

Abstract
Purpose
Angiotensin system inhibitors (ASIs) can improve prognosis in multiple cancer types, including pancreatic ductal adenocarcinoma (PDAC). However, no study has examined the effect of ASIs alone or combined with adjuvant chemotherapy in resected PDAC patients.
Experimental Design
We performed an analysis of the records of ASI users and non-user patients with PDAC seen at Massachusetts General Hospital between January 2006 and December 2010. To identify mechanisms of ASIs in PDAC, we performed RNA-Seq of resected primary lesions.
Results
794 consecutive patients were included. In 299 resected patients, ASI-users experienced longer overall survival (OS) in both univariate (median OS: 36.3 vs. 19.3 months, p=0.011) and adjusted multivariate (HR, 0.505; 95%CI, 0.339 - 0.750; p=0.001) analyses. Propensity score adjusted analysis also showed a longer median OS for chronic ASI-users. In unresected patients, the beneficial effect of ASIs was significant in patients with locally advanced disease, but not in metastatic patients. RNA-Seq analysis revealed in tumors of ASI-users (lisinopril) a normalized extracellular matrix, a reduced expression of genes involved in PDAC progression (e.g. WNT and Notch signaling) and an increased expression of genes linked with the activity of T cells and antigen-presenting cells. Finally, chronic use of ASI was associated with a gene expression signature which is predictive of survival in independent validation cohorts.
Conclusions
In patients with non-metastatic PDAC, chronic ASI use is associated with longer OS independently of chemotherapy. Our RNA-Seq analysis suggests that ASI reduce the malignant potential of cancer cells and stimulate the immune microenvironment in primary PDAC.
Keywords: patients with pancreatic ductal adenocarcinoma, angiotensin system inhibitors, overall survival, RNA-Seq analysis, immune effector cells
Introduction
The renin-angiotensin-aldosterone system (RAAS) is a well-studied hormone system. It was first recognized as a master regulator of blood pressure homeostasis and electrolyte balance. The subsequent discovery of local RAAS in various organs and tissues highlighted its significant role in basic cell biological processes such as proliferation and migration, as well as pathophysiological processes like inflammation(1). In solid tumors, angiotensin II (Ang II) enhances tumor cell proliferation and growth by acting as a paracrine and/or autocrine signal in the tumor microenvironment. It promotes the growth of stromal cells such as fibroblasts, endothelial cells, neutrophils and macrophages, leading to an increased secretion of tumor growth factors(2). Some cancer cells also utilize Ang II signaling for survival(3).
Cancer-microenvironment interactions shape the pathophysiology of pancreatic ductal adenocarcinoma (PDAC). In our attempt to understand the role of physical barriers to cancer treatment, we discovered that the collagen matrix forms a formidable obstacle which hinders the penetration of nanotherapeutics into solid tumors(4). We later found that losartan, an angiotensin receptor blocker (ARB), decreased collagen and enhanced the intratumoral distribution of nanoparticles and nanotherapeutics in breast and pancreatic cancer mice models(5). We subsequently showed that losartan and lisinopril, an angiotensin converting enzyme inhibitor (ACEi), decreased not only collagen but also hyaluronan, and enhanced the efficacy of low molecular weight chemotherapeutics in desmoplastic mice tumors(6). This benefit was in part due to the decompression of blood vessels resulting from decreased solid stress(7,8). The safety and low cost of ARBs and ACEis, together called angiotensin system inhibitors (ASIs), along with their potentiation of conventional chemotherapy make a strong case for repurposing ASI as adjuncts in cancer treatment. These insights formed the basis of a prospective trial at MGH evaluating the efficacy of losartan combined with the FOLFIRINOX cocktail and radiotherapy in patients with locally advanced PDAC (ClinicalTrials.gov identifier: NCT01821729). The preliminary results (N=25 patients) suggest that the addition of losartan to neoadjuvant FOLFIRINOX followed by radiation increases the resection rate and R0 resection. Also, the median overall survival and 2-year survival rates are longer than historically observed (9). These encouraging results agree with our pre-clinical findings and should be tested in independent prospective trials.
Several studies have associated the use of ASI with longer survival in various tumor types(2). However, no study has determined the effect of ASI use in PDAC patients independent of stage and treatment received, including surgery, chemotherapy or radiation therapy. Furthermore, no unbiased effort has been undertaken to understand the mechanisms through which ASIs confer survival benefits. To determine the role of ASI use in pancreatic cancer we conducted a retrospective analysis of all PDAC patients treated and followed at MGH during a five-year period. Through unadjusted univariate, adjusted analysis with multivariate modeling, and propensity score methods, we correlated chronic ASI-use with overall survival (OS) and recurrence-free survival (RFS) of PDAC patients. Chronic ASI use is associated with longer OS in patients with resected primary tumors as well as in locally advanced cancer patients. To gain insight into the mechanisms, we conducted RNA-Seq analysis in resected tumors from treatment naïve patients with or without chronic lisinopril use. Results suggested that ASI changed the tumor microenvironment, normalized the stroma and enhanced the activity of immune cells. Together, our results enable a deeper understanding of the biological mechanisms underlying the survival benefit associated with ASI use in PDAC.
Methods
Patient cohorts
Patient data were acquired through Massachusetts General Hospital (MGH) super-database, a research patient data registry created by Partners Healthcare. The Partners Healthcare internal review board and local ethics committee approved the retrospective analysis of patient data. Diagnosis, pathology, body mass index (BMI), treatment, medications, progression and survival were confirmed and collated by reviewing medical records. For resected patients, BMI information was collected at time of diagnosis. BMI was only available for a fraction of patients with locally advanced cancer and metastasis. Therefore, BMI was not included in the analysis of these patients. All the patients included in the analysis had a tissue diagnosis of PDAC. We used the date of tissue collection as the time of diagnosis. OS was calculated from the time of diagnosis to the time of death or last contact. RFS was calculated from the time of diagnosis to the time of recurrence in any organ or death. Organ specific time to metastasis was calculated from the time of diagnosis to the occurrence of metastasis in that organ, while occurrence to other organs were censored. Competing risk analysis were performed with first organ metastatic recurrence in liver, lung, local site, other metastasis or death were treated as competing events.
Propensity score adjusted analysis
We calculated propensity score using all parameters available. In metastatic patients: age, tumor site, chemotherapy and hypertension. In locally advanced patients: age, tumor site, tumor size, radiation therapy, chemotherapy and hypertension. In resected patients: age, tumor site, tumor size, BMI, grade, lymph node ratio, perineural invasion, vascular invasion, neoadjuvant treatment, adjuvant chemotherapy, adjuvant radiation therapy and hypertension. We calculated Cox proportional hazard ratio with the propensity score as well as the ASI status.
Fresh tumor samples
Patient samples were obtained from the Departments of Surgery and Pathology at Massachusetts General Hospital. All patients gave signed informed consent for the collection of excessive tumor samples and molecular analysis. The protocol was approved by the internal review board of Partners Healthcare. All patients had a resectable PDAC and were not treated with chemotherapy or radiation prior to surgery, but the ACEi group was treated with lisinopril for their hypertension. Fresh tumor samples were collected after surgical resection. Each fresh tumor chunk was sampled from the center of the pancreatic tumor and was snap frozen in liquid nitrogen for later use.
RNA extraction and RNA-Seq
Whole tissue RNA was extracted using a standard phenol-chloroform protocol. RNA integrity numbers (RIN) were obtained using a Bioanalyzer 2100 (Agilent), and samples with RIN >7.5 were used for subsequent steps. Sequencing libraries were prepared from 100–500 ng of total RNA using the TruSeq RNA Sample Preparation Kit v2 (Illumina). Read alignment and junction mapping were accomplished using TopHat2 v2.0.4, using a 25 bp 50 segment seed for initial mapping to map reads to the reference genome annotation, NCBI human build 37.2(10), followed by differential gene expression analysis using Cuffdiff v2.0.2.. Data were expressed as fragments per kilobase of exon per million fragments mapped (FPKM). To identify functional gene categories enriched in our differentially expressed genes, we used the Gene Ontology (GO) and REACTOME databases. We also performed gene set enrichment analysis (GSEA) in preranked analysis mode with classic enrichment method, ordering genes by their fold changes in lisinopril vs. control PDAC tumors. We used FDR q-value 0.05 as a threshold to determine significantly changed gene sets.
Validation cohorts and statistical methods
We searched for validation cohorts that have: 1) genome-wide expression measurements, 2) overall survival data and 3) normal pancreatic tissue control and 4) greater than 50 tumor samples. GEO Dataset GSE71729(11) as well as TCGA dataset were selected. Survival data and normalized expression matrices were downloaded and analyzed. RNA-Seq expression profiling was performed on fresh tumor samples as described above. Significantly differentially higher or lower expressed genes in tumors from patients with chronic ASI use vs. no ASI use were identified as two separate gene sets. These genes are PDAC-specific ASI response gene signatures and were used for pathway deregulation analysis. Briefly, we used a previously described algorithm called Pathifier to calculate a pathway deregulation score for either differentially higher or lower expressed genes in each individual patient(12). This single score is a continuous variable that represents the expression level of the entire gene set as a pathway. Patients were divided into high, medium or low expression categories based on this deregulation score, and the association of expression categories with overall survival was calculated. Kaplan-Meier and Cox proportional hazard analyses were performed using SPSS v22.
Results
944 PDAC patients visited MGH from January 2006 to December 2010. Patients were followed for periods ranging from 1 month to 9 years, with a median follow-up of 11 months. We excluded 150 patients who did not have complete follow-up information because they received follow up treatment outside of MGH. The remaining 794 Patients with complete oncological history were divided into three groups: group A patients (n=310) did not receive resection because they already presented with metastatic disease, group B patients (n=185) did not receive resection due to locally advanced disease, while group C patients (n=299) had resectable disease and successfully underwent primary tumor resection (Supplementary Figure 1). 480 of 794 patients has hypertension, among which 289 patients were chronic ASI users. 8 patients were on chronic ASI for congestive heart failure. Non-ASI users included all other patients, including patients who were normotensive and never used ASI (n=306), short term ASI users [less than 1 month or less than 50% of follow up time in medical record (n=21)], and patients treated for hypertension with non-ASI anti-hypertensives (n=170).
ASI use is not associated with survival in patients with metastatic disease
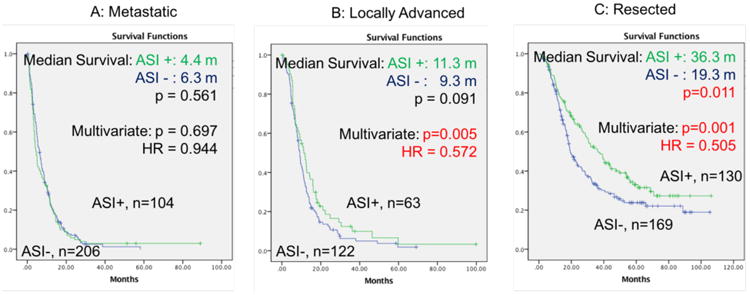
The characteristics of patients with metastasis are shown in Supplementary Table 1. Patients on ASI were significantly older (70.6 vs. 64.8) and had a higher proportion of tumor in the pancreatic head (42% vs. 27%). In the unadjusted univariate model, factors that were associated with better survival were younger age and chemotherapy treatment (Supplementary Table 1). The median survival of ASI+ and ASI- patients is 4.4 (95% CI: 3.3 - 5.5) and 6.3 (95% CI: 5.2 - 7.4) months (p=0.561), respectively (Figure 1A). In the multivariate model with adjustment for potential confounders (e.g. hypertension), tumor located in the head of the pancreas was also associated with better survival (Supplementary Table 1). However, chronic ASI use (HR=0.944 95% CI=0.705 - 1.263) was not significantly associated with overall survival in metastatic patients.
Figure 1. Unadjusted Kaplan Meier curves for overall survival in metastatic (A), locally advanced (B) and resected (C) patients. HR = Hazards ratio.
ASI use is associated with increased survival in patients with locally advanced disease
In patients with locally advanced disease, all parameters besides hypertension were comparable between chronic ASI users and ASI non-users (Supplementary Table 2). In the unadjusted univariate analysis younger age, radiotherapy treatment, and chemotherapy treatment were associated with better survival (Supplementary Table 2). The median OS of ASI+ and ASI- patients was 11.3 (95% CI: 8.1 - 14.5) vs. 9.3 (95% CI: 8.4 - 10.2) months (p=0.091), with a 5-year survival rate of 3.3±3.0% vs. 1.9±1.7%, respectively (Figure 1B). In the adjusted multivariate model, chronic ASI use was independently associated with significantly increased survival (HR, 0.572; 95%CI, 0.386-0.847; p=0.005) after adjusting for other covariates.
ASI use is independently associated with longer overall survival in resected patients
The tumor resection rate of ASI users (130/297, 43.8%) is significantly higher than for ASI naïve patients (169/497, 34%), (p<0.05). Resected ASI users are older (69.1 vs. 64.6), have a higher rate of being hypertensive and have a higher BMI compared to resected non-ASI users. All other parameters were distributed equally between chronic ASI users and non-users (Table 1). In the unadjusted univariate model, ASI use, as well as smaller tumor size, negative surgical margin, lower lymph node ratio (LNR), lower histological grade, absence of lymphovascular invasion (LVI), and absence of perineural invasion (PNI) were associated with better survival. Median survival of ASI+ and ASI- patients was 36.3 months (95% CI: 28.3 - 44.3) vs. 19.3 months (95% CI: 15.7 - 22.9) (p=0.011), respectively (Figure 1C), with a 5-year survival rate of 31.6±4.6% vs. 23.8±3.7%. ASI use is associated with lower risk for overall death (HR=0.69, 95% CI: 0.52 – 0.92, p=0.012). There was no difference in survival between patients on ARB vs. ACEi (p=0.9) (Supplementary Figure 2).
Table 1.
Patient characteristics, unadjusted univariate model, and adjusted analysis with multivariate model, of overall survival in patients with resected primary tumor.
| Patient Characteristics | Unadjusted Univariate Model | Adjusted Analysis with Multivariate Model | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| no ASI | ASI | Total | p- value | FDR q-value | n | Median Survival (months) | 95% CI | p value | FDR q-value | HR | 95% CI | p value | FDR q-value | ||
|
|
|||||||||||||||
| Age | y | 64.6 ± 10.7 | 69.1 ± 10.0 | 299 | <0.001 | 0.007 | 299 | 1.002 (HR) | 0.99 - 1.02 | 0.758 | 0.816 | 0.999 | 0.999 - 1.014 | 0.911 | 0.911 |
|
|
|||||||||||||||
| Size | cm | 2.9 ± 1.1 | 3.0 ± 1.5 | 299 | 0.625 | 0.813 | 299 | 1.14 (HR) | 1.06 - 1.23 | <0.001 | 0.005 | 1.161 | 1.061 - 1.271 | 0.001 | 0.005 |
|
|
|||||||||||||||
| BMI | kg/m̂2 | 25.4 ± 4.6 | 27.4 ± 5.7 | 299 | 0.02 | 0.0867 | 299 | 0.984 (HR) | 0.956 - 1.012 | 0.261 | 0.332 | 0.99 | 0.960 - 1.021 | 0.533 | 0.622 |
|
|
|||||||||||||||
| Site | Head | 40 (24 %) | 33 (25 %) | 73 (24 %) | 0.732 | 0.848 | 73 | 26.7 | 16.9 -36.6 | 0.941 | 0.941 | 0.96 | 0.663 - 1.389 | 0.828 | 0.892 |
| Other | 129 (76 %) | 97 (75 %) | 226 (76 %) | 226 | 26.5 | 20.6 -32.4 | |||||||||
|
|
|||||||||||||||
| Margin | - | 137 (81 %) | 107 (82 %) | 244 (82 %) | 0.783 | 0.848 | 244 | 29.9 | 23.3 -36.6 | 0.002 | 0.006 | 1.554 | 1.068 - 2.261 | 0.021 | 0.049 |
| + | 32 (19 %) | 23 (18 %) | 55 (18 %) | 55 | 18.8 | 15.9 -21.7 | |||||||||
|
|
|||||||||||||||
| LNR | 0 | 49 (29 %) | 45 (35 %) | 94 (32 %) | 0.393 | 0.754 | 94 | 49.6 | 33.8 -65.3 | <0.001 | 0.005 | 1.503 | 1.209 - 1.869 | <0.001 | 0.005 |
| <= 0.2 | 65 (39 %) | 52 (40 %) | 117 (39 %) | 117 | 24.9 | 17.4 -32.4 | |||||||||
| >0.2 | 54 (32 %) | 33 (25 %) | 87 (29 %) | 87 | 16.3 | 12.9 -19.7 | |||||||||
|
|
|||||||||||||||
| Grade | 1 & 2 | 103 (61 %) | 75 (58 %) | 178 (60 %) | 0.57 | 0.813 | 178 | 32.5 | 22.7 -42.3 | 0.002 | 0.006 | 1.524 | 1.133 - 2.050 | 0.005 | 0.018 |
| 3 & 4 | 66 (39 %) | 55 (42 %) | 121 (40 %) | 121 | 18.3 | 14.5 -22.1 | |||||||||
|
|
|||||||||||||||
| LVI | - | 72 (43 %) | 48 (37 %) | 120 (40 %) | 0.3 | 0.754 | 120 | 39.6 | 23.9 -55.4 | <0.001 | 0.005 | 1.138 | 0.810 - 1.600 | 0.456 | 0.58 |
| + | 96 (57 %) | 82 (63 %) | 178 (60 %) | 178 | 19.8 | 16.1 -23.6 | |||||||||
|
|
|||||||||||||||
| PNI | - | 20 (12 %) | 15 (12 %) | 35 (12 %) | 0.908 | 0.908 | 35 | 54.1 | NA | 0.004 | 0.009 | 1.519 | 0.892 - 2.587 | 0.124 | 0.193 |
| + | 147 (88 %) | 115 (88 %) | 262 (88 %) | 262 | 23.5 | 17.9 -29 | |||||||||
|
|
|||||||||||||||
| Naj Tx | - | 128 (76%) | 107 (82 %) | 235 (79 %) | 0.17 | 0.553 | 235 | 25.4 | 19.4 -31.4 | 0.22 | 0.308 | 0.796 | 0.530 - 1.196 | 0.272 | 0.381 |
| + | 41 (24%) | 23 (18 %) | 64 (21 %) | 64 | 28.7 | 20.8 -36.6 | |||||||||
|
|
|||||||||||||||
| Adj RT | - | 112 (66 %) | 82 (63 %) | 194 (65 %) | 0.566 | 0.813 | 194 | 21.7 | 15 -28.4 | 0.124 | 0.217 | 0.712 | 0.503 - 1.007 | 0.055 | 0.096 |
| + | 57 (34 %) | 48 (37 %) | 105 (35 %) | 105 | 31.7 | 23 -40.4 | |||||||||
|
|
|||||||||||||||
| Adj Chemo | - | 36 (21 %) | 33 (25 %) | 69 (23 %) | 0.406 | 0.754 | 69 | 16.2 | 8.8 -23.6 | 0.202 | 0.308 | 0.597 | 0.398 - 0.896 | 0.013 | 0.036 |
| + | 133 (79 %) | 97 (75 %) | 230 (77 %) | 230 | 28.3 | 23.1 -33.5 | |||||||||
|
|
|||||||||||||||
| HTN | - | 114 (67 %) | 2 (2 %) | 116 (39 %) | <0.001 | 0.007 | 116 | 21.6 | 13.8 -29.4 | 0.535 | 0.624 | 1.507 | 1.010 - 2.249 | 0.044 | 0.088 |
| + | 55 (33 %) | 128 (98 %) | 183 (61 %) | 183 | 28.7 | 21.3 -36.1 | |||||||||
|
|
|||||||||||||||
| ASI | - | 169 | 19.3 | 15.6 -22.9 | 0.011 | 0.022 | 0.505 | 0.339 - 0.750 | 0.001 | 0.005 | |||||
| + | 130 | 36.2 | 28.2 -44.3 | ||||||||||||
BMI=body mass index, LNR=lymph node ratio, LVI=lymphovascular invasion, PNI=perineural invasion, Naj=neoadjuvant, Adj=adjuvant, RT=radiotherapy, Chemo=chemotherapy, HTN=hypertension, ASI=angiotensin system inhibitors.
Cox proportional hazard analysis (Table 1) adjusted for potential confounding covariates showed that ASI use (HR, 0.505; 95%CI, 0.339 - 0.750; p=0.001) as well as smaller tumor size, negative surgical margin, lower LNR, lower grade, and adjuvant chemotherapy were associated with increased survival. Notably, our adjusted analysis with multivariate modeling revealed ASI use as an independent factor associated with overall survival with the lowest hazards ratio, followed by chemotherapy. Chronic ASI use, alone or combined with chemotherapy, provided unique survival benefit after adjustment for all parameters in the Cox model.
We then performed stratified analysis based on treatment. In patients that did not receive adjuvant chemotherapy after resection – most commonly because patients refused chemotherapy, the median survival was of 39.2 for ASI users and 9.3 months for ASI naïve patients (p = 0.01). In patients treated with chemotherapy, median OS of ASI users and ASI naïve patients was of 28.7±9.8 and 12.3±4.9 months (p=0.048), respectively (Supplementary Figure 3 and Supplementary Table 3). Additional stratified analysis showed that ASI provide a significant survival benefits in patients with BMI at diagnosis <25 (n=139, median survival ASI 44 vs. no-ASI 17 months, p=0.005), while in patients with BMI > 25 (n=160) the median survival of ASI users (34 months) versus ASI-naïve patients (21 months) was also longer but not significant (Supplementary Figures 4). In summary, our data suggest a strong independent benefit of ASI use in patients with resected disease.
Propensity score analysis
We performed propensity-adjusted analysis and inverse probability weighted analysis in all three groups of patients included in our analysis. There was no benefit of ASI use in patients with metastasis, while ASI use correlated with longer survival in locally advanced and resected patients (Supplementary Table 4A). We also performed the inverse probability weighted analysis which showed similar results indicating a significant benefit of ASI use in non-metastatic patients (Supplementary Table 4B).
In resected patients, ASI use is associated with longer time to recurrence
Additionally, we found that chronic ASI use correlated with a longer time to recurrence. The prolongation of time to recurrence is mainly due to longer time to distant metastasis, rather than to a reduction of local recurrence (Supplementary Figure 5). We performed competing risk analysis, treating the local recurrence, recurrence in the liver, lung, other distant site (peritoneal and ascites) as well as death as competing risks. We found that the hazards ratio of lung recurrence was not affected with ASI use (Supplementary Table 5). We continued with cause specific survival analysis, treating recurrence in each site as a separate event in distinctive analysis. Chronic ASI use was strongly correlated with longer time to recurrence in the liver in both unadjusted univariate and multivariate analyses (Supplementary Figure 5C and Supplementary Table 6). Time to recurrence in the lung and other sites was not correlated with ASI use, likely because liver is the most common site for PDAC metastasis, and thus there are high censor rates for these other sites.
RNA-Seq identifies pathways altered by chronic ASI use
To gain mechanistic insight into the association between ASI use and longer survival in resected PDAC patients, we prospectively collected treatment-naïve PDAC samples (4 lisinopril-treated patients vs. 4 controls) and performed RNA-Seq. We chose lisinopril because it is the most commonly used ASI in our cohort. With 4 samples in each condition, we estimated a 70% power to detect 50% of the genes that are differentially expressed with a 2.5× fold change(13). A total of 148 genes were differentially expressed (FDR q-value < 0.05) in PDAC lesions of lisinopril-users versus non-ASI users (Supplementary Table 7). The expression of 80 genes was higher and 68 genes lower in the PDAC lesions of lisinopril-users. To systematically analyze our RNA-Seq results, we performed gene annotation enrichment analysis, Gene ontology (GO) (http://geneontology.org/page/go-enrichment-analysis). In the cellular component category, gene sets associated with intermediate filaments and ECM remodeling were less expressed in lisinopril versus non-ASI lesions (Table 2A). Similarly, analysis of REACTOME gene sets(http://www.reactome.org) revealed that differentially expressed genes were involved in ECM remodeling and organization (Table 2B). For example, ECM (TNC, COL4A5, HAPLN1) and matrix degrading enzyme (MMP10, MMP13) genes were expressed at significantly lower levels in the lesions of lisinopril than non-ASI patients (Table 3). In lesions of lisinopril patients the WNT signaling ligand WNT10a, which is known to enhance fibrosis(14), was also less expressed, while WISP2 – which plays a role in the WNT-1 signaling pathway, and inhibits fibrosis and invasion – was highly expressed (Table 3). Complete results of the GO and REACTOME analysis are presented in Supplementary Tables 8 and 9. Our results indicate that ASI/lisinopril can induce a normalization of the tumor stroma.
Table 2. GO and REACTOME analysis for differentially expressed genes.
| A: PANTHER Overrepresentation Test, using GO Ontology database, on genes that are differentially downregulated with ACEi use in human PDAC tissue | ||
|---|---|---|
| Gene Ontology enrichment analysis | ||
| GO cellular component complete | P-Value | FDR q value |
| intermediate filament (GO:0005882) | 0.0053 | 0.0092 |
| intermediate filament cytoskeleton (GO:0045111) | 0.0181 | 0.0253 |
| proteinaceous extracellular matrix (GO:0005578) | 0.0254 | 0.0296 |
| extracellular matrix (GO:0031012) | 0.0486 | 0.0486 |
| extracellular space (GO:0005615) | <0.0001 | 0.0002 |
| extracellular region part (GO:0044421) | <0.0001 | 0.0002 |
| extracellular region (GO:0005576) | <0.0001 | 0.0002 |
| B: REACTOME pathway enrichment analysis | ||
| REACTOME pathway analysis | ||
|
| ||
| Pathway name | P-Value | FDR q value |
|
| ||
| Keratinization | 1.11E-16 | 7.77E-16 |
| Formation of the cornified envelope | 1.11E-16 | 7.77E-16 |
| Developmental biology | 1.53E-12 | 7.14E-12 |
| Collagen degradation | 3.03E-06 | 1.06E-05 |
| Oxidative stress Induced senescence | 7.11E-06 | 1.99E-05 |
| Activation of matrix metalloproteinases | 1.65E-05 | 3.42E-05 |
| Assembly of collagen fibrils and other multimeric structures | 1.71E-05 | 3.42E-05 |
| Extracellular matrix organization | 2.74E-05 | 4.80E-05 |
| Oncogene Induced senescence | 3.49E-05 | 5.17E-05 |
| Collagen formation | 3.69E-05 | 5.17E-05 |
| Degradation of the extracellular matrix | 4.72E-05 | 6.01E-05 |
| Regulation of pyruvate dehydrogenase (PDH) complex | 6.67E-05 | 7.78E-05 |
| Cellular senescence | 3.64E-04 | 3.92E-04 |
| Pyruvate metabolism | 7.77E-04 | 7.77E-04 |
Table 3. Representative genes differentially expressed in human PDAC with lisinopril use.
| Name | Descriptor | Control | Lisinopril | Fold | FDR q-value |
|---|---|---|---|---|---|
| TNC | tenascin C | 21.5 | 3.7 | -5.8 | 0.0093 |
| HAPLN1 | hyaluronan and proteoglycan link protein 1 | 9.56 | 0.27 | -5.14 | 0.009 |
| COL4A5 | collagen type IV alpha 5 chain | 8.4 | 1.6 | -5.3 | 0.0093 |
| MMP10 | matrix metalloproteinase-10 | 13.7 | 1.1 | -12.5 | 0.0093 |
| MMP13 | matrix metalloproteinase-13 | 11 | 0.4 | -27.5 | 0.0145 |
| WNT10A | Wnt family member 10A | 16.87 | 2.6 | -2.7 | 0.0345 |
| WISP2 | WNT1-inducible-signaling pathway protein 2 | 1.64 | 64.73 | 5.3 | 0.0093 |
| CCl4 | chemokine ligand 4 | 5 | 35.5 | 7.1 | 0.028 |
| CD209 | DC-SIGN | 0.7 | 5.4 | 7.7 | 0.0145 |
| CCL21 | chemokine ligand 21 | 5.2 | 42.1 | 8.2 | 0.0145 |
| IRF8 | Interferon regulatory factor 8 | 6 | 28.4 | 4.7 | 0.0093 |
| WT1 | Wilm's tumor protein | 0.8 | 8.7 | 10.9 | 0.0093 |
| HLA-DQB1 | MHC class II DQ beta 1 | 54.8 | 154.5 | 2.8 | 0.056 |
| TNFRSF8 | TNF Rc superfamily member 8 | 0.39 | 4.12 | 10.6 | 0.028 |
Finally, we performed gene set enrichment analysis (GSEA) using the complete expression data set. The GSEA results in PDAC-lesions of lisinopril-users versus ASI-naïve patients showed that gene sets linked to integrin signaling, Notch, WNT, and the cell cycle were under-expressed, while pathways linked to oxidative phosphorylation, PPAR signaling, normal pancreas function and anti-tumor immune response were over-expressed (Figure 2). In lesions of lisinopril-users we found an enrichment for gene sets linked to T cell activity, and antigen processing and presentation (Figure 2 and Supplementary Table 10). Several genes that were differentially expressed are also associated with the functional activity of T cells and antigen presenting cells (APCs). In PDAC lesions of ASI-users, we found a higher expression of gene transcripts for CCL4 – a chemokine which stimulates the recruitment of immature dendritic cells (DCs) and Th1-polarized T cells (15), the DC marker CD209, and CCL21 and IRF8, two genes that play significant roles in the differentiation / maturation of DCs (Table 3) (16,17). Furthermore, lisinopril increased the expression of the WT1 gene – a tumor-associated antigen – and MHC class II gene HLA-DQB1 expressed by APCs (Table 3). The increased DC/APC activity in lisinopril-treated PDAC lesions was associated with a higher expression of TNFRSF8 – expressed by activated T-cells and B-cells – which promotes the survival of memory T cells (18). The complete GSEA analysis including GO, BIOCARTA, KEGG, PID, and REACTOME pathways are included in Supplementary Table 11. Collectively, our results suggest that lisinopril use normalizes the PDAC microenvironment, reduces PDAC progression and increases anti-tumor immunity.
Figure 2.
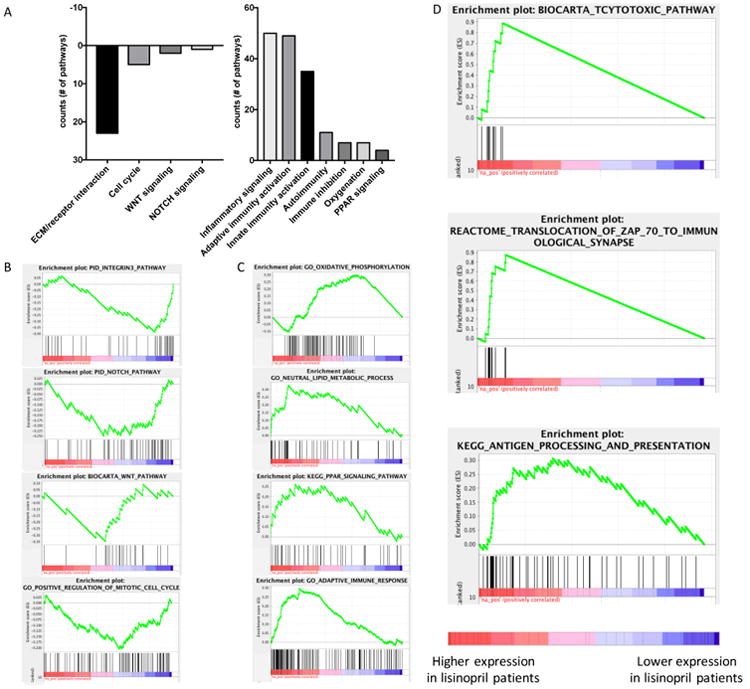
A: Number of GO, BIOCARTA, KEGG, PID, and REACTOME gene sets that are significantly changed (FDR<0.05) in our GSEA analysis, grouped by biological function. (Complete GSEA results are shown in Supplementary Table 9). Gene Set Enrichment Analysis (GSEA) of human PDAC comparing ACEi treated tumors vs. control tumors. B: Decrease in the activity of integrin beta 3, NOTCH, WNT and the cell cycle. C: Increase in oxidative phosphorylation, improvement in lipid metabolism, PPAR signaling, and adaptive immune response. D: Increase in cytotoxic activities, immuno-synapse and antigen presentation pathways (Detailed enrichment score in Supplementary Table 11).
Expression signature induced by ASI use alone is associated with longer overall survival
The survival advantage associated with chronic ASI use in non-metastatic patients as well as the gene expression changes induced by lisinopril prompted further analysis in independent patient cohorts. We intersected our RNA-Seq results with publicly available primary PDAC gene expression data that also included survival information. Two data sets are used in our study: TCGA (n=178) and UNC datasets(11) (n=125). First, we investigated in our RNA-Seq data the expression of genes differentially lower expressed in PDAC lesions of lisinopril-users (Supplementary Table 7), in these two independent cohorts. Using the algorithm Pathifier(12), we calculated a deregulation score – collapsing the expression level of all lower expressed ASI genes into one measurement – for each patient. Next, we divided patients in each cohort into three groups (low, medium, high) based on their deregulation score. In the UNC (Figure 3A) and TCGA (Figure 3B) cohorts, patients in the low category – those with the lowest expression of genes which also had lower expression in lisinopril using patients – lived significantly longer than patients with high or mid-level expression. In the TCGA dataset, which was the only data set to also provide other clinical parameters, the low expression category remained significant after correcting for tumor size, lymph node status, and other potential confounders (Supplementary Table 12). Stratification of patients based on genes that were expressed higher in ASI treatment in our RNA-Seq data set did not reach statistical significance (Supplementary Table 12). This indicates that genes associated with pancreatic tumor progression and extracellular matrix production, which has a lower expression in lisinopril treated patients, likely play a significant role in the observed survival benefit.
Figure 3.
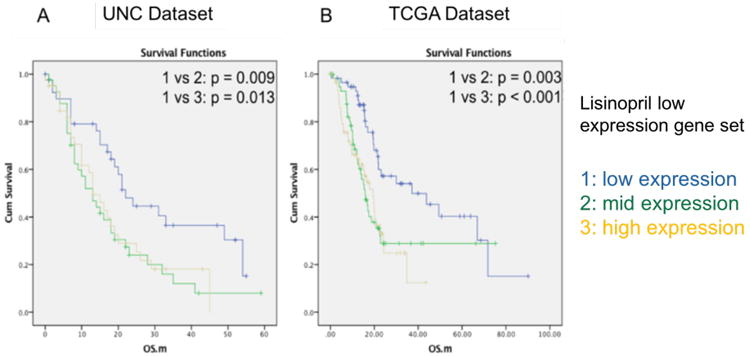
Kaplan Meier analysis on ASI down regulated gene pathway and overall survival. ASI low pathway expression level: 1 = low, 2 = med, 3 = high. A: UNC dataset. B: TCGA dataset, overall survival. UNC Dataset overall Kaplan Meier analysis p=0.017, HR =0.513, 95% CI = 0.292 - 0.900. TCGA Dataset overall Kaplan Meier analysis p=0.002, HR =0.422, 95% CI = 0.248 - 0.718.
Discussion
We retrospectively analyzed the effect of chronic ASI use on survival in patients with all stages of PDAC. Adjusted analysis with multivariate modeling showed that chronic ASI use was an independent factor associated with longer overall survival in PDAC patients with resected disease or locally advanced disease, but not in patients with metastatic disease. The benefit provided by ASI is independent of chemotherapy or radiotherapy, as well as other pathological features known to be associated with survival. Propensity score matched analysis showed longer OS in chronic ASI users as well. To determine the biological mechanisms of ASI action, we performed RNA sequencing and identified functional gene categories altered by ASI. Our results reveal that the ASI/ACEi lisinopril therapy is associated with normalization of the tumor stroma, reduced tumor progression and anti-tumor immunity. We also identified a signature of genes downregulated by lisinopril, which is significantly associated with patient survival in independent validation cohorts.
Previous retrospective analyses of patients with locally advanced and metastatic PDAC treated with gemcitabine revealed that ASI-therapy increased overall survival from 8.9 months to 15.1 months(19). A prospective phase I trial comparing gemcitabine with or without candesartan in patients with locally advanced or metastatic PDAC reported encouraging survival data(20). However, the subsequent phase II trial did not show a survival advantage for candesartan combined with gemcitabine versus gemcitabine alone(21). These conflicting results might have resulted from heterogeneous patient cohorts, which included patients with locally advanced as well as metastatic disease. Our results do not conflict with those studies, showing that patients with locally advanced PDAC benefit from RAAS inhibition, while there is no survival advantage for patients with metastasis.
In patients who underwent primary tumor resection, ASI use is also associated with a significant increase in median survival of 17 months (19.3 vs 36.3 months) in unadjusted univariate analysis, and a HR as low as 0.505 (0.339 - 0.750) in adjusted analysis with multivariate modeling. Furthermore, our subgroup analysis suggests that ASI therapy with or without chemotherapy improves the survival of PDAC patients who underwent pancreatectomy. In a small number of hypertensive patients using ASI (N=22) that did not receive adjuvant chemotherapy following surgery we found that median survival was of 39.2 months, which is surprising given that the other recorded parameters between ASI users and naïve patients were comparable (Table 1). In comparison, the median survival for patients who underwent surgery alone has been reported to vary between 13.0 and 20.2 months(22,23), while in patients who received adjuvant chemotherapy after surgery median survival can vary between 22.6 and 23.6 months(22,23).
Our transcriptome analysis suggests that the ASI lisinopril can induce changes in ECM remodeling and organization, increase oxidative phosphorylation and reduce the activity of pro-fibrotic pathways like RAAS and WNT. Pro-fibrotic pathways are known to promote tumor desmoplasia, which acts as a physical barrier to drug delivery and immune cell infiltration(24-26), thus reducing the efficacy of chemotherapy and immunotherapy. Desmoplasia compresses blood vessels and promotes a hypoxic tumor microenvironment which further aggravates immunosuppression(6,27,28). In murine models, we have shown that inhibition of the RAAS reduces tumor desmoplasia and improves vascular perfusion and drug delivery(6). Consequently, hypoxia is relieved and the efficacy of chemotherapeutic drugs, oncolytic viruses or vaccine therapy is significantly enhanced(5,6,29). Thus, our findings suggest that changes in ECM remodeling and organization could explain the increase in oxidative phosphorylation and the improved efficacy of chemotherapy in ASI users.
The survival advantage of ASI use by itself – revealed by our adjusted multivariate model analysis – could also be related to anti-tumor immunity, and altered proliferation or aggressiveness of cancer cells. Our GSEA analysis showed that cell cycle gene sets were under-represented in PDAC lesions of lisinopril-users versus ASI-naïve patients. A reduction in cell proliferation induced by ASI is consistent with previous reports that the angiotensin II receptor type 1 (AT1R) blocker losartan inhibits the growth of tumor cells overexpressing AT1R(3). Angiotensin receptors are also expressed in human PDAC and pancreatic cancer cell lines(30), and ASI can directly inhibit pancreatic cancer growth in vitro and in vivo(31). Other studies suggest an anti-angiogenic mechanism of ASI in PDAC models. In PDAC lesions of lisinopril-users our GSEA results also showed a lower expression of gene sets associated with integrin-mediated interaction of cells with the ECM, and WNT and Notch signaling. In experimental PDAC models, WNT and Notch signaling promote early tumorigenesis(32,33). Furthermore, the expression of WNT signaling proteins (β-catenin, WNT2) in PDAC lesions correlates with reduced survival of patients with a pancreatectomy(34). Additionally, the activation of WNT signaling increases the formation of lymph node and liver metastases, but does not induce metastatic seeding in the lung(34). Thus, ASI inhibition of WNT signaling could inhibit metastatic spread to the liver. This is consistent with our data showing that metastatic recurrence in the liver – but not in the lung – was significantly reduced in chronic ASI users.
In PDAC the intratumoral infiltrations of myeloid cells, macrophages, B cells, neutrophils and Tregs contributes to the immunosuppressive microenvironment (35-37), while the poor recruitment of CD8-positive T-cells and antigen presenting DCs correlates with poor prognosis (38). The RAAS can stimulate the immunosuppressive function of monocytes, tumor-associated macrophages and neutrophils(39-41). Our results suggest that ASI therapy stimulates anti-tumor immunity. The resected PDAC samples of lisinopril-users were enriched for genes sets linked to antigen processing and presentation, and activity of T cells. We have recently shown in obese mice that losartan decreases the recruitment and immunosuppressive effects of neutrophils, and increases the recruitment of CD8-positive T cells in PDAC(40). In another study, Vanpouille-Box et al showed that local radiation and blockade of TGF-β, which is downstream of RAAS pathway, can be used to enhance autovaccination in PDAC(42). These findings suggest that inhibition of TGF-β or RAAS signaling could stimulate the efficacy of immunotherapy in PDAC.
Hypertension is a common co-morbidity for this disease. ASIs are also widely used for the treatment of hypertension. Other commonly encountered medical conditions or medications have been shown to correlate with survival in PDAC patients. For example, long-term but not recent diabetes (less than 4 years before diagnosis) has a negative impact on survival(43). Similarly, the use of metformin, a drug used to treat diabetes, increased survival in preclinical PDAC models(44) and correlated with increased survival in retrospective analyses(45). However, in other retrospective studies, metformin use did not show a survival advantage(46). A randomized controlled phase II trial testing metformin in patients with advanced PDAC treated with gemcitabine and erlotinib showed no benefit(47). Diabetes in PDAC is more complicated compared to other cancer types, since the pancreas is a vital organ that maintains blood glucose levels. Diabetes in PDAC patients can be unrelated to pancreatic cancer; caused by pancreatic cancer; or caused by pancreatectomy.
Due to the known effect of obesity on PDAC progression and survival, we also addressed the combined effect of BMI and ASI on PDAC survival. Our stratified analysis showed that ASI provide a significant survival benefit in patients with BMI < 25, while in ASI users with a BMI > 25 the survival was longer but not significant. ASI may still have beneficial effects in obese patients. In a mice model we found that losartan-inhibition of the RAAS decreased obesity-induced inflammation, fibrosis, and tumor growth(40,48). In our pre-clinical study on obesity, we did not assess the effect of losartan on survival. The time of BMI assessment could also affect our findings. In other patient cohorts, baseline BMI measured two to several years prior to PDAC diagnosis correlates with overall survival(49). However, BMI at the time of diagnosis or after treatment is less clearly associated with survival, especially since low body weight associated with cachexia is also a known risk factor of poor survival. Further analysis of how obesity, diabetes and metformin affect the treatment outcome of ASI users should be evaluated in prospective pancreatic cancer trials.
Our results advocate for an early and prolonged angiotensin system inhibition in PDAC patients. With the changing paradigm in favoring early systemic treatment, and the promising efficacy of neo-adjuvant FOLFIRINOX and chemoradiation therapy in locally advanced/borderline disease(50), ASI could be added to the pre-operative cytotoxic therapy. In our ongoing phase II clinical trial at Massachusetts General Hospital (ClinicalTrials.gov identifier: NCT01821729), we are currently testing the efficacy of the AT1R blocker losartan combined with FOLFIRINOX followed by chemoradiation in patients with locally advanced PDAC.
For patients with resectable disease, prospective trials should be designed to test the benefit of the addition of ASI to neo-adjuvant chemotherapy and the continuous administration of ASI following surgical resection. The results of our retrospective analysis suggest that ASI improves survival in non-metastatic patients but not in metastatic patients. Thus, it will be critical to determine in experimental models of primary versus metastatic PDAC how ASIs alone or combined with cytotoxic agents or immunotherapies affect RAAS signaling, the tumor microenvironment, adaptive and innate immune cells, and tumor response. A translational bench to bedside approach will likely identify how to best combine ASI with other agents for the treatment of PDAC patients.
Limitations
Our retrospective analysis is based on unselected single institute experience combined with other independent retrospective cohorts. The retrospective nature of this study means it is prone to selection bias. In our cohort, the percentage of patients who presented with resectable disease was higher compared to national statistics. For the adjusted multivariate modeling, we included all available parameters. We also performed propensity score analysis. However, there may still be other un-recognized confounders.
Second, the duration of ASI usage before the time of diagnosis was difficult to assess. However, a significant fraction of patients was on ASI because of hypertension, which is a chronic condition. Very few patients switched medications during the follow-up. We thus assumed that all patients who repeatedly had ASI in their active medication were long-term ASI users. We also used propensity score weighted analysis to adjust for the potential bias regarding the probability of patients receiving ASI treatment. Our conclusions remained the same after propensity score analysis.
Third, the sample size of the prospective clinical tumor collection for RNA-seq anlysis is relatively small. During the time of prospective sample collection, a neoadjuvant chemotherapy trial was ongoing, which explains in part the relatively low number of treatment naive PDAC cases included in our analysis. Even though the sample size was small, our transcriptome analysis identified key gene sets and pathways that confirmed known effects of ASI on RAAS inhibition, and revealed novel pathways/mechanisms, which we will explore in greater depth in future studies.
Conclusions
In summary, our retrospective analysis shows that the chronic use of ASI is associated with significantly longer overall survival in PDAC patients with non-metastatic disease, independent of anti-cancer treatment or tumor characteristics. ASI-use likely has microenvironment normalizing and immunostimulatory effects in PDAC patients, and is also associated with an expression signature predictive of patient survival. Prospective randomized trials are needed to confirm the efficacy of ASI use in PDAC.
Supplementary Material
Translational Relevance.
Angiotensin system inhibitors (ASIs) are widely used to treat hypertension. ASIs have additional cardiac and renal protective effects, due to their modulation of the immune system and wound healing. In this study, we examined the effect of long-term ASI use on the survival of patients with pancreatic ductal adenocarcinoma (PDAC), and explored its potential mechanisms. Our findings indicate that chronic ASI use is independently associated with longer overall survival in non-metastatic PDAC patients. Unbiased gene expression profiling suggested that the improved survival associated with ASI therapy might be due to normalization of the extracellular matrix, inhibition of tumor progression and enhanced anti-tumor immunity. Our results advocate for prospective clinical trials to assess specific ASIs as adjuncts to primary tumor resection in PDAC. With the increasing use of neoadjuvant chemotherapy in PDAC, reprogramming of the extracellular matrix and immune microenvironment with ASIs has a great potential.
Acknowledgments
We would like to thank Ivy Chen, Mengyang Di, Meenal Datta, Jelena Grahovac, and Giorgio Seano for their insightful comments on this manuscript. The results here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Financial Support: This work was supported by grants from NCI P01CA080124 (RKJ, YB), the Lustgarten Foundation (RKJ), and the Erwin Schroedinger fellowship from the Austrian Science Fund (FWF; project number: J 3747-B28) to MP. R.M.E. and M.D. were supported in part by a Stand Up to Cancer- Dream Team Translational Cancer Research Grant (Grant Number SU2C-AACR-DT0509). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. R.M.E is an investigator of the Howard Hughes Medical Institute and March of Dimes Chair in Molecular and Developmental Biology at the Salk Institute and is supported by a grant from the Lustgarten Foundation Ipsen/Biomeasure and a gift from the Freeborn foundation.
Footnotes
Conflict of Interest Disclosure statement: R.K.J. received consultant fees from Ophthotech, SPARC, SynDevRx and XTuit. R.K.J owns equity in Enlight, SPARC, SynDevRx and XTuit, and serves on the Board of Directors of XTuit and Boards of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, Tekla Healthcare Opportunities Fund and Tekla World Healthcare Fund. Y.B. received consultant fees from XTuit. M.P. received travel grants and speaker fees from Bayer. No reagents or funding from these companies were used in these studies. R.K.J only had access to de-identified data.
Author contributions: R.K.J and Y.B. designed the studies. V.D, Y.B. and R.K.J provided IRB protocols for fresh tumor collection; Clinical database and medical records were accessed from retrospective protocols by J.M and C.R.F; H.Liu, M.P, J.I, A.J, T.M, D.S and A.Z. reviewed clinicopathological information; biostatistics analysis were perform by H.Liu, H.Lee and K.S with critical input from K.D.L., C.F.C., C.R.F., T.S.H., J.W.C., J.E.M., and D.P.R.; K.D.L., C.F.C. and V.D. provided fresh clinical samples; RNA extraction was performed by H.Liu, J.C. and W.H.; RNA sequencing was performed by M.D. and R.E.; RNA-Seq analysis was performed by H.Liu, Y.B. and K.N.; H.Liu, K.N., Y.B., and R.K.J. wrote the manuscript. All authors approved the final version of the manuscript.
References
- 1.Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. 2010;2:247–57. doi: 10.1002/emmm.201000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.George AJ, Thomas WG, Hannan RD. Nat Rev Cancer. Vol. 10. Nature Publishing Group; 2010. The renin–angiotensin system and cancer: old dog, new tricks; pp. 745–59. [DOI] [PubMed] [Google Scholar]
- 3.Rhodes DR, Ateeq B, Cao Q, Tomlins SA, Mehra R, Laxman B, et al. AGTR1 overexpression defines a subset of breast cancer and confers sensitivity to losartan, an AGTR1 antagonist. Proceedings of the National Academy of Sciences. 2009;106:10284–9. doi: 10.1073/pnas.0900351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Research. 2000;60:2497–503. [PubMed] [Google Scholar]
- 5.Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci USA National Acad Sciences. 2011;108:2909–14. doi: 10.1073/pnas.1018892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4:2516. doi: 10.1038/ncomms3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proceedings of the National Academy of Sciences. 2012;109:15101–8. doi: 10.1073/pnas.1213353109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nia HT, Liu H, Seano G, Datta M, Jones D, Rahbari N, et al. Nat biomed eng. Vol. 1. Nature Publishing Group; 2016. Solid stress and elastic energy as measures of tumour mechanopathology; p. 0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy JE, Wo JY, Ferrone CR, Wenqing J, Yeap B, Blaszkowsky LS, et al. TGF-B1 inhibition with losartan in combination with FOLFIRINOX (F-NOX) in locally advanced pancreatic cancer (LAPC): Preliminary feasibility and R0 resection rates from a prospective phase II study. | 2017 Gastrointestinal Cancers Symposium | Virtual Meeting | Meeting Library [Internet] [cited 2017 Jan 22];J Clin Oncol. 2017 35(suppl 4S; abstract 386) Available from: http://meetinglibrary.asco.org/content/176740-195. [Google Scholar]
- 10.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols. 2012;7:562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, et al. Nat Genet. Nature Publishing Group; 2015. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma; pp. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drier Y, Sheffer M, Domany E. Pathway-based personalized analysis of cancer. Proceedings of the National Academy of Sciences. 2013;110:6388–93. doi: 10.1073/pnas.1219651110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Busby MA, Stewart C, Miller CA, Grzeda KR, Marth GT. Scotty: a web tool for designing RNA-Seq experiments to measure differential gene expression. Bioinformatics. 2013;29:656–7. doi: 10.1093/bioinformatics/btt015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuma A, Yamada S, Wang KY, Kitamura N, Yamaguchi T, Iwai Y, et al. Role of WNT10A-expressing kidney fibroblasts in acute interstitial nephritis. PLoS ONE. 2014;9:e103240. doi: 10.1371/journal.pone.0103240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukaida N, Sasaki SI, Baba T. Chemokines in cancer development and progression and their potential as targeting molecules for cancer treatment. Mediators Inflamm. 2014;2014:170381. doi: 10.1155/2014/170381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marsland BJ, Bättig P, Bauer M, Ruedl C, Lässing U, Beerli RR, et al. CCL19 and CCL21 induce a potent proinflammatory differentiation program in licensed dendritic cells. Immunity. 2005;22:493–505. doi: 10.1016/j.immuni.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 17.Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseño CG, Iwata A, et al. Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol. 2016;34:93–119. doi: 10.1146/annurev-immunol-032713-120204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabbagh L, Snell LM, Watts TH. TNF family ligands define niches for T cell memory. Trends in Immunology. 2007;28:333–9. doi: 10.1016/j.it.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 19.Nakai Y, Isayama H, Ijichi H, Sasaki T, Sasahira N, Hirano K, et al. Inhibition of renin-angiotensin system affects prognosis of advanced pancreatic cancer receiving gemcitabine. British Journal of Cancer. 2010;103:1644–8. doi: 10.1038/sj.bjc.6605955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakai Y, Isayama H, Ijichi H, Sasaki T, Kogure H, Yagioka H, et al. Phase I trial of gemcitabine and candesartan combination therapy in normotensive patients with advanced pancreatic cancer: GECA1. Cancer Sci. 2012;103:1489–92. doi: 10.1111/j.1349-7006.2012.02311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakai Y, Isayama H, Ijichi H, Sasaki T, Takahara N, Ito Y, et al. A multicenter phase II trial of gemcitabine and candesartan combination therapy in patients with advanced pancreatic cancer: GECA2. Invest New Drugs. 2013 doi: 10.1007/s10637-013-9972-5. [DOI] [PubMed] [Google Scholar]
- 22.Horowitz DP, Hsu CC, Wang J, Makary MA, Winter JM, Robinson R, et al. Adjuvant chemoradiation therapy after pancreaticoduodenectomy in elderly patients with pancreatic adenocarcinoma. Int J Radiat Oncol Biol Phys. 2011;80:1391–7. doi: 10.1016/j.ijrobp.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oettle H, Neuhaus P, Hochhaus A, Hartmann JT, Gellert K, Ridwelski K, et al. Adjuvant Chemotherapy With Gemcitabine and Long-term Outcomes Among Patients With Resected Pancreatic Cancer. Jama-J Am Med Assoc. 2013;310:1473. doi: 10.1001/jama.2013.279201. [DOI] [PubMed] [Google Scholar]
- 24.Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. Journal of Clinical Oncology. 2013;31:2205–18. doi: 10.1200/JCO.2012.46.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watt J, Kocher HM. The desmoplastic stroma of pancreatic cancer is a barrier to immune cell infiltration. Oncoimmunology. 2013;2:e26788. doi: 10.4161/onci.26788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whatcott CJ, Han H, Hoff Von DD. Orchestrating the Tumor Microenvironment to Improve Survival for Patients With Pancreatic Cancer: Normalization, Not Destruction. Cancer J. 2015;21:299–306. doi: 10.1097/PPO.0000000000000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jain RK, Martin JD, Stylianopoulos T. The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng. 2014;16:321–46. doi: 10.1146/annurev-bioeng-071813-105259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26:605–22. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proceedings of the National Academy of Sciences. 2012;109:17561–6. doi: 10.1073/pnas.1215397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arafat HA, Gong Q, Chipitsyna G, Rizvi A, Saa CT, Yeo CJ. Antihypertensives as novel antineoplastics: angiotensin-I-converting enzyme inhibitors and angiotensin II type 1 receptor blockers in pancreatic ductal adenocarcinoma. J Am Coll Surg. 2007;204:996–1005. doi: 10.1016/j.jamcollsurg.2007.01.067. discussion1005–6. [DOI] [PubMed] [Google Scholar]
- 31.Fujimoto Y, Sasaki T, Tsuchida A, Chayama K. Angiotensin II type 1 receptor expression in human pancreatic cancer and growth inhibition by angiotensin II type 1 receptor antagonist. FEBS Letters. 2001;495:197–200. doi: 10.1016/s0014-5793(01)02377-8. [DOI] [PubMed] [Google Scholar]
- 32.Avila JL, Kissil JL. Notch signaling in pancreatic cancer: oncogene or tumor suppressor? Trends in Molecular Medicine. 2013;19:320–7. doi: 10.1016/j.molmed.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Morris JP, Yan W, Schofield HK, Gurney A, Simeone DM, et al. Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Research. 2013;73:4909–22. doi: 10.1158/0008-5472.CAN-12-4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sano M, Driscoll DR, DeJesus-Monge WE, Quattrochi B, Appleman VA, Ou J, et al. Activation of WNT/β-Catenin Signaling Enhances Pancreatic Cancer Development and the Malignant Potential Via Up-regulation of Cyr61. Neo. 2016;18:785–94. doi: 10.1016/j.neo.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Research. 2007;67:9518–27. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 36.Clark CE, Beatty GL, Vonderheide RH. Immunosurveillance of pancreatic adenocarcinoma: insights from genetically engineered mouse models of cancer. Cancer Letters. 2009;279:1–7. doi: 10.1016/j.canlet.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 37.Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016;6:270–85. doi: 10.1158/2159-8290.CD-15-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. British Journal of Cancer. 2013;108:914–23. doi: 10.1038/bjc.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cortez-Retamozo V, Etzrodt M, Newton A, Ryan R, Pucci F, Sio SW, et al. Angiotensin II drives the production of tumor-promoting macrophages. Immunity. 2013;38:296–308. doi: 10.1016/j.immuni.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016;6:852–69. doi: 10.1158/2159-8290.CD-15-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447–62. doi: 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 42.Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, et al. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Research. 2015;75:2232–42. doi: 10.1158/0008-5472.CAN-14-3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan C, Rubinson DA, Qian ZR, Wu C, Kraft P, Bao Y, et al. Survival among patients with pancreatic cancer and long-standing or recent-onset diabetes mellitus. Journal of Clinical Oncology. 2015;33:29–35. doi: 10.1200/JCO.2014.57.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Incio J, Suboj P, Chin SM, Vardam-Kaur T, Liu H, Hato T, et al. Metformin Reduces Desmoplasia in Pancreatic Cancer by Reprogramming Stellate Cells and Tumor-Associated Macrophages. PLoS ONE. 2015;10:e0141392. doi: 10.1371/journal.pone.0141392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amin S, Mhango G, Lin J, Aronson A, Wisnivesky J, Boffetta P, et al. Metformin Improves Survival in Patients with Pancreatic Ductal Adenocarcinoma and Pre-Existing Diabetes: A Propensity Score Analysis. Am J Gastroenterol. 2016;111:1350–7. doi: 10.1038/ajg.2016.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ambe CM, Mahipal A, Fulp J, Chen L, Malafa MP. Effect of Metformin Use on Survival in Resectable Pancreatic Cancer: A Single-Institution Experience and Review of the Literature. PLoS ONE. 2016;11:e0151632. doi: 10.1371/journal.pone.0151632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kordes S, Pollak MN, Zwinderman AH, Mathôt RA, Weterman MJ, Beeker A, et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015;16:839–47. doi: 10.1016/S1470-2045(15)00027-3. [DOI] [PubMed] [Google Scholar]
- 48.Incio J, Tam J, Rahbari NN, Suboj P, McManus DT, Chin SM, et al. PlGF/VEGFR-1 Signaling Promotes Macrophage Polarization and Accelerated Tumor Progression in Obesity. Clin Cancer Res. 2016;22:2993–3004. doi: 10.1158/1078-0432.CCR-15-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan C, Bao Y, Wu C, Kraft P, Ogino S, Ng K, et al. Prediagnostic body mass index and pancreatic cancer survival. Journal of Clinical Oncology. 2013;31:4229–34. doi: 10.1200/JCO.2013.51.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferrone CR, Marchegiani G, Hong TS, Ryan DP, Deshpande V, McDonnell EI, et al. Radiological and surgical implications of neoadjuvant treatment with FOLFIRINOX for locally advanced and borderline resectable pancreatic cancer. Ann Surg. 2015;261:12–7. doi: 10.1097/SLA.0000000000000867. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.