI] [PubMed] [Google Scholar]
- de Vanssay A, Bougé AL, Boivin A, Hermant C, Teysset L, Delmarre V, Antoniewski C, Ronsseray S. 2012. Paramutation in Drosophila linked to emergence of a piRNA-producing locus. Nature 490: 112–115. [DOI] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC. 2007. A slicer-mediated mechanism for repeat-associated siRNA 5′ end formation in Drosophila. Science 315: 1587–1590. [DOI] [PubMed] [Google Scholar]
- Homolka D, Pandey RR, Goriaux C, Brasset E, Vaury C, Sachidanandam R, Fauvarque MO, Pillai RS. 2015. PIWI slicing and RNA elements in precursors instruct directional primary piRNA biogenesis. Cell Rep 12: 418–428. [DOI] [PubMed] [Google Scholar]
- Hur JK, Luo Y, Moon S, Ninova M, Marinov GK, Chung YD, Aravin AA. 2016. Splicing-independent loading of TREX on nascent RNA is required for efficient expression of dual-strand piRNA clusters in Drosophila. Genes Dev 30: 840–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizu H, Iwasaki YW, Hirakata S, Ozaki H, Iwasaki W, Siomi H, Siomi MC. 2015. Somatic primary piRNA biogenesis driven by cis-acting RNA elements and trans-acting Yb. Cell Rep 12: 429–440. [DOI] [PubMed] [Google Scholar]
- Jensen S, Gassama MP, Heidmann T. 1999. Cosuppression of I transposon activity in Drosophila by I-containing sense and antisense transgenes. Genetics 153: 1767–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josse T, Teysset L, Todeschini AL, Sidor CM, Anxolabéhère D, Ronsseray S. 2007. Telomeric trans-silencing: an epigenetic repression combining RNA silencing and heterochromatin formation. PLoS Genet 3: 1633–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalik KM, Shimada Y, Flury V, Stadler MB, Batki J, Bühler M. 2015. The Paf1 complex represses small-RNA-mediated epigenetic gene silencing. Nature 520: 248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, Narlikar GJ. 2017. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547: 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Thomas A, Rogers AK, Webster A, Marinov GK, Liao SE, Perkins EM, Hur JK, Aravin AA, Tóth KF. 2013. Piwi induces piRNA-guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev 27: 390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Thomas A, Stuwe E, Li S, Du J, Marinov G, Rozhkov N, Chen YC, Luo Y, Sachidanandam R, Toth KF, et al. 2014. Transgenerationally inherited piRNAs trigger piRNA biogenesis by changing the chromatin of piRNA clusters and inducing precursor processing. Genes Dev 28: 1667–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksimov DA, Laktionov PP, Posukh OV, Belyakin SN, Koryakov DE. 2017. Genome-wide analysis of SU(VAR)3–9 distribution in chromosomes of Drosophila melanogaster. Chromosoma 10.1007/s00412-017-0647-4. [DOI] [PubMed] [Google Scholar]
- Marie PP, Ronsseray S, Boivin A. 2017. From embryo to adult: piRNA-mediated silencing throughout germline development in Drosophila. G3 (Bethesda) 7: 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Sienski G, Handler D, Brennecke J. 2014. The rhino-deadlock-cutoff complex licenses noncanonical transcription of dual-strand piRNA clusters in Drosophila. Cell 157: 1364–1379. [DOI] [PubMed] [Google Scholar]
- Muerdter F, Olovnikov I, Molaro A, Rozhkov NV, Czech B, Gordon A, Hannon GJ, Aravin AA. 2012. Production of artificial piRNAs in flies and mice. RNA 18: 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olovnikov I, Ryazansky S, Shpiz S, Lavrov S, Abramov Y, Vaury C, Jensen S, Kalmykova A. 2013. De novo piRNA cluster formation in the Drosophila germ line triggered by transgenes containing a transcribed transposon fragment. Nucleic Acids Res 41: 5757–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post C, Clark JP, Sytnikova YA, Chirn GW, Lau NC. 2014. The capacity of target silencing by Drosophila PIWI and piRNAs. RNA 20: 1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radion E, Ryazansky S, Akulenko N, Rozovsky Y, Kwon D, Morgunova V, Olovnikov I, Kalmykova A. 2017. Telomeric retrotransposon HeT-A contains a bidirectional promoter that initiates divergent transcription of piRNA precursors in Drosophila germline. J Mol Biol 429: 3280–3289. [DOI] [PubMed] [Google Scholar]
- Rangan P, Malone CD, Navarro C, Newbold SP, Hayes PS, Sachidanandam R, Hannon GJ, Lehmann R. 2011. piRNA production requires heterochromatin formation in Drosophila. Curr Biol 21: 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhkov NV, Hammell M, Hannon GJ. 2013. Multiple roles for Piwi in silencing Drosophila transposons. Genes Dev 27: 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotta G, Ebert A, Krauss V, Fischer A, Hoffmann J, Rea S, Jenuwein T, Dorn R, Reuter G. 2002. Central role of Drosophila SU(VAR)3-9 in histone H3-K9 methylation and heterochromatic gene silencing. EMBO J 21: 1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpiz S, Olovnikov I, Sergeeva A, Lavrov S, Abramov Y, Savitsky M, Kalmykova A. 2011. Mechanism of the piRNA-mediated silencing of Drosophila telomeric retrotransposons. Nucleic Acids Res 39: 8703–8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpiz S, Lavrov S, Kalmykova A. 2014a. Combined RNA/DNA fluorescence in situ hybridization on whole-mount Drosophila ovaries. Methods Mol Biol 1093: 161–169. [DOI] [PubMed] [Google Scholar]
- Shpiz S, Ryazansky S, Olovnikov I, Abramov Y, Kalmykova A. 2014b. Euchromatic transposon insertions trigger production of novel Pi- and endo-siRNAs at the target sites in the Drosophila germline. PLoS Genet 10: e1004138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienski G, Batki J, Senti KA, Dönertas D, Tirian L, Meixner K, Brennecke J. 2015. Silencio/CG9754 connects the Piwi-piRNA complex to the cellular heterochromatin machinery. Genes Dev 29: 2258–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmer F, Buscaino A, Kos-Braun IC, Kagansky A, Boukaba A, Urano T, Kerr AR, Allshire RC. 2010. Hairpin RNA induces secondary small interfering RNA synthesis and silencing in trans in fission yeast. EMBO Rep 11: 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH. 2017. Phase separation drives heterochromatin domain formation. Nature 547: 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe AM, Horowitz H, Grafer CM, Jackson SM, Berg CA. 2001. Drosophila rhino encodes a female-specific chromo-domain protein that affects chromosome structure and egg polarity. Genetics 159: 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J, Lee KS, Park JS, Yu K, Paik SG, Kang YK. 2008. dSETDB1 and SU(VAR)3-9 sequentially function during germline-stem cell differentiation in Drosophila melanogaster. PLoS One 3: e2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang J, Xu J, Zhang Z, Koppetsch BS, Schultz N, Vreven T, Meignin C, Davis I, Zamore PD, et al. 2012. UAP56 couples piRNA clusters to the perinuclear transposon silencing machinery. Cell 151: 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wang J, Schultz N, Zhang F, Parhad SS, Tu S, Vreven T, Zamore PD, Weng Z, Theurkauf WE. 2014. The HP1 homolog rhino anchors a nuclear complex that suppresses piRNA precursor splicing. Cell 157: 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.