

Abstract
Adeno-associated virus (AAV) vector delivery of an Apaf-1-dominant negative inhibitor was tested for its antiapoptotic effect on degenerating nigrostriatal neurons in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of Parkinson's disease. The wild-type caspase recruitment domain of Apaf-1 was used as a dominant negative inhibitor of Apaf-1 (rAAV-Apaf-1-DN-EGFP). An AAV virus vector was used to deliver it into the striatum of C57 black mice, and the animals were treated with MPTP. The number of tyrosine hydroxylase-positive neurons in the substantia nigra was not changed on the rAAV-Apaf-1-DN-EGFP injected side compared with the noninjected side. We also examined the effect of a caspase 1 C285G mutant as a dominant negative inhibitor of caspase 1 (rAAV-caspase-1-DN-EGFP) in the same model. However, there was no difference in the number of tyrosine hydroxylase-positive neurons between the rAAV-caspase-1-DN-EGFP injected side and the noninjected side. These results indicate that delivery of Apaf-1-DN by using an AAV vector system can prevent nigrostriatal degeneration in MPTP mice, suggesting that it could be a promising therapeutic strategy for patients with Parkinson's disease. The major mechanism of dopaminergic neuronal death triggered by MPTP seems to be the mitochondrial apoptotic pathway.
The neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) causes a parkinsonian syndrome in humans and primates after selective uptake of its metabolite, MPP+, into dopaminergic neurons. MPP+ is concentrated in the mitochondria according to the electrochemical gradient, where it selectively inhibits complex I of the electron transport chain. Complex I inhibition results in the inhibition of ATP production and loss of the mitochondrial membrane potential, leading to the death of nigral neurons (1).
MPP+ also induces opening of the mitochondrial transition pore (MTP) and the release of cytochrome c via a complex I-dependent, free radical-mediated process, which also may lead to cell death (2). Various signals mediating cell death may be initiated through the release of cytochrome c by mitochondrial damage (3). This pathway requires Apaf-1, which is responsible for the recruitment of procaspase-9. In the presence of dATP and cytochrome c, a 1:1 complex forms between Apaf-1 and procaspase-9, leading to the oligomerization-induced activation of caspase-9 (4, 5) and the subsequent activation of downstream caspases (6).
Transgenic mice expressing a dominant negative mutation of caspase-1 show resistance to MPTP toxicity and neuronal death in amyotrophic lateral sclerosis or Huntington's disease models (7–9), suggesting that the caspase-1 cascade may be one of the pathways responsible for neuronal death in various pathological conditions. We have been interested in determining the major pathway of MPTP toxicity among several apoptotic pathways. In the present study, we establish in vivo models of caspase cascade inhibition by using adeno-associated virus (AAV) vectors (T. Tuganezawa, M.M., H.M., and T.S., unpublished data; refs. 10 and 11). We have already established an AAV vector that produces persistent high levels of local expression after injection, with the intention of using it for further in vivo studies and for possible antiapoptotic gene therapy in patients with Parkinson's disease (PD) (12–14).
For inhibition of the mitochondrial apoptotic cascade in this study, we generated a recombinant AAV vector that contained the caspase-recruitment domain (CARD) of Apaf-1 to block the Apaf-1/caspase-9 pathway of cell death via dominant negative interference with the formation of a functional Apaf-1–caspase 9 complex (15). For inhibition of the caspase-1 cascade, we used a caspase-1 C285G mutant as a dominant negative inhibitor of caspase-1 (7–9).
Methods
Recombinant AAV Vector.
To construct a plasmid bearing the dominant negative truncated Apaf-1 transgene (Apaf-1-DN), the DNA fragment corresponding to the CARD of mouse Apaf-1 (1–97) was obtained by reverse transcription–PCR. After digesting the amplified fragment with BamHI and XhoI, the resulting fragment was ligated into a multiple cloning site (BamHI/XhoI) of the pCDNA3-FLAG expression vector. The ICE dominant negative construct (pS33; mouse ICE C285G FLAG construct) was a kind gift from J. Yuan at Harvard Medical School, Boston.
The plasmid containing the complete AAV genome (psub201) and the AAV packaging plasmid (pAAV/Ad) have been described (10). pAAV-CAG-CARD-B19-EGFP or pAAV-CAG-caspase-1-DN-B19-EGFP was constructed with a cassette containing a chicken β-actin promoter/cytomegalovirus enhancer (CAG promoter), CARD cDNA as an Apaf-1-dominant negative inhibitor or ICE dominant negative cDNA, a B19 promoter, enhanced green fluorescent protein (EGFP), and β-globin poly(A) inserted between the AAV inverted terminal repeats of psub201 (10).
For generation of the recombinant AAV vector, subconfluent HeLa cells were cotransfected with the vector plasmid and the AAV helper plasmid (pAAV/Ad) by using the calcium phosphate method. Cells were then infected with wild-type adenovirus 5 at a multiplicity of infection of 10 in 10 ml of DMEM with 2% FCS for 1 h. After subsequent incubation for 72 h, cells were harvested and lysed with four freeze-thaw cycles. The lysate was centrifuged at 12,000 × g for 10 min to remove cell debris and heated at 56°C for 30 min to inactivate adenovirus. Then, the lysate was applied to a sulfonated cellulose column (Seikagaku Kogyo, Tokyo) to concentrate the vectors as described previously (T. Tuganezawa, M.M., H.M., and T.S., unpublished data; ref. 11). After washing the column with PBS, the AAV vector was eluted with 10 mM phosphate buffer (pH 7.2) containing 1.0 M NaCl. The final titer of vectors was 5 × 1010 viral particles/ml.
In Vitro Assay of Apoptosis Using rAAV-Apaf-1-DN-EGFP.
Cells (293 cells at 2.5 × 106/ml) were cultured in 2 ml of DMEM with 10% FCS/100 units/ml penicillin/100 μg/ml streptomycin at 37°C under humidified air/5% CO2, and apoptosis was induced with 1 μM staurosporine, a protein kinase inhibitor. To assess viability, the cells were stained by trypan blue stain (Sigma) and counted by a blinded operator. Nonviable, apoptotic blue cells were small and round, whereas viable cells were flat (16). The percentage of apoptotic blue cells relative to the total number of cells was calculated.
Assay of in Vitro Caspase-3 Activation.
Procaspase-3-FLAG was synthesized in vitro by using a TNT T7 quick-coupled transcription/translation system kit (Promega) and pM130 as a template (600 ng/reaction). Procaspase-3-FLAG was incubated with S100 extracts prepared from 293 cells as indicated (Fig. 1d). Each sample was subjected to 15% SDS/PAGE, followed by Western blotting by using an anti-FLAG M2 mAb (Sigma), an anti-α-tubulin antibody (Santa Cruz Biotechnology), horseradish peroxidase-conjugated anti-mouse IgG (Amersham Pharmacia), and ECL-PLUS (Amersham Pharmacia).
Figure 1.

(a) Schematic representation of the AAV vectors. For Western blot analysis, samples of 293 cells were prepared 24 h after infection with rAAV-Apaf-1-DN-EGFP. (b) Western blotting showed that CARD (12 kDa) and EGFP were expressed in the same sample. (c) Cell viability was determined by using trypan blue staining. Results are expressed as a percentage of the control. Statistical analysis was performed by using ANOVA followed by Scheffé's post hoc test. (d) Apaf1-CARD inhibits caspase-3 activation induced by cytochrome c and dATP. Procaspase-3-FLAG (3 μl) was incubated with 16.5 μg/μl normal 293 cell extract (N) or AAV-Apaf1-CARD-FLAG transfected 293 cell extract (DN) for 1 h at 30°C in the presence or absence of cytochrome c (100 ng), dATP (1.4 mM), or both, as indicated. Samples were then analyzed by SDS/PAGE and Western blotting. Capsase-3 activation was examined by the decrease of the procaspase-3 band. Western blotting of α-tubulin was used as an internal control.
Immunoblotting.
Proteins separated on 15% Tris/Tricine gel were transferred to a nitrocellulose membrane and incubated with the primary antibody followed by the horseradish peroxidase-conjugated secondary antibody. Then, the reaction products were visualized by using a chemiluminescence reagent (Amersham Pharmacia). The primary antibodies were anti-FLAG and anti-EGFP antibodies.
Intrastriatal Injection.
Male C57/BL mice (8 weeks old, 18–20 g; Charles River Breeding Laboratories) were used. All stereotaxic surgical procedures were performed aseptically under anesthesia with pentobarbital (10 mg/kg) and ketamine hydrochloride (6 mg/kg). After each mouse was placed in a stereotaxic frame, 4 μl of the vector suspended in PBS was injected into the striatum (AP + 1.0, medial lateral 2.0, DV −3.0) over 5 min by using a 5-ml Hamilton syringe.
Two weeks after vector injection, the mice received four i.p. injections of MPTP-HCl (30 mg/kg; Research Biochemicals, Natick, MA) in saline at 24-h intervals.
Behavioral Analysis.
One week after MPTP injection in each mouse, full-body turns were monitored for 30 min after the injection of apomorphine (0.5 mg/kg, i.p.).
Histology.
Each mouse was perfused via the aorta with PBS followed by ice-cold 4% paraformaldehyde while under deep pentobarbital anesthesia. Then, 30-μm nigral sections were cut and were immunostained with a mAb for tyrosine hydroxylase (TH) (Chemicon), or with a mAb for GFP. In addition, sections were stained for anti-FLAG or anti-activated caspase-3 (PharMingen) with polyclonal antibodies. The corresponding secondary antibodies [donkey anti-mouse FITC or Texas red and donkey anti-rabbit FITC or Texas red (Jackson ImmunoResearch); 1:250] were pooled, and sections were incubated with these antibodies for 2 h at room temperature, followed by washing in PBS. The sections were then examined by confocal scanning laser microscopy, with the collected signals undergoing digital color enhancement before superimposition.
For cell counting, the substantia nigra was cut into serial sections, and every third section was subjected to immunostaining for TH with a polyclonal antibody (a kind gift from I. Nagatsu, Fujita Health University, Aichi, Japan). The number of viable TH-positive neurons was assessed by manual counting by using coded slides for blinding.
Results
In Vitro Antiapoptotic Effect of rAAV-Apaf-1-DN-EGFP.
For in vitro experiments, we first created rAAV-Apaf-1-DN-EGFP using a pAAV-CAG-CARD-B19-EGFP vector (Fig. 1a, ①). Then, we confirmed successful Apaf-1-DN expression and detection of the EGFP transgene after infection (5 × 1010 viral particles per ml) into 293 cells. We then used Western blotting to confirm the expression of CARD and EGFP in the cells 24 h after transduction (Fig. 1b).
To assess antiapoptotic activity, 293 cells were pretreated with rAAV-Apaf-1-DN-EGFP and incubated with 2 μM staurosporine. When cell viability was determined after 24 h by using trypan blue staining, treatment with rAAV-Apaf-1-DN-EGFP was shown to significantly inhibit cell death caused by staurosporine. A similar antiapoptotic effect of Apaf-1-DN was reported by Liu (17) (Fig. 1c).
We also examined the inhibitory effect of Apaf1-CARD-FLAG protein on cytochrome c-mediated caspase activation. S100 lysates containing Apaf1-CARD-FLAG were prepared, and then the effect on dATP/cytochrome c-dependent activation of caspase-3 was tested. As shown in Fig. 1d, addition of Apaf1-CARD-FLAG significantly reduced the processing of procaspase-3 by cytochrome c and dATP.
Striatal Expression of EGFP and Apaf-1-DN Genes.
Mice received an injection of the AAV vector encoding Apaf-1-DN–EGFP (rAAV-Apaf-1-DN-EGFP) into the striatum and were killed after 2 weeks. As a control, we injected rAAV-EGFP into the striatum of other mice (n = 3). All injections were localized to the putamen, as confirmed by standard staining procedures. All mice receiving rAAV-Apaf-1-DN-EGFP displayed prominent immunoreactivity for EGFP and Apaf-1-DN-FLAG in the left striatum (the injected side) (Fig. 2 a–c). In contrast, none of the mice receiving AAV-EGFP displayed specific FLAG immunoreactivity in the left striatum.
Figure 2.

Expression of CARD (Apaf-1) and EGFP in the striatum after injection. (a) Laser confocal images of EGFP immunostaining in the striatum. (b) FLAG immunostaining as a marker of CARD (Apaf-1-DN) in the same section as a. (c) Superimposed image. (d) Hematoxylin and eosin staining of the striatum. (a–c, ×200; d, ×40.)
Hematoxylin and eosin-stained sections of the striatum and substantia nigra revealed a normal cytoarchitecture without significant evidence of cytotoxicity. Macrophages were occasionally observed within the needle tracks. Gliosis was similar in all treated groups and was principally confined to the region immediately surrounding the needle track (Fig. 2d).
Retrograde Transport of Recombinant AAV.
Many cells of various neuronal subtypes expressed EGFP at the primary injection site. However, EGFP expression was also detectable in distant secondary brain areas, including the substantia nigra. Fibers labeled for EGFP were found in the nigrostriatal pathway, and EGFP-positive cells were double-labeled for Apaf-1-DN-FLAG immunoreactivity in the ipsilateral substantia nigra (Fig. 3 a–c). Confocal microscopy after staining with other antibodies revealed cells expressing TH in the pars compacta of the substantia nigra, which is characterized by a high content of dopaminergic cells (Fig. 3 d–f). These findings suggested that the AAV vector was transported to areas distant from the injection site.
Figure 3.
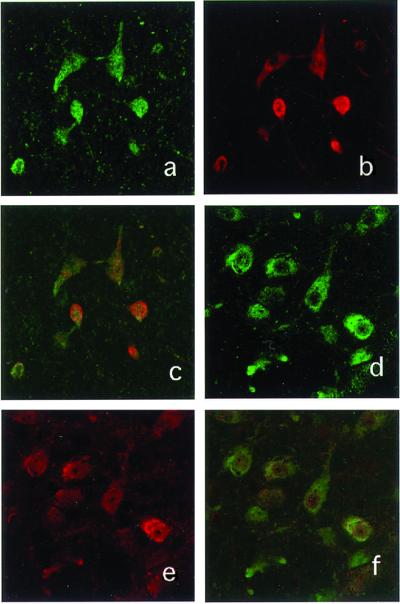
(a) Laser confocal images of EGFP immunostaining in the left substantia nigra. (b) Immunostaining for FLAG as a marker of CARD (Apaf-1) in the same section as a. (c) Superimposed image. (d) Laser confocal image of TH immunostaining in the left substantia nigra. (e) FLAG immunostaining as a marker of CARD (Apaf-1) in the same section as d. (f) Superimposed image. (Magnifications: ×400.)
Apaf-1-DN Prevents MPTP-Induced Dopaminergic Cell Death.
Delivery of AAV-Apaf-1-DN prevented MPTP-induced dopaminergic neuronal cell death within the left substantia nigra (injected side) when compared with the right side (noninjected side) (Fig. 4a). On the contralateral side to vector injection, all mice displayed dramatic loss of TH-immunoreactive neurons within the right substantia nigra (Fig. 4b). In contrast, the left substantia nigra showed evidence of neuroprotection in all four mice injected with AAV-Apaf-1-DN-EGFP (Fig. 4c). AAV-EGFP was injected into the left striatum (Fig. 4d) of control mice (n = 3), and TH-positive neurons were counted in the left substantia nigra. We also counted TH-positive neurons in the substantia nigra on the AAV-Apaf-1-DN-EGFP-injected and noninjected sides, revealing that the number of dopaminergic neurons was significantly greater on the injected side than on the noninjected side (Fig. 4d). Chronic MPTP administration induced activated caspase-3-positive cells and pyknotic neurons in the substantia nigra (Fig. 4 f–i). On the noninjected side, a few antiactivated caspase-3 and TH-positive cells (Fig. 4 f–i) were observed, but there were no activated caspase-3-positive cells on the injected side (Fig. 4 j–m).
Figure 4.

(a) Photomicrographs of TH immunostaining in the substantia nigra of an MPTP-treated AAV-Apaf-1-DN-injected (arrow) mouse. (b) The noninjected side shows neuronal loss. (c) The injected side shows nos. of neurons compared with the noninjected side. (d) Photomicrographs of TH immunostaining in the substantia nigra of an AAV-EGFP-injected mouse (control infection) (arrow). Ratio of TH-positive cells between the noninjected and injected sides. The total number of TH-positive neurons was counted in three sections each from four different mice. Statistical analysis was performed by using ANOVA followed by Scheffé's post hoc test. Laser confocal image of TH immunostaining on the substantia nigra at the noninjected side (f) and the injected side (l). Laser confocal image of antiactivated caspase-3 staining on the substantia nigra at the noninjected side (g) and the injected side (k). DNA condensation can be detected by Hoechst 33258 staining. (h) Hoechst 33258 staining reveals an intact nucleus (l). Superimposed image (j and m). (n) Graph demonstrating the mean number of amphetamine-induced full body turns per minute. Left bar, MPTP-treated mice with injection of the vehicle; Right bar, MPTP-treated mice with injection of rAAV-Apaf-1-DN injection. **, P < 0.01. (a, ×20; b and c, ×100; d, ×10; f–m, ×600.)
We also directly showed the behavior analysis as the clinical improvement by the injection of AAV-Apaf-1-DN-EGFP (Fig. 4n).
Caspase-1-DN Did Not Prevent MPTP-Induced Neuronal Cell Death in Vivo.
To evaluate the relationship between the caspase-1 cascade and MPTP-induced neuronal death, we also established an overexpression system for a caspase-1 C285G mutant as a caspase-1 dominant negative inhibitor by using the same AAV vector and the same method as for rAAV-Apaf-1-DN-EGFP (Fig. 1a, ②). Overexpression of this caspase-1 C285G mutant prevents trophic factor withdrawal-induced apoptosis in vitro (18) and ischemia-induced brain injury in vivo (19).
Expression of the caspase-1 mutant in 293 cells was demonstrated by immunoblotting (Fig. 5a). We then injected this vector into the left striatum and treated the animals with MPTP (n = 3) as in the first experiment. When the expression of caspase-1-DN and EGFP in neurons of the striatum and substantia nigra was examined using immunohistochemistry (Fig. 5b), caspase-1-DN showed similar expression to Apaf-1-DN, but the number of TH-positive cells did not differ between the injected and noninjected sides (Fig. 5 c–e). We also detected EGFP and caspase-1 mutant expression in the dopaminergic neurons on the injected side (Fig. 5 f–h).
Figure 5.

(a) Western blotting showing that the caspase-1-dominant negative inhibitor (50 kDa) and EGFP are expressed in the same sample. (b) Photomicrographs of TH immunostaining in the substantia nigra. (c) Noninjected side. (d) Injected side (arrow head). There was no significant difference between the noninjected and injected sides. (e) The ratio of the number of TH-positive cells on the noninjected and injected sides was determined by using the same procedure. Statistical analysis was performed by using ANOVA followed by Scheffé's post hoc test. (f) Laser confocal image of TH immunostaining in the substantia nigra. (g) FLAG immunostaining as a marker of mutant caspase-1 in the same section as d. (h) Superimposed image. (b, ×20; c and d, ×100; f–h, ×400.)
Discussion
In this study, we generated overexpression of Apaf-1 CARD as an Apaf1-dominant negative inhibitor in MPTP parkinsonian mice and clearly showed that inhibition of this major mitochondrial apoptotic cascade rather than the caspase-1 cascade could prevent MPTP toxicity in vivo.
In humans and nonhuman primates, MPTP causes a severe and irreversible PD-like syndrome. There is increasing evidence implicating apoptosis as a major mechanism of cell death after exposure to MPP+, an in vitro parkinsonian model (20, 21). It has also been demonstrated that chronic administration of MPTP is associated with evidence of apoptotic cell death in the substantia nigra (22). To inhibit such cell death, we focused on the major cascade of apoptosis mediated by MPTP. The MTP has been suggested to be a mediator of cell injury and death via a major apoptotic cascade (3). The MTP opens in response to various stimuli, including oxygen radicals and inhibition of the electron transport chain. It is well known that MPP+ inhibits mitochondrial complex I and generates free radicals (1). MPP+ is reported to release cytochrome c and open the MTP by inhibition of complex I and the production of reactive oxygen species (2). Also, protease-activated receptor 4 (Par-4) levels are increased in the dopaminergic neurons of monkeys and mice exposed to MPTP (23). Par-4 is a protein that contains both the leucine zipper and death domains. It is up-regulated in prostate tumor cells undergoing apoptosis and has been shown to promote mitochondrial dysfunction (23, 24). The cell death cascade may be initiated by the release of cytochrome c after mitochondrial damage (3). Apaf-1 subsequently aggregates caspase-9 in the presence of dATP and cytochrome c, leading to the activation of caspase-9, and then caspase-3 is activated by caspase-9 to induce apoptosis.
The effects of various recombinant Apaf-1 CARD domains on the activation of procaspase-9 have been investigated, and it has been shown that the recombinant wild-type CARD domain inhibits proteolytic cleavage of procaspase-9 (15). When Apaf-1 CARD is overexpressed, Apaf-1 binding with caspase-9 still occurs, but subsequent aggregation does not take place. Overexpression of Apaf-1 CARD thus acts as a dominant negative inhibitor. Our results indicate that inhibition of Apaf-1/caspase-9 pathway by overexpression of Apaf-1 CARD might also provide a method for in vivo interruption of the apoptotic pathway. Because this specific caspase dominant negative inhibitor provided significant protection against MPTP toxicity rather than the caspase-1 dominant negative inhibitor, it seems likely that the mitochondrial apoptotic cascade is involved in MPTP-related cell death.
Transgenic mice expressing caspase-1-DN are reported to show resistance to MPTP toxicity (7). However, an AAV-derived caspase-1 dominant negative inhibitor did not inhibit MPTP toxicity in our mouse model. There are some possible reasons for these differences. IFN-γ or IL-1β derived from glial cells may induce neuronal death. The neuron-specific enolase promoter driven by dominant negative caspase-1 may be also expressed by glial cells in transgenic mice and may inhibit the production of IL-1β as well as IFN-γ. In our model mouse, most of the recombinant gene was expressed in the neurons, not in glial cells, so it did not prevent the production of cytotoxic cytokines by glial cells. Because we found a difference of the antiapoptotic effect against MPTP-mediated cell death between Apaf-1-DN and caspase-1-DN despite their similar levels of expression, it seems likely that the mitochondrial apoptotic pathway is the main pathway of neuronal death related to MPTP.
EGFP and FLAG immunoreactivity were observed in the neurons of the substantia nigra after striatal injection of the AAV vector. Such positive staining was clearly distinct from that at the injection site and was consistent with the known striatal target structures, suggesting that AAV injection resulted in retrograde transport of CARD and EGFP. Because we used AAV vectors expressing the EGFP and Apaf-1-DN gene driven by a CAG promoter (composed of a cytomegalovirus immediate early enhancer and a chicken beta-actin promoter) (25), anterograde transport would be detected as AAV gene transfer if the CAG promoter induced sufficient expression in vivo. We have previously shown that employment of a strong promoter allows the effective and long-term expression of transferred genes using this system (T. Tuganezawa, M.M., H.M., and T.S., unpublished results). More important, the combination of transgene expression at the primary injection site and in secondary projection areas via retrograde transport may allow transduction of cells in target regions that are difficult to approach directly.
The role of apoptosis in PD is still controversial. We and other groups have found evidence of apoptosis based on morphological criteria or in situ end labeling (26–28), whereas other investigators have not (29, 30). It is very difficult to determine whether neuronal death occurs by apoptosis or necrosis in postmortem studies on PD patients. However, neuronal apoptosis seems to have an important role in PD, as several reports have suggested a correlation between apoptotic molecules and nigral cell death in this disease (31, 32). Mitochondrial respiratory failure and oxidative stress also seem to be two major contributors to nigral neuronal death in PD. Our present results suggest that Apaf-1-DN might be a potentially useful treatment for PD as antimitochondrial apoptotic gene therapy (12–14).
Acknowledgments
The ICE dominant negative construct (pS33; mouse ICE C285G FLAG construct) was a kind gift from J. Yuan at Harvard Medical School. We thank M. Yoshikawa and T. Saito for their technical assistance. This study was supported in part by a High Technology Research Center grant and a grant in aid for exploratory research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Abbreviations
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- TH
tyrosine hydroxylase
- MTP
mitochondrial transition pore
- AAV
adeno-associated virus
- CARD
caspase-recruitment domain
- EGFP
enhanced green fluorescent protein
- PD
Parkinson's disease
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Mizuno Y, Ikebe S, Hattori N, Nakagawa-Hattori Y, Mochizuki H, Tanaka M, Ozawa T. Biochim Biophys Acta. 1995;24:265–274. doi: 10.1016/0925-4439(95)00038-6. [DOI] [PubMed] [Google Scholar]
- 2.Cassarino D S, Parks J K, Parker W D, Jr, Bennett J P., Jr Biochim Biophys Acta. 1999;6:49–62. doi: 10.1016/s0925-4439(98)00083-0. [DOI] [PubMed] [Google Scholar]
- 3.Kroemer G, Reed J C. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 4.Zou H, Henzel W J, Liu X, Lutschg A, Wang X. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 5.Zou H, Li Y, Liu X, Wang X. J Biol Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 6.Srinivasula S M, Ahmad M, Fernandes-Alnemri T, Al-nemri E S. Mol Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- 7.Klevenyi P, Andreassen O, Ferrante R J, Schleicher J R, Jr, Friedlander R M, Beal M F. NeuroReport. 1999;25:635–638. doi: 10.1097/00001756-199902250-00035. [DOI] [PubMed] [Google Scholar]
- 8.Friedlander R M, Brown R H, Gagliardini V, Wang J, Yuan J. Nature (London) 1997;388:31. doi: 10.1038/40299. [DOI] [PubMed] [Google Scholar]
- 9.Ona V O, Li M, Vonsattel J P, Andrews L J, Khan S Q, Chung W M, Frey A S, Menon A S, Li X J, Stieg P E, et al. Nature (London) 1999;20:263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- 10.Kaplitt M G, Leone P, Samulski R J, Xiao X, Pfaff D W, O'Malley K L, During M J. Nat Genet. 1994;8:148–154. doi: 10.1038/ng1094-148. [DOI] [PubMed] [Google Scholar]
- 11.Tamayose K, Hirai Y, Shimada T. Hum Gene Ther. 1996;7:507–513. doi: 10.1089/hum.1996.7.4-507. [DOI] [PubMed] [Google Scholar]
- 12.Robertson G S, Crocker S J, Nicholson D W, Schulz J B. Brain Pathol. 2000;10:283–292. doi: 10.1111/j.1750-3639.2000.tb00262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bjorklund A, Lindvall O. Nat Med. 2000;6:1207–1208. doi: 10.1038/81291. [DOI] [PubMed] [Google Scholar]
- 14.Nicholson D W. Nature (London) 2000;407:810–816. doi: 10.1038/35037747. [DOI] [PubMed] [Google Scholar]
- 15.Qin H, Srinivasula S M, Wu G, Fernandes-Alnemri T, Alnemri E S, Shi Y. Nature (London) 1999;10:549–557. doi: 10.1038/21124. [DOI] [PubMed] [Google Scholar]
- 16.Miura M, Zhu H, Rotello R, Hartwieg E A, Yuan J. Cell. 1993;19:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- 17.Liu X, Kim C N, Yang J, Jemmerson R, Wang X. Cell. 1996;12:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 18.Gagliardini V, Fernandez P A, Lee R K, Drexler H C, Rotello R J, Fishman M C, Yuan J. Science. 1994;263:826–828. doi: 10.1126/science.8303301. [DOI] [PubMed] [Google Scholar]
- 19.Friedlander R M, Gagliardini V, Hara H, Fink K B, Li W, MacDonald G, Fishman M C, Greenberg A H, Moskowitz M A, Yuan J. J Exp Med. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mochizuki H, Nakamura N, Nishi K, Mizuno Y. Neurosci Lett. 1994;170:191–194. doi: 10.1016/0304-3940(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 21.Hartmann A, Hunot S, Michel P P, Muriel M P, Vyas S, Faucheux B A, Mouatt-Prigent A, Turmel H, Srinivasan A, Ruberg M, et al. Proc Natl Acad Sci USA. 2000;97:2875–2880. doi: 10.1073/pnas.040556597. . (First Published February 25, 2000; 10.1073/pnas.040556597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tatton N A, Kish S J. Neuroscience. 1997;77:1037–1048. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
- 23.Duan W, Zhang Z, Gash D M, Mattson M P. Ann Neurol. 1999;46:587–597. [PubMed] [Google Scholar]
- 24.Mattson M P. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 25.Niwa H, Yamamura K, Miyazaki J. Gene. 1991;15:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 26.Mochizuki H, Goto K, Mori H, Mizuno Y. J Neurol Sci. 1996;137:120–123. doi: 10.1016/0022-510x(95)00336-z. [DOI] [PubMed] [Google Scholar]
- 27.Anglade P, Vyas S, Javoy-Agid F, Herrero M T, Michel P P, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch E C, Agid Y. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 28.Tatton N A, Maclean-Fraser A, Tatton W G, Perl D P, Olanow C W. Ann Neurol. 1998;44:S142–S148. doi: 10.1002/ana.410440721. [DOI] [PubMed] [Google Scholar]
- 29.Kosel S, Egensperger R, von Eitzen U, Mehraein P, Graeber M B. Acta Neuropathol. 1997;93:105–108. doi: 10.1007/s004010050590. [DOI] [PubMed] [Google Scholar]
- 30.Banati R B, Daniel S E, Blunt S B. Movement Disorders. 1998;13:221–227. doi: 10.1002/mds.870130205. [DOI] [PubMed] [Google Scholar]
- 31.Yuan J, Yankner B A. Nature (London) 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 32.Beal M F. Trends Neurosci. 2000;23:298–304. doi: 10.1016/s0166-2236(00)01584-8. [DOI] [PubMed] [Google Scholar]