

Abstract
While classically considered a survival mechanism employed during nutrient scarcity, the autophagy pathway operates in multiple scenarios wherein a return to homeostasis or degradative removal of an invader is required. Now recognized as a pathway with vast immunoregulatory power, autophagy can no longer serve as a “one size fits all” term, as its machinery can be recruited to different pathogens, at different times, with different outcomes. Both canonical autophagy and the molecularly- related, yet divergent pathways non-canonical autophagy are key players in proper host defense and allow us an opportunity to tailor infectious disease intervention and treatment to its specific pathway.
Introduction
In 2016, Nobel Assembly at Karolinska Institutet awarded Yoshinori Ohsumi with the Nobel Prize in Medicine and Physiology for his groundbreaking work unraveling the molecular mechanisms that underlie the tightly regulated catabolic process of macroautophagy (herein referred to as autophagy). We now recognize that the reach of autophagy extends far beyond nutrient deprivation, into cellular quality control and host defense against internalized pathogens. While canonical autophagy likely evolved as a homeostatic response to cellular stress and/or nutrient deprivation, non-canonical autophagic functions are unified in the ancient theme of containment and suppression of inflammation. Similarly, efferocytosis, the immunotolerant clearance of dying host cells by tissue phagocytes, has recently been shown to rely upon recruitment of autophagy effectors to the phagosome through a non-canonical autophagic pathway called LC3-associated phagocytosis (LAP). Taken together, emerging evidence indicates that autophagy, through both canonical and non-canonical pathways, has diversified into a host defense mechanism, capable of confronting immunological and pathogenic stress and mediating immunological self-tolerance to both intracellular and extracellular threats.
Canonical Autophagy
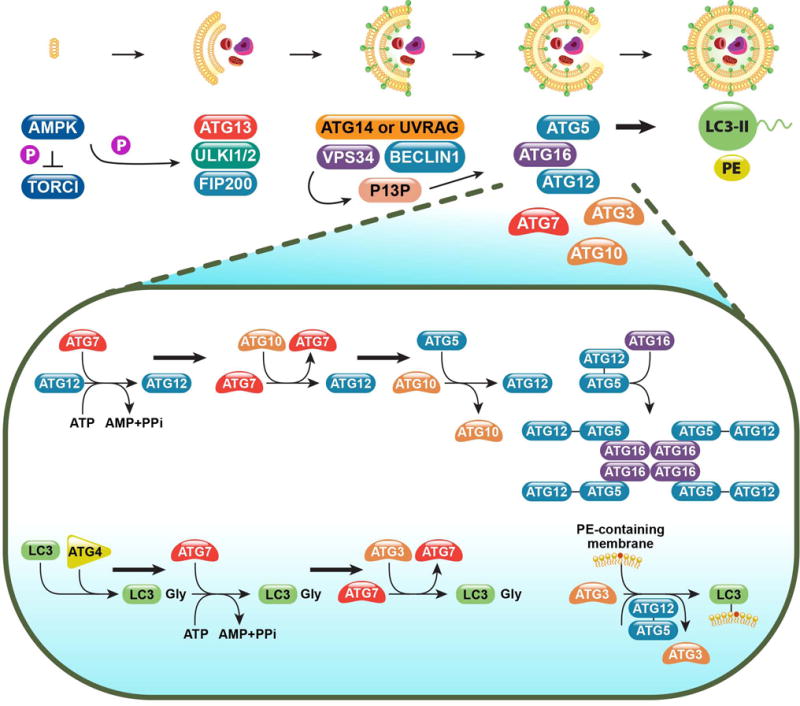
Autophagy is the highly conserved process by which eukaryotic cells scavenge their own cytoplasmic contents through sequestration into a phagophore and subsequent fusion with a lysosome for degradation. This process of “self-eating” is classically thought of as non-selective in response to nutrient deprivation and is largely orchestrated by the ATG family of proteins [1]. Upon starvation, autophagy progresses in 6 stages: inactivation of mTOR and pre-initiation complex formation, vesicle/phagophore nucleation, vesicle elongation, autophagosome formation, lysosome fusion, and component degradation [2] (Figure 1).
Figure 1. The molecular mechanisms of canonical autophagy.
Normally held in check by mTOR, autophagy-inducing signals (such as nutrient deprivation) triggers the activation of AMPK, whose kinase activity simultaneously inhibits mTOR and activates the pre-initiation complex (ULK1/2, ATG13, FIP200). This complex then activates the Class III PI3K complex, composed of VPS34 and Beclin 1, along with either ATG14 or UVRAG. The Class III PI3K complex produces phosphatidylinositol 3-phosphate (PI3P), which acts as recruitment signal for the downstream ubiquitin-like conjugation systems, the ATG12-5 system and the LC3-PE system. The activity and coordination of these two systems facilitates the curvature and sealing of the autophagosome, as well as the lipidation and embedding of LC3-PE into the autophagosomal membrane.
Extensive research has shown AMP-activated kinase (AMPK) to be the main energy sensing rheostat regulating the cell’s response to ATP/AMP imbalance [3]. When ATP levels decrease and AMP levels rise, AMPK becomes activated and inhibits mTOR complex 1(mTORC1) activity [4], leading to nuclear localization of TFEB and Gln3, two autophagy-related transcription factors [5, 6]. AMPK directly controls autophagy factors ULK1 (ATG1) and ATG13 through phosphorylation and sequestration [4]. Once free and active, ULK1 forms the autophagy pre-initiation complex with ATG13, FIP200, and ULK2 and phosphorylates ATG9 within nearby phospholipid membranes [7].
The Beclin-1-binding partner, Ambra1, directly connects the activity of this preinitiation complex, considered the most upstream regulator of the autophagic process, to the Class III PI3K complex. Ambra1 binds the core components of the Class III PI3K complex, Beclin 1 and VPS34, at the cytoskeleton through an interaction with the dynein motor complex. Upon autophagy induction, ULK1 phosphorylates Ambra1, allowing it and its bound partners to re-localize to the ER and initiate vesicle nucleation. The activity and localization of the Ambra1 complex further supports the role of the ER in autophagosome formation [8, 9]. Interestingly, Ambra1 can act in an mTORC1-sensitive positive-feedback loop to promote K63-linked ubiquitination of ULK1 through recruitment of the E3-ubiquitin ligase TRAF6 [10].
In addition to Beclin 1 and VPS34, the Class III PI3K complex consists of ATG14 or UVRAG in a mutually exclusive manner [11]. VPS34, the class III PI3 kinase in the complex, generates PI3P (phosphatidylinositol 3-phosphate), which serves as a critical recruitment signal for the two downstream ubiquitin-like conjugation systems. These two systems, the ATG5-12 system and the LC3-PE system, are required for vesicle nucleation, elongation, and curvature of the forming autophagosomes [2]. E3-ligase complex ATG7 and ATG10 mediates the conjugation of ATG5 to ATG12 in association with ATG16L1 to form a multimeric complex. Subsequently, this ATG5/12/16L1 complex is critical for the generation of LC3-PE (or LC3-II), the lipidated form of LC3 (or LC3-I). Cytosolic LC3-I is cleaved by ATG4, and conjugated to phosphatidylethanolamine (PE) via the activity of ATG7 and ATG3 [12]. This lipidated LC3-PE is bound to the autophagosomal membrane and is require for subsequent fusion to lysosomes, wherein the autophagosomal contents are degraded and recycled [13, 14].
Traditionally, autophagy is considered a cell survival process, however it is important to note that the autophagy machinery can serve as a switch from survival to death. Beclin 1 can bind pro-survival members of the BCL2 family, specifically BCL2, thus preventing its inhibition of BAX and allowing apoptosis to proceed. Importantly, BCL2-bound Beclin 1 cannot participate in autophagy [15]. Similarly, ATG proteins (ATG5, ATG3, ATG4D), cleaved by calpain or caspases have been shown to be pro-apoptotic, and mutation of the cleavage sites in these same proteins prevents the pro-apoptotic effect [16].
While the ability to self-eat evolved as a cellular response to metabolic stress and a need to return to intracellular homeostasis, autophagy has diverged in to combat infection and is a pivotal regulator of the inflammatory response. In both unicellular and multicellular organisms, autophagy can regulate different steps of the immune response, with immune signaling pathways eliciting an autophagic response to aid in defense [17–20]. Autophagy functions not only as a response to cellular stress, but is also important for pathogen recognition, pathogen degradation, antigen presentation, and regulation of pathways for cytokine production. Unsurprisingly, defects in the autophagic pathway have been strongly associated with inflammatory and autoimmune disorders, as well as infectious susceptibility [21]. However, we now recognize that the autophagic machinery serves many non-canonical functions that are critical for host defense.
Non-canonical Autophagy
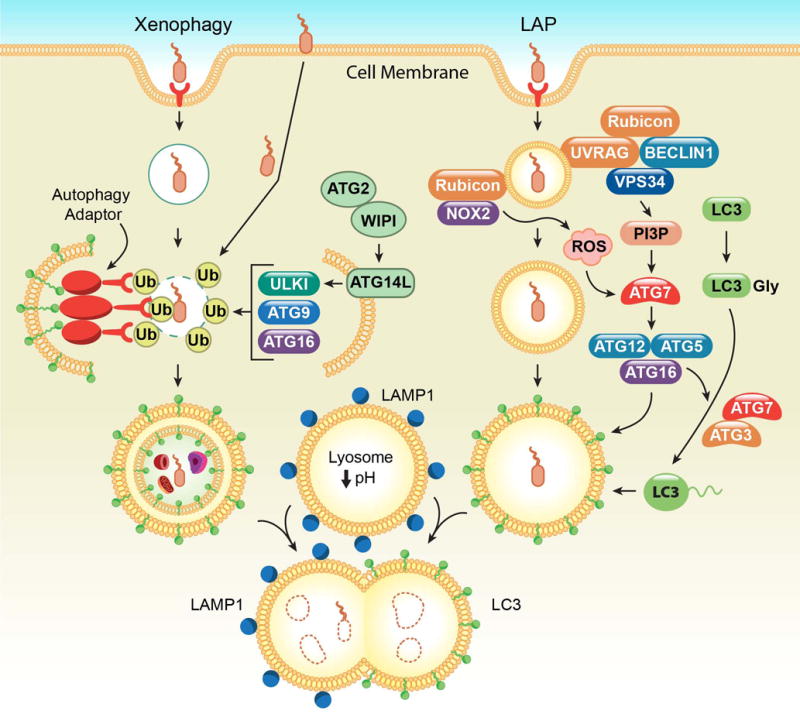
While canonical autophagy is considered a non-specific process that sequesters and degrades cytoplasmic contents in bulk, the autophagy machinery can also be selectively targeted to internal cellular substrates. Selective autophagy can be triggered for a variety of stimuli, such as damaged organelles (mitophagy for mitochondria) [22], macromolecules (lipophagy for lipids) [23], aggregated proteins (aggrephagy) [24], intracytoplasmic microbes (xenophagy), or phagocytosed particles such as dying cells or extracellular pathogens (LC3-associated phagocytosis or LAP) (Figure 2) [25–27].
Figure 2. Xenophagy versus LC3-associated phagocytosis.
(Left) During xenophagy, rupture of the pathogen-containing vesicle triggers the recruitment of ubiquitin to endosomal proteins or the pathogen itself. subsequently, autophagy adaptors, like p62, OPTN, and NDP52, are recruited and link these ubiquitinated pathogen substrates to the LC3-containing autophagosome. In addition, ATG proteins and other autophagy components are recruited via ubiquitin to mediate autophagosome formation. (Right) During LC3-associated phagocytosis (LAP), engagement of the PRRs during uptake of a pathogen triggers the recruitment of the Class III PI3K complex, comprised of VSP34, Beclin 1, UVRAG, and Rubicon, to the single membraned LAPosome. This complex is required for sustained and localized production of PI3P, which is needed for the recruitment of the downstream LAP machinery (like ATG5, ATG12, ATG16L, and ATG7) and stabilization of the NOX2 complex for ROS production. Both ROS and PI3P are required for successful LC3-PE decoration of the LAPosome. In both scenarios, LC3-PE is required for fusion to the lysosome and subsequent degradation of its contents.
Xenophagy
Hosts have evolved to utilize the autophagy machinery to detect and eliminate intracellular pathogens, such as viruses, bacteria and protozoa [17, 28]. Xenophagy (from the Greek for "strange" and "eating") is a selective form of non-canonical autophagy wherein pathogens are targeted and directed to the autophagosome for subsequent degradation via the autophagolysomal pathway [18, 19]. In addition to cytosolic detection by autophagic elements, some pathogens, such as Mycobacterium tuberculosis (Mtb) and Salmonella enterica serovar Typhimurium, can be eliminated by the fusion of the pathogen-containing vesicles to the autophagolysosome [17, 19, 20, 29].
Xenophagy is initiated by the ubiquitination of either the pathogen substrate, thus sealing its fate. Either the pathogen itself or the ruptured pathogen-containing vacuole can be ubiquitinated, as occurs during Salmonella ser. Typhimurium infection [30–33]. The process is mediated by a family of ubiquitinating enzymes comprised of the ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3). Leucine Rich Repeat and Sterile Alpha Motif Containing 1 2 3 5 RING-Type E3 Ubiquitin Transferase (LRSAM1) and Parkin are two E3 ubiquitin ligases involved in xenophagy, suggesting that the host autophagy machinery has devised a mechanism against the invading bacteria without a prompt from the pathogen [20, 34, 35].
Once ubiquitinated, the pathogen substrate is now capable to connecting to LC3-containing autophagosomal membranes via recruitment by autophagy receptors, namely SQSTM1 (p62), NDP52, NBR1, and optineurin (OPTN), which contain both a ubiquitin-binding domain and LC3-interacting region (LIR), thus bridging the ubiquitinated substrate to the autophagy machinery [19, 20]. The receptors p62, NBR1, and OPTN bind to LC3 isoforms through their LIR [36–38]. Some LC3 isoform specificity exists, as p62 selectively binds to LC3B and NDP52 binds to LC3C [36, 38, 39]. LC3 itself is a ubiquitin-like protein that conjugates with PE on the autophagosomal membrane [38, 39]. NDP52 is critical to binding ruptured endosomal membranes, as it binds to galectin-8, a β-galactose binding lectin that translocates to the endosomal membrane after its rupture [20]. The mechanisms that govern their function in xenophagy, however, are still under investigation.
Ubiquitinated pathogen substrates also recruit autophagic proteins like ULK1, ATG9L1, ATG16L1, ATG14L, and however, the mechanisms involved are currently not well understood [40]. This recruitment can trigger the formation of an autophagosomal membrane around pathogens or pathogen-containing vacuoles, even in the absence of LC3-bound membranes, indicating that xenophagy can occur independently of the autophagy receptors [40, 41]. Collectively, the goal of xenophagy is targeting of pathogens with ubiquitin, the assembly of the autophagy machinery at the autophagosomes, and degradation of the cargo [20].
The autophagy machinery can also orchestrate the capture of viral components for the removal of the both RNA and DNA viruses [42, 43], a process termed virophagy. Viral receptors such as endosomal toll-like receptors (TLR3, 7–9) or cytosolic nucleic acid sensors (RIG-I, STING, DAI, etc.) are essential for detection of a variety of viral pathogens like measles virus, human herpesvirus 6, adenovirus, and bovine viral diarrhea virus (BVDV) and triggering autophagy in antigen presenting cells [43]. In addition, there exists crosstalk between viral recognition and the autophagy machinery. For example, the lentivirus-encoded protein Nef binds Beclin 1 and induces autophagy [20, 44, 45].
There exist autophagy-independent roles for ATG proteins during host defense. Pathogens such as Listeria monocytogenes, Shigella flexneri, Mycobacterium marinum, HIV, herpesviruses, and Influenza A virus have developed strategies to circumvent detection by the host autophagy apparatus and remain hidden in the cytosol or in the vacuole [17, 18, 20, 46]. In the case of S. flexneri, the endosomal membrane is ubiquitinated after the bacteria has escaped the endosome, resulting in delivery of an empty vacuole to the autophagic machinery [30–32]. Other bacteria like Listeria monocytogenes, Mycobacterium marinum, and Francisella tularensis disrupt the formation of initial phagosome and enter the cytosol without host detection [18, 20, 32]. Strikingly, Mycobacterium tuberculosis (Mtb) colocalizes with multiple autophagy proteins, like ATG5, ATG12, ATG16L1, p62, NDP52, Beclin 1, and LC3, yet only ATG5 in polymorphic mononuclear cells (neutrophils) is required for resistance to Mtb in vivo, suggesting a novel autophagy-independent role for ATG5 in tuberculosis pathology and Mtb replication [47].
Moreover, bacteria have evolved to advantageously utilize the autophagy machinery for survival [18]. Brucella abortus recruits ULK1, Beclin 1, and ATG14L to form LC3-negative Brucella-containing vacuoles [48, 49]. Like bacteria, viruses (such as herpesviruses, HIV, and influenza A virus) have also adapted to opportunistically commandeer the autophagy machinery for their own purposes [42, 43, 50–52]. For example, Sindbis virus can degrade autophagy sensors/proteins like STQSM1 and ATG5, thereby evading detection [44]. As the autophagosome may or may not be decorated by LC3, careful consideration of the processes at play during host defense should be considered before characterizing the outcome as autophagy [20].
LC3-associated Phagocytosis
Whereas xenophagic processes are initiated once the pathogen is within the cell, the autophagic machinery can be actively recruited upon phagocytosis of a pathogen via signaling and sensing by an extracellular receptor. LC3-associated phagocytosis (or LAP) is a form of non-canonical autophagy that is initiated by the engagement of an extracellular receptor, such as Toll-like receptors (TLR), by a pathogen during phagocytosis. LAP can also be triggered by the uptake of dying cells (via phosphatidylserine receptors [PtdSer-R]) or immune complexes (via FcR), therefore LAP can be viewed as a conserved mechanism for mediating control and tolerance over exogenous threats. Receptor signaling results in the recruitment of some, but not all, of the autophagy machinery to the cargo-containing, single-membraned vesicle, which facilitates its decoration with lipidated LC3-PE [14, 25, 27]. The LC3-decorated, cargo-containing structure, or LAPosome, then fuses to lysosomes to mediate the rapid destruction of the cargo and modulation of the pursuant immune response [14].
While LAP and other autophagic immune responses share much of the same machinery, LAP is a process molecularly and functionally distinct from both canonical autophagy and xenophagy. Firstly, LAP results in a single-membraned LAPosome, whereas canonical autophagy and xenophagy create a double-membraned autophagosomes. Furthermore, the pre-initiation complex, described above, Ambra1, and WIPI2 are critical mediators of autophagy and xenophagy, yet completely dispensable for LAP [14, 25, 53]. The most upstream autophagic players required for successful execution of LAP are the components of the Class III PI3K complex (Beclin1, VPS34, and VPS15) [27, 54]. Whereas the Class III PI3K complex can contain with ATG14 or UVRAG during autophagy, LAP exclusively utilizes the UVRAG-containing Class III PI3K complex [14]. Similar to canonical autophagy, Class III PI3K complex-mediated PI(3)P on the LAPosome facilitates downstream recruitment of the ubiquitin-like conjugation systems, the ATG5-12 and LC3-PE conjugation systems required for successful LAP. LC3 bound to the LAPosome is required for subsequent fusion to the lysosome and degradation of the engulfed cargo [14].
Rubicon (RUN domain protein as Beclin-1 interacting and cysteine-rich containing) was recently identified as a protein required for LAP, yet not required for canonical autophagy. Rubicon acts as an inhibitor of autophagy, via its negative regulation of VPS34 [55, 56] or GTPase Rab7 activation [57]. Rubicon associates constitutively with the UVRAG-containing Class III PI3K complex and seems to serve two critical roles during LAP both – promoting the generation and localization of PI(3)P by the Class III PI3K complex and the production of ROS via the NOX2 complex, the major NADPH oxidase in phagocytes which is also required for LAP [14, 58, 59]. Rubicon stabilizes NOX2 by interacting with the p22phox subunit, and PI(3)P generated via Rubicon’s activity on VPS34 binds the p40phox subunit of NOX2 for optimal ROS production [60]. Further cementing the role for NOX2 in LAP are its interactions with Beclin1 via its CCD domain [58] and VPS34 via its RUN domain [61]. Collectively, Rubicon promotes the localization and activity of the Class III PI3K complex with the LAPosome and stabilizes the active NOX2 complex to promote optimal ROS production, which is also required for successful LAP [14]. As LAP is a recently described phenomenon, work uncovering its role in host defense is ongoing. Clearance of Saccharomyces cerevisiae [27], Listeria monocytogenes [58], and Aspergillus fumigatus is severely compromised in LAP-deficient macrophages and animals [14]. In response to fungal β-glucans and engagement of Dectin-1, Rubicon associates with CARD9 to displace it from the NF-κB complex needed for antifungal immunity. Similar scenarios exist during viral infection, wherein RNA viruses, such as VSV, Sendai virus, and influenza A (IAV), engage RIG-I and result in Rubicon disrupting the CARD9- NF-κB complex [62]. During HBV, IAV, and VSV infection, Rubicon can negatively regulate type I interferon signaling by interacting with IRF3 and IRF 7 [63, 64]. Furthermore, Rubicon binds to NEMO and suppresses ubiquitination, thus enhancing viral replication [64]. Hence, in these pathogenic scenarios, Rubicon-deficiency affords a survival advantage.
While LAP certainly plays a critical role in the degradation of engulfed pathogens, LAP is also an important mediator of the immunotolerant response. Cells and animal models with Rubicon deficiency produce significantly increased levels of pro-inflammatory cytokines, such as IL-6, IL-1β, and IL-12, in response to a variety of pathogens [14, 58, 62]. Strikingly, this increase in inflammation is observed in response to pathogens that require LAP for clearance (such as Aspergillus fumigatus [14], Listeria monocytogenes [58]) and pathogens that do not (fungal β-glucans, Sendai virus, HBV, IAV, and VSV [62–64]. The role of LAP in maintaining the immunotolerant state is exemplified during the clearance of dying cells, a process termed efferocytosis. Animals with LAP deficiency (i.e. Rubicon deficiency), but not canonical autophagy only-deficiency (i.e. ULK1 deficiency), develop lupus-like pathology with age, with increased serum levels of pro-inflammatory cytokines, autoantibodies, and kidney dysfunction [26]. Therefore, LAP represents a conserved cellular rheostat for shaping the appropriate immune response to engulfed pathogens and dying cells.
Mitophagy
The autophagic machinery can also target damaged organelles, such as mitochondria, for degradation, highlighting the quality control function that autophagy plays. In this sense, the degradative clearance of endosymbiont mitochondria (mitophagy) mirrors the clearance of intracellular pathogens (xenophagy) in that mitochondrial components, such as mtDNA, can mimic bacterial molecules and elicit autoinflammatory activation of cells. Both involve the ubiquitination of autophagy substrates resulting in the recruitment of autophagy machinery. Many of the adaptors utilized by xenophagy are also used in Parkin-dependent mitophagy [22].
Several mechanisms for mitophagy have been describe in the literature, all of which involve recognition of mitochondrial distress signals (depolarization, exposed cardiolipin, ubiquitination), and targeting of distressed mitochondria to LC3-containing phagophores via autophagy receptors, such as p62, OPTN, and NDP52 [65]. The most well characterized mitophagy pathway utilizes the kinase PINK1 and the E3 ubiquitin ligase Parkin. After depolarization of the mitochondrial membrane, PINK1 translocates and is stabilized on the outer mitochondrial membrane, wherein it phosphorylates ubiquitin chains that have tagged the damaged mitochondria and elicits the recruitment of Parkin from the cytosol. Here, Parkin is transformed into an active phospho-Ub-dependent E3 ligase that ubiquitinates itself as well as many different mitochondrial substrates. These ubiquitinated substrates are subsequently phosphorylated by PINK1, which fueling a feed forward amplification cycle of further PARKIN recruitment and activation [66]. These ubiquitinated, damaged mitochondria are then delivered to the LC3+ autophagosomes via autophagy receptors [22, 66].
In the absence of Parkin, several mitophagy receptors have been shown to facilitate the clearance of mitochondria during hypoxic stress in an LIR-dependent manner. LIR containing proteins NIX1 (also involved in Parkin-dependent mitophagy), BNP31, and FKBP8 have been shown to recruit lipidated LC3A to damaged mitochondria [67, 68]. FUNDC1 ubiquitination by MARCH5, a E3 ubiquitin ligase, on the outer membranes of mitochondria also targets mitochondria for mitophagy [69].
While clearance of defective mitochondria is a tool to limit unwanted inflammation and maintain homeostasis, mitochondria themselves play a crucial role in host defense by producing ROS, generating the necessary energy for an immune response, and providing a platform for host defense [70]. Studies involving RIPK2 have shown that mitophagy prevents hyperactivation of the NLRP3 inflammasome during infection [71]. In the absence of mitophagy, apoptotic mitochondria release mtDNA which can also trigger NLRP3 inflammasome activation [72] and promotes genomic instability and tumorigenesis [73]. The mitochondrial antiviral signaling (MAVS) protein localizes on the outer mitochondrial membrane where it interacts with RIG-I and MDA5 to activate downstream NF-κB and IRF signaling pathways for pro-inflammatory cytokine and type I IFN production. This mitochondrial localization makes MAVS a target for regulation via mitophagy. Ubiquitin ligases associated with mitophagy, like Smurf1 and Gp78, have been shown to also negatively modulate MAVS activity via both ubiquitin-dependent and -independent mechanisms, and in the absence of mitophagy, infection with VSV, a MAVS agonist, results in hyperstimulation [70]. Interestingly, other studies [74] have demonstrated that that healthy mitochondria are required to promote MAVS activity.
Conclusion
It is well established that defects in the autophagic machinery have been associated with aberrant host defense, inflammatory disease, and age-related disorders [21]. While initial interpretation implicates canonical autophagy in these pathologies, it is possible that the defect lies with non-canonical autophagic processes, such as LAP, rather than traditional autophagy. This is an emerging field in host defense and immunity, and our ability to discriminate between these related, yet distinct processes will have far-reaching applications in our approach to tumorigenesis, autoimmunity, and infectious disease. As broad inhibition of autophagic processes could be more harmful than beneficial, selective manipulation of specific canonical or non-canonical autophagy pathways could prove to be an invaluable tool for immunomodulation. Therefore, it is imperative that the molecular mechanisms that distinguish these processes are differentiated, as well as the physiological scenarios in which each is required, thus allowing for the design of anti-inflammatory therapeutics that specifically target the appropriate pathway, while maintaining the quality control mechanisms of canonical autophagy.
Table 1.
Pathogens associated with the autophagic machinery
| Bacteria | Autophagic machinery | Description | References |
|---|---|---|---|
| Mycobacterium Tuberculosis (Mtb) | ATG5, miRNA125a | Induction of autophagy promotes clearance; unique role for ATG5, but not other autophagy proteins, in PMN during infection in vivo | [20, 21, 47, 75] |
| MtB infection elevates expression of miRNA-125a-3p (miR-125a) and targets UVRAG to inhibit autophagy and phagosomal maturation | |||
| Mycobacterium bovis | ATG5 | Induction of autophagy promotes clearance | [20, 75] |
| Burkholderia pseudomallei | LC3 | LAP required for clearance | [76, 77] |
| Bacterial Bsa T3SS effector proteins, bopA and bipD, increases bacterial survival by decreasing LC3 accumulation | |||
| Group A Streptococcus (GAS) | ATG5, NDP52, p62, NDR1 | LC3 decorated autophagosome containing SpeB cysteine protease degrades the ubiquitin-LC3 adaptor proteins NDP52, p62, and NDR1 | [17, 20, 77, 78] |
| Listeria monocytogenes | p62, ATG5, Rubicon | Macroautophagy and LAP induced; mediates inflammatory responses to pathogen | [58, 79, 80] |
| LLO blocks maturation of autophagosome and evades into cytosol by releasing ActA | |||
| Bacteroides fragilis | ATG16L1, Rubicon | Outer membrane vesicles (OMVs) activate LAP for protection from colitis | [52] |
| Salmonella enterica serovar Typhimurium | NDP52, TBK1, OPTN, ATG9, ATG16L1 | Delivery of ubiquitinated bacteria or bacterial substrates for degradation | [28, 37] |
| Shigella flexneri | ATG5, NBR1, NDP52 | Secretes VirG which binds to ATG5 and activates autophagy; delivery of ubiquitinated bacteria or bacterial substrates for degradation | [19, 77, 80] |
| IcsB secreted by Shigella competes with ATG5 | |||
| Legionella pneumophila | Beclin 1, LC3 | Inhibition of Beclin 1 restricts autophagosome initiation and elongation. | [17, 81, 82] |
| RavZ and LegA9 secreted from T4SS uncouples LC3 from autophagosome membrane and inhibits autophagosome elongation and maturation. | |||
| L. pneumophila secretes effector protein, SGPL1 targets host sphingolipid metabolism, inhibit autophagosome formation and causes starvation-induced autophagy for intracellular survival | |||
| Adherent & invasive Escherichia coli (AIEC) | ULK1, LC3 | HIF1α-mediated retention in LC3-II+ vesicles and induces phosphorylation of ULK1 | [83] |
| Uropathogenic E. coli | ATG16L1 | Required for clearance | [84] |
| Yersinia Pseudotubercul osis | ATG5 | Defect in acidification of the LC3+ autophagosome-like vacuoles containing pathogen. | [17, 20] |
| Yersinia pestis | LC3 | Resides in LC3+ vesicles, yet prevents vacuole acidification | [17, 85] |
| Citrobacter rodentium | ATG16L1 | Required for clearance | [86] |
| Pseudomonas aeruginosa | ATG7, Beclin 1 | Autophagy mediated clearance | [87] |
| Klebsiella pneumoniae | ATG7 | Autophagy mediated clearance | [88] |
| Franscisella tularensis | NOX2, Beclin 1, ATG5, ATG12, ATG16L, ATG7, ATG4 | Disruption of NOX2-mediated ROS; downregulation of autophagy genes | [89] |
| Coxiella burnetii | Varies | Recruits autophagosomes to acquire nutrients or other factors that may trigger differentiation, and delays fusion with lysosomes for viral replication | [90, 91] |
| Brucella abortus | ULK1, Beclin 1, ATG14, VPS34 | Selectively recruits autophagy proteins to subvert clearance | [48, 49] |
| Viruses | Autophagic machinery | Description | References |
| HBV | Rubicon, ATG5 | Rubicon reduces IFN production and binds to NEMO to suppress ubiquitination, delays autophagosome maturation and allows viral replication; autophagy inhibits viral clearance | [64] |
| VSV | Rubicon, ATG5, ATG12 | Rubicon reduces IFN production and binds to NEMO to suppress ubiquitination and allows viral replication; autophagy inhibits antiviral response | [63, 64, 92] |
| IAV | Beclin 1, Rubicon | Influenza virus matrix protein 2 causes inhibition of beclin1 restricts autophagosome initiation and elongation. | [50, 63, 64] |
| Rubicon reduces IFN production and bind to NEMO to suppress ubiquitination and allows viral replication | |||
| HSV-1 | Beclin 1 | HSV inhibition of Beclin 1 to restrict autophagy | [50, 93] |
| Kaposi’s sarcoma herpes virus | Beclin 1 | BCL2-like proteins cause inhibition of Beclin 1 and restricts autophagy | [50, 94] |
| HIV | Beclin 1 | Accessory protein Nef binds/inhibits Beclin1 restricts autophagosome initiation and elongation | [50, 95] |
| Zika Virus | mTOR | NS4A and NS4B destabilize mTOR signaling | [96] |
| Sindbis Virus | ATG5, Beclin 1 | Defects in ATG5 impairs CNS clearance of Sindbis virus capsid | [50, 97] |
| Ectopic Beclin 1 expression in Sindbis virus-infected neurons suppresses viral replication in the brain and reduces mouse mortality. | |||
| Fungi | Autophagic machinery | Description | References |
| Aspergillus fumigatus | Rubicon, NOX2, LAP machinery | LAP-mediated degradation of and immune response to A. fumigatus | [14, 98] |
| Candida albicans | ATG5, Rubicon, NOX2 | Rubicon binds to CARD9 and NEMO to suppress ubiquitination and allows for increased fungal burden | [62, 99] |
| Saccharomyces cerevisiae | ATG7 | LAP required for fungal clearance | [27] |
| Cryptococcus neoformans | ATG5 | ATG5 aids in delivering C. neoformans in LC3+ autolysosome | [99] |
| Parasites | Autophagic machinery | Description | References |
| Toxoplasma gondii | ATG14, ATG9, ATG5, ATG7, ATG12, ATG16L1 | Autophagy is required for targeting and degradation of T. gondii | [28, 77] |
| Plasmodium vivax | Beclin 1, VPS34, ATG5 | LAP required for parasite control | [100] |
Pathogens (Bacteria, viruses, fungi, and parasites) with known links to components of the canonical and non-canonical autophagic machinery. Descriptions highlighted in green represent scenarios where pathogen clearance requires components of the autophagic machinery. Descriptions highlighted in red represent scenarios where autophagic components impede pathogen clearance.
Highlights.
Whereas the stress induced during infection can induce canonical autophagy, invading organisms and viruses can be specifically targeted for degradation by the autophagic machinery in a process broadly termed selective autophagy.
Xenophagy and LC3-associated phagocytosis (LAP) represent two forms of non-canonical selective autophagy. During xenophagy, intracellular pathogens are targeted for removal via ubiquitination and delivered to the LC3+ autophagosomes via autophagy receptors. During LAP, engagement of extracellular pathogen recognition receptors (PRR) trigger the recruitment of autophagy machinery and Rubicon to the pathogen-containing LAPosome for degradation.
Autophagic processes function not only in the physical removal of pathogens, but also in the modulation of the subsequent immune response.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Seminars in cell & developmental biology. 2010;21(7):683–90. doi: 10.1016/j.semcdb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. doi: 10.1007/978-3-642-00302-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hardie DG, et al. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shanware NP, et al. The PI3K, metabolic, and autophagy networks: interactive partners in cellular health and disease. Annu Rev Pharmacol Toxicol. 2013;53:89–106. doi: 10.1146/annurev-pharmtox-010611-134717. [DOI] [PubMed] [Google Scholar]
- 5.Beck T, Hall MN. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 1999;402(6762):689–92. doi: 10.1038/45287. [DOI] [PubMed] [Google Scholar]
- 6.Cardenas ME, et al. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev. 1999;13(24):3271–9. doi: 10.1101/gad.13.24.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7*.Joachim J, et al. Activation of ULK Kinase and Autophagy by GABARAP Trafficking from the Centrosome Is Regulated by WAC and GM130. Mol Cell. 2015;60(6):899–913. doi: 10.1016/j.molcel.2015.11.018. The authors demonstrate a unique requirement for the Golgi apparatus proteins, WAC and GM130, with control the subcellular localization of the LC3 homolog, GABARAP. GABARAP, but not other LC3 homologs, is required for ULK1 activation dependent on the ULK1 LIR motif and starvation-induced activation of autophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fimia GM, et al. Unleashing the Ambra1-Beclin 1 complex from dynein chains: Ulk1 sets Ambra1 free to induce autophagy. Autophagy. 2011;7(1):115–7. doi: 10.4161/auto.7.1.14071. [DOI] [PubMed] [Google Scholar]
- 9.Fimia GM, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447(7148):1121–5. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 10.Nazio F, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15(4):406–16. doi: 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- 11.Itakura E, et al. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19(12):5360–72. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ichimura Y, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408(6811):488–92. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 13.Wild P, et al. The LC3 interactome at a glance. J Cell Sci. 2014;127(Pt 1):3–9. doi: 10.1242/jcs.140426. [DOI] [PubMed] [Google Scholar]
- 14**.Martinez J, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17(7):893–906. doi: 10.1038/ncb3192. This publication describes the generation of the Rubicon−/− mouse model, and its absolute requirement in LC3-associated phagocytosis (LAP) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Maiuri MC, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 16.Martinez J, et al. The Relationship between Metabolism and the Autophagy Machinery during the Innate Immune Response. Cell Metab. 2013;17(6):895–900. doi: 10.1016/j.cmet.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cemma M, Brumell JH. Interactions of pathogenic bacteria with autophagy systems. Curr Biol. 2012;22(13):R540–5. doi: 10.1016/j.cub.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Knodler LA, Celli J. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol. 2011;13(9):1319–27. doi: 10.1111/j.1462-5822.2011.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mostowy S, Cossart P. Bacterial autophagy: restriction or promotion of bacterial replication? Trends Cell Biol. 2012;22(6):283–91. doi: 10.1016/j.tcb.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Shibutani ST, et al. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. 2015;16(10):1014–24. doi: 10.1038/ni.3273. [DOI] [PubMed] [Google Scholar]
- 21.Levine B, et al. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22**.Lazarou M, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. doi: 10.1038/nature14893. The authors delineate the roles of five autophagy receptors in the mitophagy and characterize the role of PINK1 in the recrutiment of OPTN and NDP52 during mitophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013;20(1):3–11. doi: 10.1038/cdd.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. 2012;2012:736905. doi: 10.1155/2012/736905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez J, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(42):17396–401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez J, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533(7601):115–9. doi: 10.1038/nature17950. This publication is the first to demonstrate the role of LAP, but not canonical autophagy, in the mediation of immunotolerance. The authors demonstrate that Rubicon−/− mice, as well as others, develop SLE-like autoimmunity with age as a result of defective efferocytosis. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Sanjuan MA, et al. Toll-like receptor signaling in the lysosomal pathways. Immunol Rev. 2009;227(1):203–20. doi: 10.1111/j.1600-065X.2008.00732.x. [DOI] [PubMed] [Google Scholar]
- 28.Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016;16(11):661–675. doi: 10.1038/nri.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandra P, Kumar D. Selective autophagy gets more selective: Uncoupling of autophagy flux and xenophagy flux in Mycobacterium tuberculosis-infected macrophages. Autophagy. 2016;12(3):608–9. doi: 10.1080/15548627.2016.1139263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Birmingham CL, Brumell JH. Methods to monitor autophagy of Salmonella enterica serovar Typhimurium. Methods Enzymol. 2009;452:325–43. doi: 10.1016/S0076-6879(08)03620-3. [DOI] [PubMed] [Google Scholar]
- 31.Birmingham CL, Brumell JH. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy. 2006;2(3):156–8. doi: 10.4161/auto.2825. [DOI] [PubMed] [Google Scholar]
- 32.Birmingham CL, et al. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006;281(16):11374–83. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 33.Drecktrah D, et al. Salmonella trafficking is defined by continuous dynamic interactions with the endolysosomal system. Traffic. 2007;8(3):212–25. doi: 10.1111/j.1600-0854.2006.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manzanillo PS, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501(7468):512–6. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huett A, et al. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella Typhimurium. Cell Host Microbe. 2012;12(6):778–90. doi: 10.1016/j.chom.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–96. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wild P, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333(6039):228–33. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shpilka T, Elazar Z. Essential role for the mammalian ATG8 isoform LC3C in xenophagy. Mol Cell. 2012;48(3):325–6. doi: 10.1016/j.molcel.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 39.von Muhlinen N, et al. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell. 2012;48(3):329–42. doi: 10.1016/j.molcel.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujita N, et al. Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol. 2013;203(1):115–28. doi: 10.1083/jcb.201304188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kageyama S, et al. The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol Biol Cell. 2011;22(13):2290–300. doi: 10.1091/mbc.E10-11-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Munz C. Macroautophagy–friend or foe of viral replication? EMBO Rep. 2013;14(6):483–4. doi: 10.1038/embor.2013.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong X, Levine B. Autophagy and viruses: adversaries or allies? J Innate Immun. 2013;5(5):480–93. doi: 10.1159/000346388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shoji-Kawata S, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–6. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Munz C. C Viruses. 2011;3(7):1166–78. doi: 10.3390/v3071166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Checroun C, et al. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci U S A. 2006;103(39):14578–83. doi: 10.1073/pnas.0601838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47**.Kimmey JM, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015;528(7583):565–9. doi: 10.1038/nature16451. The authors demonstrate for the first time that Atg5 plays a unique, yet indispensable, role in the control of M. tuberculosis by preventing PMN-mediated immunopathology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Starr T, et al. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe. 2012;11(1):33–45. doi: 10.1016/j.chom.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Starr T, et al. Brucella intracellular replication requires trafficking through the late endosomal/lysosomal compartment. Traffic. 2008;9(5):678–94. doi: 10.1111/j.1600-0854.2008.00718.x. [DOI] [PubMed] [Google Scholar]
- 50.Dreux M, Chisari FV. Viruses and the autophagy machinery. Cell Cycle. 2010;9(7):1295–1307. doi: 10.4161/cc.9.7.11109. [DOI] [PubMed] [Google Scholar]
- 51.Munz C. The Autophagic Machinery in Viral Exocytosis. Front Microbiol. 2017;8:269. doi: 10.3389/fmicb.2017.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52*.Chu H, et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science. 2016;352(6289):1116–20. doi: 10.1126/science.aad9948. In this publication, the authors describe the crosstalk between genetic (ATG16L1/NOD2) and environmental (microbiome) factors to promote mucosal tolerance via LAP-dependent mechanisms. This discovery of microbiome interactions with genetic programs to promote beneficial immunity provided novel insights into the pathogenesis of IBD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Henault J, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37(6):986–97. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin SJ, et al. Phosphatidylserine externalization during CD95-induced apoptosis of cells and cytoplasts requires ICE/CED-3 protease activity. J Biol Chem. 1996;271(46):28753–6. doi: 10.1074/jbc.271.46.28753. [DOI] [PubMed] [Google Scholar]
- 55.Matsunaga K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nature cell biology. 2009;11(4):385–96. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 56.Zhong Y, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nature cell biology. 2009;11(4):468–76. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun Q, et al. Rubicon controls endosome maturation as a Rab7 effector. Proc Natl Acad Sci U S A. 2010;107(45):19338–43. doi: 10.1073/pnas.1010554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang CS, et al. Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell Host Microbe. 2012;11(3):264–76. doi: 10.1016/j.chom.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang J, et al. Activation of antibacterial autophagy by NADPH oxidases. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(15):6226–31. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ueyama T, et al. Cooperation of p40(phox) with p47(phox) for Nox2-based NADPH oxidase activation during Fcgamma receptor (FcgammaR)-mediated phagocytosis: mechanism for acquisition of p40(phox) phosphatidylinositol 3-phosphate (PI(3)P) binding. J Biol Chem. 2011;286(47):40693–705. doi: 10.1074/jbc.M111.237289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun Q, et al. The RUN domain of rubicon is important for hVps34 binding, lipid kinase inhibition, and autophagy suppression. J Biol Chem. 2011;286(1):185–91. doi: 10.1074/jbc.M110.126425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang CS, et al. The autophagy regulator Rubicon is a feedback inhibitor of CARD9-mediated host innate immunity. Cell Host Microbe. 2012;11(3):277–89. doi: 10.1016/j.chom.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63*.Kim JH, et al. Rubicon Modulates Antiviral Type I Interferon (IFN) Signaling by Targeting IFN Regulatory Factor 3 Dimerization. J Virol. 2017;91(14) doi: 10.1128/JVI.00248-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64*.Wan Y, et al. Inducible Rubicon facilitates viral replication by antagonizing interferon production. Cell Mol Immunol. 2017;14(7):607–620. doi: 10.1038/cmi.2017.1. These two publications describe a unique role for Rubicon in the suppression of the type I interferon response to viral infection. These findings establish Rubicon as a negative regulator of the antiviral immune response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stolz A, et al. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16(6):495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 66.Durcan TM, Fon EA. The three ‘P’ of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015;29(10):989–99. doi: 10.1101/gad.262758.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67**.Bhujabal Z, et al. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017;18(6):94–961. doi: 10.15252/embr.201643147. Using a yeast two-hybrid screen, the authors identify FKBP8, a protein localized to the outer mitochondrial membrane, as a mitophagy receptor capable of specifically binding LC3A and inducing Parkin-independent mitophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hamacher-Brady A, Brady NR. Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci. 2016;73(4):775–95. doi: 10.1007/s00018-015-2087-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69**.Chen Z, et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017;18(3):495–509. doi: 10.15252/embr.201643309. In this publication, the authors describe the role of MARCH5, a mitochondrial E3 ligase, in the control of hypoxia-induced mitophagy. MARCH5 ubiquitinates the mitophagy receptor, FUNDC1, thereby sensitizing hypoxia-damaged mitochondria to mitophagy, revealing a feedback regulatory mechanism to modulate mitochondrial quality. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lazarou M. Keeping the immune system in check: a role for mitophagy. Immunol Cell Biol. 2015;93(1):3–10. doi: 10.1038/icb.2014.75. [DOI] [PubMed] [Google Scholar]
- 71.Lupfer C, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013;14(5):480–8. doi: 10.1038/ni.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17(6):363–375. doi: 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73**.Ichim G, et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell. 2015;57(5):860–72. doi: 10.1016/j.molcel.2015.01.018. In this publication, the authors describe the consequences of minority mitochondrial outer membrane permeabilization (MOMP), wherein a fraction of mitochondria can undergo MOMP in a stress-regulated manner. This phenomenon can leads to caspase activation and subsequent DNA damage, genomic instability, cellular transformation, and tumorigenesis. Collectively, these findings have important implications oncogenic treatments that engagement the apoptotic pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koshiba T, et al. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci Signal. 2011;4(158):ra7. doi: 10.1126/scisignal.2001147. [DOI] [PubMed] [Google Scholar]
- 75.Gutierrez MG, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119(6):753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 76.Gong L, et al. The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PLoS One. 2011;6(3):e17852. doi: 10.1371/journal.pone.0017852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell. 2014;54(2):224–33. doi: 10.1016/j.molcel.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 78.Nakagawa I, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306(5698):1037–40. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 79.Birmingham CL, et al. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy. 2007;3(5):442–51. doi: 10.4161/auto.4450. [DOI] [PubMed] [Google Scholar]
- 80.Mostowy S, et al. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem. 2011;286(30):26987–95. doi: 10.1074/jbc.M111.223610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81*.Rolando M, et al. Legionella pneumophila restrains autophagy by modulating the host’s sphingolipid metabolism. Autophagy. 2016;12(6):1053–4. doi: 10.1080/15548627.2016.1166325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82*.Rolando M, et al. Legionella pneumophila S1P-lyase targets host sphingolipid metabolism and restrains autophagy. Proc Natl Acad Sci U S A. 2016;113(7):1901–6. doi: 10.1073/pnas.1522067113. These two publications describe the ability of Legionella pneumophilia to regulate sphingolipid metabolism to inhibit autophagy and promote intracellular survival. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mimouna S, et al. HIF1A regulates xenophagic degradation of adherent and invasive Escherichia coli (AIEC) Autophagy. 2014;10(12):2333–45. doi: 10.4161/15548627.2014.984275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Clyne M. Urinary tract infections: autophagy gene mutation confers protection against uropathogenic E. coli infection. Nat Rev Urol. 2012;9(8):410. doi: 10.1038/nrurol.2012.140. [DOI] [PubMed] [Google Scholar]
- 85.Pujol C, et al. Yersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect Immun. 2009;77(6):2251–61. doi: 10.1128/IAI.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Inoue J, et al. Autophagy in the intestinal epithelium regulates Citrobacter rodentium infection. Arch Biochem Biophys. 2012;521(1–2):95–101. doi: 10.1016/j.abb.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 87.Deng Q, et al. Pseudomonas aeruginosa Triggers Macrophage Autophagy To Escape Intracellular Killing by Activation of the NLRP3 Inflammasome. Infect Immun. 2015;84(1):56–66. doi: 10.1128/IAI.00945-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ye Y, et al. Atg7 deficiency impairs host defense against Klebsiella pneumoniae by impacting bacterial clearance, survival and inflammatory responses in mice. Am J Physiol Lung Cell Mol Physiol. 2014;307(5):L355–63. doi: 10.1152/ajplung.00046.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Celli J, Zahrt TC. Mechanisms of Francisella tularensis intracellular pathogenesis. Cold Spring Harb Perspect Med. 2013;3(4):a010314. doi: 10.1101/cshperspect.a010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gutierrez MG, et al. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol. 2005;7(7):981–93. doi: 10.1111/j.1462-5822.2005.00527.x. [DOI] [PubMed] [Google Scholar]
- 91.Moffatt JH, et al. Coxiella burnetii: turning hostility into a home. Cell Microbiol. 2015;17(5):621–31. doi: 10.1111/cmi.12432. [DOI] [PubMed] [Google Scholar]
- 92.Jounai N, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A. 2007;104(35):14050–5. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.O’Connell D, Liang C. Autophagy interaction with herpes simplex virus type-1 infection. Autophagy. 2016;12(3):451–9. doi: 10.1080/15548627.2016.1139262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jang GH, et al. Suppression of lytic replication of Kaposi’s sarcoma-associated herpesvirus by autophagy during initial infection in NIH 3T3 fibroblasts. Arch Virol. 2016;161(3):595–604. doi: 10.1007/s00705-015-2698-2. [DOI] [PubMed] [Google Scholar]
- 95.Dinkins C, et al. Roles of autophagy in HIV infection. Immunol Cell Biol. 2015;93(1):11–7. doi: 10.1038/icb.2014.88. [DOI] [PubMed] [Google Scholar]
- 96**.Liang Q, et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell. 2016;19(5):663–671. doi: 10.1016/j.stem.2016.07.019. The authors demonstrate that infection of human fetal neural stem cells with Zika virus (ZIKV) results in increased activation of autophagy and defective neurogenesis Moreover, they identified 2 ZIKV proteins, NS4A and NS4B, that cooperatively suppress the mTOR pathway, highlighting them as possible targets for therapeutic intervention. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liang XH, et al. Resistance of interleukin-1beta-deficient mice to fatal Sindbis virus encephalitis. J Virol. 1999;73(3):2563–7. doi: 10.1128/jvi.73.3.2563-2567.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98*.Akoumianaki T, et al. Aspergillus Cell Wall Melanin Blocks LC3-Associated Phagocytosis to Promote Pathogenicity. Cell Host Microbe. 2016;19(1):79–90. doi: 10.1016/j.chom.2015.12.002. In this publication, the authors describe the role of the A. fumigatus PAMP, melanin, as a factor that inhibits LAP via blocking NOX2 activity and promote fungal survival. These findings identify a potential therapeutic target with broad physiological implications for LAP-associated pathologies. [DOI] [PubMed] [Google Scholar]
- 99.Nicola AM, et al. Macrophage autophagy in immunity to Cryptococcus neoformans and Candida albicans. Infect Immun. 2012;80(9):3065–76. doi: 10.1128/IAI.00358-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100*.Boonhok R, et al. LAP-like process as an immune mechanism downstream of IFN-gamma in control of the human malaria Plasmodium vivax liver stage. Proc Natl Acad Sci U S A. 2016;113(25):E3519–28. doi: 10.1073/pnas.1525606113. Here, the authors demonstrate that the role of IFN-γ in the control of liver-stage Plasmodium vivax is the induction of a non-canonical autophagy similar to LAP, wherein IFN-γ aided in the recruitment of LC3 onto P. vivax-containing vacuoles. [DOI] [PMC free article] [PubMed] [Google Scholar]